

Abstract
Spirodiketopiperazine-based CCR5 antagonists, showing improved pharmacokinetic profiles without reduction in antagonist activity, were designed and synthesized. We also demonstrate the anti-HIV activity of a representative compound 12, as measured in a p24 assay.
Keywords: CCR5, Chemokine, Anti HIV-1
Chemokines, which were initially identified as chemoattractants in the context of leukocyte trafficking to sites of inflammation, exert a variety of biological activities by binding to their receptors on the surface of specific cells. Chemokines are a large family of small cytokines that selectively control the adhesion, chemotaxis, and activation of various leukocyte populations and are known to be involved in the initiation and progress of inflammation and allergic disease.1 They are classified into two main groups-CC chemokines and CXC chemokines-based on their conserved N-terminal cysteine residues. Additionally, CC chemokine receptor 5 (CCR5) and CXC chemokine receptor 4 (CXCR4) have attracted substantial interest because they form portals of cellular entry for human immunodeficiency viruses (HIV-1 and HIV-2) and related simian or feline retroviruses.2 Thus, identifying the CCR5 receptor antagonist, which inhibits HIV from binding to the specific receptor, is one of the most promising approaches for the treatment of AIDS, especially at the early stage of infection.3 Due to its novel mode of action, the CCR5 antagonist could be one of the final agents utilized in salvage therapy in combination with other active antiviral agents. Maraviroc is the only approved CCR5 receptor antagonist on the market for treating HIV-1 infection.4 In this study, we report the optimization process to find 4-(4-{[(3S)-1-butyl-3-(cyclohexylmethyl)-2,5-dioxo-1,4,9-triaza-spiro[5.5]undec-9-yl]methyl}phenoxy)benzoic acid hydrochloride (12) with the aim of improving the antiviral activity and pharma-cokinetics of the newly found chemical leads 1a and 1b (Fig. 1).
Figure 1.
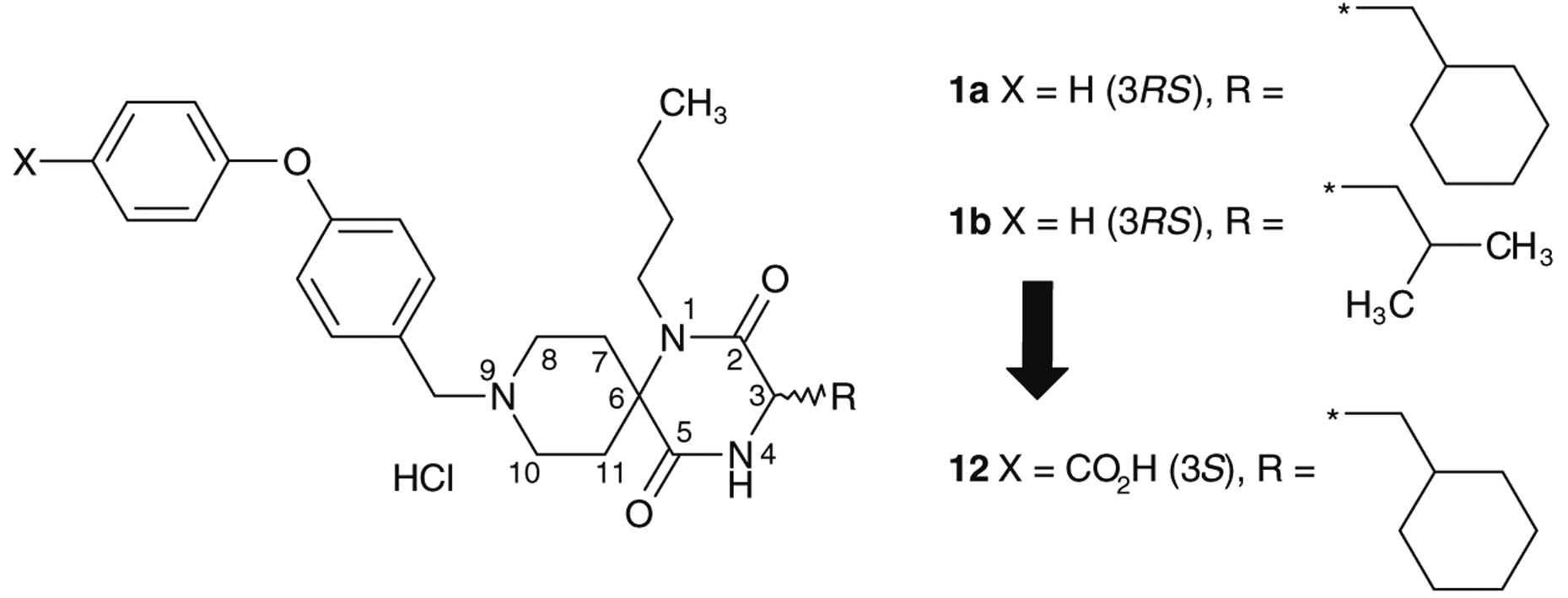
Discovery of an orally active chemical lead.
Compounds 1a, 1b, 2a, 2b, 3b and 4a were synthesized by the previously reported solid-phase method5a and compounds 6–12 were synthesized by the solution-phase method described in Scheme 1. A mixture of 1N-benzylpiperidin-4-one, n-butylamine, N-Boc-amino acid and 2-(morpholin-1-yl)ethylisocyanide in methanol was stirred at 55 °C.6 The Boc protecting group of an amino acid was removed by treatment with concentrated HCl without isolation of the Ugi product,6 cyclization of which, by heating in toluene in the presence of acetic acid at 80 °C, afforded 13. Removal of the benzyl group of 13 by catalytic hydrogenation produced the cyclized spirodiketopiperazine 14, which was isolated as HCl salt in 60–70% yield in four steps. Reductive alkylation of compound 14 resulted in a good yield (50–90%) of the desired products. Synthesis of 5a is described in Scheme 2. N-Alkylation of 14 with a tosylate, which was prepared by the tosylation of 4-phenoxyphenylethyl alcohol with polymer-supported tosyl chloride, afforded 5a. These basic compounds were isolated as their HCl salts.
Scheme 1.
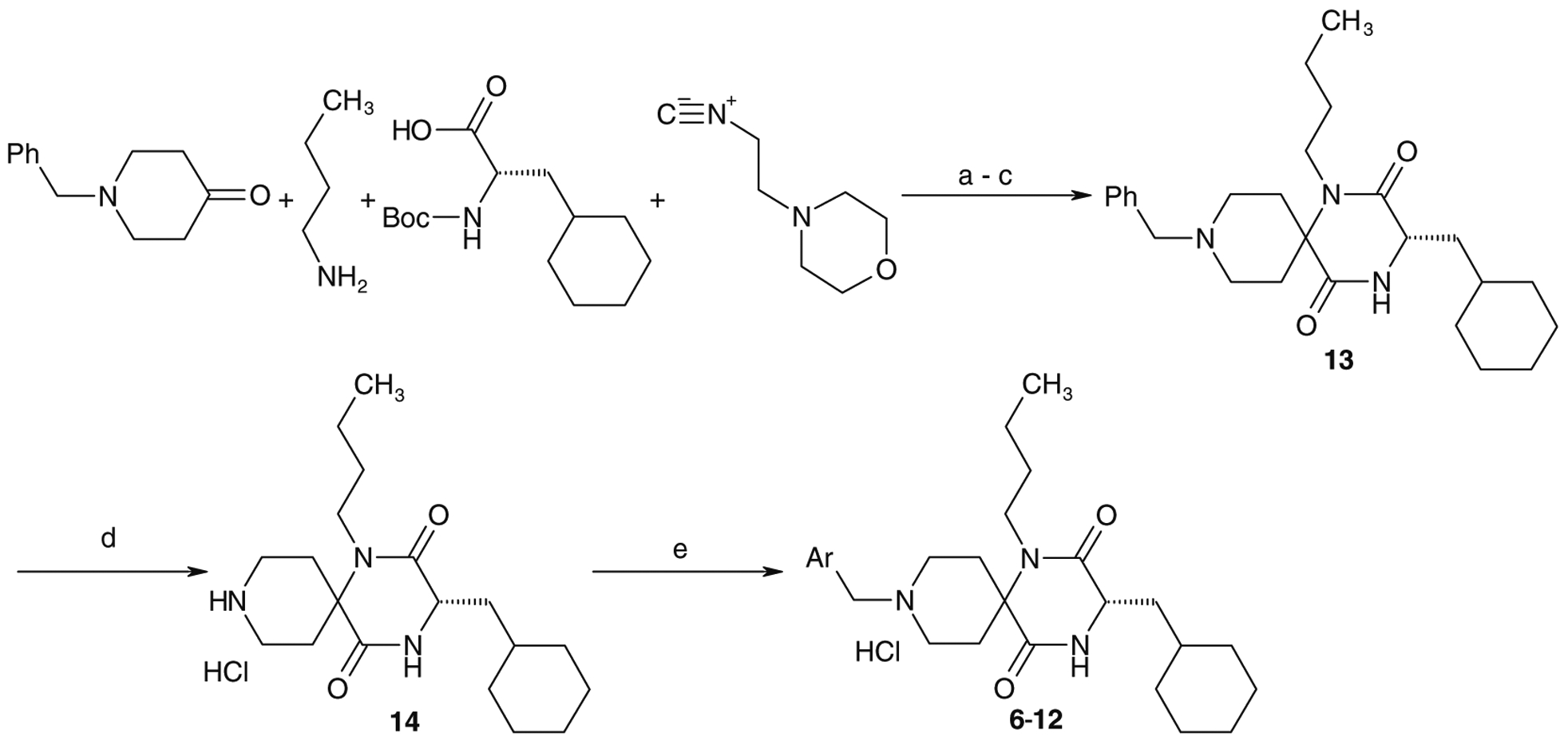
Typical synthetic route of spirodiketopiperazines. Reagents and conditions: (a) MeOH, 55 °C; (b) concd HCl, 55 °C; (c) AcOH, toluene, 80 °C; (d) H2, Pd(OH)2/C, EtOH, 55 °C then 4 N HCl/AcOEt (60–70% in four steps); (e) Ar-CHO, NaBH(OAc)3, AcOH, DMF, then 4 N HCl/AcOEt (50–90%).
Scheme 2.
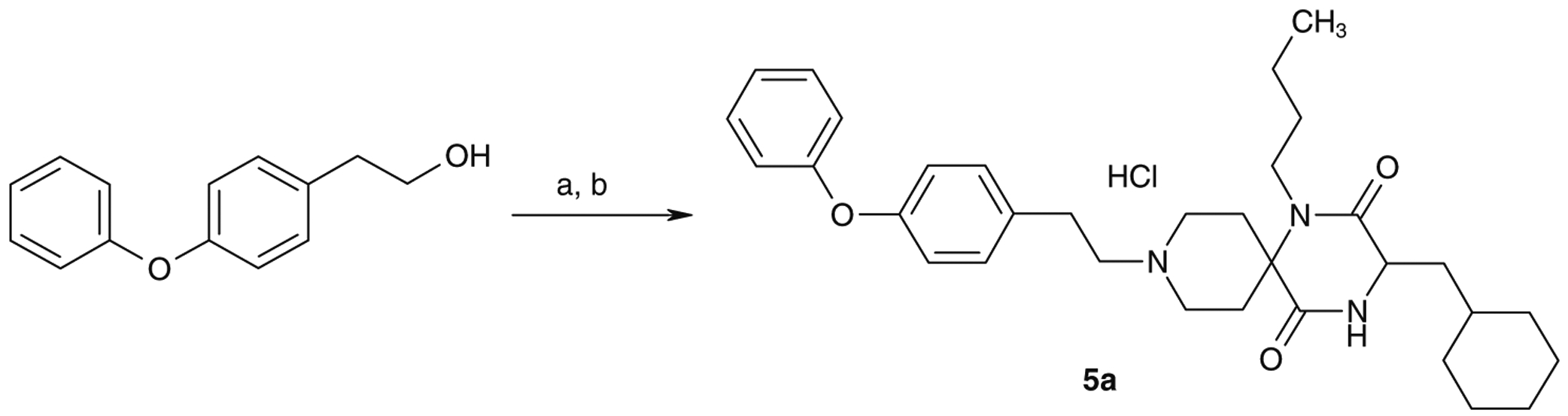
Synthesis of 5a. Reagents and conditions: (a) polystyrene-supported tosylchloride, pyridine, CH2Cl2; (b) 14, iPr2NEt, MeCN, then 4 N HCl/AcOEt (48% in two steps).
Compounds listed in Tables 1–4 were evaluated for their inhibitory activities against calcium mobilization of human CCR5 overexpressed CHO cells (hCCR5/CHO) stimulated by MIP-1α (Ca assay).7
Table 1.
Hits from the newly designed G-protein coupled receptor (GPCR) directed library
 | ||
|---|---|---|
| Compds | R1 | IC50 (nM) Ca assay |
| 1a | 28 | |
| 1b | 94 | |
| 2a | 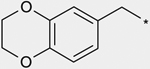 |
50 |
| 2b | 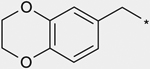 |
180 |
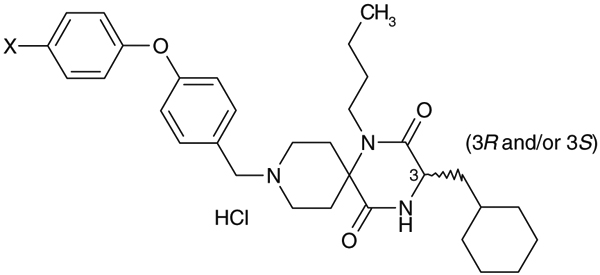
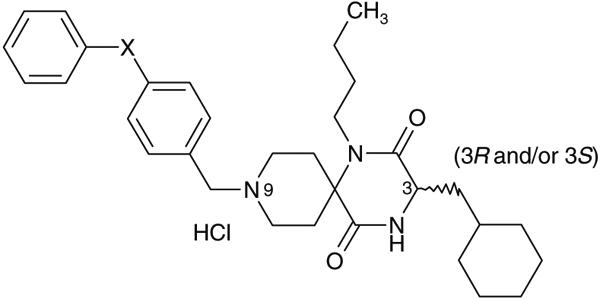
Table 4.
Effect of the p-substituent of the biphenyl ether residue on activity profiles
 | ||||
|---|---|---|---|---|
| Compds | X | IC50 (nM) Ca assay | C3 configuration (3R and/or 3S) | Stability in rat liver microsomes% remaining at 15 min |
| 8 | Me- | 79 | (3S) | 48 |
| 9 | MeO- | 130 | (3S) | 34 |
| 10 | HO- | 42 | (3S) | 22 |
| 11 | F- | 92 | (3S) | 53 |
| 12 | HO2C- | 13 | (3S) | 22 |
| 1a | H- | 28 | (3RS) | 33 |
We previously reported the discovery of spirodiketopiperazines 1a, 1b, 2a, and 2b (Table 1), as the novel chemical leads of CCR5 antagonist from our combinatorial library targeting G-protein coupled receptors (GPCRs).5 Analogues 1a and 2a tended to show stronger activity compared with the corresponding 3-isobutyl analogues 1b and 2b, respectively. Although 1a, 1b, 2a, and 2b showed from potent to moderate antagonist activity in vitro, they were not expected to display potent activity in vivo because of their unfavorable PK data, including poor area under the concentration-time curve (AUC), high clearance (CL) and distribution values (Vss), as shown in Table 5. Accordingly, structural optimization was focused not only on increased activity but also on the improvement of these PK values. Our optimization process was initiated with chemical modification of the N-substituents at the 9-position using readily available spirodiketopiperazine 14 as a key intermediate.
Table 5.
Pharmacokinetic data for compounds 1a and 11 in rats
| Compds | 30 mg/kg, po | 30 mg/kg, iv | ||||||
|---|---|---|---|---|---|---|---|---|
| Cmax ng/mL | T1/2 min | AUC ng h/m (0-∞) | BA% | AUC ng h/mL (0-∞) | T1/2 min | CL mL/min/mL | Vss mL/kg | |
| 1a | 16.7a | 103 | 74.4a | 1.9 | 400 | 19.9 | 113 | 2542 |
| 12 | 7200 | 48.4 | 10,532 | 34.1 | 3091 | 11.1 | 16 | 145 |
Dose normalized AUC and Cmax to 30 mg/kg.
As shown in Table 2, reductive amination of 14 with commercially available benzaldehyde and p-methoxybenzaldehyde provided 3b and 4a, respectively. Although reduction of the molecular weight was expected to be a promising strategy for improving oral absorption, this approach resulted in reduced activity. N-Alkylation of 14 with 4-phenoxyphenylethyltosylate afforded 5a, also with remarkable reduction in activity relative to 1a. As a result, the 9-N-phenylmethyl group was found to be one of the structural requirements for antagonist activity. Introduction of a p-phenoxy substituent into the 9-N-phenylmethyl group of 3b tended to show increased activity, as illustrated by the increased potency of 1b relative to 3b.
Table 2.
Effect of the spirodiketopiperazines 9-N-substituents on activity profiles
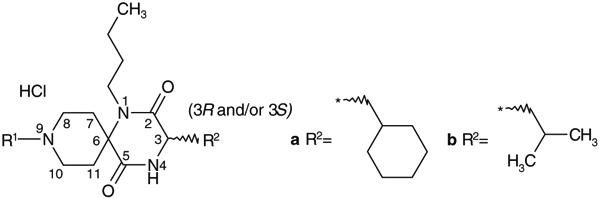 | |||
|---|---|---|---|
| Compds | R1 | C3 configuration (3R and/or 3S) | IC50 (nM) Ca assay |
| 3b | 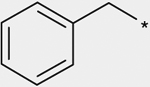 |
(3RS) | 900 |
| 4a | 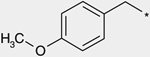 |
(3RS) | 120 |
| 1a | (3RS) | 28 | |
| 1b | 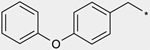 |
(3RS) | 94 |
| 5a | (3S) | ca. 10,000 | |
As shown in Table 3, the effect of chemical modification of the linker X on activity profiles was investigated. Replacement of the 9-N-4-phenoxyphenylmethyl moiety of 1a with 9-N-4-phenylthio-phenylmethyl afforded a diphenyl sulfide analogue 6 with less potent activity. Replacement of the sulfide moiety of 6 with sulfone afforded 7, which showed slightly less potent activity relative to the corresponding ether analogue 1a. For this reason, the following optimization was focused on the synthetic work of diphenyl ether derivatives.
Table 3.
Effect of chemical modification of a linker X on activity profiles
 | |||
|---|---|---|---|
| Compds | R1 | C3 configuration (3R and/or 3S) | IC50 (nM) Ca assay |
| 6 | 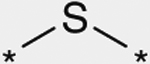 |
(3S) | 170 |
| 7 | 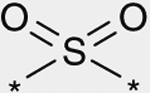 |
(3S) | 86 |
| 1a | 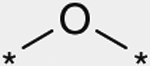 |
(3RS) | 28 |
Effect of a p-substitution of the predicted metabolic site of the terminal phenyl moiety of 1a on stability in rat liver microsomes was investigated.8 As shown in Table 4, introduction of p-methyl group and p-methoxy group as electron-donating substituents into the terminal phenyl moiety of 1a afforded 4-(p-methylphenoxy)phenylmethyl and 4-(p-methoxyphenoxy)phenylmethyl analogues 8 and 9, respectively with a tendency of showing slightly less potent activity relative to 1a, while demethylation of 9 afforded an analogue 10 with a tendency of showing slightly more potent activity relative to the corresponding methoxy analogue 9. Introduction of a p-fluoro group as an electron-withdrawing group instead of p-methyl and p-methoxy groups afforded 11 with a slightly less potent activity relative to 1a. Introduction of a carboxylic acid group as an electron-withdrawing and hydrophilic substituent as a p-substituent afforded 12 with an increased activity. Stability of these test compounds in the rat liver microsomes was investigated but these in vitro data did not indicate a significant improvement in metabolic stability in rat liver microsomes.9
PK data obtained after single-dose oral administration of the initial chemical lead 1a and the representative compound 12 to rats are presented in Table 5. As described previously, the initial lead 1a showed very poor bioavailability (1.9%). Other PK values such as the maximum plasma concentration (Cmax), plasma elimination half-life (T1/2) and AUC were also very poor. The probable reasons for such poor PK values of 1a were the large clearance (CL = 113 mL/min/kg) and distribution volumes (Vss = 2542 mL/kg) which were unfavorable for drugs which show efficacy in the blood, such as anti-HIV drugs. However, benzoic acid analogue 12 showed significantly improved PK values such as Cmax (7200 ng/mL), oral exposure (AUC = 10532 ng h/mL) and bioavailability (BA = 34.1%) after its oral dosing. Remarkable improvement of solubility (26 μM)9 and Caco2 permeability (26.4 × 10−6 cm/s)9 of 12 relative to 1a (solubility: less than 5 μM and Caco2 permeability: not detected) was estimated to be one of the most plausible reasons. The marked reduction in clearance (CL = 16 mL/min/kg) and distribution volume (Vss = 145 mL/kg) after intravenous dosing was considered to be another plausible reason for the improved AUC and BA. The marked reduction of CL of 12 strongly suggested in vivo metabolic stabilization, although in vitro studies did not indicate a significant improvement in metabolic stability.
Furthermore the representative compound 12, PK profiles of which were significantly improved relative to the initial lead 1b without reduction of the potent antagonist activity, was investigated for its anti-HIV activity using a launched reverse transcriptase inhibitor Zidovudine as a positive control. Results are summarized in Table 6. Compound 12 showed an IC50 value of 39 nM in an anti-HIV-1 p24 assay (using PBMC as the target cells7).
Table 6.
Anti-HIV activity of the representative compounds
| Compds | Anti-HIV-1 activity in p24 assay HIV-1Ba-L (R5) IC50 (nM) |
|---|---|
| 1b | 160 |
| 12 | 39 |
| Zidovudinea | 60 |
| Nelfinavirb | 12 |
Zidovudine is reverse transcriptase inhibitor.
Nelfinavir is HIV-1 protease inhibitor.
In conclusion, starting with the initial hit compounds 1a and 1b which showed unfavorable PK profiles, we identified compound 12 which showed significant improvement in bioavailability and oral exposure (AUC) without reduction in activity. Compound 12 was produced by introducing a carboxylic acid group into the p-position of the terminal phenyl moiety. Although the role of carboxylic acid is still unclear, compound 12 is thought to show improved Cmax, AUC and BA after oral dosing because of its much improved solubility and Caco2 permeability. However, its oral absorption process has not yet been elucidated. The significant reduction of CL and Vss of compound 12 was also considered to be another plausible reason for the increased Cmax, AUC and BA. As such, introduction of carboxylic acid into the p-position of the terminal phenoxy moiety was found to be effective not only to block metabolic deactivation but also to improve PK profiles. The representative compound 12 showed more potent activity than 1b in the p24 assay (with the BAL strain of HIV). Further optimization of compound 12 to improve its activity and PK profile, will be reported in near future. These findings will contribute further to the development of CCR5 antagonists for clinical use.
References and notes
- 1.(a) Schwarz MK; Wells TNC Nat. Rev. Drug Disc 2002, 1, 347; [DOI] [PubMed] [Google Scholar]; (b) Gerard C; Rollins BJ Nat. Immunol 2001, 2, 108. [DOI] [PubMed] [Google Scholar]
- 2.(a) Feng Y; Broder CC; Kennedy PE; Berger EA Science 1996, 272, 872;8629022 [Google Scholar]; (b) Deng HK; Liu R; Ellmeier W; Choe S; Unutmaz D; Burkhart M; Marzio PD; Marmon S; Sutton RE; Hill CM; Davis CB; Peiper SC; Schall TJ; Littman DR; Landau NR Nature 1996, 381, 661; [DOI] [PubMed] [Google Scholar]; (c) Dragic T; Litwin V; Allaway GP; Martin SR; Huang Y; Nagashima KA; Cayanan C; Maddon PJ; Koup RA; Moore JP; Paxton WA Nature 1996, 381, 667; [DOI] [PubMed] [Google Scholar]; (d) Alkhatib G; Combadiere C; Broder CC; Feng Y; Kennedy PE; Murphy PM; Berger EA Science 1996, 272, 1955; [DOI] [PubMed] [Google Scholar]; (e) Berger EA; Murphy PM; Farber JM Annu. Rev. Immunol 1999, 17, 657; [DOI] [PubMed] [Google Scholar]; (f) Caldwell DJ; Evans JD Exp. Opin. Pharmacother 2008, 9, 3231. [DOI] [PubMed] [Google Scholar]
- 3.Hoffmann C; Mulcahy F Overview of Antiretroviral Agents In HIV Medicine 2006; Hoffmann C, Rockstroh JK, Kamps BS, Eds.; Flying: Paris, 2006; p 94 FlyingPublisher.com. [Google Scholar]
- 4.Leonard JT; Roy K Curr. Med. Chem 2006, 13, 91. [DOI] [PubMed] [Google Scholar]
- 5.(a) Habashita H; Kokubo M; Hamano S; Hamanaka N; Toda M; Shibayama S; Tada H; Sagawa K; Fukushima D; Maeda K; Mitsuya HJ Med. Chem 2006, 49, 4140; [DOI] [PubMed] [Google Scholar]; (b) Nishizawa R; Nishiyama T; Hisaichi K; Matsunaga N; Minamoto C; Habashita H; Takaoka Y; Toda M; Shibayama S; Tada H; Sagawa K; Fukushima D; Maeda K; Mitsuya H Bioorg. Med. Chem. Lett 2007, 17, 727. [DOI] [PubMed] [Google Scholar]
- 6.Domling A; Ugi I Angew. Chem., Int. Ed 2000, 39, 3168. [DOI] [PubMed] [Google Scholar]
- 7.(a) Maeda K; Yoshimura K; Shibayama S; Habashita H; Tada H; Sagawa K; Miyakawa T; Aoki M; Fukushima D; Mitsuya HJ Biol. Chem 2001, 276, 35194; [DOI] [PubMed] [Google Scholar]; (b) Maeda K; Nakata H; Koh Y; Miyakawa T; Ogata H; Takaoka Y; Shibayama S; Sagawa K; Fukushima D; Moravek J; Koyanagi Y; Mitsuya HJ Virol. 2004, 78, 8654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kalfutkar AS; Gardner I; Obach RS; Shaffer CL; Callegari E; Henne KR; Mutlib AE; Dalvie DK; Lee JS; Nakai Y; O’Donnell JP; Boer J; Harriman SP Curr. Drug Metabol 2005, 6, 161. [DOI] [PubMed] [Google Scholar]
- 9.Full details of the experimental will be reported very soon in the full paper which we are preparing. [Google Scholar]