

Abstract
In this study, we probed the importance of O-GlcNAc transferase (OGT) activity for the survival of tamoxifen-sensitive (TamS) and tamoxifen-resistant (TamR) breast cancer cells. Tamoxifen is an antagonist of estrogen receptor (ERα), a transcription factor expressed in over 50% of breast cancers. ERα-positive breast cancers are successfully treated with tamoxifen; however, a significant number of patients develop tamoxifen-resistant disease. We show that in vitro development of tamoxifen-resistance is associated with increased sensitivity to the OGT small molecule inhibitor OSMI-1. Global transcriptome profiling revealed that TamS cells adapt to OSMI-1 treatment by increasing the expression of histone genes. This is known to mediate chromatin compaction. In contrast, TamR cells respond to OGT inhibition by activating the unfolded protein response and by significantly increasing ERRFI1 expression. ERRFI1 is an endogenous inhibitor of ERBB-signaling, which is a known driver of tamoxifen-resistance. We show that ERRFI1 is selectively downregulated in ERα-positive breast cancers and breast cancers driven by ERBB2. This likely occurs via promoter methylation. Finally, we show that increased ERRFI1 expression is associated with extended survival in patients with ERα-positive tumors (p = 9.2e−8). In summary, we show that tamoxifen-resistance is associated with sensitivity to OSMI-1, and propose that this is explained in part through an epigenetic activation of the tumor-suppressor ERRFI1 in response to OSMI-1 treatment.
Subject terms: Glycobiology, Breast cancer, Predictive markers, Biochemistry, Cancer, Biomarkers, Molecular biology, Chromatin, Epigenetics, Transcription
Introduction
Breast cancer is the most common cancer in women and over half of all breast cancers express estrogen receptor α (ERα), a nuclear hormone receptor1. Standard treatment in this patient group includes anti-ERα therapies, such as tamoxifen2. Resistance to this type of treatment is a significant challenge in the clinical setting, and results in the development of a more aggressive disease. One of the best understood mechanisms of tamoxifen-resistance is increased activity of receptor tyrosine kinases, most notably epidermal growth factor receptor (EGFR) and erb-b2 receptor tyrosine kinase 2 (ERBB2, Her2)3–5. Despite significant efforts, discovery of targeted therapies against tamoxifen-resistant breast cancer remains a significant challenge6,7.
O-GlcNAc transferase (OGT) has emerged as a candidate drug target in breast cancer. OGT is overexpressed in breast cancer patient tumor samples8–11, while genetic silencing of OGT decreases breast cancer cell proliferation both in vitro and in vivo12,13. In addition, it has been shown that OGT overexpression promotes tumor initiation in mouse models of breast cancer14. Conversely, OGT is not essential for the survival of post-mitotic normal mammalian cells15–18. These features position OGT as a candidate target for anti-breast cancer therapy.
OGT functions as a signaling hub that integrates nutrient status of the cell and modifies target proteins accordingly19,20. OGT’s co-substrate is produced via hexosamine biosynthetic pathway, which consumes glucose, glutamine, acetyl-CoA and ATP, the key metabolites involved in proliferation21. Depending on nutrient availability and other cues, OGT glycosylates intracellular proteins on serine and threonine residues, competing with protein kinases for substrates22. However, unlike hundreds of kinases, OGT is the solo enzyme catalyzing all protein glycosylation in the nucleus and cytoplasm. This unique role positions OGT as a major signaling hub.
Pharmacological inhibition of OGT represents a potential strategy to target this enzyme for cancer therapy. Several groups have developed compounds that alter O-GlcNAc signaling, but many of the compounds such as Alloxan (uracil mimic)23 and BAGDP (N-acetylgalactosamine mimic)24 require utilization of millimolar doses. An intriguing strategy is to use a synthetic carbohydrate precursor (Ac4-5SGlcNAc) that is converted into glycosyltransferase inhibitor within the cell25. This compound decreases aggressive phenotype of breast cancer cells26,27. However, a major limitation of this compound is that it additionally targets other carbohydrate processing enzymes. OSMI-1 is an OGT small molecule inhibitor that does not significantly affect other glycosyltransferases and is active in low micromolar doses28. We recently used OSMI-1 to demonstrate that triple-negative breast cancer cells are sensitive to OGT inhibition29. Overall, OSMI-1 represents an excellent tool compound to study OGT biology in vitro, and to discover possible strategies for anti-breast cancer therapy.
In this study, we used OSMI-1 to probe the importance of OGT activity for the survival of tamoxifen-resistant breast cancer cells in an isogenic cell line pair: Tamoxifen sensitive ERα-positive MCF7 cell line (TamS), and its tamoxifen-resistant derivative (TamR). We find that TamR cells show increased sensitivity to OSMI-1 relative to the parental line. Further, global transcriptome profiling shows that OSMI-1 treatment induces expression of the endogenous inhibitor of RTK-signaling, ERRFI1. The ERRFI1 promoter is hyper-methylated in breast cancer patient samples, which is associated with low expression levels and poor prognosis. Our findings suggest a link between OGT and ERRFI1 signaling, pointing to OGT as a possible therapeutic target in a significant subset of breast cancer patients.
Results and discussion
Tamoxifen resistant breast cancer cells are sensitive to OSMI-1
Here we compare the role of OGT in tamoxifen-sensitive MCF7 breast cancer cell line (TamS), and a previously established MCF7-derived tamoxifen-resistant cell line (TamR)5. First, we confirmed the tamoxifen-resistant phenotype of the TamR cells using relative cell number (DNA content) as a read-out (Suppl. Figure 1A). Notably, we did not observe significant differences in tamoxifen-sensitivity when using an MTS assay (relies on the activity of the NAD(P)H-dependent dehydrogenase enzymes, Suppl. Figure 1B). Next, we evaluated the overall O-GlcNAcylation and noted that both TamS and TamR have similar levels (Suppl. Figure 2A). The OGT small molecule inhibitor (OSMI-1) decreased total-O-GlcNAc by 30% in both cell lines (Fig. 1A). This effect was similar to treatment with another OGT inhibitor ST04584930. We also noted that OGT expression was increased in response to OSMI-1 treatment (Suppl. Figure 2B). Upregulation of OGT in response to OGT inhibition has been previously observed, and likely represents one of the compensation mechanisms that cells employ to restore O-GlcNAc homeostasis31–33. These data demonstrate on-target inhibition by both OSMI-1 and ST045849 in breast cancer cells.
Figure 1.

OGT inhibition reduces viability of breast cancer cells. (A) Total protein O-GlcNAcylation in TamS and TamR breast cancer cells after 24 h of treatment with OGT inhibitors—ST045849 or OSMI-1. Densitometry was used to determine the intensity of the signal. (B) Cell viability measured with MTS assay, following 72 h of treatment with increasing doses of either ST045849 or OSMI-1. An average of at least 3 biological replicates with SEM are shown. Significance of the data was evaluated using unpaired t-test, *-p ≤ 0.05, **-p ≤ 0.01. (C) RT-qPCR based evaluation of mRNA abundance following 24 h of OGT inhibition with either ST045849 or OSMI-1 at 40 μM dose. Average of three biological replicates with SEM are shown.
Next, we assessed the importance of high OGT activity for proliferation of TamS and TamR cells using MTS assay relying on metabolic activity of cells and live-cell imaging to generate proliferation curves. OSMI-1 and ST045849 dose-dependently decreased viability of both cell lines as determined using the MTS assay (Fig. 1B). Interestingly, TamR cells were significantly more sensitive to both OSMI-1 and ST0145849 than the parental TamS cells, with OSMI-1-EC50 value of ~15 µM in TamR and ~ 40 µM in TamS. Live-cell imaging showed that OSMI-1 treatment blocks proliferation of TamS and TamR cells (Suppl. Figure 2C). Based on these data, we conclude that the metabolic activity of TamR cells is more sensitive to OGT inhibition, and moved on to explore the basis for this differential sensitivity.
OGT inhibition has been shown to significantly alter the transcriptional program of cells31,34, and we determined whether the differential sensitivity of TamR cells against OSMI-1 is reflected at the mRNA levels. OGT inhibition can suppress cancer cell proliferation in part through induction of DNA damaged-induced transcript 3 (DDIT3, also known as CHOP)13 and downregulation of mRNAs encoding for cell specific transcription factors that support cell proliferation33. One of the most prominent effects of OGT inhibition is compensatory upregulation of the gene encoding for OGT itself31–33,35. In accordance with these reports, we found that OSMI-1 causes increased expression of OGT and DDIT3 in both TamS and TamR cells while ERα is downregulated (Fig. 1C). These effects are more prominent in TamR cells, suggesting that changes in the transcriptional program could underpin the differential sensitivity to OSMI-1.
OSMI-1 induces transcriptional program indicative of chromatin silencing in TamS cells
To further explore the mechanism of increased sensitivity of TamR cells to OSMI-1 treatment, we performed a microarray transcriptomic analysis. We used RT-qPCR to validate the expression of genes that were among the top most downregulated (Ribonucleotide Reductase Regulatory Subunit M2 (RRM2), Thymidine Kinase 1 (TK1) and Deoxyuridine Triphosphatase (DUT)) and most upregulated (Asparagine Synthetase (ASNS), Aldolase, Fructose-Bisphosphate C (ALDOC) and Solute Carrier Family 3 Member 2 (SLC3A2), Suppl. Table 1). Expression of all six genes was differentially regulated by two to sixfold in response to OSMI-1 treatment, validating the microarray data (Fig. 2A).
Figure 2.

OGT inhibitor OSMI-1 decreases the percentage of breast cancer cells in S-Phase. (A) Validation of the microarray data using RT-qPCR. Cells were treated as indicated for 24 h, mRNA was isolated and used for RT-qPCR with select primers. Data shown is an average of four biological replicates with SEM and is normalized to DMSO treated control. (B) KEGG55–57 pathway enrichment analysis for the top most downregulated genes in TamS and TamR cells (Log2(FC) atleast − 0.5). (C) Cell cycle distribution of TamS and TamR cells after treatment with 40 μM OSMI-1 for 24 h. Data shown is an average of three biological replicates with SEM. Statistical analysis was performed using t-test, *-p ≤ 0.05.
Having established the quality of the microarray data, we performed pathway enrichment analysis to identify the key processes affected by the OSMI-1 treatment. OSMI-1 caused similar changes in the transcriptome of both cell lines and the top most upregulated pathways included steroid hormone biosynthesis and lysosomal degradation, which likely represent pro-survival adaptations to OSMI-1 treatment (Suppl. Table 1). Both TamS and TamR cells shared a number of significantly downregulated pathways including cell cycle, DNA replication and RNA processing (Suppl. Table 1, adjusted p-value < 0.05). In particular, we found the transcripts related to cell cycle progression to be highly enriched (Fig. 2B and Suppl. Table 1). To verify the impact of OGT inhibition on cell cycle regulation, we performed cell cycle analysis. OSMI-1 decreased the number of cells in S-Phase and also caused a modest, but not significant, accumulation of cells in the G2-M phase (Fig. 2C). These effects are in agreement with a previous study describing the importance of OGT for successful progression through mitosis36. However, cell cycle distribution in response to OSMI-1 was similar in both cell lines, and therefore cannot explain the increased sensitivity of TamR cells to the compound.
To explain the differential sensitivity of the cell lines to OGT inhibition, we focused on the mRNAs that were exclusively affected either in the TamS or TamR cells. We noted prominent downregulation of pathways related to DNA repair in TamS, and to a lesser extend also in TamR cells in response to OSMI-1 (Suppl. Table 1). The TamR-specific downregulated pathways were predominantly associated with metabolic processes such as amino acid degradation and fatty acid elongation (Suppl. Table 1). We have previously shown that inhibiting OGT leads to metabolic vulnerabilities by suppressing glycolytic activity, which sensitized cancer cells to inhibitors of oxidative phosphorylation33. This suggests that the enhanced sensitivity of TamR cells to OSMI-1 may be explained, in part, through the impaired ability to compensate for the metabolic stress induced by OGT inhibition. Tamoxifen treatment is known to impose a selection pressure for metabolic adaptations that support the emergence of resistance37,38, and in addition to targeting ERα, tamoxifen also suppresses oxygen consumption by blocking complex 1 of the electron transport chain, increases glycolysis and alters lipid metabolism39. It is possible that the increased sensitivity of TamR cells to OGT inhibitors is in part a side effect of these adaptations.
Next, we focused on the genes that were selectively upregulated in either of the cell lines as these could explain the differential sensitivity towards OSMI-1. We noted a strong activation of the transcriptional networks related to endoplasmic reticulum stress specifically in TamR cells (Suppl. Table 1). The most enriched pathway in TamS cells was ‘Chromatin silencing’, which was completely absent in TamR cells (Fig. 3A and Suppl. Table 1). When inspecting the mRNAs that explain the enrichment of this pathway, it became obvious that increased expression of the histone genes in response to OSMI-1 constitutes almost the entire gene list. Out of 54 histone mRNAs detected, 49 were increased in the TamS cells but remained largely unchanged in the TamR cells (Fig. 3B,C). This transcriptional response is the most striking differential response to OGT inhibition between TamS and TamR cells, and its mechanistic implications warrant further interrogation.
Figure 3.
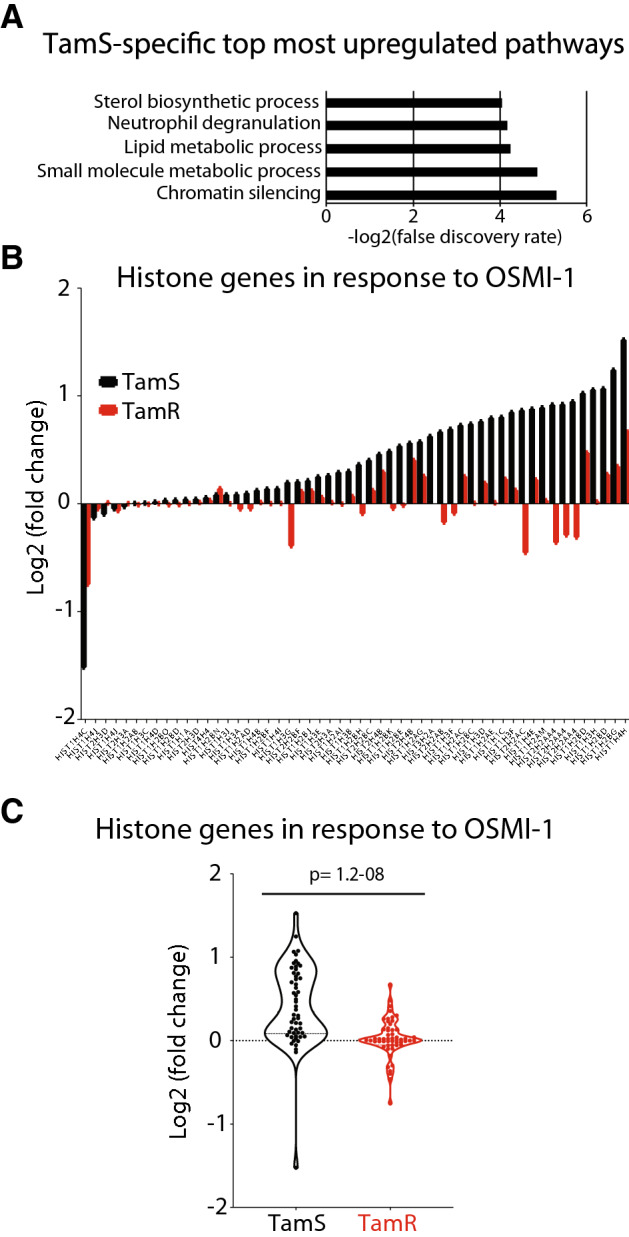
OGT inhibitor OSMI-1 induces transcriptional program indicative of chromatin silencing in tamoxifen-sensitive breast cancer cells. (A) KEGG55–57 pathway enrichment analysis for the mRNAs that were upregulated exclusively in TamS (Log2(FC) atleast ± 0.5). (B) Histone gene expression in TamS and TamR cells after OGT inhibition with 40 μM OSMI-1 for 24 h representing an average of three biological replicate experiments analyzed using microarrays. (C) Fold change in the expression of all detected histone genes after OGT inhibition in TamS and TamR cells (average of three biological replicate experiments analyzed using microarrays).
Increased expression of histone mRNAs has been previously demonstrated in response to bromodomain inhibitor-induced decrease in chromatin accessibility40. We hypothesized that if chromatin accessibility is altered in response to OGT inhibition in TamS cells, genes driven by super-enhancers would be most acutely affected by this treatment. To test this, we selected all super-enhancers in MCF7 cells 41 and assessed if the expression of associated mRNAs changes in response to OGT inhibition. Genes driven by super-enhancers were preferentially downregulated in response to OGT inhibition in the TamS cells when compared to the TamR cells (Suppl. Table 1). We have previously shown that chromatin O-GlcNAcylation overlaps with sites of active transcription and that OGT inhibition results in decreased chromatin accessibility in prostate cancer cells42. To fully elucidate the effects of OGT inhibition on chromatin accessibility in breast cancers cells, more direct assays such as FAIRE- or ATAC-seq are needed. While these experiments are essential to perform in follow-up studies, microarray data demonstrates that OGT inhibition results in a distinctive transcriptional response in TamS and TamR cells. By focusing on the differentially induced mRNAs, we may be able to identify those that have biologically meaningful functions in our model system and can additionally serve as prognostic breast cancer biomarkers.
OSMI-1 induces expression of a negative regulator of receptor tyrosine kinase signaling in TamR cells
Next, we identified the mRNAs that are selectively induced in TamR cells in response to OGT inhibition to identify potential tumor-suppressor genes that could function as disease biomarkers. Among five mRNAs that were most strongly induced in the TamR but not affected in TamS cells, we identified two genes, ERRFI1 (ERBB Receptor Feedback Inhibitor 1) and Ras Related Dexamethasone Induced 1 (RASD1) (Suppl. Table 1). OGT is important for the transcriptional regulation of certain genes43, and we therefore assessed if ERRFI1 and RASD1 genes carry an O-GlcNAc mark on their promoters using publicly available datasets42,44. Only ERRFI1 gene had a robust O-GlcNAc mark on its promoter (Suppl. Figure 3). Next, we used RT-qPCR to confirm that OSMI-1 treatment increases ERRFI1 expression in TamR cells in a dose-dependent manner up to fivefold but does not have any effect in TamS cells (Fig. 4A). In addition, treatment with ST045849 and knockdown of OGT induced a significant increase in the ERRFI1 expression in TamR cells (Suppl. Figure 4). ERRFI1 encodes a protein that functions as an endogenous inhibitor of ERBB-signaling—a well-established mechanism of tamoxifen-resistance3–5. In mouse models, deletion of ERRFI1 results in the development of spontaneous tumors45,46 while in human cells ERRFI1 decreases proliferation of cells that express high levels of epidermal growth factor receptor47. To gain further evidence of the tumor suppressor function of ERRFI1, we analyzed ERRFI1 gene dependency score using data available on the Cancer Dependency Map48,49. Depletion of ERFFI1 via CRISPR-mediated knockout increases the proliferation of almost all of the cancer cell lines included in this database (Suppl. Figure 5). Both the literature and CRISPR-data are consistent with ERRFI1 being a protein that negatively regulates proliferation of cells. However, additional experiments that go beyond the scope of this manuscript, characterization of TamS and TamR cells response to OGT inhibition, are required to assess if ERRFI1 is a bona fide tumor-suppressor. Nevertheless, these data suggest that the loss of ERFFI1 expression may be associated with the development of aggressive breast cancer.
Figure 4.

High expression of ERFFI is associated with positive prognosis in breast cancer patients. (A) RT-qPCR based evaluation of ERFFI1 expression after 24 h treatment with either DMSO or OSMI-1. Data shown is an average of four biological replicates with SEM and is normalized first to a house-keeping gene and then relative to DMSO treated control. Significance of the data was evaluated using unpaired t-test. (B, C) Expression and promoter methylation of ERFFI1 in breast cancer subclasses. The graphs were generated using UALCAN tool58 and the TCGA dataset. (D) Survival in breast cancer patients with low or high expression of ERFF1 mRNA. The plot was generated using Kaplan–Meier Plotter (mRNA gene chip, all probe sets for the gene, autoselect best cutoff and other settings as default)59.
We used publicly available datasets to evaluate ERRFI1 expression and association with cancer progression. ERRFI1 mRNA was downregulated in 17 separate breast cancer gene expression datasets available through Oncomine50, but was not prominently altered in other cancers, suggesting that this gene may be specifically associated with breast cancer (Suppl. Figure 6). Interestingly, ERRFI1 expression was significantly lower in ERα-positive (luminal) breast cancer patient samples when compared to either normal tissue or triple-negative breast cancer (TNBC) samples (Fig. 4B). In addition, we noted that ERRFI1 is downregulated in ERBB2-positive breast cancers, a disease driven by this receptor tyrosine kinase51. Overall, these data imply that ERRFI1 functions as a tumor suppressor in breast cancers driven by ERα and/or ERRB-signaling.
We investigated the mechanistic basis for the loss of ERRFI1 expression in breast cancer patient samples. ERRFI1 expression can be controlled via DNA methylation51 and OGT is known to modify proteins that regulate chromatin accessibility20. ERRFI1 promoter methylation was significantly higher in normal breast cancer tissue and TNBC patient samples when compared to the luminal breast cancer and ERBB2-positive breast cancer samples (Fig. 4C). High promoter methylation was associated with decreased ERRFI1 expression (Fig. 4B,C). Next, we asked if the differential expression of ERRFI1 has a prognostic value for breast cancer patients. We found that in luminal type A breast cancer patients, high expression of ERRFI1 was very strongly associated with a positive outcome (9.2e−08, Fig. 4D). In addition, increased expression of ERRFI1 was also significantly associated with good prognosis in patients treated with tamoxifen (Suppl. Figure 7).
To conclude, we report that tamoxifen-resistance is associated with increased sensitivity to OGT inhibition by OSMI-1 (Fig. 1B). We have previously demonstrated that TamS cells can be sensitized to OSMI-1 by tamoxifen29, and in this study we show that tamoxifen-resistant breast cancer cells are also dependent on high OGT activity. Even though we are not able to describe the specific mechanism, activation of the anti-proliferative transcriptional program specifically in endocrine-resistant breast cancer cells represents an intriguing strategy for cancer therapy. It has become apparent that OGT inhibitors can sensitize cancer cells to targeted therapies, such as inhibitors of phosphoinositide 3-kinase signaling52, glycolysis53 and proteasome54. It is not clear why a decrease in OGT activity sensitizes cancer cells to different treatments, and mechanistic understanding of this will enable more rational design of combination therapies with compounds targeting OGT. Results presented here suggest that targeting OGT may enhance the efficacy of ERBB receptor tyrosine kinase (RTK) inhibitors in cancer cells dependent on these RTKs. Based on our findings and previous data from others, we conclude that OGT functions as a context-dependent regulator of transcription.
Materials and methods
Cell culture
MCF7 cell line (referred to as TamS in this manuscript) was purchased from ATCC (Rockville, MD). TamS was cultured in DMEM (Sigma Aldrich) supplemented with 10% fetal bovine serum (FBS). TamR cells were a kind gift from Dr Julia Gee and Professor Robert I Nicholson (University of Cardiff), and have been previously described5. TamR cells were propagated in DMEM without phenol red (Thermo Fischer), supplemented with 5% charcoal stripped FBS. Cell lines were maintained in humidified incubators at 37 °C and were routinely tested for mycoplasma contamination.
Western blotting
For western blotting analysis, TamS and TamR cells were seeded into 6-well plates, using 1.8 × 105 cells per well in their respective media. Cells were treated with increasing concentrations of ST045849 (obtained from TimTec) and OSMI-1 (gift from Professor Suzanne Walker, Harvard Medical School, or obtained from Sigma Aldrich) the next day and collected on ice 24 h after the start of the treatment. Immunoblotting was performed using RL2 antibody (for total O-GlcNAcylation) (Abcam, ab2739), OGT (Cell Signalling, #5368) and β-actin antibody (Cell Signalling, #4967). Band intensity was quantified using ImageJ software.
Cell cycle analysis
TamS and TamR cells were collected on ice following 24 h of treatment with 40 µM dose of OSMI-1 compound, or DMSO. Samples were immediately fixed in 100% ice-cold methanol and placed in − 20 C° for storage. Samples were washed in cold PBS and stained with 1.5 µg/ml Hoechst 33258 in PBS for 30 min at 37 °C. Cell cycle analysis was then performed on LSR II flow cytometer (BD Bioscience, San Jose, CA).
MTS viability and crystal violet assays
Tamoxifen (Sigma, Prod. No. T5648) was freshly prepared just prior to the experiment. MTS (CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay, Promega) was added to the cells at 1:5 dilution in growth media for 1–2 h at 37 °C. Absorbance at 490 nm was then measured using a Multilabel Counter Wallac plate reader. Results were corrected for background and normalized to DMSO-treated controls. For Crystal violet assay, cells were washed twice with PBS, fixed with ice cold 70% MeOH for 2 min followed by ice cold 100% MeOH for 10 min. After the cells dried out 0.05% crystal violet solution was applied for 10 min. After staining, cells were washed twice with distilled water and let to dry. For Crystal violet dissolution and measurement, 50 µL/well of 10% acetic acid was added, followed by 15 min de-stain on a plate shaker and absorbance was measured at 590 nM by the plate reader.
Microarray data
Microarray profiling was performed at the NTNU Genomics Core Facility using Illumina HT-12 BeadChip arrays in biological triplicates.
RNA isolation/RT-qPCR
Cells were collected following 24 h of treatment. mRNA was isolated using Illustra RNA spin mini kit (GE healthcare, Chicago, IL). cDNA was prepared using the qScript cDNA Synthesis Kit in accordance with the supplier’s instructions (Quanta Biosciences, Gaithersburg, MD). qPCR reactions were set up using 2.5 µl of 10 ng/µl stock cDNA, 2.5 µl of 1 µM forward and reverse primer mix and 5 µl of Fast SYBR Green master mix (Thermo Fischer Scientific). The following primers were used for gene expression analysis: OGT forward: CAGCATCCCAGCTCACTT, reverse: CAGCTTCACAGCTATGTCTTC; OGA forward: CGAGTGAACATTCCCATCACT, reverse: CCCAAAGGAGCACAGATGTT; DDIT3 forward: CTGGGGAATGACCACTCTGT, reverse: CTTGGCTGACTGAGGAGGAG; ERα forward: TGGGCTTACTGACCAACCTG, reverse: CCTGATCATGGAGGGTCAAA; RRM2 forward: TTTAGTGAGCTTAGCACAGCGGGA, reverse: AAATCTGCGTTGAAGCAGTGAGGC; TK1 forward: GCCGATGTTCTCAGGAAAAAGC, reverse: GCGAGTGTCTTTGGCATACTTG; DUT forward: CTATGGAGAAAGCTGTTGTGAAA, reverse: TTGCAGCCAAGCCTGACC; ASNS forward: ATCACTGTCGGGATGTACCC, reverse: CTTCAACAGAGTGGCAGCAA; ALDOC forward: CGTCCGAACCATCCAGGAT, reverse: CACCACACCCTTGTCAACCTT; SLC3A2 forward: ATTGGCCTGGATGCAGCTGC, reverse: ACAGCCCCTGGGATGTCAGG; ERRFI1 forward: TGCTGATGTGACCTCTGGAA, reverse: CCTTGTGTTGCTGGTTCCTA; TBP forward: GCCAGCTTCGGAGAGTTCTG, reverse: GCACGAAGTGCAATGGTCTTT. qPCR was run on a Bio-Rad CFX Connect Real Time PCR machine (Bio-Rad, Hercules, CA).
Knockdown of OGT using siRNAs
Reverse siRNA knockdowns were performed using two OGT siRNAs: s16094 and s16095 (Thermo Fisher Scientific, Rockford, IL). 2.5 µL of 10 nM siRNA was mixed with 2 µl of Lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA) in 360 µl Opti-MEM reduced serum medium (Thermo Fisher Scientific) per well. The mixture was incubated for 20 min in room temperature and placed into 6-well culture plates. The cells were added to the plates in 2 ml of respective cell culture media, using 1 × 105 cells per well. Cells were collected for RNA extraction 72 h later.
Supplementary information
Acknowledgements
AB and SAM are funded through the Norwegian Research Council (Grant #239940). HMI and IGM were supported by Norwegian Cancer Society (Project nr. 4521627). HMI is additionally supported by the Academy of Finland Grants (Decision nr. 331324 and nr. 335902). The authors would like to thank Professor Suzanne Walker (Harvard Medical School), who provided us with the OSMI-1 compound. We are grateful to Dr Julia Gee and Professor Robert I Nicholson (Cardiff University) for TamR cells. The authors would also like to acknowledge the Genomics Core Facility at the Norwegian University of Science and Technology in Trondheim, Norway (NTNU), where the microarray analysis was performed. We would especially like to thank Sten Even Erlandsen and Arnar Flatberg for their assistance.
Author contributions
Experimental execution (A.B.), data analysis and interpretation (A.B., L.P., I.G.M., S.A.M., H.M.I.), design of the project, supervision and preparation of the manuscript (H.M.I.). Reading and editing of the manuscript (A.B., L.P., I.G.M., S.A.M., H.M.I.). Response to reviewer comments (A.B., K.S., I.G.M., H.M.I.).
Data availability
The data has been deposited to GEO with the accession number GSE148186.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-020-74083-z.
References
- 1.Di Cosimo, S. & Baselga, J. Management of breast cancer with targeted agents: Importance of heterogeneity. [corrected]. Nat. Rev. Clin. Oncol.7, 139–147. 10.1038/nrclinonc.2009.234 (2010). [DOI] [PubMed]
- 2.Zhang MH, Man HT, Zhao XD, Dong N, Ma SL. Estrogen receptor-positive breast cancer molecular signatures and therapeutic potentials (Review) Biomed. Rep. 2014;2:41–52. doi: 10.3892/br.2013.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Massarweh S, et al. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Can. Res. 2008;68:826–833. doi: 10.1158/0008-5472.CAN-07-2707. [DOI] [PubMed] [Google Scholar]
- 4.Shou J, et al. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J. Natl. Cancer Inst. 2004;96:926–935. doi: 10.1093/jnci/djh166. [DOI] [PubMed] [Google Scholar]
- 5.Knowlden JM, et al. Elevated levels of epidermal growth factor receptor/c-erbB2 heterodimers mediate an autocrine growth regulatory pathway in tamoxifen-resistant MCF-7 cells. Endocrinology. 2003;144:1032–1044. doi: 10.1210/en.2002-220620. [DOI] [PubMed] [Google Scholar]
- 6.El Sayed R, et al. Endocrine and targeted therapy for hormone-receptor-positive, HER2-negative advanced breast cancer: Insights to sequencing treatment and overcoming resistance based on clinical trials. Front. Oncol. 2019;9:510. doi: 10.3389/fonc.2019.00510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Masoud V, Pages G. Targeted therapies in breast cancer: New challenges to fight against resistance. World J. Clin. Oncol. 2017;8:120–134. doi: 10.5306/wjco.v8.i2.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krzeslak A, Forma E, Bernaciak M, Romanowicz H, Brys M. Gene expression of O-GlcNAc cycling enzymes in human breast cancers. Clin. Exp. Med. 2012;12:61–65. doi: 10.1007/s10238-011-0138-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Champattanachai V, et al. Proteomic analysis and abrogated expression of O-GlcNAcylated proteins associated with primary breast cancer. Proteomics. 2013;13:2088–2099. doi: 10.1002/pmic.201200126. [DOI] [PubMed] [Google Scholar]
- 10.Trinca GM, et al. O-GlcNAc-dependent regulation of progesterone receptor function in breast cancer. Horm. Cancer. 2018;9:12–21. doi: 10.1007/s12672-017-0310-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh JP, et al. O-GlcNAcase targets pyruvate kinase M2 to regulate tumor growth. Oncogene. 2020;39:560–573. doi: 10.1038/s41388-019-0975-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caldwell SA, et al. Nutrient sensor O-GlcNAc transferase regulates breast cancer tumorigenesis through targeting of the oncogenic transcription factor FoxM1. Oncogene. 2010;29:2831–2842. doi: 10.1038/onc.2010.41. [DOI] [PubMed] [Google Scholar]
- 13.Ferrer CM, et al. O-GlcNAcylation regulates cancer metabolism and survival stress signaling via regulation of the HIF-1 pathway. Mol. Cell. 2014;54:820–831. doi: 10.1016/j.molcel.2014.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Akella NM, et al. O-GlcNAc transferase regulates cancer stem-like potential of breast cancer cells. Mol. Cancer Res. 2020 doi: 10.1158/1541-7786.MCR-19-0732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lagerlof O, et al. The nutrient sensor OGT in PVN neurons regulates feeding. Science. 2016;351:1293–1296. doi: 10.1126/science.aad5494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li MD, et al. Adipocyte OGT governs diet-induced hyperphagia and obesity. Nat. Commun. 2018;9:5103. doi: 10.1038/s41467-018-07461-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shi H, et al. Skeletal muscle O-GlcNAc transferase is important for muscle energy homeostasis and whole-body insulin sensitivity. Mol. Metab. 2018;11:160–177. doi: 10.1016/j.molmet.2018.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruan HB, et al. Calcium-dependent O-GlcNAc signaling drives liver autophagy in adaptation to starvation. Genes Dev. 2017;31:1655–1665. doi: 10.1101/gad.305441.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hart GW. Nutrient regulation of signaling and transcription. J. Biol. Chem. 2019;294:2211–2231. doi: 10.1074/jbc.AW119.003226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang X, Qian K. Protein O-GlcNAcylation: Emerging mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2017;18:452–465. doi: 10.1038/nrm.2017.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Banerjee PS, Lagerlof O, Hart GW. Roles of O-GlcNAc in chronic diseases of aging. Mol. Aspects Med. 2016;51:1–15. doi: 10.1016/j.mam.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 22.Hart GW, Housley MP, Slawson C. Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature. 2007;446:1017–1022. doi: 10.1038/nature05815. [DOI] [PubMed] [Google Scholar]
- 23.Konrad RJ, et al. Alloxan is an inhibitor of the enzyme O-linked N-acetylglucosamine transferase. Biochem. Biophys. Res. Commun. 2002;293:207–212. doi: 10.1016/S0006-291X(02)00200-0. [DOI] [PubMed] [Google Scholar]
- 24.Pantaleon M, Tan HY, Kafer GR, Kaye PL. Toxic effects of hyperglycemia are mediated by the hexosamine signaling pathway and o-linked glycosylation in early mouse embryos. Biol. Reprod. 2010;82:751–758. doi: 10.1095/biolreprod.109.076661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gloster TM, et al. Hijacking a biosynthetic pathway yields a glycosyltransferase inhibitor within cells. Nat. Chem. Biol. 2011;7:174–181. doi: 10.1038/nchembio.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ferrer CM, et al. O-GlcNAcylation regulates breast cancer metastasis via SIRT1 modulation of FOXM1 pathway. Oncogene. 2017;36:559–569. doi: 10.1038/onc.2016.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sodi VL, et al. Nutrient sensor O-GlcNAc transferase controls cancer lipid metabolism via SREBP-1 regulation. Oncogene. 2018;37:924–934. doi: 10.1038/onc.2017.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ortiz-Meoz RF, et al. A small molecule that inhibits OGT activity in cells. ACS Chem. Biol. 2015;10:1392–1397. doi: 10.1021/acschembio.5b00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barkovskaya A, et al. O-GlcNAc transferase inhibition differentially affects breast cancer subtypes. Sci. Rep. 2019;9:5670. doi: 10.1038/s41598-019-42153-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gross BJ, Kraybill BC, Walker S. Discovery of O-GlcNAc transferase inhibitors. J. Am. Chem. Soc. 2005;127:14588–14589. doi: 10.1021/ja0555217. [DOI] [PubMed] [Google Scholar]
- 31.Itkonen HM, et al. O-GlcNAc transferase integrates metabolic pathways to regulate the stability of c-MYC in human prostate cancer cells. Can. Res. 2013;73:5277–5287. doi: 10.1158/0008-5472.CAN-13-0549. [DOI] [PubMed] [Google Scholar]
- 32.Martin SES, et al. Structure-based evolution of low nanomolar O-GlcNAc transferase inhibitors. J. Am. Chem. Soc. 2018;140:13542–13545. doi: 10.1021/jacs.8b07328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Itkonen HM, et al. Inhibition of O-GlcNAc transferase activity reprograms prostate cancer cell metabolism. Oncotarget. 2016;7:12464–12476. doi: 10.18632/oncotarget.7039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen PH, et al. Glycosylation of KEAP1 links nutrient sensing to redox stress signaling. EMBO J. 2017;36:2233–2250. doi: 10.15252/embj.201696113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Itkonen HM, et al. Inhibition of O-GlcNAc transferase renders prostate cancer cells dependent on CDK9. Mol. Cancer Res. MCR. 2020 doi: 10.1158/1541-7786.MCR-20-0339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Capotosti F, et al. O-GlcNAc transferase catalyzes site-specific proteolysis of HCF-1. Cell. 2011;144:376–388. doi: 10.1016/j.cell.2010.12.030. [DOI] [PubMed] [Google Scholar]
- 37.Tomkova V, Sandoval-Acuna C, Torrealba N, Truksa J. Mitochondrial fragmentation, elevated mitochondrial superoxide and respiratory supercomplexes disassembly is connected with the tamoxifen-resistant phenotype of breast cancer cells. Free Radic. Biol. Med. 2019;143:510–521. doi: 10.1016/j.freeradbiomed.2019.09.004. [DOI] [PubMed] [Google Scholar]
- 38.Fiorillo M, Sotgia F, Sisci D, Cappello AR, Lisanti MP. Mitochondrial, "power" drives tamoxifen resistance: NQO1 and GCLC are new therapeutic targets in breast cancer. Oncotarget. 2017;8:20309–20327. doi: 10.18632/oncotarget.15852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Daurio NA, et al. AMPK activation and metabolic reprogramming by tamoxifen through estrogen receptor-independent mechanisms suggests new uses for this therapeutic modality in cancer treatment. Cancer Res. 2016;76:3295–3306. doi: 10.1158/0008-5472.CAN-15-2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Urbanucci A, et al. Androgen receptor deregulation drives bromodomain-mediated chromatin alterations in prostate cancer. Cell Rep. 2017;19:2045–2059. doi: 10.1016/j.celrep.2017.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khan A, Zhang X. dbSUPER: A database of super-enhancers in mouse and human genome. Nucleic Acids Res. 2016;44:D164–171. doi: 10.1093/nar/gkv1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Itkonen HM, et al. High OGT activity is essential for MYC-driven proliferation of prostate cancer cells. Theranostics. 2019;9:2183–2197. doi: 10.7150/thno.30834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ranuncolo SM, Ghosh S, Hanover JA, Hart GW, Lewis BA. Evidence of the involvement of O-GlcNAc-modified human RNA polymerase II CTD in transcription in vitro and in vivo. J. Biol. Chem. 2012;287:23549–23561. doi: 10.1074/jbc.M111.330910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deplus R, et al. TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J. 2013;32:645–655. doi: 10.1038/emboj.2012.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anastasi S, et al. Feedback inhibition by RALT controls signal output by the ErbB network. Oncogene. 2003;22:4221–4234. doi: 10.1038/sj.onc.1206516. [DOI] [PubMed] [Google Scholar]
- 46.Ferby I, et al. Mig6 is a negative regulator of EGF receptor-mediated skin morphogenesis and tumor formation. Nat. Med. 2006;12:568–573. doi: 10.1038/nm1401. [DOI] [PubMed] [Google Scholar]
- 47.Cairns, J. et al. Differential roles of ERRFI1 in EGFR and AKT pathway regulation affect cancer proliferation. EMBO Rep19, 10.15252/embr.201744767 (2018). [DOI] [PMC free article] [PubMed]
- 48.Meyers RM, et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 2017;49:1779–1784. doi: 10.1038/ng.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dempster JM, Rossen J, Kazachkova M, Pan J, Kugener G, Root DE, Tsherniak A. Extracting biological insights from the project achilles genome-scale CRISPR screens in cancer cell lines. BioRxiv. 2019 doi: 10.1101/720243. [DOI] [Google Scholar]
- 50.Rhodes DR, et al. ONCOMINE: A cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6:1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Waks AG, Winer EP. Breast cancer treatment: A review. JAMA. 2019;321:288–300. doi: 10.1001/jama.2018.19323. [DOI] [PubMed] [Google Scholar]
- 52.Kwei KA, Baker JB, Pelham RJ. Modulators of sensitivity and resistance to inhibition of PI3K identified in a pharmacogenomic screen of the NCI-60 human tumor cell line collection. PLoS ONE. 2012;7:e46518. doi: 10.1371/journal.pone.0046518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang B, et al. Bitterness in sugar: O-GlcNAcylation aggravates pre-B acute lymphocytic leukemia through glycolysis via the PI3K/Akt/c-Myc pathway. Am. J. Cancer Res. 2017;7:1337–1349. [PMC free article] [PubMed] [Google Scholar]
- 54.Sekine, H. et al. O-GlcNAcylation signal mediates proteasome inhibitor resistance in cancer cells by stabilizing NRF1. Mol. Cell Biol.38, 10.1128/MCB.00252-18 (2018). [DOI] [PMC free article] [PubMed]
- 55.Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kanehisa M, Sato Y, Furumichi M, Morishima K, Tanabe M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019;47:D590–D595. doi: 10.1093/nar/gky962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kanehisa M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019;28:1947–1951. doi: 10.1002/pro.3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chandrashekar DS, et al. UALCAN: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia. 2017;19:649–658. doi: 10.1016/j.neo.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nagy A, Lanczky A, Menyhart O, Gyorffy B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci. Rep. 2018;8:9227. doi: 10.1038/s41598-018-27521-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data has been deposited to GEO with the accession number GSE148186.