

Abstract
SARS-CoV-2 has developed a substantial number of mutations, especially in the S-protein. With the advancement of the pandemic, accumulations of further mutations at the S-protein receptor-binding domain could enhance the infectivity and pathogenicity of the virus. Prediction and evaluation of such mutations are essential for understanding the potential development of more pathogenic strains and for COVID-19 management.
Keywords: SARS-CoV-2, protein−protein interactions, RNA viruses, host−pathogen interactions, virus−host interactions, infectivity, transmission
The coronavirus disease (COVID-19), caused by the severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2), has turned the clock back by years, if not decades, in the fight against infectious diseases. Since its emergence in November 2019, COVID-19 has infected more than 24 million people, claimed more than 820 000 lives worldwide, and continues to endanger many more. SARS-CoV-2 is an enveloped virus with a single-stranded RNA genome that binds to the host receptor using its spike (S)-protein, facilitating cell entry. The S-protein consists of S1 and S2 subunits; the receptor-binding domain (RBD) present at the C-terminus of the S1 subunit plays a crucial role in virus attachment, membrane fusion, and subsequent infection. Structural, functional, and deep mutational scanning studies have shown that the RBD of SARS-CoV-2 binds to the human angiotensin-converting enzyme-2 (ACE2) receptor with high affinity (Figure 1A), thereby establishing an efficient cascade of infection and transmission.1,2 RNA viruses have extremely high mutation rates that are directly linked with enhanced virulence and evolution ability. Since its emergence, SARS-CoV-2 has undergone several mutations and is rapidly evolving. In particular, the S-protein is profoundly mutated and includes the RBD as a prominent domain with substantial mutation clusters. Although mutations are primarily concentrated in the RBD of S1 subunit, the conserved residues are located mainly on S2 subunit. So far, more than 1800 mutations (including multiple mutations at the same sites) are reported in the S-protein of SARS-CoV-2, of which 235 mutations are accumulated in the RBD. The RBD of SARS-CoV-2, consisting of ∼180 residues, has been subjected to 136 unique mutations, and the number is continuously growing. This indicates that the virus may accumulate further mutations at the RBD site to improve its interaction with ACE2 and escalate its infectivity and transmission. In this article, we have explored this scenario in the structural and evolutionary contexts of the virus and proposed an urgency to predict and rapidly evaluate such mutations for the efficient management of COVID-19 and develop strategies to deter future cross-species transmissions.
Figure 1.

Structures and interactions in RBD–ACE2 complexes. (A) Ribbon structure of SARS-CoV-2 RBD in complex with human ACE2 (Protein Data Bank (PDB): 6LZG), showing the residues of S-protein RBD interacting with ACE2 receptor. The residues interacting between RBD–ACE2 are represented in orange sticks. Intermolecular interactions in (B) an affinity-increasing and (C) an affinity-decreasing RBD–ACE2 complex. In all the panels, the RBD and ACE2 are shown in cyan and green color, respectively. The interacting residues and binding sites in panel B are shown in purple sticks, and in panel C are shown in red sticks.
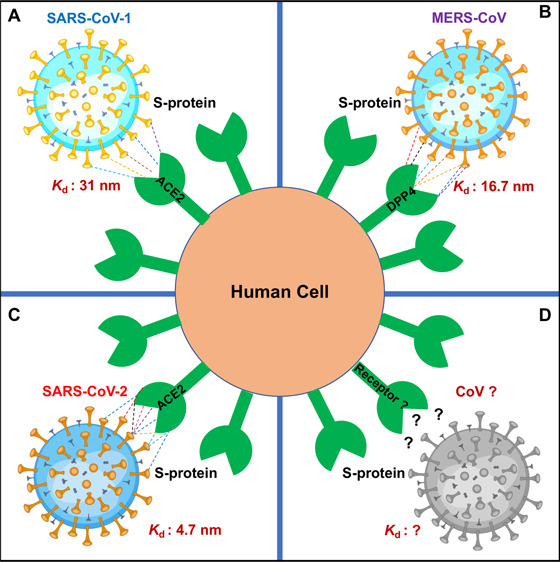
SARS-CoV-2 is the seventh member of the coronavirus (CoV) family that infects humans. It is closely related to SARS-CoV (responsible for the outbreak in 2002) and Middle East respiratory syndrome coronavirus (MERS-CoV, responsible for the outbreak in 2012). While SARS-CoV infects the epithelial cells of the lungs and enters the host cell by binding to ACE2, MERS-CoV uses dipeptidyl peptidase 4 (DPP4) to enter host cells.3 Both SARS-CoV and MERS-CoV likely originated from bats and are highly pathogenic and transmissible. When the current pandemic started, it was initially believed that SARS-CoV-2 is similar to SARS-CoV and MERS-CoV in origin and transmission ability, but later, the transmission ability of the novel virus SARS-CoV-2 was found to be much higher. Moreover, while SARS-CoV-2 shares ∼76% sequence identity with SARS-CoV and ∼43% sequence identity with MERS-CoV, it exhibits a striking ∼96% sequence similarity to a CoV isolated from a bat, Rhinolophus affinis, in Yunnan Province, China.3 These findings indicate that SARS-CoV-2 has inherited patches of amino acids from known CoVs and acquired new unique amino acid residues from unknown sources that facilitated its widespread infection capability. CoVs have a robust genomic proofreading mechanism which keeps the virus free from developing mutations that could weaken it, thus rendering them even more pathogenic. Comparison of amino acid sequences between SARS-CoV-2 and other CoVs suggests that although they have similar structures they have variations at critical residues of RBD. The S-protein of SARS-CoV-2 interacts with its human receptor with a much higher affinity (∼4.7 nM) than do SARS-CoV (∼31 nM) and MERS-CoV (∼16.7 nM).1 Evolutionary genetics data indicate that the S-protein of SARS-CoV-2 has acquired distinct mutations to enhance its binding toward ACE2 by ∼15-fold compared to that of SARS-CoV. High-throughput genomic analysis studies have further shown that several virus lineages (clades) have emerged since the original SARS-CoV-2 outbreak in China.4,5 The phylogenetic clades have been broadly classified into O, B, B1, B2, B4, A3, A6, A7, A1a, A2, and A2a types. The majority of the S-protein mutations, including the well-known D614G mutation, are classified into the A2, A1a, and A2a clades.4,5 In addition, SARS-CoV-2 has acquired mutations under the effects of natural selection and evolutionary pressure that conferred stability to the virus particle. As viruses undergo positive selection and emerge into more transmissible forms, SARS-CoV-2 may accumulate further mutations (specifically at the RBD of the S-protein) due to positive selective pressure to enhance its infectivity and transmission ability. To continuously track the amino acid changes in the evolving SARS-CoV-2 genomes, a CoV-GLUE database has been developed. It contains up-to-date information on replacement, insertion, and deletion mutations observed in SARS-CoV-2 sequences (http://cov-glue.cvr.gla.ac.uk/#/replacement).
A remarkable ability of viruses is that they quickly adapt to new hosts, environments, and drugs through the accumulation of mutations in a short time.6 The mutation rates of single-stranded RNA viruses are much higher than those of double-strand viruses and DNA viruses. However, the mutation rates evolve in response to evolutionary, immune, and drug pressures. There are more than 180 species of RNA viruses that can infect humans, and they consistently undergo recombination between humans and other hosts, including mammals and birds. Therefore, about 90% of the human-infecting species are zoonotic and considered zoonotic in origin.7 RNA viruses tend to constantly evolve and cross the species barrier to permanently select and adapt in a host for their life cycle and infection. For example, when human immunodeficiency virus (HIV) emerged, it was highly susceptible to the nucleoside analog drug azidothymidine (AZT). However, within a short time, it developed mutations that led to AZT-resistance and subsequent failure of AZT monotherapy. SARS-CoV-2 may also adapt to humans as the most prevalent host in the due course of time. For survival under selective pressures, it may acquire mutations at the S-protein RBD to increase its affinity toward ACE2 via strengthening of RBD–ACE2 interactions for maintaining its infectivity and spread (Figure 1B,C). Studies have shown that SARS-CoV-2 with the D614G mutation is presently one of the most prevalent mutant forms of the virus, with enhanced the cell entry and accelerated infectivity and spread of the pandemic globally.8 This mutation in the S-protein is present outside the RBD. Another study analyzed the genomes of several SARS-CoV-2 strains from across the world and reported 32 mutations in RBD. The RBD with mutations at N354D, D364Y, V367F, and W436R presented significantly increased affinity to human ACE2, indicating that these mutants may have acquired enhanced infectivity.9 In another work, two hot-spot residues that fall in the RBD of the S-protein (G476S and V483A) were frequently mutated in a SARS-CoV-2 sample isolated from North America.9,10 These reports suggest that the virus may emerge into a highly transmissible form. However, it is essential to note that the rate of mutation of SARS-CoV-2 is presently not significantly high as compared to that of other CoVs, suggesting that its evolution is relatively slow. Nonetheless, SARS-CoV-2 has a vast scope to mutate and increase its transmission capability in the future as the patients carry a high viral load (∼100 000 copies per mL of saliva), suggesting that throughout the infection every probable mutation may develop.11 Also, since the virus spreads rapidly, this provides ample scope for selection to act upon rare but favorable mutations. All these traits make SARS-CoV-2 a virus highly probable to mutate and evolve to enhance infectivity and transmission, posing a risk of antigenic drift and the accumulation of immunologically relevant mutations across diverse ethnic groups. SARS-CoV-2 can establish a higher rate of infectivity if it acquires combinatorial mutations at the RBD-ACE2 interface region. However, it should be noted that it is difficult to predict how a virus may evolve, particularly during a nascent outbreak. Detailed studies on genomic epidemiology, evolutionary, and selection pressures will enable predictions into possible mutations.
As recombination plays a vital role in the natural selection and evolution of viruses, a thorough inspection and prediction of the “hot-spot” residues of the virus (specifically at the RBD–ACE2 interface) and comprehension of the mutational landscape should be studied as an urgent mission. Knowledge of the direction of mutational pressure and elucidating the mutations that help enhance RBD/ACE2 affinity are vital to choose “cold-spots” residues as targets for developing effective therapeutic strategies12 (Figure 2). Statistical and evolutionary data show that the present rate of synonymous mutations in SARS-CoV-2 is greater than that of non-synonymous ones. This indicates that the currently known circulating strains are under negative selection pressure and may become more contagious when subjected to significant immune or drug pressure by more rapidly accumulating mutations. The resulting viral phenotype may also develop drug resistance and could evade the host more easily. High-throughput computational strategies that can predict such mutational profiles would be extremely useful in modulating the affinity-increasing and -decreasing mutations in RBD–ACE2 complexes. The availability of more genomes will help in the identification of potential RBD–ACE2 interface residues that upon mutation render the virus more pathogenic. Using robust molecular technologies, the experimental validation of such mutations will be crucial for (i) understanding the potential development of more pathogenic strains for COVID-19 management, i.e., prepare for the future of the pandemic, (ii) understanding the clinical pathogenesis of the virus, (iii) development of specific RBD–ACE2-interaction-inhibiting compounds, (iv) design of antibodies targeting RBD, and (v) development of strategies to prevent future cross-species transmissions.
Figure 2.

Schematic representation of the viewpoint. S-protein RBD can undergo mutations under positive selection pressure to generate variations to increase affinity with ACE2, creating strains with increased infectivity. Here, the hot-spot residues responsible for improved interactions can be identified. The RBD can also undergo mutations under negative selection pressure to generate variations to decrease affinity with ACE2, producing less pathogenic strains. Here, the cold-spot residues responsible for decreased interactions can be identified. The hot- and cold-spot residues can be used to design therapeutic strategies for COVID-19.
Acknowledgments
A.K.P. acknowledges the Japan Society for the Promotion of Science (JSPS), Govt. of Japan, for the postdoctoral research fellowship.
Author Contributions
A.K.P. and T.T. conceived the idea and wrote the manuscript.
The authors declare no competing financial interest.
References
- Wang Q.; Zhang Y.; Wu L.; Niu S.; Song C.; Zhang Z.; Lu G.; Qiao C.; Hu Y.; Yuen K. Y.; et al. (2020) Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 181 (4), 894–904. 10.1016/j.cell.2020.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr T. N., Greaney A. J., Hilton S. K., Ellis D., Crawford K. H. D., Dingens A. S., Navarro M. J., Bowen J. E., Tortorici M. A., and Walls A. C., et al. (2020) Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell 10.1016/j.cell.2020.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P.; Yang X. L.; Wang X. G.; Hu B.; Zhang L.; Zhang W.; Si H. R.; Zhu Y.; Li B.; Huang C. L.; Chen H. D.; Chen J.; Luo Y.; Guo H.; Jiang R. D.; Liu M. Q.; Chen Y.; Shen X. R.; Wang X.; Zheng X. S.; Zhao K.; Chen Q. J.; Deng F.; Liu L. L.; Yan B.; Zhan F. X.; Wang Y. Y.; Xiao G. F.; Shi Z. L. (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579 (7798), 270–273. 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama T.; Platt D.; Parida L. (2020) Variant analysis of SARS-CoV-2 genomes. Bull. World Health Organ 98 (7), 495–504. 10.2471/BLT.20.253591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercatelli D.; Giorgi F. M. (2020) Geographic and Genomic Distribution of SARS-CoV-2 Mutations. Front. Microbiol. 11, 1800. 10.3389/fmicb.2020.01800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy S. (2018) Why are RNA virus mutation rates so damn high?. PLoS Biol. 16 (8), e3000003. 10.1371/journal.pbio.3000003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolhouse M. E. J.; Adair K.; Brierley L. (2014) RNA Viruses: A Case Study of the Biology of Emerging Infectious Diseases. One Health 83–97. 10.1128/9781555818432.ch6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korber B.; Fischer W. M.; Gnanakaran S.; Yoon H.; Theiler J.; Abfalterer W.; Hengartner N.; Giorgi E. E.; Bhattacharya T.; Foley B. (2020) Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 182, 812–827. 10.1016/j.cell.2020.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou J., Zhou Z., Dai R., Zhang J., Lan W., Zhao S., Wu J., Seto D., Cui L., Zhang G., and Zhang Q.. Emergence of RBD mutations in circulating SARS-CoV-2 strains enhancing the structural stability and human ACE2 receptor affinity of the spike protein. bioRxiv (Microbiology), March 17, 2020, https://doi.org/10.1101/2020.03.15.991844.
- Kim J. S.; Jang J. H.; Kim J. M.; Chung Y. S.; Yoo C. K.; Han M. G. (2020) Genome-Wide Identification and Characterization of Point Mutations in the SARS-CoV-2 Genome. Osong Public Health Res. Perspect 11 (3), 101–111. 10.24171/j.phrp.2020.11.3.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- To K. K.; Tsang O. T.; Leung W. S.; Tam A. R.; Wu T. C.; Lung D. C.; Yip C. C.; Cai J. P.; Chan J. M.; Chik T. S.; Lau D. P.; Choi C. Y.; Chen L. L.; Chan W. M.; Chan K. H.; Ip J. D.; Ng A. C.; Poon R. W.; Luo C. T.; Cheng V. C.; Chan J. F.; Hung I. F.; Chen Z.; Chen H.; Yuen K. Y. (2020) Temporal profiles of viral load in posterior oropharyngeal saliva samples and serum antibody responses during infection by SARS-CoV-2: an observational cohort study. Lancet Infect. Dis. 20 (5), 565–574. 10.1016/S1473-3099(20)30196-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padhi A. K., Kalita P., Zhang K. Y. J., and Tripathi T.. High Throughput Designing and Mutational Mapping of RBD-ACE2 Interface Guide Non-Conventional Therapeutic Strategies for COVID-19. bioRxiv (Biophysics), May 19, 2020, https://doi.org/10.1101/2020.05.19.104042.