

Abstract
Since the first human respiratory syncytial virus (HRSV) genotype classification in 1998, inconsistent conclusions have been drawn regarding the criteria that define HRSV genotypes and their nomenclature, challenging data comparisons between research groups. In this study, we aim to unify the field of HRSV genotype classification by reviewing the different methods that have been used in the past to define HRSV genotypes and by proposing a new classification procedure, based on well-established phylogenetic methods. All available complete HRSV genomes (>12,000 bp) were downloaded from GenBank and divided into the two subgroups: HRSV-A and HRSV-B. From whole-genome alignments, the regions that correspond to the open reading frame of the glycoprotein G and the second hypervariable region (HVR2) of the ectodomain were extracted. In the resulting partial alignments, the phylogenetic signal within each fragment was assessed. Maximum likelihood phylogenetic trees were reconstructed using the complete genome alignments. Patristic distances were calculated between all pairs of tips in the phylogenetic tree and summarized as a density plot in order to determine a cutoff value at the lowest point following the major distance peak. Our data show that neither the HVR2 fragment nor the G gene contains sufficient phylogenetic signal to perform reliable phylogenetic reconstruction. Therefore, whole-genome alignments were used to determine HRSV genotypes. We define a genotype using the following criteria: a bootstrap support of 70 per cent for the respective clade and a maximum patristic distance between all members of the clade of ≤0.018 substitutions per site for HRSV-A or ≤0.026 substitutions per site for HRSV-B. By applying this definition, we distinguish twenty-three genotypes within subtype HRSV-A and six genotypes within subtype HRSV-B. Applying the genotype criteria on subsampled data sets confirmed the robustness of the method.
Keywords: human respiratory syncytial virus, classification, genotypes
1. Introduction
Human respiratory syncytial virus (HRSV) is worldwide the most common viral cause of acute respiratory tract infections in children up to the age of 5 years (Shi et al. 2017). Currently, there is no licensed vaccine available, and treatment options are scarce (Heylen et al. 2017). Mechanisms to evade the host immune responses and the young age of the main target patient group are only two examples of the challenges related to the development of HRSV antiviral therapies and vaccines (Simoes et al. 2015; Battles and McLellan 2019; Rossey and Saelens 2019). Furthermore, HRSV diversity is more shaped by temporal than by geographical distribution with rapid global spread of new variants (Pangesti et al. 2018). In temperate climates, HRSV circulates according to a reoccurring seasonal pattern with peaks during the late fall or early winter, whereas the peak in tropical climates occurs in the late summer months (Ramaekers et al. 2017; Li et al. 2019). To support ongoing developments of new therapies, mapping the genetic diversity of HRSV remains of critical importance.
HRSV has since 2018 been reclassified under the species name Human orthopneumovirus within the family of Pneumoviridae (Rima et al. 2017). The genome of HRSV is single-stranded negative sense RNA with a length of 15.2 kb and is composed of ten genes which code for eleven proteins: three transmembrane glycoproteins (F, G, SH), two matrix proteins (M, M2), three proteins associated with the nucleocapsid (N, P, L), and two nonstructural proteins (NS1, NS2) (Battles and McLellan 2019) (Fig. 1). The attachment protein (G) and the fusion protein (F) are responsible for the attachment and entry of the viral particle into the host cell and are therefore important targets of the host immune responses (Collins and Melero 2011).
Figure 1.

Genome structure of HRSV. The HRSV-B reference genome (AF013254) is 15,225 nt long and comprises ten genes, coding for eleven proteins.
Two subtypes of HRSV (A and B) have been distinguished by using monoclonal antibodies against the G, F, M, NP, and P proteins (Anderson et al. 1985; Mufson et al. 1985; Gimenez et al. 1986). This subdivision was later confirmed by different genetic analyses (Sullender 2000). The F gene sequence is relatively conserved between the two subtypes of HRSV (Battles and McLellan 2019), which makes it the main focus for vaccine development (Rossey and Saelens 2019). The G protein has a cytoplasmic tail, a transmembrane domain, and an ectodomain, which has been reported to be the most variable part of the HRSV genome (Tan et al. 2012). Within the ectodomain, a central conserved domain with four cysteine residues is flanked by two hypervariable regions (HVR1 and HVR2) and a heparin-binding domain (Battles and McLellan 2019) (Fig. 1). Duplication events in the HVR2 region of the ectodomain of the G gene have generated HRSV strains that rapidly became dominant all over the world, suggesting a selective advantage (Trento et al. 2006; Eshaghi et al. 2012; Pangesti et al. 2018). Because of its high variability, evaluation of HRSV genetic diversity has historically relied most often on the G gene (Cane et al. 1991; Garcia et al. 1994).
Since the first HRSV genotype classification by Peret et al. (1998), several research groups have shown their interest in HRSV genetic diversification, resulting in the discovery of additional genotypes. Inconsistent conclusions have been drawn regarding the number of genotypes, based on different parts of the HRSV genome. Moreover, different criteria have been used to define genotypes, whereas similarly defined genotypes have been named inconsistently: either based on the subtype and gene that was studied, based on the country or the city where the genotypes were first described, and based on their phylogenetic clustering or sometimes even seemingly arbitrary.
1.1 Genotype classification based on the HVR2 region of the G gene
The first classification system of HRSV genotypes, proposed in 1998, relied on sequencing information of the second HVR2 of the G gene. Based on visual inspection of a phylogenetic tree, seven genotypes could be distinguished for HRSV-A and four genotypes for HRSV-B. Bootstrap support (BS) values of 78 per cent or higher were observed for the relevant clusters. The genotypes were named based on the gene used for classification (G), followed by the HRSV subtype (A or B) and an ascending number: GA1–GA7 and GB1–GB4 (Peret et al. 1998, 2000) (Table 1).
Table 1.
Overview of criteria and nomenclature used in literature to define HRSV genotypes.
| Reference | Genotyping region | Genotyping definitions | Genotype nomenclature | Genotypes identified HRSV-A | Genotypes identified HRSV-B |
|---|---|---|---|---|---|
| Peret et al. (1998) | HVR2 | BS ≥ 78% | Gene + subtype + number | GA1–GA5 | GB1–GB4 |
| Peret et al. (2000) | HVR2 | BS ≥ 70% | Gene + subtype + number | GA6–GA7 | / |
| Choi and Lee (2000) | G gene (HRSV-A) or partial HVR2 (HRSV-B) | Restriction analysis G gene | Gene + P + subtype + number | GP-A1–GP-A24 | GP-B1–GP-B6 |
| Venter et al. (2001) | HVR2 | BS ≥70%, p distance <0.07 | Country + subtype + number | SAA1 | SAB1–SAB3 |
| Trento et al. (2003) | G gene | / | / | / | No genotype, but “BA viruses” with 60 nt duplication |
| Zlateva et al. (2004) | Ectodomain | BS > 77% | Country + subtype + number | BE/A1 | / |
| Zlateva et al. (2005) | Ectodomain | BS > 77% | Gene + subtype + number | / | GB5–GB13 |
| Blanc et al. (2005) | HVR2 | BS ≥ 70%, p distance <0.07 | Country + subtype + number | / | URU1–URU2 |
| Trento et al. (2006) | HVR2 | BS ≥ 70% | City | / | BA (subclades I–VI) |
| Shobugawa et al. (2009) | HVR2 | BS ≥ 70%, p distance | City + subtype + number | NA1–NA2 | / |
| Dapat et al. (2010) | HVR2 | BS > 50% | Clustering + number | / | BA7–BA10 |
| Arnott et al. (2011) | HVR2 | BS ≥ 70%, p distance <0.07 | Clustering + number | / | SAB4 |
| Baek et al. (2012) | G gene | BS ≥ 70%, p distance <0.07 | City + subtype or clustering | CB-A | CB-B, BA11 |
| Eshaghi et al. (2012) | HVR2 | BS ≥ 70%, p distance <0.07 | City + number | ON1 | / |
| Cui et al. (2013) | HVR2 | BS ≥ 70%, p distance <0.07 | Clustering or unsystematic (CB-1) | NA3–NA4 | BA-C, CB-1 |
| Khor et al. (2013) | HVR2 | / | Clustering + number | / | BA12 cluster in BA genotype |
| Agoti et al. (2014) | G gene | For clades: BS ≥ 60%, average genetic distance cutoff 1.5% | Genotype of origin | / | BA1–BA12 → BA, subclades BA 2.1–2.4 |
| Liu et al. (2014) | HVR2 | BS > 85% | Clustering + number | / | BA1–BA10 |
| Trento et al. (2015) | G gene | p distance < 0.049 | Gene + subtype + number | NA1, NA2, NA4, ON1 → GA2 | / |
| Agoti et al. (2015b) | Ectodomain | Cluster identity threshold | Clustering + number | SAB1 variants V1–V10 | BA variants V1–V36 |
| Gimferrer et al. (2016) | HVR2 | BS ≥ 70%, p distance <0.07 | Clustering + number | / | BA13 |
One year later, Venter et al. used a similar approach to expand the number of HRSV genotypes. The method of classification described by Peret et al. was refined by including genetic distance as a metric to define clusters. If a group of sequences would cluster together with BS values of 70 per cent or more and if characterized with a pairwise distance of <0.07 nucleotide (nt) substitutions per site to all other members part of the same phylogenetic cluster, a genotype was distinguished. In addition to introducing a new genotype definition, the nomenclature system was altered, including now the country of discovery (i.e. SA for South Africa) when naming HRSV genotypes. This method resulted in four new genotypes: SAA1 within subtype HRSV-A and SAB1–SAB4 within subtype HRSV-B (Venter et al. 2001). In line with the method of Venter et al., two new genotypes were described in Uruguay (URU1 and URU2) within subgroup HRSV-B (Blanc et al. 2005). Over the course of several years, the genotype definition of Venter et al. was used to distinguish additional genotypes in Argentina (Trento et al. 2006), Cambodia (Arnott et al. 2011), Canada (Eshaghi et al. 2012), and Spain (Gimferrer et al. 2016). However, the naming of the genotypes did not adhere to the same subtype- and country-based nomenclature system of Venter et al. because some genotypes were named according to the city where they were first described. Within subtype HRSV-B, genotype Buenos Aires (BA) was distinguished, with subclades BAI–BAVI (Trento et al. 2006). Within subtype HRSV-A, new genotypes NA1 and NA2 (Niigata) (Shobugawa et al. 2009) and ON1 (Ontario) were described, the latter containing a 72 nt duplication in the HVR2 region (Eshaghi et al. 2012). Furthermore, two new genotypes were named according to their respective clustering in the phylogenetic tree: SAB4 within subtype HRSV-B (Arnott et al. 2011) and BA13 within subtype HRSV-A (Gimferrer et al. 2016). Similarly, four new genotypes were defined in China by Cui et al., based on the rules of Venter et al., however, named according to their clustering with previously described genotypes: NA3, NA4 (within subgroup HRSV-A) and BA-C (within subgroup HRSV-B) or named seemingly unsystematically as CB-1, unrelated to a previously described genotype CB-B (Cui et al. 2013) (Table 1).
In addition to the approaches of Peret et al. and Venter et al., alternative definitions have been formulated to distinguish genotypes and their subclades based on the HVR2 region of the G gene. For example, Khor et al. considered all previously described BA strains as one genotype and defined a new cluster within genotype BA, based on a bootstrap value of 81 per cent and nt similarity of 93–95 per cent with other BA strains (Khor et al. 2013). Alternatively, Liu et al. defined a new genotype in case the following criteria were met: “sequences are unable to cluster together with any reference strain and form an independent cluster with a bootstrap value >85 per cent.” Additionally, both the BA clade and its sublineages were now named “genotypes,” further adding to the discrepancies in terminology used for the BA genotype and its subclades (Liu et al. 2014) (Table 1).
1.2 Genotype classification based on the ectodomain of the G gene
Zlateva et al. introduced a genotype classification system in 2004, based on phylogenies of slightly larger alignments of the G gene, including both HVRs and the central conserved domain. Separate genotypes were defined based on BS values higher than 77 per cent for clades. The use of the original genotype nomenclature of Peret et al. (considering the gene and subtype) was combined with the country-based nomenclature system of Venter et al. Accordingly, a new genotype BE/A1 was described within subtype HRSV-A (Zlateva et al. 2004) and eight new genotypes (GB5–GB13) within subtype HRSV-B (Zlateva et al. 2005). New HRSV-B variants, described for the first time by Trento et al. as “BA viruses” with a 60 nt duplication in the HVR2 region of the ectodomain (Trento et al. 2003), clustered together in genotype GB13 (Zlateva et al. 2004). However, the genotypes described in this study were not taken into account in later publications in which HRSV-B genotypes GB5–GB13 were grouped into one overlapping genotype BA, with subclades BAI–BAVI, based on the HVR2 region (Trento et al. 2006). As the emergence of strains with a duplicated region, some research groups considered the BA subclades as being separate genotypes and therefore added more genotypes under the BA group (Dapat et al. 2010; Baek et al. 2012; Gimferrer et al. 2016), whereas others considered them as subclades within the BA genotype (Cui et al. 2013b; Khor et al. 2013) or within the GB13 genotype (Tan et al. 2012; Houspie et al. 2013) (Table 1).
Agoti et al. followed the method of Peret et al. and Venter et al. for the classification of HRSV-B strains, grouping BA strains into one genotype. Moreover, a new system based on cluster identity was created to classify strains into genotypes and subsequently into variants, using alignments of the ectodomain region. According to this new method, genotype SAB1 could be divided into ten variants (V1–V10) and genotype BA into thirty-six variants (V1–V36) (Agoti et al. 2015b) (Table 1).
1.3 Genotype classification based on the entire G gene
In parallel to the work of Peret et al. and Venter et al., an alternative classification system has been developed by Choi and Lee. Based on restriction mapping and nt sequencing of the entire G gene (HRSV-A) and the C-terminal end of the G gene (HRSV-B), twenty-four genotypes were distinguished within subgroup HRSV-A: GP-A1–GP-A24 and six genotypes for HRSV-B: GP-B1–GP-B6 (Choi and Lee 2000). As these genotypes could not be well distinguished using phylogenies, the proposal has not been taken up by the HRSV research community.
Three new genotypes were published by Baek et al., applying the genotype definition of Venter et al. on phylogenies of entire G gene alignments. Two genotypes were named after the city of first detection (Chongbuk) and the respective subtype (A or B), because they clustered separately from previously described genotypes in the phylogenetic tree: CB-A and CB-B. A third genotype clustered close to the BA genotypes/sublineages within subtype HRSV-B and was therefore named BA11 (Baek et al. 2012).
Upon discovery of new HRSV strains by Agoti et al., a proposal was made to unify the HRSV classification system below the genotype level. The authors suggested to use a system based on the H5N1 influenza virus classification approach of WHO, in which clade names would be derived from the names of the genotype they originate from. Additionally, clades would be defined based on BS values of 60 per cent or higher and with an average genetic distance of at least 1.5 per cent to other clades and <1.5 per cent within the clade (WHO/OIE/FAO H5N1 Evolution Working Group 2008; Agoti et al. 2014). As such, all BA strains were classified under one genotype BA, dismissing previous divisions of BA1–BA12 and grouping them into four new subclades BA (2.1) to (2.4) (Agoti et al. 2014). Soon after however, a genotype BA13 classification was used that did not follow this proposal (Gimferrer et al. 2016). Furthermore, a proposal was written in which the previously described HRSV-A genotypes NA1, NA2, NA4, and ON1 were reclassified into the GA2 genotype (Trento et al. 2015). The highest intragenotypic p distance was used, being 0.049, as the minimal threshold to define genotypes, leading to a reduction in the number of genotypes from fourteen (GA1–GA7, SAA1–SAA2, NA1–NA4, and ON1) to seven (GA1–GA7) (Trento et al. 2015). It remains unclear where strains from genotypes BA/A1 and CB-A would cluster in this classification, as representative members of these genotypes were not included in the analysis (Table 1).
1.4 Classification based on other parts of the genome
Although the majority of research has focused on the G gene for the classification of HRSV, there have been few proposals based on other parts of the HRSV genome. Based on limited nt sequencing of the SH gene for instance, Cane et al. identified six lineages within subgroup HRSV-A (SHL1–SHL6) (Cane and Pringle 1991; Cane et al. 1992). The SHL lineages can be linked to the classification of Peret et al. as follows: lineages SHL1, SHL3, and SHL4 correspond to genotype GA3, lineage SHL2 to GA5, and SHL5 to GA1 (Sullender 2000). When genotype ON1 was first described, phylogenetic trees were reconstructed based on the HVR2 region of the G gene as well as using a partial F gene sequence. In both phylogenies, the authors were able to prove separate clustering of ON1 strains from other circulating strains (Eshaghi et al. 2012).
1.5 Genotype classification based on the full genome
Strains with identical G gene sequences have shown significant differences in other regions of the genome, supporting the benefit of switching from a short fragment to full genome analysis for evolutionary studies on HRSV (Agoti et al. 2015a). Similarly, differences in classification following the use of different regions of the viral genome have been reported for other viruses, such as Dengue, for which the use of full genome information ensures to have a more complete understanding of the evolution of circulating strains (Cuypers et al. 2018). For the classification of Hepatitis B virus, full genome sequences have been used for classification since many years (Bartholomeusz and Schaefer 2004).
The multitude of parallel classification systems has left us with an abundance of genotypes and a lack of consistency across the field. Research groups have repeatedly expressed the need to wipe out the inconsistency (Cane 2001; Agoti et al. 2014; Trento et al. 2015; Pangesti et al. 2018). With the prospect of marketed HRSV vaccines (Rossey and Saelens 2019), a coherent surveillance of circulating strains is urgently needed in order to predict their long-term efficacy (Otieno et al. 2016) and guide potential improvements in the content to increase their impact on the HRSV epidemiology.
With this manuscript, we aim to unify the field by reviewing the different methods that have been used in the past to define HRSV genotypes and by proposing a new classification procedure, based on well-established phylogenetic methods. This project is part of a joined effort embedded within the GeNom consortium which aims to improve the global surveillance of HRSV by creating well-defined guidelines for the genotyping and nomenclature of HRSV strains.
2. Methodology
2.1 Compilation of a full-length HRSV genome data set
All available complete HRSV genomes (>12,000 bp) were downloaded from GenBank on January 15, 2019. The resulting 2,212 sequences were first confirmed as HRSV genomes, using CD-HIT v4.5.4 at a sequence identity threshold of 80 per cent (Li and Godzik 2006). The two largest clusters were retained and clustered into HRSV-A and HRSV-B, using the default 90 per cent sequence identity threshold of CD-HIT v4.5.4. Upon removal of inadequate strains for phylogenetic analysis (i.e. mutants, patented sequences, duplicates, cell culture strains, and partial genomes with gaps of 100 bp or more), alignments were generated for each subtype, using the default options of MAFFT v7 (Katoh and Standley 2013). Alignments were visually inspected and edited in AliView v1.23 (Larsson 2014).
The resulting alignments were screened for sequences with potential recombination events, using the detection methods RDP, GENECONV, MaxChi, BootScan, and SiScan as implemented in the Recombination Detection Program RDP4 (Martin et al. 2015). After additional exclusion of all strains flagged as potential recombinants, final alignments of, respectively, 861 sequences for subgroup HRSV-A and 492 sequences for subgroup HRSV-B were used for further analysis.
2.2 Evaluation of phylogenetic signal
Starting from whole-genome alignments, we extracted the regions that correspond to the open reading frame (ORF) of the Glycoprotein G and the second HVR2 of the ectodomain, either with or without inclusion of the duplication within the HVR2 region. For the resulting partial alignments, the phylogenetic signal within each fragment was assessed, using the likelihood mapping function as implemented in Tree-Puzzle v5.3 (Schmidt et al. 2002).
2.3 Phylogenetic tree reconstruction
The best-fitting nt substitution model for each data set was identified by comparing eighty-eight candidate models using jModeltest v2.1.10 (Guindon and Gascuel 2003; Darriba et al. 2012). Phylogenetic trees of the complete genome alignments were reconstructed using RAxML v8.2.12, using the Generalized Time Reversible substitution model with a gamma model of rate heterogeneity and taking invariable sites into account (GTR + GAMMA + I) (Stamatakis 2014). Branch support was evaluated by bootstrapping based on 1,000 pseudoreplicates. Nt sequences of both HRSV subtypes (HRSV-A vs. HRSV-B) do not align unambiguously and are therefore not closely enough related to be used as a meaningful outgroup for each other (Salemi 2009). Therefore, trees were midpoint rooted using the R package phytools v0.6 (Revell 2012), and rendered with increasing node order using the R package ape v5.3 (Paradis and Schliep 2019).
2.4 Defining HRSV genotypes using patristic distance and BS
Patristic distances, that is the shortest distance between two tips, measured as the sum of the branch lengths, were calculated between all tips of the phylogenetic trees based on the alignment of the whole-genome lacking the duplicated region, using the adephylo v1.1 package in R (Jombart and Dray 2010). A density plot of the resulting patristic distances was created using the ggplot2 package in R (Wickham 2016) and used to visually determine a cutoff value at the lowest point following the major distance peak (Prosperi et al. 2011), respectively, for HRSV-A and HRSV-B.
Two criteria were used to distinguish genotypes in each subtype of HRSV: 1. the maximum patristic distance (PatDist_max) between all tips within the clade is below the cutoff value of the whole tree and 2. the bootstrap value of the parent node of the clade is 70 per cent or higher. The R packages ape v 5.3 (Paradis and Schliep 2019) and adephylo v1.1 (Jombart and Dray 2010) were used to define genotypes, whereas visualizations were created with ggtree v1.16.0 (Yu et al. 2017, 2018), and ggplot2 v3.1.1 (Wickham 2016).
2.5 Method robustness
Finally, to correct for the overrepresentation of recently sampled strains, the analysis was repeated with data sets in which the overrepresented group was reduced. A subset of ten sequences, representing the large majority of the genetic diversity embedded within the overrepresented cluster, was determined for both data sets (HRSV-A and HRSV-B), using the software package Phylogenetic Diversity Analyzer (PDA v1.0.3) (Chernomor et al. 2015). Phylogenetic tree reconstruction and genotype demarcation were repeated on the reduced data sets, comprising of 261 and 70 taxa, respectively, as described earlier.
3. Results
3.1 Artificial recombination in both subtypes
Although recombination within the HRSV genome has not been described in natural strains (Tan et al. 2012), we tested our data set for the presence of possible recombination events prior to proceeding with phylogenetic analyses. In both HRSV subtypes, potential recombination events were flagged in the data set. These events are most likely artificial recombination events as a result of errors during the assembly of shorter sequencing fragments. Especially when whole-genome sequences have been obtained by the use of metagenomics, caution is needed to avoid mistakes during de novo assembly (Tan et al. 2012; Simmonds et al. 2017).
3.2 The G gene and HVR2 region are not suitable for phylogenetic analysis
Quality assessment of 2,212 genome sequences from GenBank resulted in final data sets consisting of 861 sequences for HRSV-A and 493 for HRSV-B. Genome fragments of the G gene ORF (967 nt/951 nt) and the HVR2 region (409 nt/399 nt) were extracted from the whole-genome nt alignments (14,953 nt/14,949 nt) for subtype HRSV-A and HRSV-B, respectively. The phylogenetic signal of each fragment, with and without the duplicated region present, was assessed, using the likelihood-mapping algorithm implemented in Tree-Puzzle (Supplementary Fig. S1). To allow reliable reconstruction of a phylogenetic tree, a fragment should have at least 90 per cent phylogenetic support, determined as the percentage of resolved phylogenies in Tree-Puzzle (Schmidt et al. 2002). The alignments of the shorter fragments (G gene and HVR2) do not meet this criterion and are therefore not suitable to proceed to phylogenetic analysis, with exception of the HRSV-B G gene alignment without the duplicated region. The whole-genome alignments were supported well beyond the 90 per cent cutoff for both subtypes, regardless of inclusion or exclusion of the duplicated nt stretch in the HVR2 region (Table 2).
Table 2.
Percentages of resolved phylogenies in different fragments of the HRSV genome.
| Alignment | Unresolved (%) |
Conflict (%) |
Resolved (%) |
|||
|---|---|---|---|---|---|---|
| HRSV-A | HRSV-B | HRSV-A | HRSV-B | HRSV-A | HRSV-B | |
| HVR2 region w/o duplication | 16.2 | 12.7 | 2.6 | 4.2 | 81.2 | 83.1 |
| HVR2 region w/ duplication | 16.4 | 12.5 | 2.4 | 4 | 81.2 | 83.5 |
| G gene w/o duplication | 12 | 6.6 | 3 | 3.2 | 85.0 | 90.2 |
| G gene w/ duplication | 11.5 | 7.7 | 3 | 3.3 | 85.5 | 89.0 |
| Full genome w/o duplication | 1.1 | 0.7 | 1.9 | 1.4 | 97.0 | 97.9 |
| Full genome w/ duplication | 1.3 | 0.6 | 1.8 | 1.1 | 96.9 | 98.3 |
With exception of the HRSV-B G gene without duplication, for both subtypes only the whole-genome alignments show sufficient phylogenetic signal (defined as resolved phylogenies for at least 90% of the quartets, in bold) for reliable phylogenetic analysis (Supplementary Fig. S1).
3.3 Patristic distances and BS values define HRSV genotypes
ML trees were reconstructed for both subtypes based on whole-genome alignments excluding the duplicated region. The patristic distances for all pairs of taxa were calculated in each phylogeny and summarized as a density plot. Cutoff values were determined objectively by identifying the lowest point following the major distance peak, which resulted in distances of 0.018 nt substitutions per site for HRSV-A and 0.026 subst./site for HRSV-B (Fig. 2).
Figure 2.

Whole-genome phylogenies and density distributions of patristic distances for HRSV-A and HRSV-B. Patristic distances were calculated between all tips of the ML trees of whole-genome alignments of HRSV-A (A) and HRSV-B (C) and cut-off values were chosen at the lowest point after the major peak in the density plot, determined at 0.018 subst./site for HRSV-A (B) and slightly higher at 0.026 subst./site for HRSV-B (D).
Genotypes within each subtype were defined based on the patristic distance cutoff value and a BS of at least 70 per cent. For HRSV-A, twenty-three clades (A1–A23) were defined based on our genotype criteria (BS 70%, PatDist_max ≤0.018 subst./site), whereas six genotypes (B1–B6) were distinguished for HRSV-B (BS 70%, PatDist_max ≤0.026 subst./site) (Fig. 3). The accession numbers of all members per genotype are listed in Supplementary Table S1.
Figure 3.

Genotypes defined within subtypes HRSV-A and HRSV-B. Based on the genotype criteria of BS 70 per cent, PatDist_max ≤0.018 subst./site (HRSV-A), or ≤0.026 subst./site (HRSV-B), we distinguish twenty-three and six genotypes for subtype HRSV-A (A) and HRSV-B (B), respectively. Underneath each tree, the evolutionary distance scale is indicated, expressed as nt subst./site.
3.4 Robust method for application on smaller data sets
By applying these genotype criteria to the phylogenetic tree reconstructed for a subsampled HRSV-A alignment, in which the presence of recent strains was downsized, we could discriminate twenty-two instead of twenty-three HRSV-A genotypes. Taxa that cluster together in the phylogeny of the full data set (n = 861) also cluster together in the phylogeny of the subset (n = 261), with the exception of taxa of genotypes A22 and A23 in the full data set analysis that are now combined into one group (Fig. 4). For subtype HRSV-B, the number of genotypes was identical, that is, six, whether the genotype criteria were applied on the full data set alignment (n = 493) or on the subsampled alignment (n = 70), with 100 per cent of the taxa clustering together in the same way (data not shown).
Figure 4.
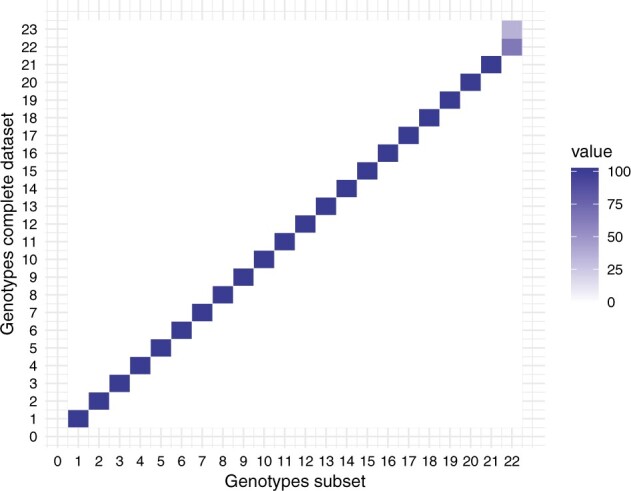
Genotype co-occurrence matrix HRSV-A. The heat map shows for each genotype defined using the full data set (y axis), the percentage of taxa that corresponds to the genotypes defined using a subset of the data set (x axis). The correlation of the taxa is 100 per cent for all genotypes, with the exception of the taxa within genotypes A22 and A23 that are merged into one genotype when using a subset of the data.
4. Discussion
Human respiratory syncytial virus is one of the most important causes of acute respiratory tract infections and is estimated to cause 76,600 annual deaths, primarily in young children under the age of five and in the elderly population (Li et al. 2019). We are now on the verge of having a licensed HRSV vaccine on the market and several antivirals are under development (Higgins et al. 2016; Heylen et al. 2017; PATH 2019; Rossey and Saelens 2019). A systematic monitoring of genetic diversity of the circulating strains is essential for a good understanding of the long-term effectiveness of vaccines (Otieno et al. 2016). However, due to lack of consistency in the methodology and nomenclature of HRSV classification, several genotype definitions are being used in parallel, creating confusion and leading to difficulties in comparing data from different areas in the world. Furthermore, the correlation between disease severity and genotype has been under discussion for several years due to conflicting results (Anderson et al. 2019; Vos et al. 2019). All these reasons call for a unified classification system (Cane 2001; Agoti et al. 2014; Trento et al. 2015; Pangesti et al. 2018).
In recent years, several research groups have attempted to update the classification methodology, but so far no one succeeded to encourage fellow researchers to follow their approach (Agoti et al. 2014, 2015b, 2017; Liu et al. 2014; Trento et al. 2015). Based on the highest intragenotypic p-distance as the minimal threshold to define a genotype, a suggestion has been proposed to reduce the number of genotypes within subtype HRSV-A from fourteen to seven (Trento et al. 2015). The proposal failed to become the new reference in the HRSV field possibly due to its restriction to only one out of two HRSV subtypes. The suggestion to use an influenza-like system for genotype and subclade classification meets this limitation but has not been adopted beyond the proposing research group either (Agoti et al. 2014). In addition to suggesting a well-founded and robust classification method, receiving support from several authorities in the field proves to be essential in order to make a change in the current practice. Therefore, the GeNom consortium attempts to reach harmony within HRSV strain nomenclature and genotype classification by combining the efforts of several authorities in the field. In this study, we thoroughly reviewed all methods currently used with respect to the classification of HRSV genotypes. Over the last two decades, we could appreciate a tremendous improvement in laboratory techniques and software development, which makes comparison of existing methods challenging. Therefore, instead of making a choice between existing methods, we decided to use a bottom-up approach by starting from the available sequence data in GenBank. With this approach, we aim to propose a classification system based on patterns in the data, rather than historical preferences for a prevailing research track, tradition or practical considerations.
The most commonly used definition to classify HRSV strains into genotypes is still the original definition, formulated by Peret et al. in 1998 and refined by Venter et al. in 2001, in which a genotype is distinguished based on a phylogenetic cluster with BS of at least 70 per cent and a pairwise distance of <0.07 subst./site to all other members of the cluster (Peret et al. 1998; Venter et al. 2001). This definition was formulated at a time when genetic sequencing was a time-consuming and costly process and was therefore applied to phylogenies of the HVR2 region of the G gene, which is a highly variable region of the genome and thus expected to be informative for phylogenetic analysis and classification. Technological advances have made nt sequencing of larger fragments, and even complete genomes, easier and cheaper, eliminating the need to use short fragments for genotype classification (Thomson et al. 2016; Goya et al. 2018). Additionally, studies in mice have indicated the importance of the F protein and of the central conserved domain of the G protein in the development and severity of the associated disease (Tripp et al. 2001; Hotard et al. 2015; Currier et al. 2016; Boyoglu-Barnum et al. 2017). As a consequence of the persistent focus on the HVR2 region of the G gene for genotyping, an association between virulence and viral genotype may have been missed (Anderson et al. 2019). In order to assess the loss of information when using the HVR2 region of the G gene, we evaluated the phylogenetic signal in this fragment, as well as in the G gene in comparison to the whole-genome alignment. Although the HVR2 region has been proposed as a good proxy to assess the variability for the whole genome (Peret et al. 1998), our analysis shows that neither the HVR2 fragment nor the G gene contains sufficient phylogenetic signal to perform reliable phylogenetic reconstruction. Whole-genome data from all over the world are required in order to perform extensive analyses on the distribution and transmission dynamics of HRSV (Di Giallonardo et al. 2018). Currently, novel real-time surveillance tools, such as Nextstrain, limit themselves to subtype HRSV-A due to a shortage of whole-genome strains for the subtype HRSV-B (Hadfield et al. 2018). Further motivated by the large interest of antiviral (Heylen et al. 2017) and vaccine (Rossey and Saelens 2019) research in parts of the HRSV genome other than the G gene, we suggest using whole-genome phylogenies as the basis for a future genotype classification system.
In our method, we chose, in addition to BS values, patristic distances as a parameter to distinguish genotypes rather than pairwise distances. Patristic distance is a tree-based estimation of the genetic distance, measured as the shortest distance over the branch lengths between two tips of the phylogenetic tree. Therefore, patristic distances reflect the information from the evolutionary model that was chosen to build the phylogenetic tree and result in a better estimation of the true genetic distances represented in the data set compared with pairwise distances (Lemey et al. 2009). Patristic distances were calculated between all pairs of tips of the phylogenetic trees of the whole-genome alignments of HRSV-A and HRSV-B. The resulting matrix was visualized in a density plot and the cutoff to distinguish a genotype was chosen at the lowest point after the major peak (Prosperi et al. 2011).
We define a clade as a genotype when the following criteria are met: the respective clade has a BS of 70 per cent and the maximum patristic distance between all members of the clade is ≤0.018 subst./site for HRSV-A or ≤0.026 subst./site for HRSV-B. By applying this definition, we distinguish twenty-three genotypes within subtype HRSV-A (A1–A23) and six genotypes within subtype HRSV-B (B1–B6). The genotypes were temporarily named by ascending numbers until a suitable nomenclature can be defined. In order to test the robustness of our proposed genotype definition, we redefined the genotypes on subsets of the initial HRSV-A and HRSV-B data sets. Twenty-two out of twenty-three HRSV-A genotypes and all six HRSV-B genotypes were distinguished, with the same taxa clustering together, confirming the robustness of our method to a high extent.
The evolution of HRSV strains is a continuous process, with relatively rapid sequential replacement of dominating strains about every 7 years (Otieno et al. 2016). Consequently, HRSV classification, including the cutoffs used, may need further updating in the future with the prospect of increasing population turnover and sequence sampling. With our approach based on data patterns in complete genomes, we aim to formulate a strategy for future HRSV genotype classification.
Funding
The National Reference Center for Respiratory Pathogens received funding from Sciensano, the Institute of Public Health.
Data availability
Data are publicly available on GenBank.
Supplementary data are available at Virus Evolution online.
Conflict of interest: None declared.
Supplementary Material
Contributor Information
Kaat Ramaekers, KU Leuven, Department of Microbiology, Immunology and Transplantation, Rega Institute for Medical Research, Laboratory of Clinical and Epidemiological Virology, Herestraat 49 box 1040, BE-3000 Leuven, Belgium.
Annabel Rector, KU Leuven, Department of Microbiology, Immunology and Transplantation, Rega Institute for Medical Research, Laboratory of Clinical and Epidemiological Virology, Herestraat 49 box 1040, BE-3000 Leuven, Belgium.
Lize Cuypers, KU Leuven, Department of Microbiology, Immunology and Transplantation, Rega Institute for Medical Research, Laboratory of Clinical and Epidemiological Virology, Herestraat 49 box 1040, BE-3000 Leuven, Belgium; University Hospitals Leuven, Department of Laboratory Medicine and National Reference Centre for Respiratory Pathogens, Herestraat 49, BE-3000 Leuven, Belgium.
Philippe Lemey, KU Leuven, Department of Microbiology, Immunology and Transplantation, Rega Institute for Medical Research, Laboratory of Clinical and Epidemiological Virology, Herestraat 49 box 1040, BE-3000 Leuven, Belgium.
Els Keyaerts, KU Leuven, Department of Microbiology, Immunology and Transplantation, Rega Institute for Medical Research, Laboratory of Clinical and Epidemiological Virology, Herestraat 49 box 1040, BE-3000 Leuven, Belgium; University Hospitals Leuven, Department of Laboratory Medicine and National Reference Centre for Respiratory Pathogens, Herestraat 49, BE-3000 Leuven, Belgium.
Marc Van Ranst, KU Leuven, Department of Microbiology, Immunology and Transplantation, Rega Institute for Medical Research, Laboratory of Clinical and Epidemiological Virology, Herestraat 49 box 1040, BE-3000 Leuven, Belgium; University Hospitals Leuven, Department of Laboratory Medicine and National Reference Centre for Respiratory Pathogens, Herestraat 49, BE-3000 Leuven, Belgium.
References
- Agoti C. N. et al. (2014) ‘Examining Strain Diversity and Phylogeography in Relation to an Unusual Epidemic Pattern of Respiratory Syncytial Virus (RSV) in a Long-Term Refugee Camp in Kenya’, BMC Infectious Diseases, 14: 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agoti C. N. et al. (2017) ‘Transmission Patterns and Evolution of Respiratory Syncytial Virus in a Community Outbreak Identified by Genomic Analysis’, Virus Evolution, 3: vex006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agoti C. N. et al. (2015. a) ‘Local Evolutionary Patterns of Human Respiratory Syncytial Virus Derived from Whole-Genome Sequencing’, Journal of Virology, 89: 3444–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agoti C. N. et al. (2015. b) ‘Successive Respiratory Syncytial Virus Epidemics in Local Populations Arise from Multiple Variant Introductions, Providing Insights into Virus Persistence’, Journal of Virology, 89: 11630–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson L. J. et al. (1985) ‘Antigenic Characterization of Respiratory Syncytial Virus Strains with Monoclonal Antibodies’, The Journal of Infectious Diseases, 151: 626–33. [DOI] [PubMed] [Google Scholar]
- Anderson L. J. et al. (2019) ‘RSV Strains and Disease Severity’, The Journal of Infectious Diseases, 219: 514–6. [DOI] [PubMed] [Google Scholar]
- Arnott A. et al. (2011) ‘A Study of the Genetic Variability of Human Respiratory Syncytial Virus (HRSV) in Cambodio Reveals the Existence of a New HRSV Group B Genotype’, Journal of Clinical Microbiology, 49: 3504–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek H. Y. et al. (2012) ‘Prevalence and Genetic Characterization of Respiratory Syncytial Virus (RSV) in Hospitalized Children in Korea’, Archives of Virology, 157: 1039–50. [DOI] [PubMed] [Google Scholar]
- Bartholomeusz A. Schaefer S. (2004) ‘Hepatitis B Virus Genotypes: Comparison of Genotyping Methods’, Reviews in Medical Virology, 14: 3–16. [DOI] [PubMed] [Google Scholar]
- Battles M. B. McLellan J. S. (2019) ‘Respiratory Syncytial Virus Entry and How to Block It’, Nature Reviews Microbiology, 17: 233–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc A. et al. (2005) ‘Genotypes of Respiratory Syncytial Virus Group B Identified in Uruguay’, Archives of Virology Virol, 150: 603–9. [DOI] [PubMed] [Google Scholar]
- Boyoglu-Barnum S. et al. (2017) ‘Mutating the CX3C Motif in the G Protein Should Make a Live Respiratory Syncytial Virus Vaccine Safer and More Effective’, Journal of Virology, 91: e02059–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cane P. A. (2001) ‘Molecular Epidemiology of Respiratory Syncytial Virus’, Reviews in Medical Virology, 11: 103–16. [DOI] [PubMed] [Google Scholar]
- Cane P. A. et al. (1991) ‘Identification of Variable Domains of the Attachment (G) Protein of Subgroup A Respiratory Syncytial Viruses’, Journal of General Virology, 72: 2091–6. [DOI] [PubMed] [Google Scholar]
- Cane P. A. et al. (1992) ‘Analysis of Relatedness of Subgroup A Respiratory Syncytial Viruses Isolated Worldwide’, Virus Research, 25: 15–22. [DOI] [PubMed] [Google Scholar]
- Cane P. A. Pringle C. R. (1991) ‘Respiratory Syncytial Virus Heterogeneity during an Epidemic: Analysis by Limited Nucleotide Sequencing (SH Gene) and Restriction Mapping (N Gene)’, The Journal of General Virology, 72: 349–57. [DOI] [PubMed] [Google Scholar]
- Chernomor O. et al. (2015) ‘Split Diversity in Constrained Conservation Prioritization Using Integer Linear Programming’, Methods in Ecology and Evolution, 6: 83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi E. H. Lee H. J. (2000) ‘Genetic Diversity and Molecular Epidemiology of the G Protein of Subgroups A and B of Respiratory Syncytial Viruses Isolated over 9 Consecutive Epidemics in Korea’, The Journal of Infectious Diseases, 181: 1547–56. [DOI] [PubMed] [Google Scholar]
- Collins P. L. Melero J. A. (2011) ‘Progress in Understanding and Controlling Respiratory Syncytial Virus: Still Crazy after All These Years’, Virus Research, 162: 80–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui G. et al. (2013a) ‘Emerging Human Respiratory Syncytial Virus Genotype ON1 Found in Infants with Pneumonia in Beijing’, Emerging Microbes & Infections, 2: 1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui G. et al. (2013b) ‘Genetic Variation in Attachment Glycoprotein Genes of Human Respiratory Syncytial Virus Subgroups A and B in Children in Recent Five Consecutive Years’, PLoS One, 8: e75020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currier M. G. et al. (2016) ‘EGFR Interacts with the Fusion Protein of Respiratory Syncytial Virus Strain 2-20 and Mediates Infection and Mucin Expression’, PLoS Pathogens, 12: e1005622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuypers L. et al. (2018) ‘Time to Harmonize Dengue Nomenclature and Classification’, Viruses, 10: 569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dapat I. C. et al. (2010) ‘New Genotypes within Respiratory Syncytial Virus Group B Genotype BA in Niigata’, Journal of Clinical Microbiology, 48: 3423–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darriba D. et al. (2012) ‘JModelTest 2: More Models, New Heuristics and Parallel Computing’, Nature Methods, 9: 772–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giallonardo F. et al. (2018) ‘Evolution of Human Respiratory Syncytial Virus (RSV) over Multiple Seasons in New South Wales, Australia’, Viruses, 10: 476–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshaghi A. et al. (2012) ‘Genetic Variability of Human Respiratory Syncytial Virus a Strains Circulating in Ontario: A Novel Genotype with a 72 Nucleotide G Gene Duplication’, PLoS One, 7: e32807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L. et al. (2012) ‘CD-HIT: Accelerated for Clustering the Next-Generation Sequencing Data’, Bioinformatics (Oxford, England)), 28: 3150–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia O. et al. (1994) ‘Evolutionary Pattern of Human Respiratory Syncytial Virus (Subgroup A): Cocirculating Lineages and Correlation of Genetic and Antigenic Changes in the G Glycoprotein’, Journal of Virology, 68: 5448–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimenez H. B. et al. (1986) ‘Antigenic Variation between Human Respiratory Syncytial Virus Isolates’, Journal of General Virology , 67: 863–70. [DOI] [PubMed] [Google Scholar]
- Gimferrer L. et al. (2016) ‘Circulation of a Novel Human Respiratory Syncytial Virus Group B Genotype during the 2014–2015 Season in Catalonia (Spain)’, Clinical Microbiology and Infection, 22: 97.e5–97.e8. [DOI] [PubMed] [Google Scholar]
- Goya S. et al. (2018) ‘An Optimized Methodology for Whole Genome Sequencing of RNA Respiratory Viruses from Nasopharyngeal Aspirates’, PLoS One, 13: e0199714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S. Gascuel O. (2003) ‘A Simple, Fast, and Accurate Algorithm to Estimate Large Phylogenies by Maximum Likelihood’, Systematic Biology, 52: 696–704. [DOI] [PubMed] [Google Scholar]
- Hadfield J. et al. (2018) ‘NextStrain: Real-Time Tracking of Pathogen Evolution’, Bioinformatics, 34: 4121–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heylen E. et al. (2017) ‘Drug Candidates and Model Systems in Respiratory Syncytial Virus Antiviral Drug Discovery’, Biochemical Pharmacology, 127: 1–12. [DOI] [PubMed] [Google Scholar]
- Higgins D. et al. (2016) ‘Advances in RSV Vaccine Research and Development: A Global Agenda’, Vaccine, 34: 2870–75. [DOI] [PubMed] [Google Scholar]
- Hotard A. L. et al. (2015) ‘Identification of Residues in the Human Respiratory Syncytial Virus Fusion Protein That Modulate Fusion Activity and Pathogenesis’, Journal of Virology, 89: 512–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houspie L. et al. (2013) ‘Circulation of HRSV in Belgium: From Multiple Genotype Circulation to Prolonged Circulation of Predominant Genotypes’, PLoS One, 8: e60416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jombart T. Dray S. (2010) ‘Adephylo: Exploratory Analyses for the Phylogenetic Comparative Method’, Bioinformatics, 26: 1–21. [DOI] [PubMed] [Google Scholar]
- Katoh K. Standley D. M. (2013) ‘MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability’, Molecular Biology and Evolution, 30: 772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khor C. S. et al. (2013) ‘Displacement of Predominant Respiratory Syncytial Virus Genotypes in Malaysia between 1989 and 2011’, Infection, Genetics and Evolution, 14: 357–60. [DOI] [PubMed] [Google Scholar]
- Larsson A. (2014) ‘AliView: A Fast and Lightweight Alignment Viewer and Editor for Large Datasets’, Bioinformatics, 30: 3276–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemey P. Vandamme A.-M. Salemi M. (2009) The Phylogenetic Handbook. A Practical Approach to Phylogenetic Analysis and Hypothesis Testing, second edition. Cambridge University Press, Cambridge, UK. [Google Scholar]
- Li W. Godzik A. (2006) ‘Cd-Hit: A Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences’, Bioinformatics (Oxford, England)), 22: 1658–9. [DOI] [PubMed] [Google Scholar]
- Li Y. et al. (2019) ‘Global Patterns in Monthly Activity of Influenza Virus, Respiratory Syncytial Virus, Parainfluenza Virus, and Metapneumovirus: A Systematic Analysis’, The Lancet Global Health, 7: e1031.4. [DOI] [PubMed] [Google Scholar]
- Liu J. et al. (2014) ‘Genetic Variation of Human Respiratory Syncytial Virus among Children with Fever and Respiratory Symptoms in Shanghai, China, from 2009 to 2012’, Infection, Genetics and Evolution, 27: 131–6. [DOI] [PubMed] [Google Scholar]
- Martin D. P. et al. (2015) ‘RDP4: Detection and Analysis of Recombination Patterns in Virus Genomes’, Virus Evolution, 1: 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mufson M. A. et al. (1985) ‘Two Distinct Subtypes of Human Respiratory Syncytial Virus’, The Journal of General Virology, 66: 2111–24. [DOI] [PubMed] [Google Scholar]
- Otieno J. R. et al. (2016) ‘Molecular Evolutionary Dynamics of Respiratory Syncytial Virus Group A in Recurrent Epidemics in Coastal Kenya’, Journal of Virology, 90: 4990–5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pangesti K. N. A. et al. (2018) ‘Molecular Epidemiology of Respiratory Syncytial Virus’, Reviews in Medical Virology, 28: e1968. [DOI] [PubMed] [Google Scholar]
- Paradis E. Schliep K. (2019) ‘Phylogenetics Ape 5.0: An Environment for Modern Phylogenetics and Evolutionary Analyses in R’, Bioinformatics, 35: 526–8. [DOI] [PubMed] [Google Scholar]
- PATH. (2019) RSV Vaccines and MAb Snapshot. <https://vaccineresources.org/files/RSV-snapshot-2019_08_28_HighResolution_PDF.pdf> accessed 1 Dec 2019.
- Peret T. C. T. et al. (2000) ‘Circulation Patterns of Group A and B Human Respiratory Syncytial Virus Genotypes in 5 Communities in North America’, The Journal of Infectious Diseases, 181: 1891–6. [DOI] [PubMed] [Google Scholar]
- Peret T. C. T. et al. (1998) ‘Circulation Patterns of Genetically Distinct Group A and B Strains of Human Respiratory Syncytial Virus in a Community’, The Journal of General Virology, 79: 2221–9. [DOI] [PubMed] [Google Scholar]
- Prosperi M. C. F. et al. (2011) ‘A Novel Methodology for Large-Scale Phylogeny Partition’, Nature Communications, 2: 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaekers K. et al. (2017) ‘Prevalence and Seasonality of Six Respiratory Viruses during Five Consecutive Epidemic Seasons in Belgium’, Journal of Clinical Virology, 94: 72–8. [DOI] [PubMed] [Google Scholar]
- Revell L. J. (2012) ‘Phytools: An R Package for Phylogenetic Comparative Biology (and Other Things)’, Methods in Ecology and Evolution, 3: 217–23. [Google Scholar]
- Rima B., et al. (2017) ‘ICTV Virus Taxonomy Profile: Pneumoviridae’, The Journal of General Virology, 98: 2912–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossey I. Saelens X. (2019) ‘Vaccines against Human Respiratory Syncytial Virus in Clinical Trials, Where Are We Now?’ Expert Review of Vaccines, 18: 1053–67. [DOI] [PubMed] [Google Scholar]
- Salemi M. (2009) ‘Phylogenetic Inference Based on Distance Methods: Practice’, in Lemey P., Vandamme A.-M., Salemi M. (eds) The Phylogenetic Handbook. A Practical Approach to Phylogenetic Analysis and Hypothesis Testing, second edition, pp. 142–80. Cambridge University Press, Cambridge, UK. [Google Scholar]
- Schmidt H. A. et al. (2002) ‘TREE-PUZZLE: Maximum Likelihood Phylogenetic Analysis Using Quartets and Parallel Computing’, Bioinformatics, 18: 502–4. [DOI] [PubMed] [Google Scholar]
- Shi T. et al. (2017) ‘Global, Regional, and National Disease Burden Estimates of Acute Lower Respiratory Infections due to Respiratory Syncytial Virus in Young Children in 2015: A Systematic Review and Modelling Study’, The Lancet, 390: 946–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shobugawa Y. et al. (2009) ‘Emerging Genotypes of Human Respiratory Syncytial Virus Subgroup A among Patients in Japan’, Journal of Clinical Microbiology, 47: 2475–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmonds P. et al. (2017) ‘Consensus Statement: Virus Taxonomy in the Age of Metagenomics’, Nature Reviews. Microbiology, 15: 161–8. [DOI] [PubMed] [Google Scholar]
- Simoes E. A. F. et al. (2015) ‘Challenges and Opportunities in Developing Respiratory Syncytial Virus Therapeutics’, Journal of Infectious Diseases, 211: S1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. (2014) ‘RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies’, Bioinformatics, 30: 1312–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullender W. M. (2000) ‘Respiratory Syncytial Virus Genetic and Antigenic Diversity’, Clinical Microbiology Reviews, 13: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan L. et al. (2012) ‘Genetic Variability among Complete Human Respiratory Syncytial Virus Subgroup A Genomes: Bridging Molecular Evolutionary Dynamics and Epidemiology’, PLoS One, 7: e51439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson E. et al. (2016) ‘Comparison of Next-Generation Sequencing Technologies for Comprehensive Assessment of Full-Length Hepatitis C Viral Genomes’, Journal of Clinical Microbiology, 54: 2470–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trento A. et al. (2015) ‘Conservation of G-Protein Epitopes in Respiratory Syncytial Virus (Group A) Despite Broad Genetic Diversity: Is Antibody Selection Involved in Virus Evolution?’ Journal of Virology, 89: 7776–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trento A. et al. (2003) ‘Major Changes in the G Protein of Human Respiratory Syncytial Virus Isolates Introduced by a Duplication of 60 Nucleotides’, Journal of General Virology, 84: 3115–20. [DOI] [PubMed] [Google Scholar]
- Trento A. et al. (2006) ‘Natural History of Human Respiratory Syncytial Virus Inferred from Phylogenetic Analysis of the Attachment (G) Glycoprotein with a 60-Nucleotide Duplication’, Journal of Virology, 80: 975–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripp R. A. et al. (2001) ‘CX3C Chemokine Mimicry by Respiratory Syncytial Virus G Glycoprotein’, Nature Immunology, 2: 732–8. [DOI] [PubMed] [Google Scholar]
- Venter M. et al. (2001) ‘Genetic Diversity and Molecular Epidemiology of Respiratory Syncytial Virus over Four Consecutive Seasons in South Africa: Identification of New Subgroup A and B Genotypes’, Journal of General Virology, 82: 2117–24. [DOI] [PubMed] [Google Scholar]
- Vos L. M. et al. (2019) ‘High Epidemic Burden of RSV Disease Coinciding with Genetic Alterations Causing Amino Acid Substitutions in the RSV G-Protein during the 2016/2017 Season in the Netherlands’, Journal of Clinical Virology, 112: 20–6. [DOI] [PubMed] [Google Scholar]
- WHO/OIE/FAO H5N1 Evolution Working Group. (2008) ‘Toward a Unified Nomenclature System for Highly Pathogenic Avian Influenza Virus (H5N1)’, Emerging Infectious Diseases, 14: e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. (2016). Ggplot2: Elegant Graphics for Data Analysis. New York: Springer-Verlag. [Google Scholar]
- Yu G. et al. (2017) ‘Ggtree: An R Package for Visualization and Annotation of Phylogenetic Trees with Their Covariates and Other Associated Data’, Methods in Ecology and Evolution, 8: 28–36. [Google Scholar]
- Yu G. et al. (2018) ‘Two Methods for Mapping and Visualizing Associated Data on Phylogeny Using Ggtree’, Molecular Biology and Evolution, 35: 3041–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlateva K. T. et al. (2005) ‘Genetic Variability and Molecular Evolution of the Human Respiratory Syncytial Virus Subgroup B Attachment G Protein’, Journal of Virology, 79: 9157–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlateva K. T. et al. (2004) ‘Molecular Evolution and Circulation Patterns of Human Respiratory Syncytial Virus Subgroup A: Positively Selected Sites in the Attachment G Glycoprotein Molecular Evolution and Circulation Patterns of Human Respiratory Syncytial Virus Subgroup A: Positi’, Journal of Virology, 78: 4675–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are publicly available on GenBank.
Supplementary data are available at Virus Evolution online.
Conflict of interest: None declared.