

Summary
Spermatogonial stem cells (SSCs) both self-renew and give rise to progenitors that initiate spermatogenic differentiation in the mammalian testis. Questions remain regarding the extent to which the SSC and progenitor states are functionally distinct. Here we provide the first multiparametric integrative analysis of mammalian germ cell epigenomes comparable with that done for >100 somatic cell types by the ENCODE Project. Differentially expressed genes distinguishing SSC- and progenitor-enriched spermatogonia showed distinct histone modification patterns, particularly for H3K27ac and H3K27me3. Motif analysis predicted transcription factors that may regulate spermatogonial subtype-specific fate, and immunohistochemistry and gene-specific chromatin immunoprecipitation analyses confirmed subtype-specific differences in target gene binding of a subset of these factors. Taken together, these results show that SSCs and progenitors display distinct epigenetic profiling consistent with these spermatogonial subtypes being differentially programmed to either self-renew and maintain regenerative capacity as SSCs or lose regenerative capacity and initiate lineage commitment as progenitors.
Subject Areas: Developmental Genetics, Omics
Graphical Abstract
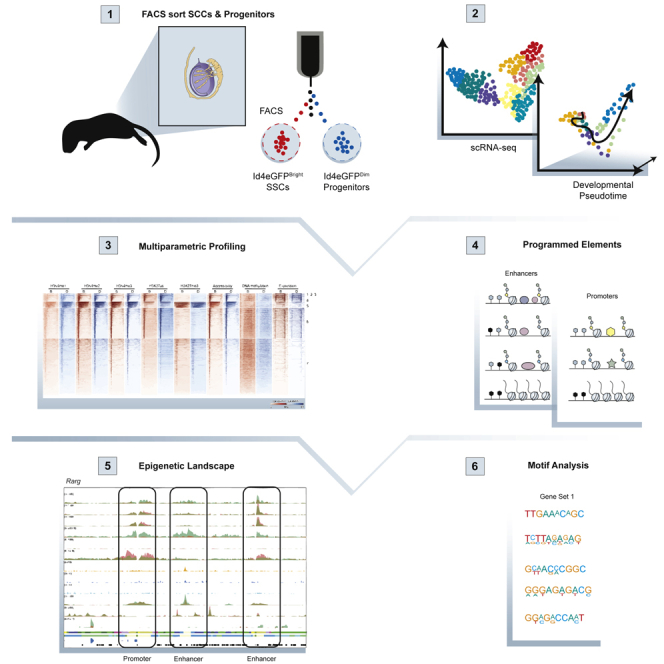
Highlights
-
•
Spermatogonial stem cells both self-renew and give rise to progenitor spermatogonia
-
•
Differential gene expression reflects stem versus differentiating cell fates
-
•
Differentially expressed genes show differential epigenetic programming
-
•
Motif analysis predicts regulators of cell type-specific epigenetic programming
Developmental Genetics; Omics
Introduction
An average adult human male produces 85–100 million sperm per day, all of which emanate from the highly proliferative seminiferous epithelium in the testis (Johnson et al., 1980). Within this epithelium spermatogenesis is sustained by spermatogonial stem cells (SSCs), progeny of which either replenish the SSC pool or contribute to the spermatogenic differentiation pathway as transit-amplifying progenitors that give rise to differentiating spermatogonia (de Rooij, 2017). SSCs can be functionally distinguished in mice on the basis of their regenerative capacity measurable by a quantifiable transplantation assay (Brinster and Avarbock, 1994; Kubota and Brinster, 2018) analogous to that reliably used for decades to identify hematopoietic stem cells (Lewis and Trobaugh, 1964).
In the postnatal mouse testis, prospermatogonia give rise to spermatogonia, exhausting the prospermatogonia population by postnatal day 5 (P5) (Drumond et al., 2011; Kluin et al., 1982; Yoshida et al., 2006). By P6-8, distinct spermatogonial subtypes (SSC, progenitor, differentiating) can be distinguished on the basis of specific marker proteins (Buaas et al., 2004; Chan et al., 2014; Valli et al., 2014), lineage tracing (Hara et al., 2014; Sun et al., 2015), bulk (Helsel et al., 2017), and single-cell (sc) (Chen et al., 2018; Ernst et al., 2019; Green et al., 2018; Grive et al., 2019; Guo et al., 2018; Hermann et al., 2018; Jung et al., 2019; Law et al., 2019; Liao et al., 2019; Sohni et al., 2019; Velte et al., 2019) RNA-sequencing (RNA-seq), or transplantation analysis (McLaren, 2003; Shinohara et al., 2000). SSCs retain regenerative capacity and divide to either self-renew or generate progenitors that lose regenerative capacity as they become primed to initiate spermatogenic differentiation giving rise to differentiating spermatogonia (Drumond et al., 2011; Kluin et al., 1982; Yang et al., 2013).
In the developing testis of Id4-eGfp transgenic mice, eGFP marks undifferentiated spermatogonia (Chan et al., 2014), and selective FACS-based recovery of the brightest (ID4-eGFPBright) and dimmest (ID4-eGFPDim) portions of the ID4-eGFP+ population significantly enriches regenerative SSCs or non-regenerative progenitors, respectively (Helsel et al., 2017). Collectively, ID4-eGFPBright spermatogonia from the P6-8 testis display characteristics of SSCs, including (1) a 5.5-fold higher colonization capacity following transplantation into a recipient testis (Helsel et al., 2017), (2) a distinct transcriptome that includes elevated expression of known SSC-expressed genes (Gfra1, Id4, Bcl6b, Etv5) as well as genes encoding factors favoring self-renewal and maintenance of a stem cell state including SSC marker transcripts such as Sall4 and genes involved in cell cycle control and replication such as Ccnb1, Cdc20, Cdk1, Mcm5, and Pcna (Helsel et al., 2017; Hermann et al., 2018; Law et al., 2019; Mutoji et al., 2016), (3) elevated expression of known SSC marker proteins (GFRA1, CDH1, ZBTB16, ID4) (Chan et al., 2014; Niedenberger et al., 2015), (4) absence of differentiation markers (KIT, STRA8, RARG, SOHLH2) (Hermann et al., 2015), and (5) insensitivity to induction of differentiation by retinoic acid (RA) (Velte et al., 2019). In contrast, ID4-eGFPDim spermatogonia at P6-8 are enhanced for characteristics of progenitors and early differentiating spermatogonia including (1) a significant depletion of transplantable colonizing capacity (Helsel et al., 2017; Hermann et al., 2018; Law et al., 2019), (2) a transcriptome featuring elevated expression of genes associated with proliferation and commitment to spermatogenic differentiation (Neurog3, Lin28a, Sohlh1, Rarg, Kit) (Helsel et al., 2017; Hermann et al., 2018; Mutoji et al., 2016), (3) elevated expression of proteins unique to spermatogonia committed to differentiation (RARG, KIT, STRA8, SOHLH2, NDRG4) (Hermann et al., 2015; Lord et al., 2018), and (4) responsiveness to induction of differentiation by RA (Velte et al., 2019).
These characteristics indicate that progeny of SSCs adopt alternative fates manifest as retention of the SSC phenotype or adoption of the progenitor phenotype based on differential gene expression. The ENCODE and NIH Roadmap Epigenomics Mapping consortia reported distinct transcriptomes accompanied by up to 15 unique cell type-specific epigenetic programming profiles at promoters and enhancers for >100 different somatic cell types in mammals (ENCODE Project Consortium, 2012; Ernst et al., 2011; Gifford et al., 2013; Kellis et al., 2014; Ram et al., 2011; Roadmap Epigenomics Consortium et al., 2015; Shema et al., 2019; Zhu et al., 2013). However, none of these studies examined germ cells. We reasoned that similarly distinguishable epigenomic programming profiles should be associated with differentially expressed genes (DEGs) in SSCs and progenitors. We used FACS to selectively recover ID4-eGFPBright “SSC-enriched” spermatogonia and ID4-eGFPDim “progenitor-enriched” spermatogonia and performed multiparametric integrative analysis of genome-wide patterns of DNA methylation, six different histone modifications, and chromatin accessibility in conjunction with subtype-specific bulk and single-cell transcriptome analyses to identify unique epigenetic landscapes associated with DEGs in each spermatogonial subtype. We then performed de novo motif analysis followed by immunohistochemistry (IHC) and chromatin immunoprecipitation (ChIP) to identify candidate factors that may either directly establish or subsequently mediate the effects of differential epigenetic programming of spermatogonial-subtype specific genes. Our results provide unprecedented insight into the multiparametric epigenetic programming associated with DEG patterns that distinguish SSC- and progenitor-enriched spermatogonia and suggest that SSCs represent a unique spermatogonial subtype epigenetically programmed to retain SSC function, whereas progenitors have transitioned to a subtype associated with lineage commitment and spermatogenic differentiation.
Results
Triplicate samples of regenerative SSC-enriched and non-regenerative progenitor-enriched spermatogonia were selectively recovered from testes of P6 Id4-eGfp transgenic mice by FACS sorting for relative eGFP epifluorescence as previously described (Helsel et al., 2017). Each epigenomic assay was run on identical aliquots of each replicate sample of ID4-eGFPBright and ID4-eGFPDim cells to assess genome-wide patterns of DNA methylation, six different histone modifications, chromatin accessibility, and gene expression, rendering results of each assay directly comparable with one another. Importantly, our previous scRNA-seq data demonstrated that FACS-based recovery of these subpopulations very effectively excluded contamination by somatic cells to <0.2% in the ID4-eGFPBright subpopulation and <1.3% in the ID4-eGFPDim subpopulation (Hermann et al., 2018).
Differential Gene Expression Distinguishes SSC- and Progenitor-Enriched Spermatogonial Subpopulations
Previous bulk and scRNA-seq analyses of ID4-eGFPBright SSC- and ID4-eGFPDim progenitor/differentiating spermatogonia-enriched (hereafter referred to as progenitor-enriched) spermatogonial subpopulations from the developing testis revealed distinct patterns of gene expression in each (Helsel et al., 2017; Hermann et al., 2018). Here, we first conducted bulk RNA-seq on aliquots of the same samples on which we also conducted bulk epigenomics analyses (Figure 1A). The bulk RNA-seq data revealed statistically significant (p < 0.01) differential gene expression patterns confirming significant enrichment of SSCs in the ID4-eGFPBright subpopulation and of progenitors in the ID4-eGFPDim subpopulation that was further corroborated by differential expression of known marker genes consistent with previous reports (Helsel et al., 2017; La et al., 2018). SSC marker transcripts including Gfra1, Lhx1, Zbtb16, Etv5, Ret, T, and Bcl6b were expressed at significantly higher levels in ID4-eGFPBright cells, whereas markers of progenitors including Sox3, Nanos3, Neurog3, Rarg, and Kit were expressed at higher levels in the ID4-eGFPDim cells (Figures 1D and S1A). Importantly, the absence of expression of genes recently reported to be markers of prospermatogonia, including Etv4, Mmp9, and Ttc28 (Tan et al., 2020), supports the contention that, by P5, essentially all prospermatogonia have transitioned to undifferentiated spermatogonia (Drumond et al., 2011; Hilscher et al., 1974; Kluin and Rooij, 1981; Roosen-Runge and Leik, 1968; Wartenberg, 1976). A complete list of genes differentially expressed in the ID4-eGFPBright and ID4-eGFPDim subpopulations is provided in Table S1.
Figure 1.

Differential Gene Expression in SSC-Enriched and Progenitor-Enriched Spermatogonial Subpopulations
(A) Differential gene expression profiling of ID4-eGFPBright and ID4-eGFPDim spermatogonia by bulk RNA-seq. Genes with >1.5 LFC (Log2 Fold Change difference >1.5, p < 0.01) were hierarchically clustered. Red–blue colors indicate high–low expression levels in Z score. Gene-specific results of bulk RNA-seq can be found in Table S1.
(B) Subpopulations among P6 ID4-eGFPBright and ID4-eGFPDim spermatogonia are revealed in this UMAP display of single-cell RNA-seq (scRNA-seq) data. Each dot represents a single cell. The cell number in each cluster is indicated via color coding.
(C) Developmental trajectory of neonatal spermatogonia. A dynamic diffusion map shows the developmental progression of ID4-eGFPBright to ID4-eGFPDim spermatogonia in pseudotime.
(D) Scaled average expression of selected marker genes and the percentage of cells within each cluster with detectable expression (dot radius) for each selected marker gene within each cell cluster. The cell cluster order was arranged by Diffusion Map from top to bottom.
(E) Scaled, normalized expression of the 2,000 most variably expressed genes in ID4-eGFPBright and ID4-eGFPDim spermatogonia. Cells are ordered along their pseudotime developmental progression shown in Figure 1C. The top bar indicates the progression from ID4-eGFPBright (red) to ID4-eGFPDim (blue) cells based on intensity of ID4-eGFP expression. The lower bar represents the cell clusters identified according to the color code shown in Figure 1B. These 2,000 most variably expressed genes were hierarchically clustered into six gene sets based on expression patterns throughout the pseudotime progression. A detailed list of variably expressed genes is provided in Table S1.
We next mined our previously described scRNA-seq data (Hermann et al., 2018) to further delineate specific spermatogonial subpopulations among either the ID4-eGFPBright or ID4-eGFPDim populations (Figure 1B). We delineated 11 different clusters, 5 of which (clusters 1–5) were enriched in ID4-eGFPBright cells and the other 6 of which (6–11) were enriched in ID4-eGFPDim cells (Figure 1B). These clusters were then ordered by pseudotime analysis, which indicated that ID4-eGFPBright cells precede ID4-eGFPDim cells developmentally (Figure 1C). This developmental progression was further confirmed by differential expression of transcripts encoding markers known to initiate expression in prospermatogonia, SSCs, progenitors, differentiating spermatogonia, or premeiotic cells (Figure 1D). Finally, in addition to identifying clusters of spermatogonial subpopulations based on our scRNA-seq data, we also identified six distinct gene sets distinguished by their expression patterns among the different spermatogonial clusters (Figure 1E). Together the various approaches to assessing variation among spermatogonia in the developing testis confirm that this is a developmentally dynamic population of cells that is beginning to subdivide into distinct spermatogonial subtypes including SSCs and progenitors.
An analysis of the representation of transcripts characteristic of different phases of the cell cycle revealed some difference in representation of the M and M/G1 phases in ID4-eGFPBright and ID4-eGFPDim cells, which could potentially reflect differences in cell division rates between SSCs and progenitors (Oatley and Brinster, 2008). However, we otherwise detected a very similar distribution of transcripts associated with the G1/S and S phases in these subpopulations (Figure S1E), consistent with previous assessments of cell cycle phase distributions in ID4-eGFPBright and ID4-eGFPDim spermatogonia in the developing testis (Mutoji et al., 2016; Hermann et al., 2018). These observations were further corroborated by a cell cycle phase-specific DNA content analyses of live P6 ID4-eGFPBright and ID4-eGFPDim spermatogonia, which confirmed that both subpopulations contained cells in all phases of the cell cycle (Figure S1E). This analysis also detected a small but statistically significant difference in the proportion of cells in G1/G0 in each subpopulation (p = 0.039). Finally, GO analyses of the six differentially expressed gene sets revealed that sets 1 and 2 were enriched with genes related to stem cell maintenance, whereas set 3 was enriched for genes associated with spermatogenesis, oogenesis, and cell cycle (Figure S1E). Gene sets 4 and 5 were also enriched for cell cycle genes as well as genes for DNA replication and DNA repair, and gene set 6 was enriched for genes involved in epigenetic modifications. Taken together, these data reveal an ordered progression between SSC- and progenitor-enriched spermatogonial subpopulations that is consistently characterized by differential gene expression patterns related to a variety of cellular functions including stem cell maintenance, cell cycle, gametogenesis, and epigenetic reprogramming, but no one function appears to be disproportionately responsible for a majority of the differential gene expression.
Multiparametric Integrative Analysis Delineates Distinct Epigenetic Landscapes in Spermatogonial Subpopulations
We analyzed aliquots of the same samples of each spermatogonial subtype by (1) ChIP-seq to detect six different histone modifications—H3K4me1,2,3, H3K9me3, H3K27ac, and H3K27me3; (2) ATAC-seq to assess chromatin accessibility; and (3) MeDIP-seq to examine DNA methylation (Figure 2A). We chose to examine all three forms of H3K4 methylation because all tend to inhibit DNA methylation in the same region, but H3K4me1&2 tend to mark enhancers, whereas H3K4me3 tends to mark promoters. We examined H3K4me3 and H3K27me3 because the simultaneous presence of these marks is indicative of poised genes (Guo et al., 2017; Lesch et al., 2013, 2016). We assessed H3K27me3 and H3K27ac because these are repressive and active marks, respectively, that can function as a switch to promote expression or repression of transcription (Katoh et al., 2018). Finally, we examined H3K9me3 because it tends to mark regions of the genome other than promoters or enhancers, especially repeat elements and heterochromatin (Becker et al., 2016). Assessment of the relationships among these parameters revealed that, as has been observed in somatic cell epigenomes (ENCODE Project Consortium, 2012; Zacher et al., 2017), patterns of H3K4me1,2,3 were highly correlated with one another in all spermatogonia, whereas H3K27me3 showed less correlation with H3K4me1,2,3 and H3K27ac showed higher correlation with H3K4me1,2,3, and high discordance with H3K27me3 (Figure 2B). Patterns of H3K9me3 peaks showed very little co-localization with any of the other histone modification peaks, presumably reflecting disparate functions of these modifications. Indeed, we found H3K9me3 peaks were enriched at repetitive elements such as LINEs, LTRs (ERV1, ERVK, ERVL) (Figures 2C, S2A, and S2C), and at 5′ AND-3′ UTRs of transposons, presumably contributing to repression of transposon mobility (Figure 2D). A majority of histone modifications were found in distal intergenic regions; however, peaks were also detected at or flanking gene promoters (Figure S2B). Within genic regions, peaks of chromatin accessibility and absence of DNA methylation correlated with peaks of H3K4me1,2,3 and H3K27ac, especially at gene promoters (Figures 2D and 2E). These data demonstrate that spermatogonia utilize complex combinations of epigenetic programming parameters to regulate gene expression in a manner similar to those reported to be used in numerous different somatic cell types by the ENCODE and NIH Roadmap Epigenomics Mapping consortia (ENCODE Project Consortium, 2012; Ernst et al., 2011; Gifford et al., 2013; Kellis et al., 2014; Ram et al., 2011; Roadmap Epigenomics Consortium et al., 2015; Shema et al., 2019; Zhu et al., 2013).
Figure 2.

Epigenetic Profiling of Genic Regions in ID4-eGFPBright and ID4-eGFPDim Spermatogonial Subpopulations
(A) A heatmap shows peaks of histone modifications (H3K4me1, H3K4me2, H3K4me3, H3K9me1, H3K27ac, and H3K27me3) deduced by ChIP-seq, chromatin accessibility deduced by ATAC-seq, and DNA methylation deduced by MeDIP-seq in ID4-eGFPBright and ID4-eGFPDim spermatogonia. Red color indicates reads from ID4-eGFPBright B cells; blue color indicates reads from ID4-eGFPDim D cells.
(B) A heatmap depicts correlations among individual histone modification patterns in ID4-eGFPBright and ID4-eGFPDim spermatogonia.
(C) Heatmaps of H3K9me3 deposition in four types of repeats—LINEs(L2) and LTRs (ERV1, ERVK, ERVL) in ID4-eGFPBright (red) and ID4-eGFPDim (blue) spermatogonia.
(D) A genome browser snapshot showing sequencing reads of six histone modifications, chromatin accessibility, DNA methylation, and transcript expression in ID4-eGFPBright (coral colored tracks) and ID4-eGFPDim spermatogonia (green colored tracks). The location of a gene that is expressed in both ID4-eGFPBright and ID4-eGFPDim spermatogonia (Fam92b) as well as that of repeat elements is shown at the bottom of this browser image.
(E) Epigenetic profiles within genic regions (TSS – TES + 3 kb upstream and 3 kb downstream) of all mouse Refseq annotated genes (n = 24,012) in ID4-eGFPBright (red tracks, B) and ID4-eGFPDim (blue tracks, D) spermatogonia together with corresponding transcript expression tracks. K-means clustering revealed seven distinct epigenetic profiles, including three that are too small to visually resolve (1–3). The order of genes represented in these tracks is shown in Table S2.
Genic Region Patterns of Chromatin Modifications Distinguish Genes Expressed in SSC- and Progenitor-Enriched Spermatogonial Subpopulations
K-means clustering of genic region data revealed seven different patterns of histone modifications (Figure 2E). Four modifications (H3K4me1,2,3 & H3K27ac) were enriched in genes expressed in one or both spermatogonial subpopulations, predominantly in promoter regions (Figure 2E, clusters 1,2,3,4,6), as has been observed in multiple somatic cell types (ENCODE Project Consortium, 2012; Zacher et al., 2017). Genes that were either not expressed or expressed at very low levels in one or both spermatogonial subpopulations (clusters 5,7) showed enrichment of the repressive H3K27me3 modification and depletion of the active H3K27ac modification, both within gene bodies (cluster 5) and at promoters (cluster 7). Repressed genes showed enrichment of H3K4me1,2,3 and H3K27me3 within gene bodies or downstream genic regions and lacked histone modification peaks at promoter regions. A GO analysis of gene promoters enriched for either H3K27ac or H3K27me3 was consistent with differential gene expression favoring enhanced maintenance of the stem cell state in SSCs and of lineage priming in progenitors (Figure S2D).
Exemplary sets of genes up-regulated in SSC-enriched spermatogonia (Tspan8, Pax7, Lhx1, Egr2, Gfra1, Id4, Tcl1) and genes up-regulated in progenitor-enriched spermatogonia (Dmrtb1, Neurog3, Rarg, Kit, Lmo1, Crabp1) illustrate differences in promoter programming associated with spermatogonial subtype-specific gene expression. Collectively, our assessments revealed that, in each respective spermatogonial subpopulation, promoters of up-regulated genes showed (1) enriched H3K4me1,2,3; (2) enriched H3K27ac; (3) depleted H3K27me3; (4) enhanced chromatin accessibility; and (5) hypomethylated DNA, whereas promoters of down-regulated genes showed (1) decreased H3K4me1,2,3; (2) depleted H3K27ac; (3) enriched H3K27me3; (4) decreased chromatin accessibility; and (5) hypomethylated DNA (Figures 3, S3A, and S3B). Thus, within genic regions, differential enrichment of H3K27ac or H3K27me3 in promoter regions correlated most closely with regulation of DEGs distinguishing ID4-eGFPBright and ID4-eGFPDim spermatogonia (Figures 3I and S3C–S3F). Notably, the correlations between differential patterns of H3K4me1/2/3 and H3K27me3 in promoters of genes that were up- or down-regulated in each spermatogonial subtype, respectively, were highly significant (p < 2.2−16) (Figures S3A and S3B).
Figure 3.

Epigenetic Profiling at Promoters in ID4-eGFPBright and ID4-eGFPDim Spermatogonia
(A–H) Scatterplots correlating H3K4me3 (x axis) and H3K27me3 (y axis) enrichments (log2RPKM) at promoters (TSS ±500 bp) in ID4-eGFPBright (A–C) and ID4-eGFPDim (E–G) spermatogonia. Color codes indicate gene expression levels (A, E), H2K27ac enrichment (B and F), and chromatin accessibility (C and G) at promoters. H3K27ac/H3K27me3 double ChIP-seq signal enrichment correlated with gene expression levels at promoters in ID4-eGFPBright and ID4-eGFPDim spermatogonia (D and H).
(I) Comparisons of histone modification, chromatin accessibility, and DNA methylation levels (RPKM, Reads Per Kilobase of sequence range per Million mapped reads) in the most differentially expressed gene sets in ID4-eGFPBright (red trace) and ID4-eGFPDim (blue trace) spermatogonia.
Intergenic Enhancers Are Differentially Programmed in SSC- and Progenitor-Enriched Spermatogonial Subpopulations
ATAC-seq peaks identify open chromatin regions indicative of promoters and enhancers (Buenrostro et al., 2013). Nearly 50% of accessible chromatin regions resided in distal intergenic regions in both ID4-eGFPBright and ID4-eGFPDim spermatogonia (Figure S2B), and nearly all intergenic ATAC-seq peaks co-localized with peaks of H3K4me1,2,3 enrichment and hypomethylated DNA (Figure 2D), indicative of enhancers (Calo and Wysocka, 2013). We integrated histone ChIP-seq, MeDIP-seq, and ATAC-seq data to identify three distinct types of enhancers: (1) active enhancers marked by H3K27ac; (2) primed enhancers marked by H3K4me1/2; and (3) poised enhancers marked by H3K4me1 and H3K27me3 (Figure 4A). As at promoters, H3K4me1,2,3 marks co-localized within enhancers (Figures 4B and 4D), whereas H3K27me3 or H3K27ac marks appeared to be mutually exclusive in both ID4-eGFPBright and ID4-eGFPDim spermatogonia (Figures 4C and 4E). For each type of enhancer, we observed significant overlap in ID4-eGFPBright and ID4-eGFPDim spermatogonia, but we also observed active, poised, and primed enhancers unique to either ID4-eGFPBright or ID4-eGFPDim cells (Figures 4F–4H). We next utilized GREAT for Gene Ontology (GREAT-GO) analysis (McLean et al., 2010) to identify categories of biological functions of genes predicted to be associated with these different types of enhancers (Figures 4I–4K). Active enhancers in ID4-eGFPBright spermatogonia were associated with genes that were enriched for stem cell maintenance functions (e.g., Dappa2, Eomes, Lin28a, Pou5f1, Nodal, and Tcl1) and other early developmental functions. In ID4-eGFPDim spermatogonia we observed enrichment of active enhancers associated with differentiation functions plus some enrichment of active enhancers associated with stem cell maintenance. As expected, genes associated with active enhancers within each spermatogonial subtype were up-regulated in that subtype (Figure S4). These results confirm that, as in somatic cells, individual genes in spermatogonia are typically regulated by a single promoter plus multiple enhancers and that epigenetic programming of enhancers is more variable than that of promoters (Shen et al., 2012). These results also indicate that differential programming of enhancers is a primary distinction between SSC-enriched ID4-eGFPBright and progenitor-enriched ID4-eGFPDim spermatogonia.
Figure 4.

Epigenetic Profiling at Enhancers in ID4-eGFPBright and ID4-eGFPDim Spermatogonia
(A) Heatmaps show profiles of each set of histone modification, chromatin accessibility, and DNA methylation peaks at sites of intergenic enhancers in ID4-eGFPBright (red) and ID4-eGFPDim (blue) spermatogonia. Peak profiles are shown for active, poised, and primed enhancers in each spermatogonial subtype. Genomic coordinates of enhancers shown in this figure are listed in Table S3.
(B–E) Scatterplots showing positive or negative combinatorial enrichment of different histone modifications at enhancers. Dashed lines indicate the minimum threshold value indicative of enrichment within each bimodal distribution. Each dot is 1 enhancer. (B) Enrichment of the H3K4me3 modification correlates positively with enrichment of the H3K4me2 and H3K4me1 modifications. (C) Enrichment of the H3K4me1 modification correlates positively with enrichment of either the H3K27me3 or the H3K27ac modifications, but enrichment of the H3K27me3 modification correlates negatively with enrichment of the H3K27ac modification, and vice versa. (D) Enrichment of the H3K4me3 modification correlates positively with enrichment of either the H3K27me3 or the H3K27ac modifications, but enrichment of the H3K27me3 modification correlates negatively with enrichment of the H3K27ac modification, and vice versa.
(F–H) Venn diagrams show proportions of active (F), poised (G), and primed (H) enhancers unique to ID4-eGFPBright spermatogonia, common to both ID4-eGFPBright and ID4-eGFPDim spermatogonia, or unique to ID4-eGFPDim spermatogonia.
(I–K) GREAT GO analysis of functions encoded by genes associated with active (I), poised (J), or primed (K) enhancers in ID4-eGFPBright or ID4-eGFPDim spermatogonia.
Genes Differentially Expressed in SSCs and Progenitor Spermatogonia Show Similarly Reduced Promoter Region DNA Methylation in Both Spermatogonial Subtypes
Our MeDIP-seq analysis of genic region DNA methylation patterns revealed constitutively hypomethylated promoters of DEGs in SSC- and progenitor-enriched spermatogonia, respectively (Figures 3A and 3J). We observed >300,000 MeDIP-seq peaks genome wide in ID4-eGFPBright and/or ID4-eGFPDim spermatogonia, of which ∼62% were common to both spermatogonial subtypes, ∼25% were unique to ID4-eGFPBright spermatogonia and the remaining ∼13% were unique to ID4-eGFPDim spermatogonia (Figure 5A). The latter two categories reflect the 1,486 differentially methylated regions (DMRs) we observed between ID4-eGFPBright and ID4-eGFPDim spermatogonia (Figure 5D). In genic regions, DNA methylation occurred within gene bodies, in both exons and introns (Figure 5B). Genome wide, a majority of the MeDIP-seq peaks occurred in distal intergenic regions or introns (Figure 5C). GREAT-GO analysis of DMRs showed that many were predicted to be associated with genes enriched in cell fate commitment and stem cell population maintenance (Figure 5E).
Figure 5.

DNA Methylation Profiles in ID4-eGFPBright and ID4-eGFPDim Spermatogonia
(A) Venn diagrams show proportions of DNA methylation peaks unique to ID4-eGFPBright spermatogonia, common to both ID4-eGFPBright and ID4-eGFPDim spermatogonia, or unique to ID4-eGFPDim spermatogonia.
(B) Genome browser snapshots showing correlations or lack thereof between DNA methylation and transcript peaks in ID4-eGFPBright (B, orange tracks) and ID4-eGFPDim (D, green or blue tracks) spermatogonia in regions encompassing a gene that is up-regulated in ID4-eGFPBright spermatogonia (Fut10) and a gene that is up-regulated in ID4-eGFPDim spermatogonia (Ccng1). Differentially methylated regions within gene bodies, but not at promoters, appear to be positively correlated with gene expression levels.
(C) The distribution of DNA methylation peaks in each spermatogonial subtype and DMRs between the two subtypes is shown. DMRs were rare at promoters but abundant in intra- and intergenic regions.
(D) DMRs were found throughout the autosomes, but, with one exception, not on the sex chromosomes. Genomic coordinates of DMRs are shown in Table S4.
(E) GREAT-GO analysis of genes associated with DMRs. The color code indicates the enrichment levels of DMR-associated genes (blue–red = low–high). Dot diameters indicate the number of DMR-associated genes within each GO term.
De Novo Motif Analysis Reveals Potential Regulators of Differential Epigenetic Programming Associated with Distinct Spermatogonial Subtypes
We performed de novo motif analysis of promoter and enhancer regions associated with genes expressed differentially or constitutively in each spermatogonial subpopulation (Figure 6). We performed de novo motif discovery for putative promoter-binding factors enriched in genes in each of the six different sets described in Figure 1E, for which consensus binding sequences are shown in Figure 6A. Examples of TF binding sites over-represented in promoters of each gene set include those for CEBPB, BACH2, BCL6B, and NF-Y in genes differentially up-regulated in SSC-enriched spermatogonia; MXI1, ZBTB3, EBF1/2, SPI1, and HLTF in genes up-regulated in progenitor-enriched spermatogonia; and NF-Y, IRF5, ZFP711, SIX4, the E2F family, RELA, and TWIST1 in genes up-regulated in both SSC- and progenitor-enriched spermatogonia (Figure 6A). Expression patterns of transcripts encoding each of these factors and others in ID4-eGFPBright and ID4-eGFPDim spermatogonia are shown in Figure S5A. Interestingly, in addition to the presence of binding motifs for factors expressed in either SSC- or progenitor-enriched spermatogonia, or both, in promoters of genes expressed in these subtypes, we also observed binding motifs for several factors that are not expressed in spermatogonia (Figure S5A). We were surprised to see evidence of expression of the Mef2a gene in spermatogonia but note that this evidence refers only to transcript-level expression and does not comment on expression at the protein level.
Figure 6.

Enrichment of Transcription Factor Binding Sites Predicts Cell Type and Gene-Specific Patterns of Expression and Binding of Specific Transcription Factors in ID4-eGFPBright and ID4-eGFPDim Spermatogonia
(A) De novo motif discovery within promoters of genes most differentially expressed in ID4-eGFPBright and ID4-eGFPDim spermatogonia.
(B–D) Differential enrichment of transcription factor binding motifs within active, poised, or primed enhancers in ID4-eGFPBright (red) and ID4-eGFPDim (blue) spermatogonia, including active enhancers (B), poised enhancers (C), and primed enhancers (D).
(E) ChIP-qPCR confirms predicted binding patterns of specific transcription factors in ID4-eGFPBright and ID4-eGFPDim spermatogonia. FOXP1 preferentially binds to promoters of Egr1, Egr2, and Etv5 in ID4-eGFPBright spermatogonia. DMRTB1 preferentially binds to promoters of Syce2 and Sohlh2 in ID4-eGFPDim spermatogonia. LHX1 preferentially binds to the Cited2 gene promoter (Cited2.p), the Cited2 proximal enhancer (Cited2.e), and the Spry4 enhancer (Spry4.e).
(F) Immunochemistry staining (IHC) of marker proteins in seminiferous cords in whole-mount sections of P6 mouse testes. GFRA1 and FOXC2 localized specifically to ID4-eGFPBright SSCs, OCT4 appeared in both ID4-eGFPBright SSCs and progenitor-enriched ID4-eGFPDim spermatogonia but was more prominent in the latter, FOXO1 and DMRTB1 localized specifically in progenitor-enriched ID4-eGFPDim spermatogonia, DMRT1 appeared in ID4-eGFPDim spermatogonia and some Sertoli cells, but not in ID4-eGFPBright SSCs, and EGR1 and REST co-localized with both ID4-eGFPBright SSCs and ID4-eGFPDim progenitor/early differentiating spermatogonia as well as in some somatic Sertoli cells.  , ID4-eGFPBright spermatogonium;
, ID4-eGFPBright spermatogonium;  , ID4-eGFPDim spermatogonium;
, ID4-eGFPDim spermatogonium;  , Sertoli cell. Scale bars, 20 μm.
, Sertoli cell. Scale bars, 20 μm.
Similarly, de novo motif analysis revealed over-representation of TF-binding sites present in active enhancers including those for FUBP1, KLF5, FOXO3, KLF6, ZBTB17, and FOXC2 in ID4-eGFPBright spermatogonia and SOX15, SOX3, SOX10, RREB1, DMRTB1, and EGR1 in ID4-eGFPDim spermatogonia (Figure 6B). TF-binding sites over-represented in poised enhancers included those for ELF5, ETS2, KLF15, and ETV5 in ID4-eGFPBright spermatogonia and ZNF281, SP3, SP2, EGR1, FOXPs, and EGR2 in ID4-eGFPDim spermatogonia (Figure 6C), and those in primed enhancers included sites for SIX4, HOXD13, and ZIC1 in ID4-eGFPBright spermatogonia and SP3, SP2, DMRT1, KLF153, ZBTB17, and DMRTB1 in ID4-eGFPDim spermatogonia (Figure 6D). Expression patterns of transcripts encoding each of these factors and others in ID4-eGFPBright and ID4-eGFPDim spermatogonia are shown in Figures S5B and S5C.
We next used immunohistochemistry to validate the predicted presence of several of these factors in SSC-enriched ID4-eGFPBright or ID4-eGFPDim spermatogonia in situ (Figure 6F), including detection of GFRA1 and FOXC2 in SSC-enriched ID4-eGFPBright spermatogonia, FOXO1 and DMRTB1 in progenitor-enriched ID4-eGFPDim spermatogonia, OCT4 in both SSC-enriched ID4-eGFPBright spermatogonia and progenitor-enriched ID4-eGFPDim spermatogonia (though more prominently in the latter), DMRT1 in progenitor-enriched ID4-eGFPDim spermatogonia and some Sertoli cells but not in ID4-eGFPBright cells, and EGR1 and REST in both ID4-eGFPBright SSCs and ID4-eGFPDim progenitor/early differentiating spermatogonia as well as in some somatic Sertoli cells. We then validated a representative sample of spermatogonial-subtype specific differential binding patterns of specific factors to specific enhancers on the basis of factor/locus-specific ChIP-qPCR, which demonstrated elevated binding of FOXP1 to enhancers of three genes, Egr1, Egr2, and Etv5, specifically in SSC-enriched ID4-eGFPBright spermatogonia, and elevated binding of DMRTB1 to enhancers of two genes, Syce2 and Sohlh2, and of LHX1 to enhancers of two genes, Cited2 and Spry4, in progenitor-enriched ID4-eGFPDim spermatogonia (Figure 6E). Interestingly, transcripts encoding these three transcription factors are also differentially expressed in ID4-eGFPBright and ID4-eGFPDim spermatogonia, respectively, with those encoding FOXP1 and LHX1 being expressed at higher levels in ID4-eGFPBright spermatogonia and those encoding DMRTB1 being expressed at higher levels in D4-eGFPDim spermatogonia. Taken together, these data demonstrate the power of multiparametric integrative analysis of epigenetic parameters as a means to reveal differential epigenetic programming of elements potentially regulating differential gene expression in SSC-enriched ID4-eGFPBright or progenitor-enriched ID4-eGFPDim spermatogonia, respectively. Confirmation of spermatogonial subtype-specific binding of specific factors for which motifs are overrepresented in these elements affords a means to identify candidate regulators that may either establish and/or mediate this differential epigenetic programming.
Differential Fates of ID4-eGFPBright and ID4-eGFPDim Spermatogonia Are Associated with Coordinated, Multiparametric Programming of Differentially Expressed Genes
Ultimately, it is specific combinations of chromatin parameters defined by epigenetic signatures and specific TF interactions that drive differential gene expression, which, in turn, establishes distinct fates of different cell types or subtypes (Ernst and Kellis, 2017). Thus, we integrated our ChIP-seq, MeDIP-seq, ATAC-seq, and RNA-seq data using multivariate Hidden Markov Model building by ChromHMM to identify and characterize 15 different chromatin states within the spermatogonial genome in agreement with ENCODE project methods (Bailey et al., 2013; Landt et al., 2012) (Figure 7A, S6A, and S6B). These 15 different states were assessed in SSC- and progenitor-enriched spermatogonia to compare the extent to which transitions among each were associated with unique fates of spermatogonial subtypes (Figure 7B). For instance, the pattern of quiescent enhancers in state 7 showed little or no difference between ID4-eGFPBright and ID4-eGFPDim spermatogonia, likely representing enhancers involved with gene expression in non-spermatogonial cell types. In contrast, a portion of enhancers displaying either an inactive (state 1) or active (state 6) status in SSC-enriched spermatogonia resolved to a quiescent status (state 5) in progenitor-enriched spermatogonia (Figure 7B).
Figure 7.

Coordinated Epigenetic Programming Correlates with Differential Gene Expression in ID4-eGFPBright and ID4-eGFPDim Spermatogonia
(A) A heatmap shows that 15 distinct chromatin states are predicted by ChromHMM analysis of unique combinational patterns of eight different epigenetic parameters.(B) A heatmap reveals spermatogonial-subtype specific transitions among the 15 chromatin states color-coded to match those shown in Figure 7A.
(C and D) Genome browser views from ID4-eGFPBright spermatogonia (coral colored tracks) and ID4-eGFPDim spermatogonia (green colored tracks) of epigenetic parameters associated with a gene (Rab4a) up-regulated in ID4-eGFPBright spermatogonia (C), and a gene (Rarg) up-regulated in ID4-eGFPDim spermatogonia (D). Note the elevated H3K27ac levels associated with both the enhancer and promoter regions of each gene in the spermatogonial subtype in which the gene is up-regulated and the elevated H3K27me3 levels associated with both the enhancer and promoter regions of each gene in the spermatogonial subtype in which the gene is down-regulated.
Parallel visualization of tracks indicative of read intensities derived from each epigenomic analysis facilitated the most direct, comprehensive, locus-specific comparisons of coordinated epigenetic programming in SSC- and progenitor-enriched spermatogonia, respectively (Figures 7C and 7D). This revealed unique epigenetic signatures associated with promoters or enhancers of DEGs in these two spermatogonial subtypes. Thus, as noted above, programming patterns associated with promoters of DEGs up-regulated in one or the other spermatogonial subpopulation included enriched H3K4me1,2,3 and H3K27ac, depleted H3K27me3, hypomethylated DNA, and elevated chromatin accessibility (Figures 7C and 7D). Interestingly, promoters of many genes showed enriched H3K4me1,2,3, hypomethylated DNA and elevated chromatin accessibility in both spermatogonial subpopulations, despite the fact that these genes were differentially expressed. However, enrichment of H3K27ac or H3K27me3 varied directly with up- or down-regulation of these genes in each subpopulation, respectively. This suggests differential enrichment of these two promoter region modifications contributes directly to differential regulation of gene expression in SSCs versus progenitors. Enhancers of DEGs showed enrichment of H3K27ac and depletion of H3K4me1 for up-regulated genes and enrichment of H3K27me3 and H3K4me1 for down-regulated genes (Figures 7C and 7D).
We augmented these data with those from published reports of genome-wide binding patterns of DMRT1 (Murphy et al., 2015), DMRTB1 (Zhang et al., 2014), and CTCF (Yue et al., 2014) in adult testis tissue. In a study by Murphy et al. in which no distinction was made between SSC- and progenitor-enriched spermatogonia, or even between spermatogenic and somatic cells, it is noteworthy that peaks of DMRT1 and DMRTB1 binding were detected at enhancers of many of the spermatogonial subtype-specific genes identified in our study (Figures 7C and 7D) (Murphy et al., 2015). In addition, in a study by Rivero-Hinojosa et al. binding motifs for CTCF were observed predominantly in distal flanking intergenic regions throughout the genome consistent with reports that CTCF plays an important role in 3-dimensional organization of the genome to mediate long-range enhancer-promoter interactions contributing to cell fate determination (Ren et al., 2017; Rivero-Hinojosa et al., 2017). Finally, using the code shown in Figure 7A, we were able to predict the arrangement of chromatin states in a linear context in each spermatogonial subtype (Figures 7C, 7D, and S6C), as well as the extent to which these states varied either between genes differentially expressed in each spermatogonial subtype or within the same genes in each spermatogonial subtype (Figures 7C and 7D). Additional data regarding the genome-wide distribution of potential regulatory elements and chromatin states identified by our ChromHMM analysis are shown in Figure S6. These results further demonstrate the correlation between spermatogonial subtype-specific differential epigenetic programming and spermatogonial subtype-specific differential gene expression.
Discussion
The ENCODE (ENCODE Project Consortium, 2012), NIH Roadmap Epigenomics Mapping Consortium (Zacher et al., 2017) and related (Ernst et al., 2011) studies characterized the variable epigenetic states of key regulatory elements throughout the genomes of >100 different somatic cell types on the basis of multiparametric integrative methodology (Ernst and Kellis, 2017) but did not examine any germ cell types. One previous study (Hajkova et al., 2008) provided an initial characterization of histone modifications in fetal mouse prospermatogonia, but did not examine postnatal germ cells, and therefore did not characterize epigenetic programming distinguishing SSCs from progenitors. Fetal and postnatal spermatogenic cell types have been assessed for poised genes, but those studies were restricted to a limited set of histone modifications (Lesch et al., 2013). Several other studies assessed one or a few different epigenetic parameters in various spermatogenic cell types, including SSCs (Gaysinskaya et al., 2018; Hammoud et al., 2015, 2014; Kubo et al., 2015; Lambrot et al., 2019; Maezawa et al., 2018a, 2018b), but these were too limited in scope to facilitate integrative profiling analogous to that developed by the ENCODE and NIH Roadmap Epigenomics Mapping consortia (ENCODE Project Consortium, 2012; Ernst et al., 2011; Gifford et al., 2013; Kellis et al., 2014; Ram et al., 2011; Roadmap Epigenomics Consortium et al., 2015; Shema et al., 2019; Zhu et al., 2013). Our simultaneous analysis of nine different parameters in aliquots of the same samples of SSC- or progenitor-enriched spermatogonial subpopulations yielded combinatorial data sufficient to distinguish the same 15 different chromatin states that were described for somatic cell types by the ENCODE study (ENCODE Project Consortium, 2012).
Results from transplantation studies have shown that regenerative SSC capacity resides in a small subpopulation of undifferentiated spermatogonia (Kubota and Brinster, 2018); however, the advent of the Id4-eGfp transgenic mouse has facilitated selective recovery of spermatogonial subpopulations highly enriched for, or significantly depleted of, this capacity (Chan et al., 2014; Helsel et al., 2017). Importantly, the fact that these different spermatogonial subpopulations are recovered from the intact testis in a similar manner but then display consistent, significant differences in regenerative capacity suggests that this reflects an inherent distinction between SSCs and progenitors rather than any sort of artifactual effect imposed by the isolation, dissociation, or transplantation of these cells. Indeed, use of the Id4-eGfp transgenic mouse and the transplantation assay to subdivide and validate regenerative capacity among different spermatogonial subpopulations or subtypes affords a robust, objective means by which to monitor regenerative capacity characteristic of SSCs and typically absent from progenitors.
Our bulk and single-cell RNA-seq data corroborate multiple recent studies confirming consistent differences in gene expression patterns in SSC- and progenitor-enriched subpopulations (Green et al., 2018; Guo et al., 2018; Helsel et al., 2017; Hermann et al., 2018; Law et al., 2019; Mutoji et al., 2016), indicating these spermatogonial subtypes represent the emergence of distinct cell fates driven by distinct transcriptomes. Here, we have extended these analyses by conducting the first comprehensive analysis of epigenetic programming associated with spermatogonial-subtype specific DEGs. This revealed distinct epigenetic landscapes associated with DEGs in SSCs and progenitors, which we further mined to identify binding motifs for specific factors that may either direct establishment of this differential epigenetic programming or mediate its effects to coordinate subsequent differential expression of genes required to either maintain the SSC phenotype or transition to the progenitor phenotype.
Among the parameters we measured, differential enrichment of H3K27me3 and H3K27ac at gene promoters and related intergenic enhancers correlated most closely with differential spermatogonial subtype-specific gene expression patterns. Specifically, elevated representation of H3K27me3 was associated with down-regulated DEGs in each spermatogonial subtype, whereas that of H3K27ac was associated with up-regulated DEGs in each case. Interestingly, several other chromatin parameters, including patterns of H3K4me1,2,3, chromatin accessibility and DNA methylation at promoters, and those of H3K4me2,3 and chromatin accessibility at enhancers, showed no significant variation among DEGs regardless of whether the gene was expressed in both or only one spermatogonial subpopulation. Collectively, this is consistent with the concept that development of distinct cell fates from a common lineage involves an ordered series of changes in epigenetic programming to first initiate and subsequently stabilize differential gene expression. Similar results regarding DNA methylation and chromatin accessibility patterns have been reported for undifferentiated and differentiating human spermatogonia (Guo et al., 2017, 2020).
Previous reports have described epigenetic poising of genes (promoters simultaneously marked with H3K4me3 and H3K27me3) in the spermatogenic lineage that appears to predispose the capacity of the paternal genome to rapidly transition to an embryonic transcriptome following fertilization (Guo et al., 2017; Lesch et al., 2013, 2016). We found that many non-poised genes expressed in SSC-enriched spermatogonia become poised and repressed in progenitor-enriched spermatogonia. This raises the intriguing possibility that, in addition to marking initiation of commitment to the spermatogenic differentiation pathway, the SSC-progenitor transition may also demarcate initiation of a final phase of epigenetic programming to prepare the paternal genome for post-fertilization functions.
Enhancers are rich in transcription factor binding sites (Stadler et al., 2011), and it has been shown that transcription factors act as key drivers of differential states of activity or inactivity at enhancers (Zentner and of Chemistry, 2012). Our de novo motif analysis revealed many binding sites common to regulatory regions in both spermatogonial subtypes. However, we also identified differential enrichment of certain motifs in promoters or enhancers regulating DEGs in ID4-eGFPbright and ID4-eGFPdim spermatogonia, and these formed the basis for testable predictions of differential expression and/or binding of specific transcription factors in these spermatogonial subpopulations. We confirmed the predicted differential expression of eight different TFs (FOXC2, OCT4, FOXO1, EGR1, REST, DMRT1, DMRTB1, EZH2) and the predicted differential, locus-specific binding of three TFs (FOXP1, DMRTB1, LHX1). Thus, we validated spermatogonial subpopulation-specific differences in TF expression at the RNA level, prevalence/intracellular location at the protein level, and binding to enhancers of target DEGs at the genomic level. Interestingly, we observed enrichment of binding motifs for six factors that were also identified by de novo motif analysis of regulatory regions in human SSCs (Guo et al., 2017) including CTCF, DMRT1, CTCFL, NFY, FOXP1, and SOX3.
Our results reveal that SSC- and progenitor-enriched spermatogonia manifest distinct patterns of epigenetic programming associated with consistent differential patterns of gene expression distinguishing these spermatogonial subtypes. We suggest this differential epigenetic programming drives cell fate divergence between SSCs and progenitors, thereby directing a significant developmental switch between retention of SSC fate and initiation of spermatogenic differentiation, respectively. We have identified specific epigenetic parameters that correlate with these states that can now be monitored within the normal developmental context of spermatogenesis.
Limitations of the Study
Our multiparametric integrative analysis showed that differential gene expression is accompanied by differential epigenetic programming in SSCs and progenitors and that SSCs maintain expression of stemness genes while progenitors initiate expression of spermatogenic differentiation genes. However, the extent to which progenitors derived from SSCs retain the potential to reverse these differences in epigenetic programming and gene expression and transition back into SSCs remains unclear, as does the identity of regulators of the differential epigenetic programming that distinguishes SSCs and progenitors. Also unknown is the extent to which differential epigenetic programming regulating changes in gene expression required to promote developmental progression from one cell fate to the next becomes primed earlier in the lineage, prior to transcriptional activation of cell fate-specific genes.
Resource Availability
Lead Contact
Further information and requests for resources and questions should be directed to and will be fulfilled by the lead contact John R McCarrey (John.McCarrey@UTSA.edu).
Materials Availability
All material used are listed in Transparent Methods section and Key Resources Table in Supplemental Information, and any further information and requests for resources and questions should be directed to and will be fulfilled by the Lead Contact.
Data and Code Availability
All datasets generated from this study have been archived in the NCBI GEO database, with the accession number GSE131657 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE131657).
P6 ID4-eGFP + single cell RNA-seq data were obtained from GSE109049 (Hermann et al., 2018). Adult mouse testis ChIP-seq data for CTCF, DMRT1, and DMRTB1 was obtained from GSM918711 (Yue et al., 2014), GSE64892 (Murphy et al., 2015), and GSM1480189 (Zhang et al., 2014), respectively.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
The authors thank Mr. Sean Vargas for assistance with procedures conducted in the UTSA Genomics Core. Funding for this research was provided by NIH grants HD078679 and HD098593 (to J.R.M.), HD090083 (to C.B.G.), HD061665 (to J.M.O.), HD090007 (to B.P.H.); as well as from the Robert J. Kleberg and Helen C. Kleberg Foundation, and the Nancy Smith Hurd Foundation. Results were generated in part with assistance from the UTSA Genomics Core supported by NIH grant G12-MD007591, NSF grant DBI-1337513, and UTSA. This work received computational support from UTSA's HPC cluster SHAMU, operated by University Technology Solutions.
Author Contributions
K.C. and J.R.M. conceived and designed the experiments. K.C. performed all RNA-seq, ChIP-seq, ATAC-seq, and MeDIP-seq experiments and analyzed all of the data from each of those experiments and prepared all of the figures. K.C. also performed the de novo motif analyses and gene-specific ChIP experiments as well as the IHC with assistance from C.-H.E.C. I.-C.C. assisted with cell sorting. J.M.O. provided the Id4-egfp transgenic mouse line and related spermatogonial transplantation results. B.J.H. and C.B.G. assisted with interpretation of the IHC results. K.C. mined single-cell transcriptome data from datasets previously prepared by K.C., B.P.H., and J.R.M. B.P.H. assisted with interpretations of the scRNA-seq data and directs the UTSA Genomics Core in which K.C. prepared libraries for sequencing. All authors contributed to interpretation of data. J.R.M. wrote the manuscript with very significant input from K.C. plus additional input from all other authors. All authors read and accepted the final version of the manuscript.
Declaration of Interests
The authors declare no conflict of interest.
Published: October 23, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101596.
Supplemental Information
References
- Bailey T., Krajewski P., Ladunga I., Lefebvre C., Li Q., Liu T., Madrigal P., Taslim C., Zhang J. Practical guidelines for the comprehensive analysis of ChIP-seq data. PLoS Comput. Biol. 2013;9:e1003326. doi: 10.1371/journal.pcbi.1003326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker J.S., Nicetto D., Zaret K.S. H3K9me3-Dependent heterochromatin: barrier to cell fate changes. Trends Genet. 2016;32:29–41. doi: 10.1016/j.tig.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinster R.L., Avarbock M.R. Germline transmission of donor haplotype following spermatogonial transplantation. Proc. Natl. Acad. Sci. U S A. 1994;91:11303–11307. doi: 10.1073/pnas.91.24.11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buaas F.W., Kirsh A.L., Sharma M., McLean D.J., Morris J.L., Griswold M.D., De Rooij D.G., Braun R.E. Plzf is required in adult male germ cells for stem cell self-renewal. Nat. Genet. 2004;36:647–652. doi: 10.1038/ng1366. [DOI] [PubMed] [Google Scholar]
- Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo E., Wysocka J. Modification of enhancer chromatin: what, how, and why? Mol. Cell. 2013;49:825–837. doi: 10.1016/j.molcel.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan F., Oatley M.J., Kaucher A.V., Yang Q.E., Bieberich C.J., Shashikant C.S., Oatley J.M. Functional and molecular features of the Id4+ germline stem cell population in mouse testes. Genes Dev. 2014;28:1351–1362. doi: 10.1101/gad.240465.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Zheng Y., Gao Y., Lin Z., Yang S., Wang T., Wang Q., Xie N., Hua R., Liu M. Single-cell RNA-seq uncovers dynamic processes and critical regulators in mouse spermatogenesis. Cell Res. 2018;28:879–896. doi: 10.1038/s41422-018-0074-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drumond A.L., Meistrich M.L., Chiarini-Garcia H. Spermatogonial morphology and kinetics during testis development in mice: a high-resolution light microscopy approach. Reproduction. 2011;142:145–155. doi: 10.1530/REP-10-0431. [DOI] [PubMed] [Google Scholar]
- ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J., Kellis M. Chromatin-state discovery and genome annotation with ChromHMM. Nat. Protoc. 2017;12:2478–2492. doi: 10.1038/nprot.2017.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J., Kheradpour P., Mikkelsen T.S., Shoresh N., Ward L.D., Epstein C.B., Zhang X., Wang L., Issner R., Coyne M. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst C., Eling N., Martinez-Jimenez C.P., Marioni J.C., Odom D.T. Staged developmental mapping and X chromosome transcriptional dynamics during mouse spermatogenesis. Nat. Commun. 2019;10:1251. doi: 10.1038/s41467-019-09182-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaysinskaya V., Miller B.F., De Luca C., van der Heijden G.W., Hansen K.D., Bortvin A. Transient reduction of DNA methylation at the onset of meiosis in male mice. Epigenetics Chromatin. 2018;11:15. doi: 10.1186/s13072-018-0186-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gifford C.A., Ziller M.J., Gu H., Trapnell C., Donaghey J., Tsankov A., Shalek A.K., Kelley D.R., Shishkin A.A., Issner R. Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell. 2013;153:1149–1163. doi: 10.1016/j.cell.2013.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green C.D., Ma Q., Manske G.L., Shami A.N., Zheng X., Marini S., Moritz L., Sultan C., Gurczynski S.J., Moore B.B. A comprehensive Roadmap of murine spermatogenesis defined by single-cell RNA-seq. Dev. Cell. 2018;46:651–667.e10. doi: 10.1016/j.devcel.2018.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grive K.J., Hu Y., Shu E., Grimson A., Elemento O., Grenier J.K., Cohen P.E. Dynamic transcriptome profiles within spermatogonial and spermatocyte populations during postnatal testis maturation revealed by single-cell sequencing. Plos Genet. 2019;15:e1007810. doi: 10.1371/journal.pgen.1007810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J., Grow E.J., Yi C., Mlcochova H., Maher G.J., Lindskog C., Murphy P.J., Wike C.L., Carrell D.T., Goriely A. Chromatin and single-cell RNA-seq profiling reveal dynamic signaling and metabolic transitions during human spermatogonial stem cell development. Cell Stem Cell. 2017;21:533–546.e6. doi: 10.1016/j.stem.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J., Grow E.J., Mlcochova H., Maher G.J., Lindskog C., Nie X., Guo Y., Takei Y., Yun J., Cai L. The adult human testis transcriptional cell atlas. Cell Res. 2018;28:1141–1157. doi: 10.1038/s41422-018-0099-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J., Nie X., Giebler M., Mlcochova H., Wang Y., Grow E.J., Kim R., Tharmalingam M., Matilionyte G., Lindskog C. The dynamic transcriptional cell atlas of testis development during human puberty. Cell Stem Cell. 2020;26:262–276.e4. doi: 10.1016/j.stem.2019.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajkova P., Ancelin K., Waldmann T., Lacoste N., Lange U.C., Cesari F., Lee C., Almouzni G., Schneider R., Surani M.A. Chromatin dynamics during epigenetic reprogramming in the mouse germ line. Nature. 2008;452:877–881. doi: 10.1038/nature06714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammoud S.S.S., Low D.H.P.H.P., Yi C., Carrell D.T.T., Guccione E., Cairns B.R.R. Chromatin and transcription transitions of mammalian adult germline stem cells and spermatogenesis. Cell Stem Cell. 2014;15:239–253. doi: 10.1016/j.stem.2014.04.006. [DOI] [PubMed] [Google Scholar]
- Hammoud S.S., Low D.H.P., Yi C., Lee C.L., Oatley J.M., Payne C.J., Carrell D.T., Guccione E., Cairns B.R. Transcription and imprinting dynamics in developing postnatal male germline stem cells. Genes Dev. 2015;29:2312–2324. doi: 10.1101/gad.261925.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K., Nakagawa T., Enomoto H., Suzuki M., Yamamoto M., Simons B.D.D., Yoshida S. Mouse spermatogenic stem cells continually interconvert between equipotent singly isolated and syncytial states. Cell Stem Cell. 2014;14:658–672. doi: 10.1016/j.stem.2014.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helsel A.R., Yang Q.-E., Oatley M.J., Lord T., Sablitzky F., Oatley J.M. ID4 levels dictate the stem cell state in mouse spermatogonia. Development. 2017;144 doi: 10.1242/dev.146928. dev.146928–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann B.P., Mutoji K.N., Velte E.K., Ko D., Oatley J.M., Geyer C.B., McCarrey J.R. Transcriptional and translational heterogeneity among neonatal mouse Spermatogonia1. Biol. Reprod. 2015;92:54. doi: 10.1095/biolreprod.114.125757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann B.P., Cheng K., Singh A., Roa-De La Cruz L., Mutoji K.N., Chen I.C., Gildersleeve H., Lehle J.D., Mayo M., Westernströer B. The mammalian spermatogenesis single-cell transcriptome, from spermatogonial stem cells to spermatids. Cell Rep. 2018;25:1650–1667.e8. doi: 10.1016/j.celrep.2018.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilscher B., Hilscher W., Bülthoff-Ohnolz B., Krämer U., Birke A., Pelzer H., Gauss G. Kinetics of gametogenesis: I. Comparative histological and autoradiographic studies of oocytes and transitional prospermatogonia during oogenesis and prespermatogenesis. Cell Tissue Res. 1974;154:443–470. doi: 10.1007/BF00219667. [DOI] [PubMed] [Google Scholar]
- Johnson L., Petty C.S., Neaves W.B. A comparative study of daily sperm production and testicular composition in humans and rats. Biol. Reprod. 1980;22:1233–1243. doi: 10.1093/biolreprod/22.5.1233. [DOI] [PubMed] [Google Scholar]
- Jung M., Wells D., Rusch J., Ahmad S., Marchini J., Myers S.R., Conrad D.F. Unified single-cell analysis of testis gene regulation and pathology in five mouse strains. ELife. 2019;8:e43966. doi: 10.7554/eLife.43966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh N., Kuroda K., Tomikawa J., Ogata-Kawata H., Ozaki R., Ochiai A., Kitade M., Takeda S., Nakabayashi K., Hata K. Reciprocal changes of H3K27ac and H3K27me3 at the promoter regions of the critical genes for endometrial decidualization. Epigenomics. 2018;10:1243–1257. doi: 10.2217/epi-2018-0006. [DOI] [PubMed] [Google Scholar]
- Kellis M., Wold B., Snyder M.P., Bernstein B.E., Kundaje A., Marinov G.K., Ward L.D., Birney E., Crawford G.E., Dekker J. Defining functional DNA elements in the human genome. Proc. Natl. Acad. Sci. U S A. 2014;111:6131–6138. doi: 10.1073/pnas.1318948111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluin Ph.M., Rooij D.G. A comparison between the morphology and cell kinetics of gonocytes and adult type undifferentiated spermatogonia in the mouse. Int. J. Androl. 1981;4:475–493. doi: 10.1111/j.1365-2605.1981.tb00732.x. [DOI] [PubMed] [Google Scholar]
- Kluin P.M., Kramer M.F., Derooij D.G. Spermatogenesis in the immature mouse proceeds faster than in the adult. Int. J. Androl. 1982;5:282–294. doi: 10.1111/j.1365-2605.1982.tb00257.x. [DOI] [PubMed] [Google Scholar]
- Kubo N., Toh H., Shirane K., Shirakawa T., Kobayashi H., Sato T., Sone H., Sato Y., Tomizawa S., Tsurusaki Y. DNA methylation and gene expression dynamics during spermatogonial stem cell differentiation in the early postnatal mouse testis. BMC Genomics. 2015;16:617–624. doi: 10.1186/s12864-015-1833-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota H., Brinster R.L. Spermatogonial stem cells. Biol. Reprod. 2018;99:52–74. doi: 10.1093/biolre/ioy077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La H.M., Mäkelä J.-A., Chan A.-L., Rossello F.J., Nefzger C.M., Legrand J.M.D., Seram M., Polo J.M., Hobbs R.M. Identification of dynamic undifferentiated cell states within the male germline. Nat. Commun. 2018;9:2819. doi: 10.1038/s41467-018-04827-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrot R., Siklenka K., Lafleur C., Kimmins S. The genomic distribution of histone H3K4me2 in spermatogonia is highly conserved in sperm. Biol. Reprod. 2019;100:1661–1672. doi: 10.1093/biolre/ioz055. [DOI] [PubMed] [Google Scholar]
- Landt S.G., Marinov G.K., Kundaje A., Kheradpour P., Pauli F., Batzoglou S., Bernstein B.E., Bickel P., Brown J.B., Cayting P. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22:1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law N.C., Oatley M.J., Oatley J.M. Developmental kinetics and transcriptome dynamics of stem cell specification in the spermatogenic lineage. Nat. Commun. 2019;10:2787. doi: 10.1038/s41467-019-10596-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesch B.J., Dokshin G.A., Young R.A., McCarrey J.R., Page D.C. A set of genes critical to deèelopment is epigenetically poised in mouse germ cells from fetal stages through completion of meiosis. Proc. Natl. Acad. Sci. U S A. 2013;110:16061–16066. doi: 10.1073/pnas.1315204110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesch B.J., Silber S.J., McCarrey J.R., Page D.C. Parallel evolution of male germline epigenetic poising and somatic development in animals. Nat. Genet. 2016;48:888–894. doi: 10.1038/ng.3591. [DOI] [PubMed] [Google Scholar]
- Lewis J.P., Trobaugh F.E. Hæmatopoietic stem cells. Nature. 1964;204:589–590. doi: 10.1038/204589a0. [DOI] [PubMed] [Google Scholar]
- Liao J.Y., Ng S.H., Luk A.C., Suen H.C., Qian Y., Lee A.W.T., Tu J.J., Fung J.C.L., Tang N.L.S., Feng B. Revealing cellular and molecular transitions in neonatal germ cell differentiation using single cell RNA sequencing. Development. 2019;146:dev174953. doi: 10.1242/dev.174953. [DOI] [PubMed] [Google Scholar]
- Lord T., Oatley M.J., Oatley J.M. Testicular architecture is critical for mediation of retinoic acid responsiveness by undifferentiated spermatogonial subtypes in the mouse. Stem Cell Rep. 2018;10:538–552. doi: 10.1016/j.stemcr.2018.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maezawa S., Yukawa M., Alavattam K.G., Barski A., Namekawa S.H. Dynamic reorganization of open chromatin underlies diverse transcriptomes during spermatogenesis. Nucleic Acids Res. 2018;46:593–608. doi: 10.1093/nar/gkx1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maezawa S., Hasegawa K., Alavattam K.G., Funakoshi M., Sato T., Barski A., Namekawa S.H. SCML2 promotes heterochromatin organization in late spermatogenesis. J. Cell Sci. 2018;131:jcs217125. doi: 10.1242/jcs.217125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren A. Primordial germ cells in the mouse. Dev. Biol. 2003;262:1–15. doi: 10.1016/s0012-1606(03)00214-8. [DOI] [PubMed] [Google Scholar]
- McLean C.Y., Bristor D., Hiller M., Clarke S.L., Schaar B.T., Lowe C.B., Wenger A.M., Bejerano G. GREAT improves functional interpretation of \textlessi\textgreatercis\textless/i\textgreater-regulatory regions. Nat. Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy M.W., Lee J.K., Rojo S., Gearhart M.D., Kurahashi K., Banerjee S., Loeuille G.A., Bashamboo A., McElreavey K., Zarkower D. An ancient protein-DNA interaction underlying metazoan sex determination. Nat. Struct. Mol. Biol. 2015;22:442–U26. doi: 10.1038/nsmb.3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutoji K., Singh A., Nguyen T., Gildersleeve H., Kaucher A.V., Oatley M.J., Oatley J.M., Velte E.K., Geyer C.B., Cheng K. TSPAN8 expression distinguishes spermatogonial stem cells in the prepubertal mouse testis. Biol. Reprod. 2016;95:117. doi: 10.1095/biolreprod.116.144220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedenberger B.A., Busada J.T., Geyer C.B. Marker expression reveals heterogeneity of spermatogonia in the neonatal mouse testis. Reproduction. 2015;149:329–338. doi: 10.1530/REP-14-0653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oatley J.M., Brinster R.L. Regulation of spermatogonial stem cell self-renewal in mammals. Annu. Rev. Cell Dev. Biol. 2008;24:263–286. doi: 10.1146/annurev.cellbio.24.110707.175355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ram O., Goren A., Amit I., Shoresh N., Yosef N., Ernst J., Kellis M., Gymrek M., Issner R., Coyne M. Combinatorial patterning of chromatin regulators uncovered by genome-wide location analysis in human cells. Cell. 2011;147:1628–1639. doi: 10.1016/j.cell.2011.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren G., Jin W., Cui K., Rodrigez J., Hu G., Zhang Z., Larson D.R., Zhao K. CTCF-mediated enhancer-promoter interaction is a critical regulator of cell-to-cell variation of gene expression. Mol. Cell. 2017;67:1049–1058.e6. doi: 10.1016/j.molcel.2017.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivero-Hinojosa S., Kang S., Lobanenkov V.V., Zentner G.E. Corrigendum: testis-specific transcriptional regulators selectively occupy BORIS-bound CTCF target regions in mouse male germ cells. Sci. Rep. 2017;7:46891. doi: 10.1038/srep46891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics Consortium, Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rooij D.G. The nature and dynamics of spermatogonial stem cells. Development. 2017;144:3022–3030. doi: 10.1242/dev.146571. [DOI] [PubMed] [Google Scholar]
- Roosen-Runge E.C., Leik J. Gonocyte degeneration in the postnatal male rat. Am. J. Anat. 1968;122:275–299. doi: 10.1002/aja.1001220208. [DOI] [PubMed] [Google Scholar]
- Shema E., Bernstein B.E., Buenrostro J.D. Single-cell and single-molecule epigenomics to uncover genome regulation at unprecedented resolution. Nat. Genet. 2019;51:19–25. doi: 10.1038/s41588-018-0290-x. [DOI] [PubMed] [Google Scholar]
- Shen Y., Yue F., McCleary D.F., Ye Z., Edsall L., Kuan S., Wagner U., Dixon J., Lee L., Lobanenkov V.V. A map of the cis-regulatory sequences in the mouse genome. Nature. 2012;488:116–120. doi: 10.1038/nature11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara T., Orwig K.E., Avarbock M.R., Brinster R.L. Spermatogonial stem cell enrichment by multiparameter selection of mouse testis cells. Proc. Natl. Acad. Sci. U S A. 2000;97:8346–8351. doi: 10.1073/pnas.97.15.8346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohni A., Tan K., Song H.W., Burow D., de Rooij D.G., Laurent L., Hsieh T.C., Rabah R., Hammoud S.S., Vicini E. The neonatal and adult human testis defined at the single-cell level. Cell Rep. 2019;26:1501–1517.e4. doi: 10.1016/j.celrep.2019.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler M.B., Murr R., Burger L., Ivanek R., Lienert F., Schöler A., Wirbelauer C., Oakeley E.J., Gaidatzis D., Tiwari V.K. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480:490–495. doi: 10.1038/nature10716. [DOI] [PubMed] [Google Scholar]
- Sun F., Xu Q., Zhao D., Chen C. Id4 marks spermatogonial stem cells in the mouse testis. Sci. Rep. 2015;5:17594. doi: 10.1038/srep17594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan K., Song H.-W., Wilkinson M.F. Single-cell RNAseq analysis of testicular germ and somatic cell development during the perinatal period. Development. 2020;147:dev183251. doi: 10.1242/dev.183251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valli H., Sukhwani M., Dovey S.L., Peters K.A., Donohue J., Castro C.A., Chu T., Marshall G.R., Orwig K.E. Fluorescence- and magnetic-activated cell sorting strategies to isolate and enrich human spermatogonial stem cells. Fertil. Steril. 2014;102:566–580.e7. doi: 10.1016/j.fertnstert.2014.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velte E.K., Niedenberger B.A., Serra N.D., Singh A., Roa-DeLaCruz L., Hermann B.P., Geyer C.B. Differential RA responsiveness directs formation of functionally-distinct spermatogonial populations at the initiation of spermatogenesis in the mouse. Development. 2019;146:dev173088. doi: 10.1242/dev.173088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wartenberg H. Comparative cytomorphologic aspects of the male germ cells, especially of the “gonia”∗. Andrologia. 1976;8:117–130. doi: 10.1111/j.1439-0272.1976.tb02120.x. [DOI] [PubMed] [Google Scholar]
- Yang Q.-E., Gwost I., Oatley M.J., Oatley J.M. Retinoblastoma protein (RB1) controls fate determination in stem cells and progenitors of the mouse male germline. Biol. Reprod. 2013;89:113. doi: 10.1095/biolreprod.113.113159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida S., Sukeno M., Nakagawa T., Ohbo K., Nagamatsu G., Suda T., Nabeshima Y. The first round of mouse spermatogenesis is a distinctive program that lacks the self-renewing spermatogonia stage. Development. 2006;133:1495–1505. doi: 10.1242/dev.02316. [DOI] [PubMed] [Google Scholar]
- Yue F., Cheng Y., Breschi A., Vierstra J., Wu W., Ryba T., Sandstrom R., Ma Z., Davis C., Pope B.D. A comparative encyclopedia of DNA elements in the mouse genome. Nature. 2014;515:355–364. doi: 10.1038/nature13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacher B., Michel M., Schwalb B., Cramer P., Tresch A., Gagneur J. Accurate promoter and enhancer identification in 127 ENCODE and Roadmap epigenomics cell types and tissues by GenoSTAN. PLoS One. 2017;12:e0169249. doi: 10.1371/journal.pone.0169249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner G.E., of Chemistry S.-P.C. The chromatin fingerprint of gene enhancer elements. J. Biol. Chem. 2012;287:30888–30896. doi: 10.1074/jbc.R111.296491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T., Murphy M.W., Gearhart M.D., Bardwell V.J., Zarkower D. The mammalian Doublesex homolog DMRT6 coordinates the transition between mitotic and meiotic developmental programs during spermatogenesis. Development. 2014;141:3662–3671. doi: 10.1242/dev.113936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J., Adli M., Zou J.Y.Y., Verstappen G., Coyne M., Zhang X., Durham T., Miri M., Deshpande V., De Jager P.L. Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell. 2013;152:642–654. doi: 10.1016/j.cell.2012.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All datasets generated from this study have been archived in the NCBI GEO database, with the accession number GSE131657 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE131657).
P6 ID4-eGFP + single cell RNA-seq data were obtained from GSE109049 (Hermann et al., 2018). Adult mouse testis ChIP-seq data for CTCF, DMRT1, and DMRTB1 was obtained from GSM918711 (Yue et al., 2014), GSE64892 (Murphy et al., 2015), and GSM1480189 (Zhang et al., 2014), respectively.