

Abstract
The interindividual variability and sexual dimorphisms in the development of nonalcoholic fatty liver disease (NAFLD) are still poorly understood. In the present study, male and female strains of Collaborative Cross (CC) mice were fed a high-fat and high-sucrose (HF/HS) diet or a control diet for 12 weeks to investigate interindividual and sex-specific variations in the development of NAFLD. The severity of liver steatosis varied between sexes and individual strains and was accompanied by an elevation of serum markers of insulin resistance, including increases in total cholesterol, low-density lipoproteins, high-density lipoproteins, phospholipids, and glucose. The development of NAFLD was associated with over-expression of critical fatty acid uptake and de novo lipogenesis genes Pparg, Mogat1, Cd36, Acaab1, Fabp2, and Gdf15 in male and female mice. The expression of Pparg, Mogat1, and Cd36 was positively correlated with liver triglycerides in male mice, and Mogat1 and Cd36 expression was positively correlated with liver triglycerides in female mice. Our results indicate the value of CC mice in combination with HF/HS-diet-induced alterations as an approach to study the susceptibility and interindividual variabilities in the pathogenesis of nonalcoholic fatty liver and early nonalcoholic steatohepatitis at the population level, uncovering of susceptible and resistant cohorts, and identifying sex-specific molecular determinants of disease susceptibility.
Keywords: Nonalcoholic fatty liver disease, collaborative cross mice, gene expression, Cd36
Introduction
Nonalcoholic fatty liver disease (NAFLD), a hepatic manifestation of the metabolic syndrome, represents several related chronic liver phenotypes ranging from nonalcoholic fatty liver (NAFL) to nonalcoholic steatohepatitis (NASH), fibrosis, and cirrhosis (1,2). Currently, NAFLD is the most prevalent form of chronic liver disease in the United States (2,3) and other countries (2,4), affecting approximately one-quarter of the world’s population (5). Accumulating evidence has demonstrated that among the epidemiological, demographic, and lifestyle risk factors for NAFLD, obesity is the most common feature that parallels not only with the occurrence of NAFLD (2,4,6) but also disease severity (7). Furthermore, with the continued increasing rates of obesity, NAFLD prevalence is predicted to increase worldwide (8). Even though obesity is strongly and progressively associated with NAFLD (6,7), there is insufficient knowledge as to how the disease develops because not all obese individuals manifest with NAFLD.
One of the important unknowns of the NAFLD epidemic is the interindividual variability in susceptibility to NAFLD (9), as well as substantial interpatient variations in the progressive development of the disease (10). Identification of those individuals who are at greatest risk of developing NAFLD and, perhaps more importantly, the early detection of NAFLD-prone patients are unmet needs in medical practice (11,12). In light of this, there is a need for an in-depth examination of the molecular basis and pathways that underlie NAFLD pathogenesis and progression to advanced forms of liver disease.
Studies of the molecular mechanisms of NAFLD, as well as identification of molecular and pathological drivers for stratification of the disease in humans, are desirable. However, clinical and epidemiological cohorts are widely heterogeneous, and great difficulties exist with early diagnosis and disease staging through non-invasive methods. In contrast, mouse models that mimic human NAFLD pathology and pathophysiology have proven to be useful tools to study molecular mechanisms of NAFLD development and progression (13,14). Among experimental models used to study NAFLD, diet-based mouse models and genetically modified animals are the most widely employed; however, until recently, no single experimental model has been shown to reflect the full spectrum of pathogenic pathways and manifestations associated with human NAFLD (13,14).
Recently, Asgharpour et al. have established a mouse model of NAFLD and NASH that recapitulates key pathophysiologic, pathohistologic, metabolic, immunologic, and transcriptomic features of the human NAFLD and NASH (15). Feeding an obesogenic high-fat diet to stable male isogenic mice that were obtained by crossing C57BL/6J and 129S1/SvImJ mice provided valuable information on the mechanisms of NAFLD pathogenesis. Unfortunately, the study does not interrogate the impact of genetic differences within a genetically-diverse population for the development of NAFLD due to insufficient genetic diversity in this model. To address this limitation, several studies have used a population-based approach to investigate the molecular pathogenesis of NAFLD induced by obesogenic high-fat and high-sucrose (HF/HS) diets in the Hybrid Mouse Diversity Panel (HMDP; 16,17) or BXD recombinant strains (18). These studies have identified fundamental molecular phenotypes and metabolic and transcriptomic pathways associated with the development and progression of NAFLD; however, the role of sex-specific alterations that contribute to the pathogenesis of human NAFLD (19) remained unanswered because only male mice were used (16–18).
Based on these considerations, we sought to extend the understanding of the underlying mechanisms and molecular pathways and determinants associated with susceptibility to NAFLD in a genetically diverse mouse population. We used male and female Collaborative Cross (CC) mice, a state-of-the-art mouse population model (20) that captures a greater genetic variability at the population level as compared to other genetic mouse panels (21–23) and is representative of the genetic diversity of the human population. In this study, we fed CC mice an obesogenic HF/HS diet and investigated (i) the inter-strain variations in physiological, histological, and molecular alterations in the susceptibility to NAFL and early NASH, and (ii) molecular determinants of disease susceptibility among strains and sexes.
Materials and Methods
Animals, diets, and experimental design
Twenty five CC mouse strains: CC001/Unc (CC001), CC002/Unc (CC002), CC003/Unc (CC003), CC004/TauUnc (CC004), CC005/TauUnc (CC005), CC006/TauUnc (CC006), CC009/Unc (CC009), CC010/GeniUnc (CC010; male mice only), CC011/Unc (CC011), CC012/GeniUnc (CC012), CC013/GeniUnc (CC013), CC019/TauUnc (CC019), CC025/GeniUnc (CC025), CC026/GeniUnc (CC026), CC032/GeniUnc (CC032), CC037/TauUnc (CC037), CC040/TauUnc (CC040), CC041/TauUnc (CC041), CC042/GeniUnc (CC042), CC043/GeniUnc, (CC043) CC051/TauUnc (CC051), CC060/Unc (CC060), CC061/GeniUnc (CC061), CC068/TauUnc (CC068), and CC080/Unc (CC080) (6 weeks of age) were obtained from Jackson Laboratory (Bar Harbor, ME, USA) and acclimated for two weeks. The mice were housed in sterilized cages in a temperature-controlled room (24°C) with a 12 h light/dark cycle and given ad libitum access to water and NIH-41 irradiated pelleted diet. At 8 weeks of age, mice from each strain were allocated randomly into either an experimental or control group (n = 3 per group per sex). Our experimental design aimed to maximize the statistical power of the study by maximizing the number of strains tested. Specifically, the power to detect variation in a diverse population is greater when testing more unique genomes rather than replicating the individual genomes (24). Mice in the experimental group were maintained on a HF/HS diet (Teklad Custom Diet - TD.88137, Envigo, Madison, WI, USA) for 12 weeks. This diet contains 42% Kcal from milkfat high in palmitic acid, 0.2% of total cholesterol, and is high in sucrose (>30%). It has been demonstrated that this diet induces obesity, metabolic syndrome, and NAFL in mice within 9 weeks of feeding (25,26). Mice in the control group received an ingredient-matched low-fat and reduced sucrose diet (Teklad Custom Diet – TD.08485, Envigo). Diets were stored at 4° C before use and given ad libitum, with a replacement once a week. Body weights of the mice were recorded weekly. Mice were euthanized with carbon dioxide 12 weeks after diet initiation. The livers were excised, and a slice of the median lobe was fixed in neutral buffered formalin for 48 h for histopathological examination. The remaining liver was snap-frozen immediately in liquid nitrogen and stored at −80°C for subsequent analyses. All experimental procedures were reviewed and approved by the National Center for Toxicological Research Animal Care and Use Committee and conducted in full accordance with the Public Health Service Policy on Humane Care and Use of Laboratory Animals.
Liver tissue processing and histological analysis
A slice of the median lobe of the liver that had been fixed in 10% neutral buffered formalin for 48 h was trimmed, processed, and embedded in infiltrating media (Surgipath Formula R®, Leica Biosystems, Richmond, IL, USA), sectioned at approximately 5 microns, mounted on a glass slide, and stained with hematoxylin and eosin. The hematoxylin- and eosin-stained sections were evaluated for steatosis, inflammation, necrosis, and hepatocellular karyocytomegaly. Additionally, liver sections were examined for lipid droplets by using an osmium tetroxide staining method; images were obtained with an Aperio Scanscope System (Leica Biosystems, Vista, CA, USA). The Positive Pixel Count Algorithm was used to evaluate the osmium tetroxide-stained area. Settings of the algorithm were adjusted to recognize osmium tetroxide-stained lipid droplets as strong positive (dark brown) and negative (dark blue) pixels. The surrounding area was recognized as weak positive (orange) and positive (light brown) pixels. The osmium tetroxide stained area (%) was calculated using the following formula:
where the total number of pixels was the sum of the strong positive, positive, weak positive, and negative pixels.
Immunohistochemistry
The level of α-smooth muscle actin (α-SMA) in formalin-fixed, paraffin-embedded liver sections was determined by immunohistochemical staining as described previously (27).
Blood biochemistry
Blood samples were collected in serum separator tubes by cardiac puncture at necropsy between 8.00 and 11.00 am. The serum tubes were kept at room temperature to allow clotting, then centrifuged at 1000 x g for 10 minutes, and the serum was removed and aliquoted in separate tubes. Clinical biochemistry analyses were conducted with a Vet Axcel® Clinical Chemistry System (Alfa Wassermann, West Caldwell, NJ, USA). Alfa Wassermann reagents were used to measure triglycerides, total cholesterol, high-density lipoprotein (HDL) cholesterol, phospholipids, and glucose concentrations and alanine aminotransferase (ALT) activity. Catachem reagents (Bridgeport, CT, USA) were used for nonesterified fatty acids (NEFA) analysis and Diazyme reagents (Poway, CA, USA) were used for low-density lipoprotein (LDL) cholesterol determination.
Insulin concentrations were quantified with a Mouse Ultrasensitive Insulin ELISA kit (ALPCO, Salem, NH, USA) on an Epoch Universal Microplate Reader (Bio-Tek, Winooski, VT, USA).
Determination of liver triglyceride content
Approximately 20-30 mg of liver tissue was homogenized in 700 µl 10% NP-40 water solution, followed by three heating (90°C)-cooling (room temperature) cycles to solubilize all the triglycerides. The level of liver triglycerides was measured with Triglyceride Quantification Assay kits (Abcam, Cambridge, MA, USA).
Determination of liver total cholesterol level
An HDL and LDL/VLDL Cholesterol Assay kit (Abcam) was used to determine the level of total cholesterol in the livers.
Quantitative reverse transcription polymerase chain reaction
Total RNA was extracted from liver tissue samples using miRNeasy Mini kits (Qiagen, Valencia, CA, USA). Total RNA (2 μg) was reverse transcribed using random primers and High Capacity cDNA Reverse Transcription kits (Life Technologies, Grand Island, NY, USA). Gene expression was determined by quantitative reverse transcription polymerase chain reaction PCR (qRT-PCR) using the TaqMan gene expression assays that are listed in Supplementary Table 1. The glyceraldehyde-3-phosphate dehydrogenase (Gapdh) gene was used as an endogenous control. The relative amount of each mRNA transcript was determined with the 2−ΔΔCt method (28). A p-value cut-off ≤ 0.05 and a fold change threshold > 1.5 were used to generate a list of differentially expressed genes (DEGs) in the livers of mice fed the HF/HS diet.
Statistical analyses
To analyze the effect of the HF/HS diet at the population level, the average values from control diet-fed mice and HF/HS diet-fed mice were evaluated for a normal distribution and analyzed using an unpaired 2-tailed Student’s t-test or Mann-Whitney Rank Sum test, as appropriate. To determine significant differences between control diet-fed mice and HF/HS diet-fed mice within individual strain (n = 3 per group per sex), an unpaired two-tailed Student’s t-test was applied. Spearman’s rank-order correlation analysis or Pearson correlation coefficient was used to determine trends. R software (version 3.4.3) was used to generate paired correlation matrices.
Results
The effect of HF/HS diet on body and liver weight
Feeding a HF/HS diet for 12 weeks resulted in a significant gain in body weight of 12% in male CC mouse strains and 16% in female CC mouse strains as compared to control diet-fed mice (Figure 1A). Importantly, food intake did not correlate with elevated body weight (Supplementary Figure 1). The body weight gain was sex- and strain- specific: three strains (CC001, CC009, and CC041) exhibited a significant increase in body weight in both sexes; three strains (CC042, CC043, and CC051) exhibited a significant increase in male mice only; and four strains (CC005, CC013, CC032, and CC060) exhibited a significant increase in female mice only (Figures 1C). Likewise, the extent of body weight gain varied across strains with the largest increase being found in male CC041 mice and in female CC032 mice. In contrast, there was no effect of the HF/HS diet on body weight gain in some strains, for example, male and female CC012 and CC068 mice (Figure 1C).
Figure 1. Body weight and liver-to-body weight ratio in Collaborative Cross (CC) mice fed a high-fat and high-sucrose diet or a control diet.
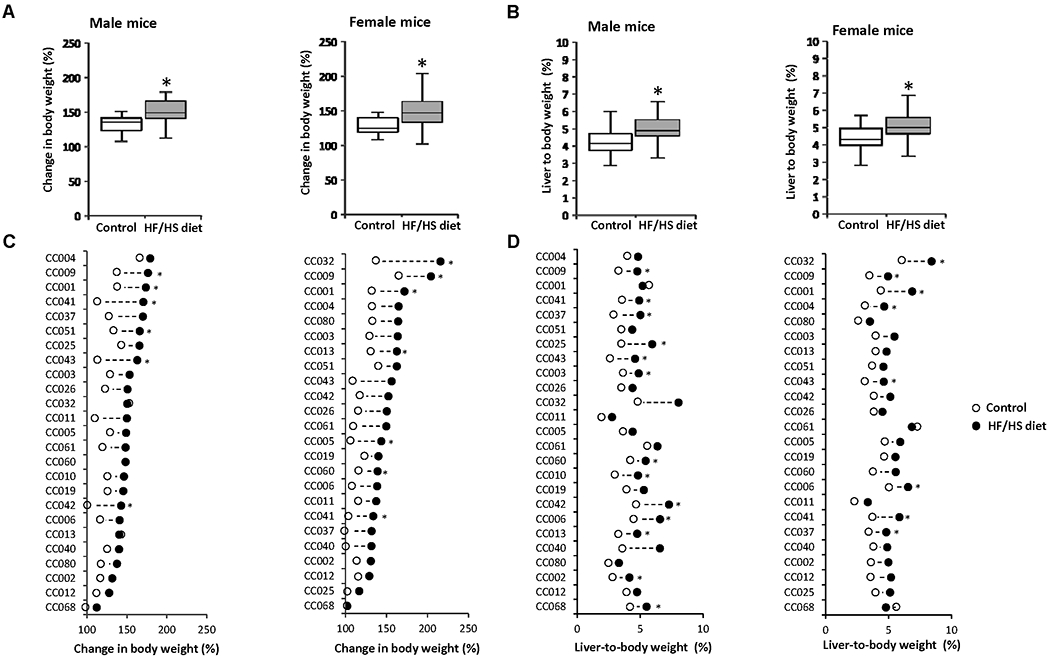
(A) Population averages for body weight and (B) liver-to-body weight ratios in male and female mice fed a control diet or HF/HS diet. (C) Average change in body weight and (D) liver-to-body weight ratios in HF/HS diet-fed mice (black dot) as compared to control diet-fed mice (white dot) in the individual mouse strains. Asterisks * denote significant difference (p < 0.05).
Feeding the HF/HS diet also resulted in a significant increase of liver-to-body weight ratio (23% in male CC mice and 17% in female CC mice; Figure 1B). The changes in liver-to-body weight ratio were also sex- and strain-specific (Figure 1D) and were not necessarily associated with an increase in body weight. For instance, nine male strains (CC002, CC003, CC006, CC010, CC013, CC025, CC037, CC060, and CC068) and four female strains (CC004, CC006, CC037, and CC043) had a significant increase in the liver-to-body weight ratio without significant changes in body weight.
The effect of HF/HS diet on lipid accumulation in the liver
Feeding a HF/HS diet for 12 weeks resulted in the development of a NAFL-like phenotype in the livers in all male and female mice. Histological examination of liver sections revealed an increased incidence and/or severity of micro- and macro-vesicular steatosis across HF/HS diet-fed mice (Supplementary Table 2). This was further evidenced by increased osmium tetroxide staining (Figures 2A and 2B) that visualizes lipid droplets containing saturated and unsaturated lipids (29). The development of a NAFL-like phenotype was confirmed by biochemical evaluation, showing increased levels of triglycerides and total cholesterol in the livers of male and female mice and elevated ALT activity in the serum of male mice (Figures 2A and 2B). In addition to hepatic lipid accumulation, hepatocyte ballooning degeneration (Supplementary Figure 2A), with an incidence of 67-100%, was found in eight male strains and six female strains (Supplementary Table 2). Inflammatory cellular infiltration was also observed, but it was of minimal severity and did not appear to be diet related (Supplementary Table 2).
Figure 2. Effect of a high-fat and high-sucrose diet on metabolic alterations and extent of steatosis across the Collaborative Cross mouse strains.
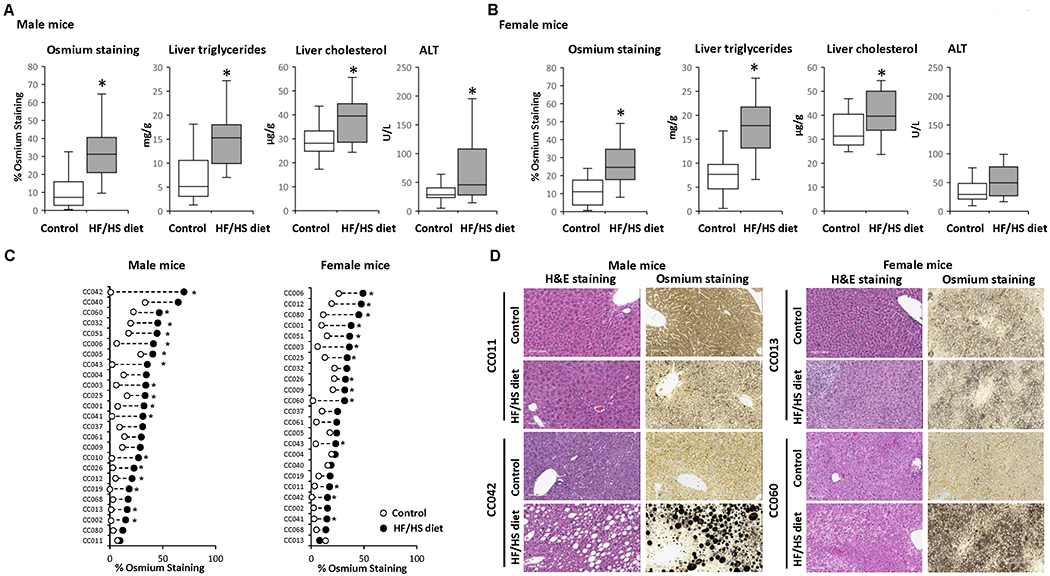
(A, B) Population averages for the percentage of osmium tetroxide staining in liver sections, liver triglycerides, liver cholesterol, and plasma ALT activity in male (A) and female (B) mice fed a control diet or HF/HS diet. (C) Strain-specific changes in osmium staining in HF/HS diet-fed mice (black dot) as compared to control diet-fed mice (white dot). Asterisks * denote significant difference (p < 0.05). (D) Representative hematoxylin and eosin and osmium staining of liver sections from CC011 and CC042 male mice and CC013 and CC060 female mice fed control or HF/HS diet.
The extent of liver steatosis, as indicated by osmium tetroxide staining, in HF/HS diet-fed mice varied between sexes and individual strains, with 68% of male strains and 58% of female strains showing significant changes in the extent of steatosis. The magnitude of inter-strain changes was 66% between control diet-fed and HF/HS diet-fed male mice, ranging from 3% to 69%, and 27% between control diet-fed and HF/HS diet-fed female mice, ranging from 4% to 31% (Figure 2C). This is further illustrated by representative hematoxylin and eosin and osmium staining images of liver sections that show differences in the severity of steatosis in male CC011 and CC042 mice and female CC013 and CC060 mice (Figure 2D).
In general, the level of steatosis coincided with increased body weight, although some strains (e.g., male CC002, CC003, CC006, CC010, CC013, CC025, and CC060) and (e.g., female CC006 and CC043) that exhibited liver fat accumulation (Figure 2C) and increased liver-to-body weight ratios (Figure 1D) had little or no changes in body weight (Figures 1C and 2C).
To investigate the association between hepatic molecular traits related to hepatic NAFL phenotype, we conducted Spearman rank correlation analysis. This analysis showed a strong positive correlation between the osmium tetroxide staining and liver triglycerides in male and female mice, r = 0.75 and r = 0.72, respectively, and a moderate correlation of the osmium tetroxide staining and liver total cholesterol and plasma ALT activity, r = 0.45 and r = 0.51, respectively, in male mice.
Serum biochemical indicators of metabolic alterations and liver injury in HF/HS diet-fed mice
The development of obesity and steatosis in HF/HS diet-fed mice was accompanied by a significant increase in the serum levels of total cholesterol, LDL, HDL, phospholipids, and glucose in male and female mice, whereas the levels of triglycerides, NEFA, and insulin were not statistically significantly different (Figures 3A and 3C). Spearman rank correlation analysis showed positive correlations (r > 0.5) between the extent of steatosis, as indicated by osmium tetroxide staining, and serum levels of total cholesterol, LDL, HDL, and ALT in male mice (Figure 3B) and between steatosis and serum levels of total cholesterol and phospholipids female mice fed the HF/HS diet (Figure 3D). Additionally, a positive correlation between the serum levels of total cholesterol, LDL, HDL, and phospholipids was found in mice of both sexes (Figures 3B and 3D).
Figure 3. Population averages and correlation analysis of serum biochemical indicators of metabolic alterations and extent of steatosis across the Collaborative Cross mouse strains.
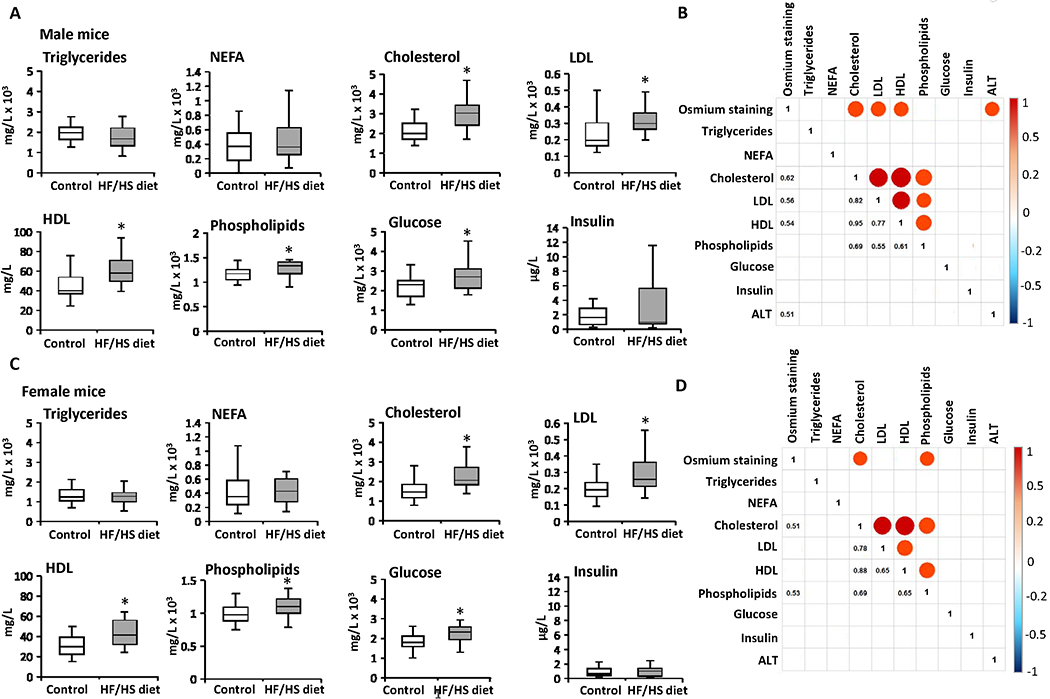
(A, C) Population averages for the serum triglycerides, non-esterified fatty acids (NEFA), total cholesterol, low-density lipoprotein (LDL), high-density lipoprotein (HDL), phospholipids, glucose, and insulin in male (A) and female (C) mice fed control diet or HF/HS diet. Asterisks * denote significant difference (p < 0.05). Spearman’s rank-order correlation matrix of the phenotypes in male (B) and female (D) mice. Each circle represents a significant correlation (p < 0.05) that was positive (red) or negative (blue). Color bar shows Spearman correlation.
Dysregulation of lipid metabolism-associated genes in the livers of mice fed HF/HS diet
To determine the underlying mechanisms of hepatic fat accumulation induced by the HF/HS diet, we investigated the expression of 22 key lipid metabolism-associated genes in the livers of HF/HS diet fed-mice and control diet-fed mice. These genes are involved in four major molecular pathways regulating hepatic lipid homeostasis (Figure 4A), including circulating lipid uptake (Fabp2, Fabp4, Fabp5, Cd36, Slc25a2, Slc27a2, Ppara, Pparg, and caveolin), de novo lipogenesis (Mogat1, Fasn, Acadm, and Srebf1), fatty acid oxidation (Acaa1a, Acaa1b, Cpt1a, Gdf15, Mlxipl, and Sirt3), and lipid export (Abcb4, ApoB100, and Mttp). Feeding the HF/HS diet resulted in marked sex- and strain-specific changes in gene expression in the livers of male and female mice, with all four hepatic lipid metabolic pathways being altered (Figures 4B and 4C).
Figure 4. Expression of lipid metabolism genes in the livers of Collaborative Cross mice fed high-fat and high-sucrose or control diet.
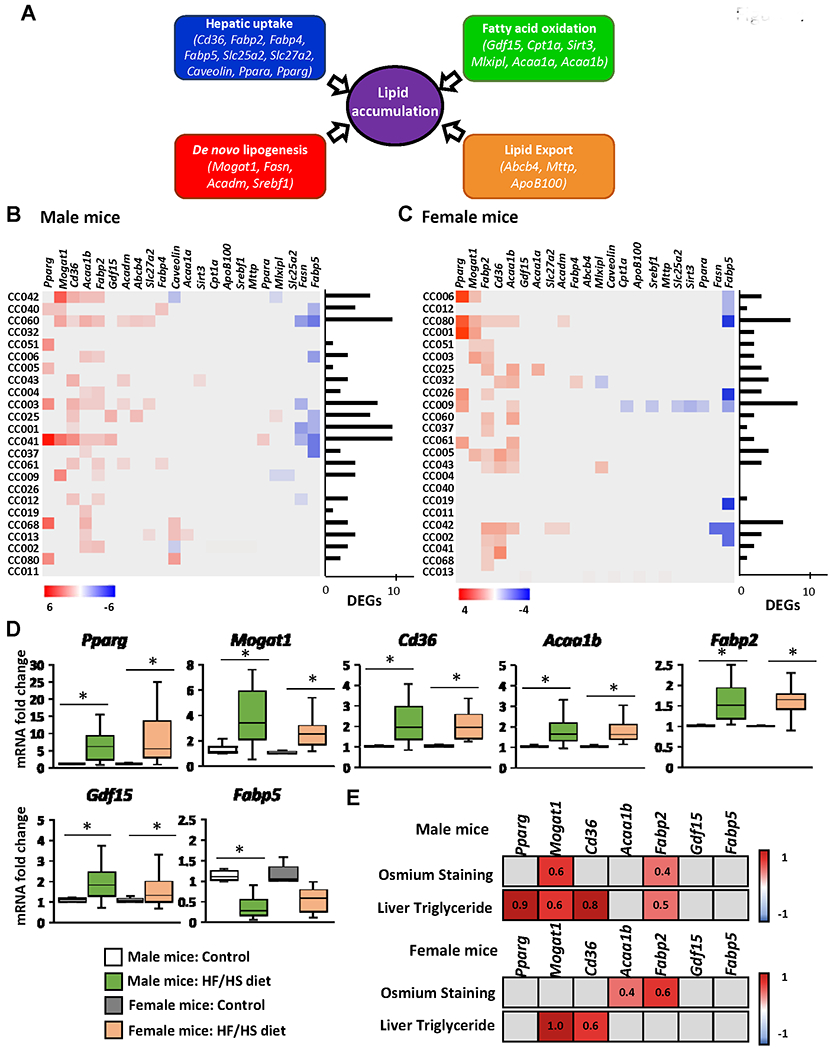
(A) Schematic of hepatic lipid metabolism pathways and genes selected for qRT-PCR gene expression analysis. Heat map fold change in the expression of lipid accumulation genes in individual mouse strains of male (B) and female (C) mice. Gene expression was measured by qRT-PCR and the relative amount of each mRNA transcript was determined using the 2−ΔΔCt method. Significant differentially expressed genes in the livers of mice fed a HF/HS diet compared to a control diet were identified by Student’s t-test (p < 0.05 considered significant). Red color denotes > 1.5-fold increase in gene expression in the livers of HF/HS diet-fed mice; blue color denotes > 0.67 decrease in gene expression in the livers of HF/HS diet-fed mice. Grey boxes denote non-significant change in gene expression. (D) Averages of genes significantly altered (p < 0.05) at the population level in the livers of male and female mice fed a HF/HS diet compared to mice fed control diet. (E) Heat map of Pearson correlation coefficients (r) of genes with population wide significant changes in gene expression of male and female mice fed a HF/HS diet in relation to osmium staining and liver triglyceride levels. p<0.05 was considered a significant correlation. Red color denotes positive correlation, blue color denotes negative correlation, grey boxes statistically not significant. Pearson r values are indicated.
The most distinct gene expression changes based on the numbers of differentially expressed genes were found in male CC042, CC060, CC003, CC001, and CC041 mice and in female CC080, CC009, and CC042 mice (Figures 4B and 4C). The most notable changes in the gene expression at the population level based on an average fold change were an over-expression of the Acaab1, Fabp2, Gdf15, and especially Pparg, Mogat1, and Cd36 genes, which were more than two-times greater in HF/HS diet-fed male and female mice than in control mice, and a down-regulation of Fabp5 by more than 50% in male mice (Figure 4D). Furthermore, a positive correlation between the expression of Pparg, Mogat1, Cd36, and Fabp2 and the levels of liver triglycerides was found in male mice, and between the expression of Mogat1 and Cd36 expression and the levels of liver triglycerides in female mice (Figure 4E).
Transcriptomic analysis of fibrosis marker genes in the livers of mice fed HF/HS diet
A hallmark of the progression of NAFL to advanced disease stages is the evidence of liver fibrosis. Therefore, expression of markers of fibrosis was analyzed in control diet-fed and HF/HS diet-fed male and female mice. The expression of the well-established liver fibrosis marker genes Col1a1, Timp, and Tnf, and the Krt8/Krt18 ratio (18,30,31) was increased in the livers of both male and female HF/HS diet-fed mice (Figures 5A and 5C); however, as observed with lipid metabolism genes, their expression changes varied between sexes and individual strains (Figures 5B and 5D). The largest changes of all four fibrosis marker indices were found in the livers of male CC042 and CC041 mice. These male mouse strains not only showed severe steatosis but also exhibited hepatocyte ballooning and positive α-SMA immunohistochemical staining (Figure 2, Supplementary Table 2, Supplementary Figure 2). In contrast, no hepatocyte ballooning or positive α-SMA staining was found in strains with a non-altered expression of fibrosis marker genes.
Figure 5. Expression of fibrosis marker genes in the livers of Collaborative Cross mice fed a high-fat and high-sucrose or control diet.
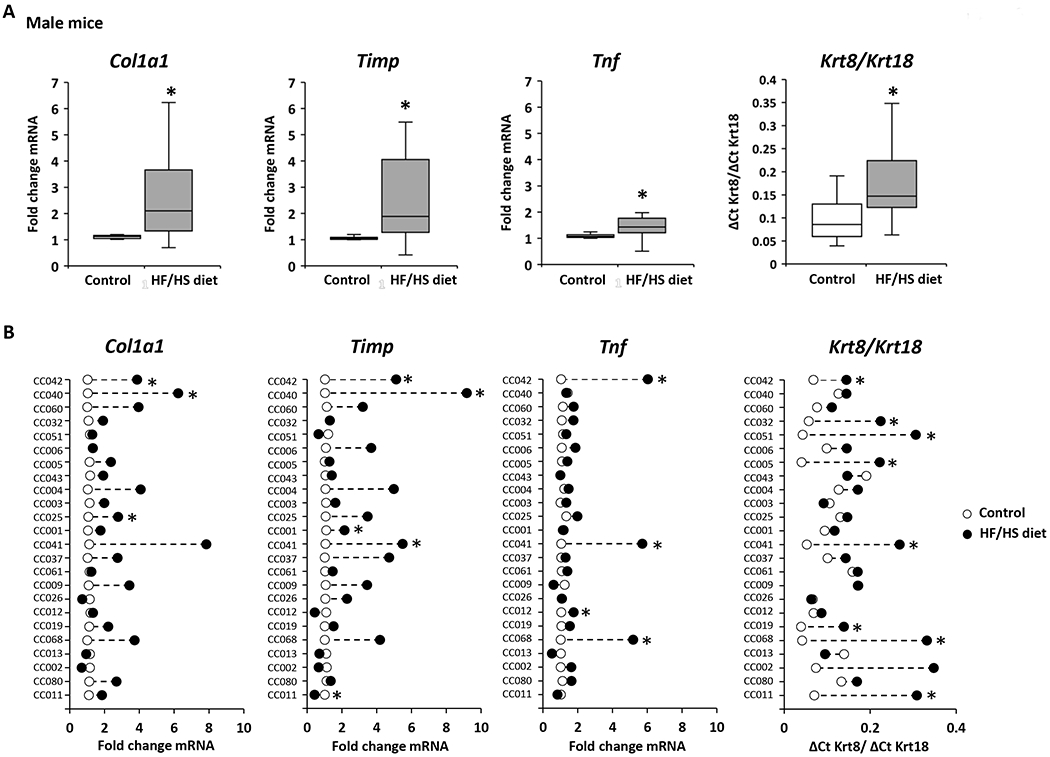
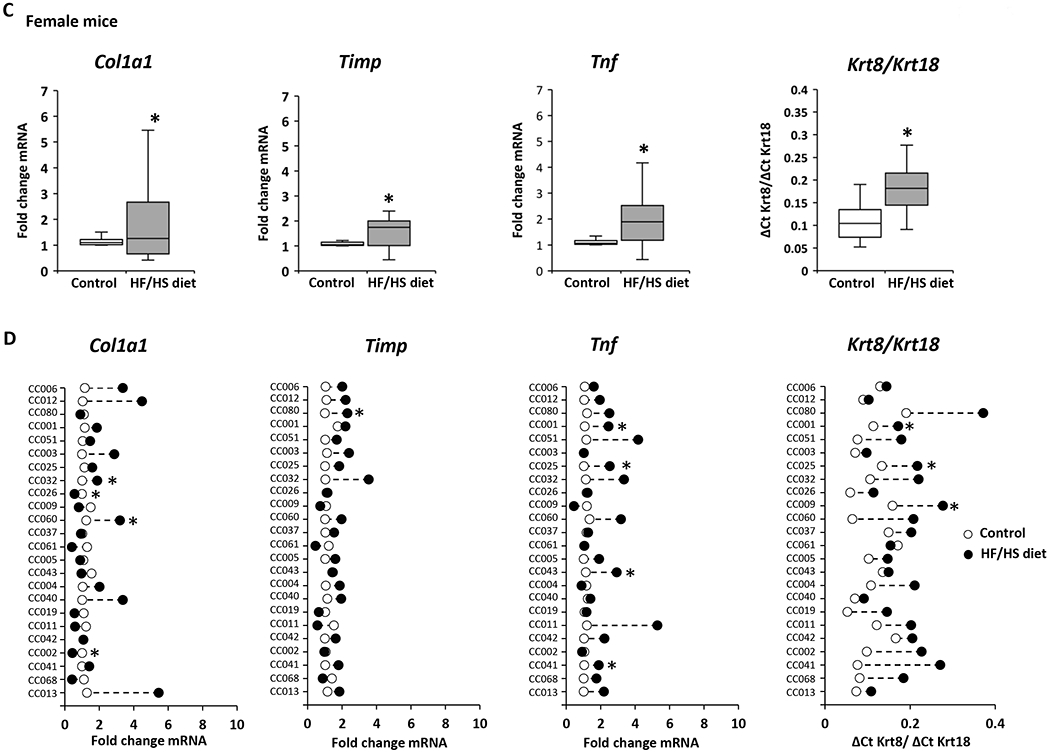
(A, C) Population averages of fibrosis marker-gene expression with significant changes (p < 0.05) in the livers of HF/HS diet-fed male (A) and female (C) compared to control diet-fed mice. (B, D) Strain-specific changes in expression of fibrosis marker-genes in the livers of HF/HS diet-fed mice (black dot) as compared to control diet-fed mice (white dot). Asterisks * denote significant difference (p < 0.05)
Discussion
The alarming rise in the incidence of NAFLD worldwide indicates an urgent need to understand better the molecular mechanisms and drivers of the disease development, and especially, molecular determinants of interindividual susceptibility to NAFLD. This understanding is critical for the discovery of disease-specific indicators that can be used in mechanism-based screening approaches in the clinic for diagnostics and disease monitoring. Until recently, most experimental studies investigating the molecular pathogenesis of NAFLD have been conducted using either outbred animals or single inbred animal strains. Despite the indisputable value of these models in discovering fundamental disease-specific pathophysiological events, investigating differences in the susceptibility to the disease and clinical variations in disease development and/or progression have not been possible. This is predominantly due to the lack of genetic diversity or the lack of reproducible genetic diversity in these models. Considering this, by using CC mice, a multiparental mouse population model with diverse and reproducible genetic backgrounds (20), we investigated interindividual- and sex-specific variations in the development of NAFLD in mice induced by a HF/HS diet, a model that replicates the key features of NAFLD found in human patients (15,32).
The results of the present study show that feeding a HF/HS diet resulted in negative health effects across all mouse strains; however, the severity of these effects was sex- and strain-specific. In particular, although the HF/HS diet increased the body weight at the population level in both sexes of mice, the extent of the body weight increases substantially differed among strains and was not associated with food consumption either between diet groups, within the diet group or between sexes. This finding is in a good agreement with previous studies reporting that food intake is not the only driving factor that impacts body weight and emphasizes the importance of other factors, e.g., microbiome, genetics, activity or energy expenditure, in obesity (33).
Histological examination of liver sections and direct measurement of hepatic triglycerides revealed the development of NAFL in both sexes of mice fed the HF/HS diet, with a greater magnitude of hepatic steatosis found in male mice. This is concordant with existing epidemiological evidence of the prevalence of NAFLD in men (19). Similar to the effect of the HF/HS diet on body weight, the extent of lipid accumulation in the livers of HF/HS diet-fed mice was strain-specific and varied greatly across strains, which is consistent with the previous observation of large variations in hepatic triglyceride accumulation in HMDP mouse strains fed a HF/HS diet for 8 weeks (16).
In general, the severity of steatosis in HF/HS diet-fed mice was associated with an increase in body weight; however, in some strains exhibiting an extensive hepatic fat accumulation, little or no changes in body weight were found. This finding corresponds to growing evidence of high prevalence of NAFLD in non-obese individuals (34).
The development of steatosis in male and female CC mice fed the HF/HS diet at the population level was accompanied by an elevation of serum NAFLD diagnostic markers. Among these, marked dyslipidemia, characterized by elevated serum total cholesterol, LDL, HDL, and phospholipids, and increased glucose levels were the most prominent in both sexes of mice. Importantly, the serum levels of LDL, HDL, and total cholesterol positively correlated with the severity of steatosis. The elevation of these indices has been reported in human and mouse NAFLD (12,15). Furthermore, an increased level of total serum cholesterol has been regarded as an independent predictor for NAFLD (35).
The regulation of intrahepatic lipid levels is controlled and maintained by the balance of lipid acquisition (e.g., uptake of circulating fatty acids and de novo lipogenesis pathways) and lipid disposal (e.g., oxidation of fatty acids and lipid export) (36). Deregulation of any of these pathways results in hepatic lipid accumulation. In the present study, we investigated the expression of key genes of these pathways. We found that the development of steatosis induced by the HF/HS diet was associated with altered gene expression in all four liver lipid metabolism pathways, with a marginally higher number of differentially expressed genes being found in the livers of male mice than in female mice, 86 vs 62 genes. The most notable gene expression changes were over-expression of genes involved in the uptake of circulating fatty acids and de novo lipogenesis pathways, especially, Pparg, Cd36, Fabp2, and Mogat1 in the livers of male and female mice fed the HF/HS diet; however, like the strain-specific diet-induced liver injury, the expression of these genes was strain-specific, with the most prominent alterations being found in strains characterized by a greater extent of NAFLD-like phenotype. This finding suggests a potential role of Pparg, Cd36, Fabp2, and Mogat1 genes as determinants of interindividual variability in NAFL susceptibility and that they can be used for the assessment of the severity of hepatosteatosis.
Pparg encodes the peroxisome-proliferator-activated receptor that is involved in the transcriptional regulation of lipid metabolism (37) and has been defined as a steatogenic factor (38). Fabp2 and Cd36 encode membrane-associated fatty acid-binding proteins (fatty acid transporters) that facilitate and regulate cellular fatty acid uptake (39), whereas Mogat1 is involved in an alternative pathway for triglyceride synthesis contributing to the development of hepatic steatosis, especially obesity-related steatosis (40,41). Consistent with our results, upregulation of Pparg, Cd36, Fabp2, and Mogat1 has been reported in the livers of mice fed a high-fat diet for weeks (38, 42–45), and over-expression of these genes has been associated with the pathogenesis of human NAFLD (44–48). Furthermore, the results of the present study show a positive correlation between the increased expression of Pparg, Cd36, Fabp2, and Mogat1 in the livers of HF/HS diet-fed male mice and Mogat1 and Cd36 in the livers of HF/HS diet-fed female mice and hepatic triglyceride content. A similar finding of a high correlation between expression of Fabp2, and especially, Cd36, with hepatic triglyceride levels, has been reported in the genetically diverse HMDP mouse strains fed a HF/HS diet (16). These two independent observations that demonstrate strain-specific differences in susceptibility to steatosis and expression of Cd36 and Fabp2 are in good agreement with human clinical studies reporting higher serum levels of CD36 and FABP2 in patients with advanced NAFLD and cirrhosis (49,50) and highlight the potential significance of Fabp2 and Cd36 as indicators of NAFLD susceptibility.
Another important outcome of our study is an increased expression of fibrogenesis-related genes in the livers of mice fed the HF/HS diet, especially in male mice with prominent liver injury. Importantly, activation of fibrogenesis-related genes in these male mouse strains was accompanied by hepatocyte ballooning, a component of the NAFLD activity score and the definitive feature of NASH (51,52), and positive immunohistochemical staining of α-SMA, a major profibrogenic factor (53). This indicates that analysis of the expression of fibrosis marker genes can be used for monitoring the progression of NAFL to NASH. Similar results of early upregulation fibrosis marker genes, including Col1a1 and Tnf, and a high Krt8/Krt18 ratio were observed in a diet-induced mouse model of hepatic fibrosis (18) and in a Krt18−/− transgenic mouse model of NASH (31).
Until recently, NAFL was generally considered as a benign form of NAFLD with a favorable outcome; however, overlooking steatosis may substantially hamper ongoing efforts to uncover the underlying mechanisms of NAFLD pathogenesis and underpin diagnostic and therapeutic targets of the disease (54). Additionally, it has been reported that approximately 30-40% of individuals with simple steatosis will develop NASH (5,55), which rarely resolves without improvement of steatosis (56). In view of this, the identification of molecular determinants driving the development and progression of NAFL is of great importance.
There is increasing reports that the intestinal microbiome plays an important role in the pathogenesis of NAFLD. Animal models and human volunteer clinical studies have provided considerable information on the association between dysbiosis and NAFLD pathogenesis (57–60). However, less is known about the mechanisms involved in the bidirectional liver-gut microbiome axis and how dysbiosis in the gastrointestinal tract contributes to the incidence of NAFLD. Although the intestinal microbiome role in NAFLD was not the primary focus of this investigation, preliminary studies using the CC mice population model indicated that the mouse strains that showed molecular features of NALFD correlated with taxonomic compositional shifts in abundance of intestinal microbiota (data not shown). Further microbiome-host interaction studies are now being planned to link intestinal microbiota composition, metabolic functional endpoints, gut permeability intestinal barrier effects, intestinal mucosal immune and inflammatory responses using CC mice to add mechanistic evidence on the role of the intestinal microbiome in NAFLD.
In this report, we show the value of the genetically diverse CC mice in combination with HF/HS diet-induced alterations to study the molecular pathogenesis of NAFLD. This combination demonstrates the strength of our approach to study the susceptibility to NAFL and early NASH and the interindividual variabilities in the pathogenesis of NAFLD at the population level, uncovering of susceptible and resistant cohorts to NAFLD, and identifying sex-specific and individual molecular determinants of disease susceptibility and disease progression.
Supplementary Material
Acknowledgements
This study was funded by the National Center for Toxicological Research, Food and Drug Administration Protocol E7635.01. This work partly supported by appointment of R. Willett and B. Borowa-Mazgaj to the Postgraduate Research Program at the National Center for Toxicological Research administered by the Oak Ridge institute for Science and Education.
Conflict of interest Arun J. Sanyal is the President of Sanyal Biotechnology, LLC, and has stock options in Genfit, Akarna, Tiziana, Indalo, and Durect. He has served as a consultant to AbVie, Astra Zeneca, Nitto Denko, Ardelyx, Conatus, Nimbus, Amarin, Salix, Tobira, Takeda, Fibrogen, Jannsen, Gilead, Boehringer, Lilly, Zafgen, Novartis, Novo Nordisk, and Pfizer. He has served as an unpaid consultant to Exhalenz, Intercept, Echosens, Immuron, Galectin, Fractyl, Northsea Pharma, Gencia, Syntlogic, Affimune, Chemomab, Nordic Bioscience Zydus, and Bristol Myers Squibb. His institution has received grant support from Gilead, Salix, Tobira, Bristol Myers, Shire, Intercept, Merck, Astra Zeneca, Malinckrodt, Cumberland, and Novartis. He receives royalties from Elsevier and UptoDate. Virginia Commonwealth University has ownership interests in Sanyal Biotechnology, LLC.
Nonstandard Abbreviations:
- CC
Collaborative Cross
- HCC
hepatocellular carcinoma
- HF/HS
high fat and high sucrose
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- NAFLD
nonalcoholic fatty liver disease
Footnotes
The views expressed in this manuscript do not necessarily represent those of the U.S. Food and Drug Administration.
References
- 1.Cohen JC, Horton JD and Hobbs HH (2011) Human fatty liver disease: old questions and new insights. Science 332, 1519–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Younossi Z, Tacke F, Arrese M, Charma BC, Mostafa I, Bugianesi E, Wong VW-S, Yilmaz Y, George J, Fan J and Vow MB (2019) Global perspectives on nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology 69, 2672–2682. [DOI] [PubMed] [Google Scholar]
- 3.Williams VF, Taubman SB and Stahlman S (2019) Non-alcoholic fatty liver disease (NAFLD), active component, U.S. Armed Forces, 2000–2017. MSMR 26, 2–11. [PubMed] [Google Scholar]
- 4.Younossi ZM (2019) Non-alcoholic fatty liver disease - A global public health perspective. J. Hepatol 70, 531–544. [DOI] [PubMed] [Google Scholar]
- 5.Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L and Wymer M (2016) Global epidemiology of nonalcoholic fatty liver disease meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64, 73–84. [DOI] [PubMed] [Google Scholar]
- 6.Li L, Liu D-W, Yan H-Y, Wang Z-Y, Zhao S-H and Wang B (2016) Obesity is an independent risk factor for non-alcoholic fatty liver disease: evidence from a meta-analysis of 21 cohort studies. Obes. Rev 17, 510–519. [DOI] [PubMed] [Google Scholar]
- 7.Lu F-B, Hu E-D, Xu L-M, Chen L, Wu J-L, Li H, Chen D-Z and Chen Y-P (2018) The relationship between obesity and the severity of non-alcoholic fatty liver disease: systematic review and meta-analysis. Expert. Rev. Gastroenterol. Hepatol 12, 491–502. [DOI] [PubMed] [Google Scholar]
- 8.Estes C, Razavi H, Loomba R, Younossi Z and Sanyal AJ (2018) Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 67, 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Valenti LVC and Baselli GA (2018) Genetics of nonalcoholic fatty liver disease: a 2018 update. Curr. Pharm. Des 24, 4566–4573. [DOI] [PubMed] [Google Scholar]
- 10.Eslam M, Valenti L and Romeo S (2018) Genetics and epigenetics of NAFLD and NASH: clinical impact. J. Hepatol 68, 268–279. [DOI] [PubMed] [Google Scholar]
- 11.Vilar-Gomez E and Chalasani N (2018) Non-invasive assessment of non-alcoholic fatty liver disease: clinical prediction rules and blood-based biomarkers. J. Hepatol 68, 305–315. [DOI] [PubMed] [Google Scholar]
- 12.Castera L, Friedrich-Rust M and Loomba R (2019) Noninvasive assessment of liver disease in patients with nonalcoholic fatty liver disease. Gastroenterology 156, 1264–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Santhekadur PK, Kumar DP and Sanyal AJ (2018) Preclinical models of non-alcoholic fatty liver disease. J. Hepatol 68, 230–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jahn D, Kircher S, Hermanns HM and Geier A (2019) Animal models of NAFLD from a hepatologist’s point of view. Biochim. Biophys. Acta. Mol. Basis Dis 1865, 943–953. [DOI] [PubMed] [Google Scholar]
- 15.Asgharpour A, Cazanave SC, Pacana T, Seneshaw M, Vincent R, Banini BA, Kumar DP, Daita K, Min H-K, Mirshahi F, Bedossa P, Sun X, Hoshida Y, Koduru SV, Contaifer D Jr., Warncke UO, Wijesinghe DS and Sanyal AJ (2016) A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J. Hepatol 65, 579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hui ST, Parks BW, Org E, Norheim F, Che N, Pan C, Castellani LW, Charugundla S, Dirks DL, Psychogios N, Neuhaus I, Gerszten R, Kirchgessner T, Gargalovic PS and Lusis AJ (2015) The genetic architecture of NAFLD among inbred strains of mice. eLife 4, e05607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jha P, McDevitt MT, Gupta R, Quiros PM, Williams EG, Gariani K, Sleiman MB, Diserens L, Jochem A, Ulbrich A, Coon JJ, Auwerx J and Pagliarini DJ (2018) Systems analyses reveal physiological roles and genetic regulators of liver lipid species. Cell Syst. 6, 722–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hui ST, Kurt Z, Tuominen I, Norheim F, Davis RC, Pan C, Dirks DL, Magyar CE, French SW, Krishnana KC, Sabir S, Campos-Pérez F, Méndez-Sánchez N, Macías-Kauffer L, León-Mimila P, Canizales-Quinteros S, Yang X, Beaven SW, Huertas-Vazquez A and Lusis AJ (2018) The genetic architecture of diet induced hepatic fibrosis in mice. Hepatology 68, 2182–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lonardo A, Nascimbeni F, Ballestri S, Fairweather D, Win S, Than TA, Abdelmalek MF and Suzuki A (2019) Sex differences in nonalcoholic fatty liver disease: state of the art and identification of research gaps. Hepatology 70, 1457–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Threadgill DW, Miller DR, Churchill GA and de Villena FP-M (2011) The Collaborative Cross: a recombinant inbred mouse population for the systems genetic era. ILAR J. 52, 24–31. [DOI] [PubMed] [Google Scholar]
- 21.Atamni HAT, Nashef A and Iraqi FA (2018) The Collaborative Cross mouse model for dissecting genetic susceptibility to infectious diseases. Mamm. Genome 29, 471–487. [DOI] [PubMed] [Google Scholar]
- 22.Salimova E, Nowak KJ, Estrada AC, Furtado MB, McNamara E, Nguyen Q, Balmer L, Preuss C, Holmes JW, Ramialison M, Morahan G and Rosenthal NA (2019) Variable outcomes of human heart attack recapitulated in genetically diverse mice. NPJ Regen. Med 4, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang P, Wang Y, Langley SA, Zhou Y-X, Jen K-Y, Sun Q, Brislawn C, Rojas CM, Wahl KL, Wang T, Fan X, Jansson JK, Celniker SE, Zou X, Threadgill DW, Snijders AM and Mao J-H (2019) Diverse tumour susceptibility in Collaborative Cross mice: identification of a new mouse model for human gastric tumourigenesis. Gut 68, 1942–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaeppler SM (1997) Quantitative trait locus mapping using sets of near-isogenic lines: relative power comparisons and technical considerations. Theor. Appl. Genet 95, 384–392. [Google Scholar]
- 25.Li ZZ, Berk M, McIntyre T and Feldstein AE (2009) Hepatic lipid partitioning and liver damage in nonalcoholic fatty liver disease: role of stearoyl-CoA desaturase. J. Biol. Chem 284, 5637–5644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Charlton M, Krishnan A, Viker K, Sanderson S, Cazanave S, McConico A, Masuoko H and Gores G (2011) Fast food diet mouse: novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am. J. Physiol. Gastrointest. Physiol 301, G825–G834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pogribny IP, Kutanzi K, Melnyk S, de Conti A, Tryndyak V, Montgomery B, Pogribna M, Muskhelishvili L, Latendresse JR, James SJ, Beland FA, and Rusyn I (2013) Strain-dependent dysregulation of one-carbon metabolism in male mice is associated with choline- and folate-deficient diet-induced liver injury. FASEB J. 27, 2233–2243. [DOI] [PubMed] [Google Scholar]
- 28.Schmittgen TD and Livak KJ (2008) Analyzing real-time PCR data by the comparative CT method. Nat. Protoc 3, 1101–1108. [DOI] [PubMed] [Google Scholar]
- 29.Nishimura R, Nakao I, Shikata N, Tsubura A and Morii S (1995) Improved lipid visualization with a modified osmium tetroxide method using ultrasonic treatment and intensification with imidazole or triazole. Biotech. Histochem 70, 28–32. [DOI] [PubMed] [Google Scholar]
- 30.Wong VW-S, Adams LA, de Lédinghen V, Wong GL-H and Sookoian S (2018) Noninvasive biomarkers in NAFLD and NASH – current progress and future promise. Nat. Rev. Gastroenterol. Hepatol 15, 461–478. [DOI] [PubMed] [Google Scholar]
- 31.Golob-Schwarzl N, Bettermann K, Mehta AK, Kessler SM, Unterluggauer J, Krassnig S, Kojima K, Chen X, Hoshida Y, Bardeesy NM, Müller H, Svendova V, Schimek MG, Diwoky C, Lipfert A, Mahajan V, Stumptner C, Thüringer A, Fröhlich LF, Stojakovic T, Nilsson KPR, Kolbe T, Rülicke T, Magin TM, Strnad P, Kiemer AK, Moriggl R and Haybaeck J (2019) High keratin 8/18 ratio predicts aggressive hepatocellular cancer phenotype. Transl. Oncol 12, 256–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Teufel A, Itzel T, Erhart W, Brosch M, Wang XY, Kim YO, von Schönfels W, Herrmann A, Brückner S, Stickel F, Dufour J-F, Chavakis T, Hellerbrand C, Spang R, Maass T, Becker T, Schreiber S, Schafmayer C, Schuppan D and Hampe J (2016) Comparison of gene expression patterns between mouse models of nonalcoholic fatty liver disease and liver tissues from patients. Gastroenterology 151, 513–525. [DOI] [PubMed] [Google Scholar]
- 33.Barrington WT, Wulfridge P, Wells AE, Rojas CM, Howe SYF, Perry A, Hua K, Pellizzon MA, Hansen KD, Voy BH, Bennett BJ, Pomp D, Feinberg AP and Threadgill DW (2018) Improving metabolic health through precision dietetics in mice. Genetics 208, 399–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Younes R and Bugianesi E (2019) NASH in lean individuals. Semin. Liver Dis 39, 86–95. [DOI] [PubMed] [Google Scholar]
- 35.Zelber-Sagi S, Salomone F, Yeshua H, Lotan R, Webb M, Halpern Z, Santo E, Oren R and Shibolet O (2014) Non-high-density lipoprotein cholesterol independently predicts new onset of non-alcoholic fatty liver disease. Liver Int. 34, e128–e135. [DOI] [PubMed] [Google Scholar]
- 36.Ipsen DH, Lykkesfeldt J and Tveden-Nyborg P (2018) Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell Mol. Life Sci 75, 3313–3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Silva AKS and Peixoto CA (2018) Role of peroxisome proliferator-activated receptors in non-alcoholic fatty liver disease. Cell Mol. Life Sci 75, 2951–2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Greenstein AW, Majumdar N, Yang P, Subbaiah PV, Kineman RD and Cordoba-Chacon J (2017) Hepatocyte-specific, PPARγ-regulated mechanisms to promote steatosis in adult mice. J. Endocrinol 232, 107–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schwenk RW, Holloway GP, Luiken JJFP, Bonen A and Glatz JFC (2010) Fatty acid transport across the cell membrane: regulation by fatty acid transporters. Prostaglandins Leukot. Essent. Fatty Acids 82, 149–154. [DOI] [PubMed] [Google Scholar]
- 40.Hayashi Y, Suemitsu E, Kajimoto K, Sato Y, Akhter A, Sakurai Y, Hatakeyama H, Hyodo M, Kaji N, Baba Y and Harashima H (2014) Hepatic monoacylglycerol O-acyltransferase 1 as a promising therapeutic target for steatosis, obesity, and type 2 diabetes. Mol. Ther. Nucleic Acids 3, e154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hall AM, Soufi N, Chambers KT, Chen Z, Schweitzer GG, McCommis KS, Erion DM, Graham MJ, Su X and Finck BN (2014) Abrogating monoacylglycerol acyltransferase activity in liver improves glucose tolerance and hepatic insulin signaling in obese mice. Diabetes 63, 2284–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Inoue M, Ohtake T, Motomura W, Takahashi N, Hosoki Y, Miyoshi S, Suzuki Y, Saito H, Kohgo Y and Okumura T (2005) Increased expression of PPARγ in high fat diet-induced liver steatosis in mice. Biochem. Biophys. Res. Commun 336, 215–222. [DOI] [PubMed] [Google Scholar]
- 43.Wang W, Xu M-J, Cai Y, Zhou Z, Cao H, Mukhopadhyay P, Pacher P, Zheng S, Gonzalez FJ and Gao B (2017) Inflammation is independent of steatosis in a murine model of steatohepatitis. Hepatology 66, 108–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leroux A, Ferrere G, Godie V, Cailleux F, Renoud M-L, Gaudin F, Naveau S, Prévot S, Makhzami S, Perlemuter G and Cassard-Doulcier A-M (2012) Toxic lipids stored by Kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis. J. Hepatol 57, 141–149. [DOI] [PubMed] [Google Scholar]
- 45.Sheedfar F, Sung MMY, Aparicio-Vergara M, Kloosterhuis NJ, Miquilena-Colina ME, Vargas-Castrillón J, Febbraio M, Jacobs RL, de Bruin A, Vinciguerra M, García-Monzón C, Hofker MH, Dyck JRB and Koonen DPY (2014) Increased hepatic CD36 expression with age is associated with enhanced susceptibility to nonalcoholic fatty liver disease. Aging (Albany NY) 6, 281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lima-Cabello E, Garciá-Mediavilla MV, Miquilena-Colina ME, Vargas-Castrillón J, Lozano-Rodríguez T, Fernández-Bermejo M, Olcoz JL, González-Gallego J, García-Monzón C and Sánchez-Campos S (2011) Enhanced expression of pro-inflammatory mediators and liver X-receptor-regulated lipogenic genes in non-alcoholic fatty liver disease and hepatitis C. Clin. Sci. (Lond) 120: 239–250. [DOI] [PubMed] [Google Scholar]
- 47.Pettinelli P and Videla LA (2011) Up-regulation of PPAR-γ mRNA expression in the liver of obese patients: an additional reinforcing lipogenic mechanism to SREBP-1c induction. J. Clin. Endocrinol. Metab 96, 1424–1430. [DOI] [PubMed] [Google Scholar]
- 48.Hall AM, Kou K, Chen Z, Pietka TA, Kumar M, Korenblat KM, Lee K, Ahn K, Fabbrini E, Klein S, Goodwin B and Finck BN (2012) Evidence for regulated monoacylglycerol acyltransferase expression and activity in human liver. J. Lipid. Res 53: 990–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.García-Monzón C, Iacono OL, Crespo J, Romero-Gómez M, García-Samaniego J, Fernández-Bermejo M, Domínguez-Díez A, Rodríguez de Cía J, Sáez A, Porrero JL, Vargas-Castrillón J, Chávez-Jiménez E, Soto-Fernández S, Díaz A, Gallego-Durán A, Madejón A and Miquilena-Colina ME (2014) Increased soluble CD36 is linked to advanced steatosis in nonalcoholic fatty liver disease. Eur. J. Clin. Invest 44, 65–73. [DOI] [PubMed] [Google Scholar]
- 50.Graupera I, Coll M, Pose E, Elia C, Piano S, Solà E, Blaya D, Huelin P, Solé C, Moreira R, de Prada G, Fabrellas N, Juanola A, Morales-Ruiz M, Sancho-Bru P, Villanueva C and Ginès P (2017) Adipocyte fatty-acid binding protein is overexpressed in cirrhosis and correlates with clinical outcomes. Sci. Rep 7, 1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bedossa P (2016) Histological assessment of NAFLD. Dig. Dis. Sci 61, 1348–1355. [DOI] [PubMed] [Google Scholar]
- 52.Farrell G, Schattenberg JM, Leclercq I, Yeh MM, Goldin R, Teoh N, and Schuppan D (2019) Mouse models od nonalcoholic steatohepatitis: toward optimization of their relevance to human nonalcoholic steatohepatitis. Hepatology 69, 2241–2257. [DOI] [PubMed] [Google Scholar]
- 53.Novo E, Cannito S, Morello E, Paternostro C, Bocca C, Miglietta A, and Parola M (2015) Hepatic myofibroblasts and fibrogenic progression of chronic liver diseases. Histol. Histopathol 30, 1011–1032. [DOI] [PubMed] [Google Scholar]
- 54.Sanyal AJ (2019) Past, present and future perspectives in nonalcoholic fatty liver disease. Nat Rev Gastroenterol. Hepatol 16, 377–386. [DOI] [PubMed] [Google Scholar]
- 55.Brunt EM, Wong VW-S, Nobili V, Day CP, Sookoian S, Maher JJ, Bugianesi E, Sirlin CB, Neuschwander-Tetri BA and Rinella ME (2015) Nonalcoholic fatty liver disease. Nat. Rev. Dis. Primers 1, 15080. [DOI] [PubMed] [Google Scholar]
- 56.Brunt EM, Kleiner DE, Wilson LA, Sanyal AJ and Neuschwander-Tetri BA (2019) Improvements in histologic features and diagnosis associated with improvement in fibrosis in nonalcoholic steatohepatitis: results from the Nonalcoholic Steatohepatitis Clinical Research Network Treatment Trials. Hepatology 70, 522–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bashiardes S, Shapiro H, Rozin S, Shibolet O and Elinav E (2016) Non-alcoholic fatty liver and the gut microbiota. Mol. Metab 5, 782–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Adolph TE, Grander C, Moschen AR and Tilg H (2018) Liver–Microbiome Axis in Health and Disease. Trends in Immunology 39, 712–723. [DOI] [PubMed] [Google Scholar]
- 59.Sharpton SR, Ajmera V and Loomba R (2019) Emerging role of the gut microbiome in nonalcoholic fatty liver disease: from composition to function. Clin. Gastroenterol. Hepatol 17, 296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schwimmer JB, Johnson JS, Angeles JE, Behling C, Belt PH, Borecki I, Bross C, Durelle J, Goyal NP, Hamilton G, Holtz ML, Lavine JE, Mitreva M, Newton KP, Pan A, Simpson PM, Sirlin CB, Sodergren E, Tyagi R, Yates KP, Weinstock GM and Salzman NH (2019) Microbiome signatures associated with steatohepatitis and moderate to severe fibrosis in children with nonalcoholic fatty liver disease. Gastroenterology 157, 1109–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.