

Abstract
Drug-induced liver injury is a major reason for drug candidate attrition from development, denied commercialization, market withdrawal, and restricted prescribing of pharmaceuticals. The metabolic bioactivation of drugs to chemically reactive metabolites (CRMs) contribute to liver-associated adverse drug reactions in humans that often goes undetected in conventional animal toxicology studies. A challenge for pharmaceutical drug discovery has been reliably selecting drug candidates with a low liability of forming CRM and reduced drug-induced liver injury potential, at projected therapeutic doses, without falsely restricting the development of safe drugs. We have developed an in vivo rat liver transcriptional signature biomarker reflecting the cellular response to drug bioactivation. Measurement of transcriptional activation of integrated nuclear factor erythroid 2-related factor 2 (NRF2)/Kelch-like ECH-associated protein 1 (KEAP1) electrophilic stress, and nuclear factor erythroid 2-related factor 1 (NRF1) proteasomal endoplasmic reticulum (ER) stress responses, is described for discerning estimated clinical doses of drugs with potential for bioactivation-mediated hepatotoxicity. The approach was established using well benchmarked CRM forming test agents from our company. This was subsequently tested using curated lists of commercial drugs and internal compounds, anchored in the clinical experience with human hepatotoxicity, while agnostic to mechanism. Based on results with 116 compounds in short-term rat studies, with consideration of the maximum recommended daily clinical dose, this CRM mechanism-based approach yielded 32% sensitivity and 92% specificity for discriminating safe from hepatotoxic drugs. The approach adds new information for guiding early candidate selection and informs structure activity relationships (SAR) thus enabling lead optimization and mechanistic problem solving. Additional refinement of the model is ongoing. Case examples are provided describing the strengths and limitations of the approach.
Keywords: drug safety, drug-induced liver injury, bioactivation, NRF2, NRF1, chemically reactive metabolites, rat liver, transcriptional biomarkers, lead optimization, drug candidate attrition
Later stage drug candidate attrition from the pharmaceutical development pipeline can result when nonclinical animal studies do not accurately recapitulate inevitable clinical toxicities. Drug-induced liver injury (DILI) is a major reason for attrition during drug development, denied commercialization, withdrawal from the marketplace and for restricted prescribing of new pharmaceuticals that may gain marketing approval. However, as noted by pharmaceutical industry collaborators unanticipated liver safety liabilities are the second most poorly predicted clinical toxicity by conventional animal studies, reportedly unable to identify roughly 50% of drugs with human DILI that are discovered later in clinical development (Olson et al., 2000). Furthermore, difficulties in differentiating the potential clinical implications of ambiguous or subtle liver safety signals seen in standard regulatory animal studies have been described (Sistare et al., 2016). Here, we describe additional gene transcriptional endpoints refining initial requisite rat studies by providing greater insights to human DILI potential early in preclinical development that can reduce drug failures and the need for additional animal studies in keeping with 3R principles. Moreover, improved prediction of DILI potential will reduce risk to patients during clinical trials, reduce the cost burden of late-stage clinical failures, and resultingly accelerate the successful development of medicines.
Recent reviews (Blomme and Will, 2016; Sistare et al., 2016) summarize current strategies that are deployed nonuniformly by pharmaceutical companies to improve earlier understanding of human DILI risk potential, and describe numerous in vitro test systems that have been variably applied to inform 4 nonexclusive DILI mechanisms including innate and acquired immune system modulation, altered bile acid homeostasis, altered mitochondrial function, and formation of chemically reactive metabolites (CRM). CRM formation of electrophiles may be the most commonly encountered core underlying mechanism of the four. CRM can result in covalent binding to pivotal proteins causing toxicity via disruption of vital cellular functions, or it can indirectly trigger liver injury via immune-mediated mechanisms likely involving hapten formation (Guengerich, 2011; Park et al., 2011). Radiochemical and trapping methods have been described for screening drug candidates for CRM formation potential and shown to poorly discriminate liver toxic from liver-safe compounds (Bauman et al., 2009; Obach et al., 2008; Usui et al., 2009). Nakayama et al. (2009) proposed that a measure of CRM formation in human hepatocytes when considered together with human daily dose results in improved discrimination, but we report here disappointing results from efforts to further validate this strategy. These chemical-based measures of assessing a drug’s CRM formation potential are subject to important limitations including the location of radiolabel placement on the molecule, potential for hydrolysis of tritium-based radiolabel before adduction, challenges of routine 14C-based labeling, choice of trapping agent, use of tissue homogenate, misinterpretation of drugs that may adduct only a specific protein in a targeted and innocuous manner, etc. Although novel more advanced in vitro models hold tremendous promise and are certain to help address this problem (Blomme and Will, 2016; Sistare et al., 2016), none of these have been adopted for regulatory purposes presently. Rather the conduct of conventional animal toxicology studies in 2 species is relied upon by regulatory authorities to evaluate human liver toxicity potential and will remain the cornerstone for the foreseeable future, as novel emerging in vitro approaches are certain to gain eventual acceptance as optional supplements.
We reasoned that a measure of a hepatocyte’s acute biological response following intracellular generation of a significant burden of electrophiles adducting a large array of proteins could add enhanced mechanistic and predictive insight of human DILI potential to animal toxicology studies already being conducted. Furthermore, in contrast to dedicated radiolabel in vitro or in vivo strategies, this approach would require only modest additional resources when incorporated into an early rat tolerability study designs, and can be translated to a complementary in vitro platform (Kang et al., 2020).
We (Podtelezhnikov et al., 2020) and others (Chia et al., 2010; Leone et al., 2014) have noted that certain drugs associated with clinical DILI and known to form electrophilic intermediates can activate the compensatory antioxidant response element/NRF2 (nuclear factor erythroid 2-related factor 2)/Keap1 (Kelch-like ECH-associated protein 1) antioxidant response pathway, presumably as a protective mechanism. We have also demonstrated using over 100 compounds (Podtelezhnikov et al., 2020) that the majority of these molecules will concurrently activate the nuclear factor erythroid 2-related factor 1 (NRF1) proteasomal endoplasmic reticulum (ER) stress pathway. With this in mind, we assessed the ability of the NRF2/Keap1 and the NRF1 coregulated gene network in rat liver to distinguish a library of known high, medium, and low covalent protein-binding (CPB) drug candidates discontinued from the MSD pipeline. After refining this drug bioactivation tissue biomarker to the most reliably responsive gene set, we assessed its ability to distinguish drugs with known clinical DILI risk from those without evidence of DILI risk, and developed thresholds and strategies for incorporating this insightful endpoint into early rat tolerability studies as a bioactivation liver response assay (BA-LRA).
MATERIALS AND METHODS
Curation of DILI Compound Training (Threshold Setting) and Test (Threshold Verification) Sets
Attempts to classify drugs on the basis of risk for DILI have yielded variable results (Chen et al., 2016; Stepan et al. 2011; Suzuki et al., 2010). Several sources were referenced to make judgments on liver safety for drugs used for this assessment. Our approach in this analysis is described below. Clinical case reports from published literature, product labels, the NIH LiverTox database, published country registries were interrogated. Marketed drugs labeled with more moderate and nonlife-threatening increases in aminotransferases, representing a reversible and adaptive response to drug treatment and not documented to be accompanied by acute liver failure indicating permanent injury to the liver, were not classified as DILI-positive clinical hepatotoxicants. True clinical hepatotoxicants were identified for testing among the marketed drugs with labeled warnings and precautions or those marketed but withdrawn compounds that have documented clinical diagnoses of significant incidences of acute liver failure. Marketed drugs that are not associated with documented instances of acute liver failure and that have had wide market exposure over prolonged time were considered liver nontoxicants. Because high daily dose is a risk, drugs were sought that have been administered safely at relatively high doses with greater likelihood to result in significant daily liver dose burdens. In addition, compounds were selected for testing that had been discontinued by pharmaceutical sponsors at clinical stages of drug development due to strong liver safety transaminase signals, but without allowing instances of acute liver failure (premarket DILI signal notation in Supplementary Table 1). For such drugs, the liver safety signals were considered sufficiently strong for sponsors to discontinue development and so are representative of the type of test candidate that needs to be identified earlier, and therefore were also characterized as true clinical DILI positives. The list of drugs, their categorical assignments, and rationale supporting their categorization have been described recently (Xu et al., 2019) and are provided in Supplementary Table 1.
Animal Studies
Male Sprague Dawley, strain Crl:CD(SD), or Wistar, strain Crl:WI(HAN), rats were obtained from Charles River Laboratories (Raleigh, North Carolina) for studies conducted in the United States, or from Charles River Laboratories (Saint Germain sur l’Arbresle, France), for studies conducted in Mirabel. Initially studies were conducted in Sprague Dawley until the departmental decision was made to switch to Wistar strain rats for all routine toxicology studies. Supplementary Table 1 describes which compounds were evaluated in each strain and the compounds that were assessed in both strains as bridging studies that ensured no significant difference in assay performance was seen between strains. Animals were 6–10 weeks of age, weighing 120–425 g. The rats were housed individually in wire mesh cages (at 18°C–26°C, relative humidity of 50 ± 20% on a 12-h light/dark cycle), fed PMI Certified Rodent Diet (SD-restricted [fed 16–22 g shortly after dosing 1× per day]; Wistar ad libitum). Animals were acclimated for at least 4 days prior to randomization into treatment groups. All animal husbandry and experimental procedures were in accordance with the 'Guide for the Care and Use of Laboratory Animals' and were approved by the Institutional Animal Care and Use Committee of the facility in which the studies were conducted. Animals were dosed daily via oral gavage with vehicle or with compounds for 4 days (daily formulation prep) at 1 of 2 dose levels and sacrificed 24 h later. For the studies conducted with the DILI Compound Set, the doses selected were generally a high dose level of 500–750 mkd, and a lower dose level of 100–300 mkd. We empirically discovered early on (described below) that daily doses exceeding 300 mkd are generally needed for rat liver to elicit a reliably detectable BA-LRA gene expression signal for human DILI-positive compounds. A standard high dose of 500–750 mkd which is roughly 2-fold above the 300-mkd threshold dose, was chosen to maximize opportunity for exposing a drug’s CRM potential with a signal that can be seen against a significant background of NRF1/NRF2 basal activation tone, and also not to be so high a dose that it would generally be sufficiently well tolerated for 4 days of dosing. The 2-fold lower dose level generally assures that some data will be generated at a tolerable dose in cases where 500–750 mkd may be poorly tolerated; it may also provide valuable insight to the shape of the dose-response curve. In some studies, such high doses were not tolerated (eg, clotrimazole, meloxicam, tacrine, olanzapine, verapamil) and effects seen at maximally tolerated lower doses are presented. At lower doses, negative BA-LRA outcomes are considered insufficiently tested (InsfT), whereas positive BA-LRA results even at such lower dose levels are considered positive evidence of CRM-mediated DILI potential. In 3 instances, results from day 1 single dose study sample collections are presented, as the preferred longer duration multiple dose study samples were not tolerated (olanzapine [DILI negative and InsfT]; clozapine [DILI positive and BA-LRA positive]; pemoline [DILI positive and BA-LRA positive]).
CPB Studies in Human Hepatocytes
The 14C-labeled pharmaceuticals were synthesized and characterized at Moravek Biochemicals, Brea, California, using literature-derived procedures. The materials were further characterized on receipt at MSD Research Laboratories (Rahway, New Jersey) to confirm purity. The radiochemicals were at least 98.5% pure with no single impurity present at > 0.5%. The position of the labels is as shown in Supplementary Table 2. Cryopreserved pooled human male hepatocytes from 10 donors (Cat No. MX008001; Lot No. SAK) were obtained from Celsis Corporation, now Bioreclamations, New York. In vitro GRO hepatocyte incubation medium (Cat No. Z99099) and in vitro GRO Krebs Henseleit buffer (Cat No. Z99074) were also purchased from Celsis Corporation. Ethanol and HPLC grade acetonitrile were purchased from Fisher Scientific, New York.
Test Compound (10 µM) incubations (approximately 0.5–1µCi/ml) were carried out in triplicate in 250 µl volume at a cell density of 1 × 106 cells/ml. Incubations were conducted in Krebs-Henseleit buffer at 37°C for 2 and 120 min. Cellular viability was at least ≥ 79% prior to initiating the incubation. Incubations were quenched using 1 ml of acetonitrile/ethanol (1:1) mixture followed by sonication, vortexing and centrifugation (3000 × g) to obtain a pellet. The next wash (acetonitrile/ethanol) cycle included sonicating, vortexing, and freezing of the pellet at −20°C for 10 min. The pellet was washed again 4 more times in this sequence and the final wash solution was counted for radioactivity after addition of scintillation cocktail (0.25:7). Typically, 5 washes reduced the counts to < 2X of that of the background values. Control cells (0.25 ml) in duplicate were quenched with 1 ml of acetonitrile/ethanol (1:1) mixture, vortexed and centrifuged to obtain the pellets. The pellets of control cells and samples were dissolved in 1 ml of 0.25 N NaOH, vortexed and left on the bench overnight. Aliquots (0.8 ml) were neutralized with 0.25 N HCl and counted for radioactivity after addition of 17 ml of cocktail. In addition, maximum controls (n = 3) containing the initial radioactivity were directly counted to define the maximal binding. Blanks were prepared so as to contain the same ratio of NaOH/HCl. The protein assay was performed by the bicinchoninic acid method using 20 µl of the pellet solutions and reading the absorbance at 562 nm. The percentage of protein recovered following the washing cycles was calculated by comparing with protein contents of the control cells prior to the washing cycles. CPB per mg protein was calculated using the equation below (disintegrations per minute DPM):
RNA Preparation and Transcriptional Data Analysis
Approximately 24 h after dosing, animals were sacrificed, livers removed, a section (approximately 50–100 mg) frozen on dry ice and stored at approximately −80°C until total RNA was isolated. Frozen liver was pulverized in TRIzol reagent, extracted with Chloroform, and further purified using the RNEasy column RNA isolation procedure (Qiagen, Chatsworth, California).
Affymetrix microarray analysis was used for the genome-wide assessment of transcriptional response to CRM for 26 compounds with measured CPB. Fifty nanograms of each purified RNA sample was amplified and labeled using a custom automated version of the NuGEN Ovation WB protocol. Hybridization to custom rat Affymetrix arrays (containing 36 991 probesets, GEO platform GPL28473), labeling and scanning were completed following the manufacturer’s recommendations and profiles were normalized using robust multiarray average as described (Irizarry et al., 2003) followed by ratio’ing to corresponding vehicle control samples within each study. The data are available under GEO accession number GSE149734.
Pathway enrichment analyses were performed by comparing input sets to GeneGo (www.genego.com), Ingenuity (www.ingenuity.com), and KEGG (www.genome.jp/kegg/) pathway sets. Bonferroni-corrected hypergeometric p values (expectation [e] values) of less than 0.1 were considered significant overlap between sets.
For assessment of BA-LRA signature performance, primarily either a 384-well microfluidic taqcard assessing 48 transcripts per sample, or a 224-well open array that contains 198 genes of interest (several wells contained duplicate assays for monitoring technical replicates) were used. Both platforms contain the 46 constituent genes of the BA-LRA signature as well as endogenous control genes for RNA loading normalization. The open array contains additional genes associated with other liver processes and functions. Each well of the taqcard or open array contains gene-specific primers and a fluorescently labeled probe for detection on the Taqman real-time PCR instrument Applied Biosystems (ABI). Using ABI reagents and procedures, total RNA was reverse transcribed at 5 ng/µl (taqcard) or 200 ng/µl (open array) using random hexamers. Samples from the reverse transcription reaction were amplified over 40 cycles of PCR and the PCR cycle where the amplified product crossed the threshold (10x the SD of background fluorescence) was captured. The Ct (384-well taqcard) or Crt (224-well open array) was used to calculate relative fold changes of all samples normalized to the pooled concurrent study control animals using the ΔΔCt calculation (Applied Biosystems User Bulletin No. 2) for specific genes of interest. An extended set of endogenous control genes (generally resistant to acute transcriptional regulation), including Hnrnpul1, Inpp5a, Ddx47, Pum1, Srrm1, Tlk2, Gusb, Rab35, Tmem183a, Rchy1, Tmed4, and 18S, was utilized as available on each array for increased measurement accuracy. Gene set signature scores were calculated by taking the unweighted average log10 ratio of the constituent genes. For a small number of studies indicated, previously obtained Affymetrix or Agilent microarray data was used to calculate BA-LRA scores which were normalized using data-derived constants (1.81X and 1.75X for Affymetrix and Agilent, respectively) determined from platform bridging studies (data not shown) to account for the consistently observed compression of fold changes in microarray data relative to Taqman.
Histopathology, Clinical Chemistry, and Liver Gene Expression Toxicity Scores
Tissues were collected and immersion-fixed for 24 h in 10% neutral buffered formalin, processed routinely to paraffin block, and 4–5 micron-thick slide-mounted histologic sections were stained with hematoxylin-eosin. For each study, primary and peer review histopathology evaluations were conducted by qualified veterinary pathologists according to procedures recommended for nonclinical safety biomarker qualification studies. Results of microscopic histologic examinations are summarized in Supplementary Table 1. Routine clinical chemistry endpoints for assessing liver toxicity (alanine aminotransferase [ALT], aspartate aminotransferase [AST], total bilirubin [T Bil], alkaline phosphatase) were collected and only those studies resulting in test article-related changes in any of these parameters are noted in Supplementary Table 1.
In addition, liver tissue toxicity scores reflecting a conserved set of tissue transcriptional responses to tissue damage were determined and are also included in Supplementary Table 1. A liver tissue toxicity score based on a gene signature that diagnoses liver degeneration/necrosis and conserved in male and female Sprague Dawley and Wistar rats was developed and qualified using a training set of known liver toxicants and liver nontoxicants using histopathology as the benchmark for injury. The development and qualification of this signature has been presented previously (Glaab et al., 2018) and will be described in greater detail elsewhere (Glaab et al., in preparation). Briefly, microarray data were used to first identify approximately 400 genes that correlated with the endpoint of histopathology in liver, and then downselected using quantitative PCR (qPCR) resulting in 12 genes that were the most consistent and robustly responding transcripts (Anxa2, Bcl2a1, Cdk1, Fcnb, Gpnmb, Olr1, Pvr, S100a4, Serpine1, Spp1, Timp1, Tnfrsf12a). A logistic regression model was used to generate an algorithm that converts these 12 gene expression responses to a single toxicity metric, scaled between 0 and 1, and a positive threshold of 0.7 selected to maximize sensitivity at 95% specificity in the test set. The subsequent performance of the signature was evaluated with an independent test set of 34 compounds (16 positive/18 negative) dosed for 7 days at 2 and 3 dose levels, demonstrating approximately 90% sensitivity and 100% specificity in diagnosing drug-induced liver degeneration/necrosis among all individual study animals.
HEK Cell CYP450-dependent Versus -independent NRF2 Stabilization Assay
Cell culture
HEK293/CYP cells were cultured in DMEM + GlutaMax media supplemented with Penicillin (50 units/ml)/Streptomycin (50 units/ml), 10% Fetal Bovine Serum (vol/vol), Hygromycin (150 µg/ml), and Blasticidin (100 µg/ml) at 37°C in the presence of 5% CO2. All culture reagents were obtained from Life Technologies/ThermoFisher Scientific.
Construction of human CYP450 expression constructs
Expression constructs for 8 human CYP450 isoforms were synthesized and inserted into expression vector pcDNA5/FRT/TO (Invitrogen/ThermoFisher Scientific) downstream of a tetracycline (doxycycline)-inducible CMV promoter. To facilitate the detection of the expressed CYP450 enzymes by immunoblot analysis, a Flag tag epitope was fused to the C-terminus of each expression construct. Expression constructs were generated for the following CYP450 enzymes: CYP1A2 (NM_000761), CYP2B6 (NM_000767), CYP2C8 (NM_000770), CYP2C9 (NM_000771), CYP2C19 (NM_000769), CYP2D6 (NM_000106), CYP2E1 (NM_000773), and CYP3A4 (NM_001202855).
Generation of HEK293/CYP450 cell lines
The HEK293 cell lines expressing each of the 8 CYP450 enzymes were generated using the Flp-In T-TEx system according to Manufacturer’s instructions (Invitrogen/ThermoFisher Scientific). Briefly, each of the expression constructs was cotransfected with pOG44 into the HEK293 Flp-In host cell line per standard protocol using Lipofectamine 2000 reagent. Following transfection, isogenic, stable expression cell lines were selected and expanded in fully supplemented DMEM media containing the antibiotics Hygromycin and Blasticidin. Expression of the Flag-tagged CYP450 enzymes was induced with doxycycline (Dox, 1 µg/ml). Functional activities of the expressed enzymes were assessed, and they were confirmed to be metabolically active toward their respective substrate by LC-MS analysis (data not shown).
Immunoblot analysis
Cells were seeded in 12-well plates in the presence or absence of Dox (1 µg/ml) and incubated for 20 h. The culture media was replaced with prewarmed, fresh media (absent Dox) containing increasing concentrations of test article (0.1% DMSO final concentration) for 6 h. Cells were rinsed 1x with PBS and harvested with the Millipore Nuclear Extraction kit. Whole cell lysates were cleared by centrifugation and subjected to electrophoresis on 10% SDS polyacrylamide gels, transferred to PVDF membranes and immunoblotted with anti-Flag (Sigma), anti-NRF2 (Nguyen et al., 2003), or anti-GAPDH (R&D System) antibodies.
Generation of NRF2 Knockout Rats
NRF2 knockout (KO) rats were custom created by SAGE Labs (now Sigma-Aldrich Co) in the Wistar Han background (Charles River Laboratories) using Zinc Finger Nucleases (ZFNs). Briefly, the ZFN target site (TCCGCCCTCAGCATGatggacTTGGAATTGCCACCGCCA, binding sites in upper case and cleavage sites in lower case) was designed to disrupt exon 1 of the rat NRF2 (NCBI Gene ID: 83619, NCBI Ref Seq: NC_005102.3, mRNA NCBI Ref Seq: NM_031789.1). A founder animal with a 1593 bp deletion (spanning 918 bp upstream to 630 bp downstream of the exon 1 coding sequence) was selected to generate homozygous knockout animals following the standard transgenic breeding scheme using heterozygotes sibling mating at Charles River Laboratories. The deletion was confirmed by DNA sequencing and genotypes of animals were verified by PCR analysis using forward primer GTGGAGGCAGGAGGATTGTA and reverse primer CAACTGATCAACAGCTCCA.
NRF2 KO rats and their wild type (WT) littermates were treated with bardoxolone (10 mg/kg), or ticlopidine (400 mg/kg) daily via oral gavage for 4 days. BA-LRA scores were assessed from the rat liver samples collected approximately 24 h after the last dose. BA-LRA scores of the drug-treated groups were normalized to their own genotype controls dosed with respective vehicles (sesame oil for bardoxolone, and 0.5% methylcellulose for ticlopidine).
In Vitro BA-LRA HepatoPac Studies
Targeted gene expression to assess the same bioactivation response mechanisms (NRF2 oxidative stress and NRF1 proteasomal stress pathways) was evaluated in a rat (male Wistar HAN) hepatocyte micropatterned coculture model (HepatoPac; BioIVT, Medford, Massachusetts) and the in vitro BA-LRA signature derivation and performance metric details are described elsewhere (Kang et al., 2020). Briefly, the manufacturer’s proprietary Maintenance Medium (containing 10% serum) was used for compound treatments. Cultures were replenished with fresh compound-containing media at study days 2, 5, 7, for a total of 9 days of treatment. At the end of the incubation period, cells were harvested for RNA extraction, QT-PCR-based transcriptional analyses completed, and conclusions from NRF1/NRF2-based in vitro BA-LRA transcriptional analyses results are described here.
RESULTS
Additional Assessment of the Value of CPB Using Human Hepatocytes for DILI Risk Prediction
We extended previously published investigations to further understand how measures of covalent modification of protein could be of use for clinical DILI prediction using radiolabeled drug and primary human hepatocytes (Nakayama et al., 2009) rather than microsomes. While originally targeting fifty compounds, we ceased after generating data for 38 compounds (25 for their ability, and 13 compounds for their documented lack of ability to cause liver injury at prescribed doses in humans) as the results showed that a predictive threshold for separating liver safe from DILI risk drugs could not be defined. There was significant overlap in the magnitude of CPB (expressed as pmol-eq/mg protein) between liver safe and DILI risk compounds (Figure 1). By considering the maximal human daily dose, an optimized sensitivity (number of true positives detected/total number of true positives tested) and specificity (number of true negatives detected/total number of true negatives tested) to predict DILI of 68% and 54%, respectively, was achieved. The high CPB results seen for DILI-safe compounds sumatriptan, atorvastatin, cimetidine, and furosemide, eg, which had not been tested previously by Nakayama et al. (2009) curtailed plans for testing additional liver-safe drugs. We concluded from the poor specificity that a measure of intrinsic chemical reactivity of CRM toward proteins in human hepatocytes or microsomes cannot adequately inform on clinical liver safety.
Figure 1.
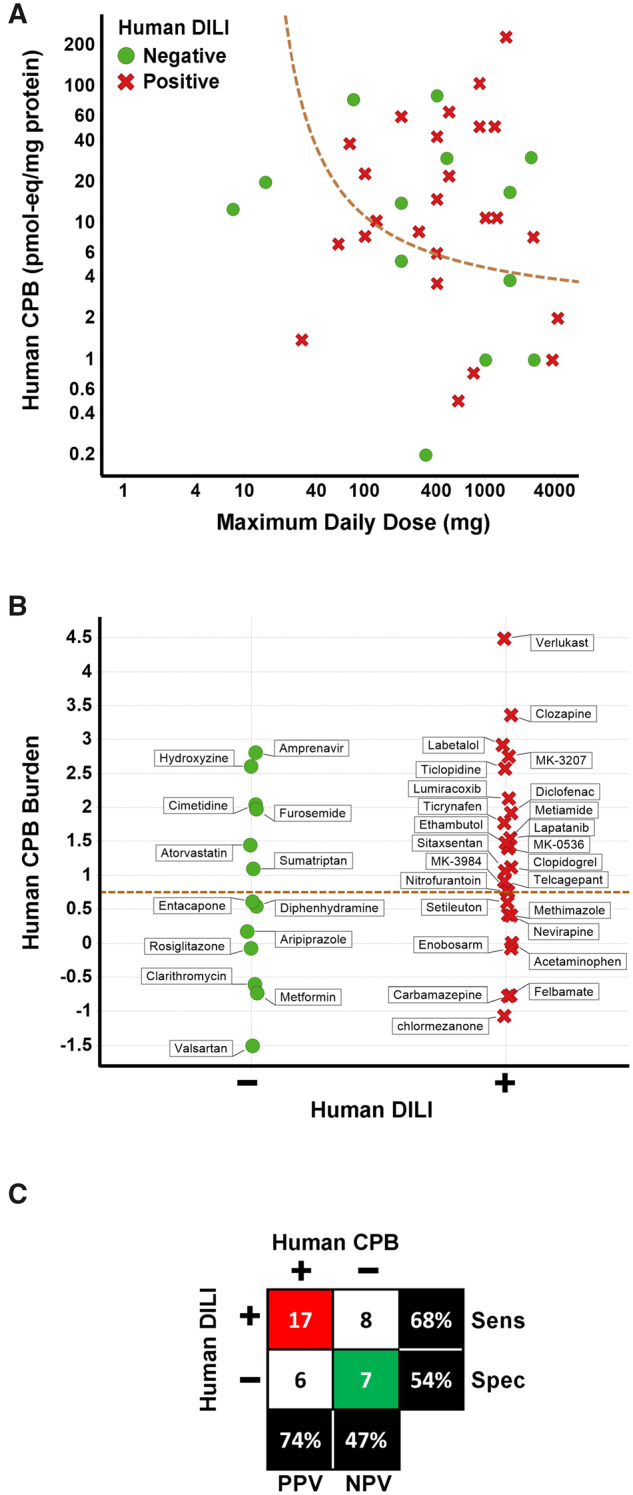
Covalent protein binding (CPB) in human hepatocytes is not predictive of drug-induced liver injury (DILI) risk. A, The relationship between CPB measured in human hepatocytes and maximal human daily dose for compounds with a lack of (green circles) or the ability to cause (red x’s) liver injury in humans. Dashed line indicates CPB burden (CPB × maximum daily clinical dose) threshold of 0.75. B, CPB burden (CPB × maximum daily clinical dose) is plotted for the same compounds in (A). Dashed line indicates CPB burden threshold of 0.75. C, Matrix (2 × 2) of the association of DILI risk and CPB burden > 0.75. Red highlights number of true positives, green the number of true negatives. Abbreviations: Sens, sensitivity; Spec, specificity.
Optimization of a Transcriptional Signature of Rat Liver Response to CRM
We hypothesized that a measure of transcriptional adaptation triggered by strong protein electrophiles in rat liver might better inform on CRM-mediated DILI risk potential in humans. Our goal was not to capture transcriptional biomarkers of all biological mechanisms of DILI, but rather to focus specifically on those transcriptional response pathways associated with bioactivation of parent drug to CRM. Our strategy was to leverage genome-wide transcriptional data to query broad biological responses to reactive metabolism including for example, the NRF2 and NRF1 rat liver signatures (Podtelezhnikov et al., 2020), and to integrate this strategy together with a rich foundation of compounds evaluated previously for covalent-binding potential within MSD (Evans et al., 2004) as a basis for probing CRM truth. Our expectation was for imperfect concordance between rat liver transcriptional response to bioactivation with human liver microsome CPB values while at the same time expecting to surface a promising data pattern and an opportunity to investigate and understand discordance.
To test this hypothesis, we first selected a training set of 26 internal MSD compounds that had been discontinued prior to clinical development (CPB training set), 21 of which were discontinued based on the signals generated using in vitro microsomal CPB assays (Evans et al., 2004). These 26 included 14 compounds with very high CPB (> 200 pmol/mg protein), 7 compounds with intermediate levels (50–200 pmol/mg protein), and 5 compounds with acceptably low levels of CPB (< 50 pmol/mg protein) (Figure 2A). The 26 compounds were administered to rats at doses ranging from 15 to 750 mkd and gene expression changes were assessed from samples of liver collected 24 h after 4 or 7 days of dosing using Affymetrix microarrays. The 4-day study design, with tissue collection 24 h after the 4th and final dose, conformed with our routine practice of an initial resource-sparing rat tolerability study supporting the early safety lead optimization phase of drug development (Glaab et al., 2018).
Figure 2.

Differentiation of compounds with varying degrees of covalent protein binding (CPB) by bioactivation liver response assay (BA-LRA). A, Heatmap of Affymetrix Log10 RNA expression ratio values in liver from compound treated versus respective vehicle rat controls for the training set of compounds with low (< 50), medium (50–200), or high (> 200) covalent binding (pmol-eq/mg protein) in human hepatocytes. Compounds are ordered by covalent-binding category. Genes are arranged by signature loading coefficients [10] for nuclear factor erythroid 2-related factor 2 (NRF2) (left side of heatmap from high to low) or nuclear factor erythroid 2-related factor 1 (NRF1) (right side of heatmap from low to high). B, Barplots of BA-LRA signature scores for the training (dark) and test (pale) compounds with low (green), medium (yellow), and high (red) human hepatocytes covalent binding. Values represent Taqman data where available (confirmatory Taqman data for CMPD P, MK-5046, and MK-3207 substituted for original Affymetrix data in A), or normalized (see Materials and Methods section) values from Affymetrix or Agilent microarrays. Red dashed line = BA-LRA threshold of 0.22. C, Receiver operating characteristic analysis of the 44 training/test dataset in (B) for BA-LRA score prediction of High/Med versus low covalent-binding compounds. Red dashed lines indicate sensitivity and specificity at a BA-LRA threshold of 0.22. D, Dose-response of BA-LRA score using the 26 compound Affymetrix dataset with low (green), medium (yellow), and high (red) human hepatocyte covalent binding. Lines connect data for same compounds at multiple doses. Abbreviation: AUC, area under the curve.
Student’s t test was used to identify probe sets differentially expressed between compounds with Med/High versus low in vitro covalent binding (Supplementary Table 3). Pathway enrichment analysis found that despite a large number of expected false discoveries due to limited statistical power, the 436 probe sets upregulated >25% with p < .05 for Med/High versus low in vitro covalent-binding compounds, were significantly enriched in CRM relevant biology including oxidative stress, and proteasomal stress genes.
The specific enrichment of anticipated CRM-related biology in the de novo gene signatures prompted a more supervised approach for biomarker gene identification focused specifically on oxidative stress pathway genes. A set of 42 genes known to be upregulated by the NRF1-/NRF2-mediated oxidative stress response based on our other efforts (Podtelezhnikov et al., 2020) and supported by the literature (Chia et al., 2010; Leone et al., 2014; Rooney et al., 2018, 2019) was compiled and assessed for association with the covalent-binding outcomes of the full training compound set. Indeed, the average gene expression change for these genes was significantly associated (t test p < .02) with the Med/High in vitro covalent-binding classification. The gene set was further refined by removing genes negatively correlated with the overall signature score using the microarray data, and by adding in genes from the genome-wide analysis with correlation >0.8 to the signature score. The resultant list of 56 genes was then subsequently reduced to the 46 most correlated genes due to taqcard size restraints to form the final BA-LRA signature gene set (Supplementary Table 4).
The subsequent analyses, summarized in Supplementary Table 4, confirmed that this signature was able to report quantitatively on cellular exposure to electrophilic CRM reacting promiscuously with proteins by activating NRF2/Keap1 and the NRF1/proteasomal pathways. Specifically, NRF2 chromatin immunoprecipitation sequencing assays and RNA sequencing were performed on livers from rats treated with vehicle or the NRF2 activator bardoxolone to identify 20 (43%) of the 46 BA-LRA signature genes as being induced by direct NRF2 binding (Tamburino et al., unpublished data). Moreover, we analyzed the genes using multiple linear regression modeling of RNA sequencing data from over 100 compounds. The loading coefficients in this model were interpreted as contributions to gene induction from different xenobiotic sensors and repressors (Podtelezhnikov et al., 2020). We showed that, out of the 46 genes, 24 and 12 genes could be described as NRF2- and NRF1-driven, respectively, due to the dominant loading coefficients for these 2 transcription factors (Supplementary Table 4).
The average gene induction of the 46 signature genes had an AUC = 0.8 and p = .02 for differentiating in vitro Med/High from low covalent-binding compounds in the training set. Performance of the BA-LRA signature was evaluated using an independent set of 18 compounds (CPB test set) having both in vitro covalent-binding data and the signature gene expression measured via Agilent microarrays (retrospective analysis of existing data) or in newly executed Taqcard studies. The maximally attained BA-LRA scores differentiated the CPB test compounds of low from Med/High in vitro covalent-binding compounds with AUC = 0.8 and p value = .02, and the results for the combined set of 44 CPB training and test compounds are shown in Figure 2 with a combined area under the curve (AUC) of 0.75 and p value of .007 (Figs. 2B and 2C). Receiver operating characteristic (ROC) analysis indicated optimal accuracy at a BA-LRA threshold of 0.22 providing 72% sensitivity and 73% specificity for predicting High/Med versus low in vitro covalent-binding results. We concluded that the degree of change in expression of the genes in this signature measured at doses intended to provide high liver exposures could provide a quantitative measure of the rat liver’s defensive response to the test drug’s capacity for bioactivation by all metabolic pathways available to the drug.
As a step toward analytical validation of this 46-gene BA-LRA signature, we identified samples for which we had generated data using both targeted 46-gene qPCR-based platforms and broad RNAeq profiling. Group BA-LRA scores were calculated using both platforms and found to correlate with a linear relationship of [qPCR BA-LRA] = 0.98 [RNA-Seq BA-LRA] + 0.022 and an R-square value of 0.85 and p = 2e−38 (data not shown). These results indicate a very high level of consistency using these 46-genes to define BA-LRA scores within our lab across these independent analytical platforms on the same samples.
Furthermore, within the Open TG-GATEs male Sprague Dawley rat toxicogenomics database (Igarashi et al., 2015) we identified samples from rats dosed similarly with compounds for which we had generated BA-LRA scores using our qPCR platform. Specifically, 20 different compounds in TG-GATEs (25 samples) were dosed within 40% of dose levels used for our studies for either 1 or 3 days. Using the 46 BA-LRA genes, BA-LRA scores were calculated from the TG-GATEs Affymetrix GeneChip data. The calculated BA-LRA scores were found to correlate with the linear relationship [TG-GATEs Affy BA-LRA] = 0.5 [MSD qPCR BA-LRA] + 0.022 and an R-square value of 0.8, and p = 2e−9 (data not shown). These results indicate a very high level of consistency using these 46-genes to define BA-LRA scores even across laboratories and across separate samples collected from independent in-life studies, whereas the slope of 0.5 reflects the well-known gene expression signal suppression of the GeneChip platform.
We also attempted to define and apply biomarkers from blood that could inform on a DILI mechanism through CRM formation, encouraged by published results showing that drugs such as APAP can generate protein biomarkers in blood of mice suggestive of CRM formation (Hu et al., 2014). Using MS-based proteomic approaches, we analyzed plasma collected from rats dosed with certain of the drugs described here that result in DILI through different mechanisms including CRM formation. Our analyses were discouraging, however, with the specificity and the sensitivity deemed insufficient (data not shown). We encourage others to further pursue qualification of such DILI mechanism-based accessible and translational biomarkers that would be a very valuable contribution to the drug development tool box.
Optimizing the Rat BA-LRA Study Design
We noted from these studies that doses exceeding 100–300 mkd were needed to reliably trigger the 46-gene BA-LRA signature above the 0.22 threshold (Figure 2D). Indeed, limiting the dataset to just those compounds dosed at 300 mkd or higher improved sensitivity to 82% at this threshold. We further evaluated the time course of BA-LRA signature response for a limited number of both DILI-positive and -negative compounds at these higher dose levels (data not shown). Examples were observed of compounds with significantly either higher or lower BA-LRA values after 4 days of dosing compared with a single dose. However, a steady-state BA-LRA score value was generally achieved by 4 days of oral dosing and this steady state level was more predictive of DILI than results from a single dose. We settled, therefore, on a standard 4-day rat study design where test compounds must be well tolerated at doses exceeding 300 mkd (eg, 500–750 mkd) to deliver sufficient drug to the liver for an adequate test of BA-LRA response. While some of the earlier learning phase studies described herein had durations shorter than 4 days and/or doses under 500 mkd, subsequent studies where designed with at least 500 mkd and 4 days of dosing.
Assessment of the Liver Response Signature and Threshold Using DILI-positive and Liver-safe Drugs
Using this paradigm, we next assessed the association of the BA-LRA signature, not with covalent-binding chemical reactivity, but with risk for clinical liver injury. Similar to the CPB assessment, we first selected a small training set of 30 drugs (Figure 3) based on their lack of (13) or their ability (17) to cause liver injury in humans (Supplementary Table 1). Importantly, traditional drug development paradigms failed to anticipate the DILI hazard for many of the DILI risk compounds (Park et al., 2011; Sistare et al., 2016). In agreement with this notion, a liver transcriptional signature corresponding to liver injury/degeneration (Glaab et al., 2018) did not differentiate clinical outcome in the DILI training set of drugs with only 2 compounds breaching an established threshold for a bona fide injury/degeneration response, and these were DILI-negative compounds (Figure 3A). In contrast, 9 of the 17 clinical DILI-positive compounds exhibited an LRA score > 0.22 (Figure 3A). Three of the remaining 8 DILI positives with LRA score <0.22 were considered insufficiently tested as we were not able to dose above 300 mkd. Thus, 9 (64% sensitivity) of the 14 sufficiently tested DILI-positive compounds were correctly identified. Lastly, 8 of the 13 DILI negatives exhibited BA-LRA scores < 0.22. As 2 of these 8 were InsfT, specificity was only 54% (Figure 3B).
Figure 3.

Bioactivation liver response assay (BA-LRA) scores of drug-induced liver injury (DILI) prediction test set. A, Plotted are BA-LRA scores for training set of 30 drugs based on a lack of (−) or ability (+) to cause liver injury in humans. Dashed red line indicates BA-LRA 0.22 threshold. Data points are colored by liver injury/degeneration-based gene expression scores with orange indicating presence of liver injury/degeneration. B, Bar plots of the BA-LRA scores for training set of 30 drugs based on a lack of (−, green) or ability (+, red) to cause liver injury in humans. Compounds deemed insufficiently tested due to either dose (< 300 mkd), are indicated by pale colors. C, Heatmap of the Taqcard data from the DILI training set of compounds tested at doses > 300 mkd. Values represent avg Log10 RNA expression ratio values in liver from compound treated versus respective study vehicle controls. Compounds are ordered by BA-LRA score. Genes are arranged by signature loading coefficients [10] for nuclear factor erythroid 2-related factor 2 (NRF2) (left side of heatmap from high to low) or nuclear factor erythroid 2-related factor 1 (NRF1) (right side of heatmap from low to high).
From these studies we also noted that nonsteroidal anti-inflammatory drugs (NSAIDs), which are known to perforate the rat GI and drive systemic release of cytokines to mount a strong systemic inflammatory response and activate liver toll-like receptors (TLRs) (Tugendreich et al., 2006), resulted in marked suppression of the BA-LRA response as well as suppression of the transcriptional responses of other major nuclear receptors (CAR, PXR, PPAR) (Podtelezhnikov et al., 2020). Indeed, >20 of the BA-LRA signature genes are consistently downregulated by the NSAID compounds naproxen, meloxicam, diclofenac, sudoxicam and lumiracoxib, whereas 5 of the genes (Hmox1, Gpx2, Nqo1, Nol3, and Sod2) are consistently upregulated by each of these compounds (Figure 3C and Podtelezhnikov et al. [2020]). These findings indicate an uncoupling of the coexpressed bioactivation gene network by NSAIDs, and the degree of uncoupling could be monitored by calculating the difference between the average of the 20 downregulated genes and the 6 upregulated genes (NSAID score). Compounds with uncoupling scores of > 0.8, including these NSAIDs, were considered uninterpretable or underestimated with respect to BA-LRA determination. Excluding these as well as the insufficiently tested compounds, a total of 13 DILI-positive compounds and 10 DILI-negative compounds were considered adequately tested with a sensitivity of 69% and specificity of 50% when considering the 0.22 threshold alone (AUC = 0.75, p value = .05) and not yet taking the clinical daily dose into consideration.
We applied the strategy described by Nakayama et al. (2009) to BA-LRA assessment of potential DILI risk; instead of just using the 0.22 BA-LRA we considered both BA-LRA strength and clinical dose. Plotting the relationship of recommended maximal daily dose and BA-LRA score for the 23 sufficiently tested DILI test compounds indicated an enrichment of DILI-positive compounds when both BA-LRA score and daily clinical doses were high (Figure 4A). Adopting the concept described by Usui et al. (2009), and multiplying the BA-LRA score by the clinical dose as a measure of BA-LRA burden similarly distinguished DILI-positive and -negative compounds with an ROC AUC of 0.8 and p value of .01 (Figure 4B). Indeed, this approach improved specificity to 90% while maintaining 69% sensitivity with the test set of 23 compounds using a BA-LRA burden threshold of 0.85. This burden threshold corresponds to the function, BA-LRA score = 0.85 (log10 Max clinical dose) shown in Figure 4A.
Figure 4.
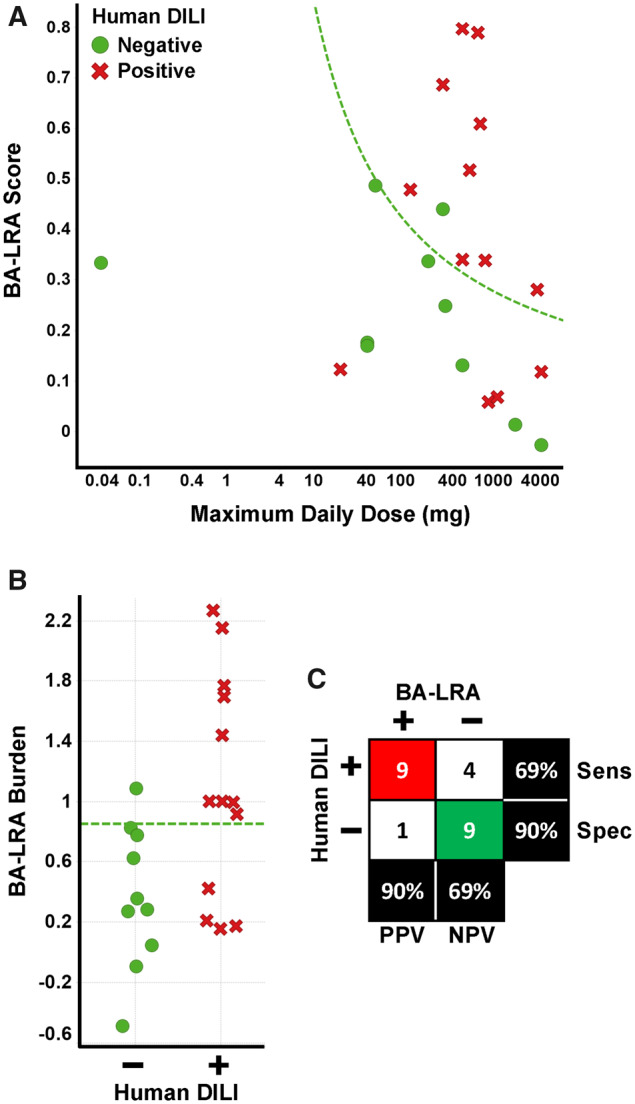
Consideration human daily dose improves performance of liver response score. A, The relationship of bioactivation liver response assay (BA-LRA) score and maximum daily clinical dose for the 23 compounds sufficiently tested and without evidence of signature uncoupling. Green circles indicate compounds with a lack of evidence for liver injury in humans, whereas red x’s indicate compounds with the ability to cause liver injury in humans. Dashed green line indicates BA-LRA burden (BA-LRA score × maximum daily clinical dose) threshold of 0.85. B, The BA-LRA burden (BA-LRA score × maximum daily clinical dose based) is plotted for the same compounds in (A). Compounds with a lack of (−) or ability (+) to cause liver injury in humans are plotted in green and red, respectively. Dashed green line indicates a BA-LRA burden threshold of 0.85. C, Matrix (2 × 2) of the association of drug-induced liver injury (DILI) risk and BA-LRA burden > 0.85. Red highlights number of true positives, green the number of true negatives. Abbreviations: Sens, sensitivity; Spec, specificity.
Testing the Performance of the BA-LRA for Assessing DILI Risk Potential Using 98 Additional Paradigm Drugs
It is important to note, that the training set was enriched in compounds suspected to cause DILI through a reactive metabolism mechanism. To test the performance of the signature in a broader and less biased set of DILI compounds, we curated a larger set of rat liver data sufficiently tested (dosed > 300 mkd to be considered BA-LRA negative) with an additional 52 drugs with a history of DILI in humans, and with 46 drugs that were considered to have a safe track record for the liver (Supplementary Table 1). Of these compounds, 5 (aripiprazole, fluoxetine, Telcagepant, nabumetone and pemoline) exhibited an NSAID-like uncoupling of the BA-LRA signature network and so BA-LRA scores were therefore considered to be underestimated and the compounds to be insufficiently tested. Results are summarized in Figure 5 for the combined set of 23 adequately tested training compounds and the 93 additional adequately tested drugs, for a total of 116 compounds (63 DILI positive and 53 DILI negative). The BA-LRA burden for the 93 test set compounds gave an AUC of 0.6 and p = .04, while the combined set of 116 training and test compounds gave an AUC of 0.7 and p = .003. The combined sensitivity was 32% and the combined specificity was 92% for identifying DILI compounds at the 0.85 BA-LRA burden threshold. These data suggest that at least 32% of the selected DILI test compounds mediate liver injury at least in part via a mechanism that relies upon CRM formation. The reduced sensitivity is expected, given a more unbiased and diverse set of DILI mechanisms represented in the full DILI positive set of compounds. Despite the lower sensitivity we conclude that this genomic-based approach provides value because liver-safe drugs are correctly called with 92% specificity, and positive predictivity of the assay exceeds 83% to flag drug candidates for structure activity relationship (SAR) modifications in order to reduce CRM potential.
Figure 5.
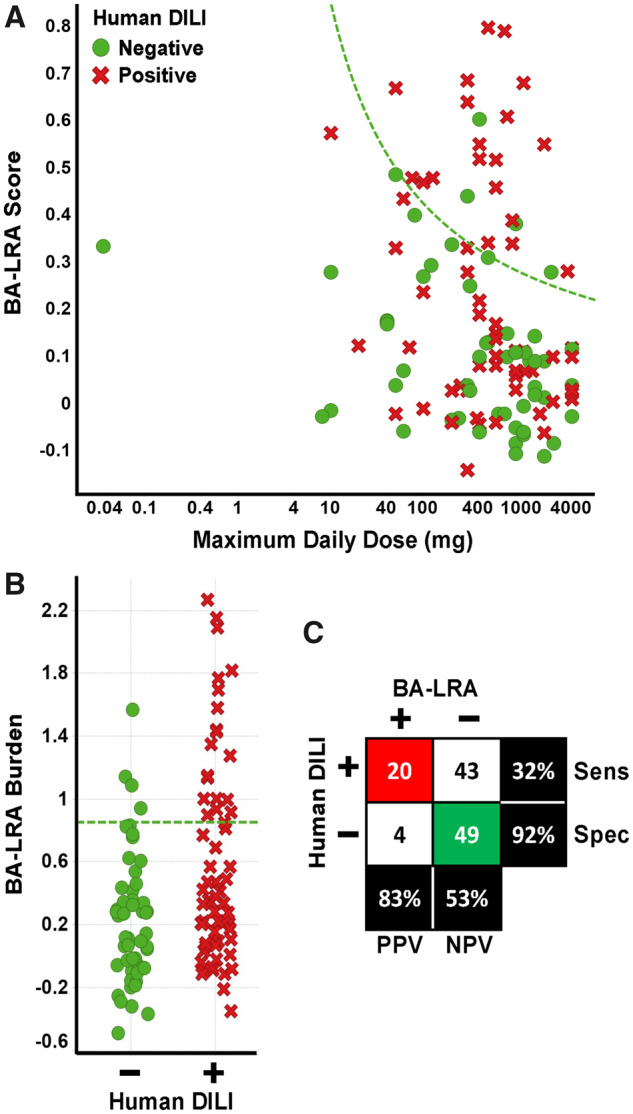
Summary of bioactivation liver response assay (BA-LRA) signature scores and association with drug-induced liver injury (DILI) risk. A, The relationship of BA-LRA score and maximum daily clinical dose for the combined 116 compound DILI training and test set. Maximum daily clinical doses, rat study doses and study durations, and resulting BA-LRA scores are listed in Supplementary Table 1. Data for compounds deemed insufficiently tested are not plotted. Green circles indicate compounds with a lack of evidence for liver injury in humans, whereas red x’s indicate compounds with the ability to cause liver injury in humans. Dashed green line indicates BA-LRA burden (BA-LRA score × maximum daily clinical dose) threshold of 0.85. B, The BA-LRA burden (BA-LRA score × maximum daily clinical dose based) is plotted for the same compounds in (A). Compounds with a lack of (−) or ability (+) to cause liver injury in humans are plotted in green and red, respectively. Dashed green line indicates a BA-LRA burden threshold of 0.85. C, Matrix (2 × 2) of the association of DILI risk and BA-LRA burden > 0.85. Red highlights number of true positives, green the number of true negatives. Abbreviations: Sens, sensitivity; Spec, specificity.
Ensuring Study Adequacy and Enhancing the Interpretation of a Liver Response Score
In an ideal scenario, each drug would be metabolized in rats by the same metabolic routes and in the exact proportional extent to all other routes of elimination as in humans. However, like any nonclinical analysis using animals, whereas significant qualitative species differences are very infrequent, quantitative differences are commonly observed. Therefore, maximizing metabolite exposure in the liver is the goal, as this increases the chance of adequately testing all metabolites, including those from metabolic pathways which are under-represented in animals. Although high doses can be administered when tolerability is not limiting, and peripheral total and unbound drug exposures of parent drug can be measured, we also sought approaches to account for nonlinear absorption of parent drug as this affects the liver’s potential exposure to metabolites.
The magnitude of the in vivo BA-LRA response is expected to result from the exposure to metabolites formed in the liver as well as their intrinsic chemical reactivities, as opposed to parent drug exposure. It is impractical to identify and measure the liver exposure to all metabolites or predict or measure their reactivities. Therefore, a detailed metabolite exposure-response relationship is improbable to develop in the fast pace of drug discovery safety testing. However, because the amount of metabolite formed under linear metabolic conditions in the liver is expected to be proportional to the fraction of the orally administered dose that is absorbed and reaches the portal vein, an estimate of this can be leveraged to define a liver metabolite(s) dose-response. Accounting for nonlinear absorption at high dose affords a better correlate to metabolite exposure in the liver than simply the administered dose. This allows high doses to be administered to maximize parent/metabolite exposure in the liver, while evaluating the dose-response or threshold for adequate testing based only on the drug reaching the liver.
Evaluating Drug Exposure Metrics to Understand Dose-response Relationship of the BA-LRA Score
Consistent with the hypothesis that the liver exposure to CRM defines the BA-LRA score and potential for liver injury risk, we observed from separate studies conducted for several (n = 6) compounds that the strength of the liver response score was correlated with parent drug (and therefore nonquantified metabolite) concentration(s) in liver, residence time in the liver, and route of administration. Oral administration could be seen, for example, to generate higher BA-LRA scores after 4 days than subcutaneous doses achieving the same peripheral exposures (data not shown). Consequently, we determined a resource-sparing approach to estimate the dose of drug absorbed that reaches the liver called the “hepatic available dose” (HAD) to define a dose-response and minimum threshold required for sufficient testing.
We defined the HAD as [dose administered] × (Fa × Fg), where Fa is fraction of dose that was absorbed and Fg is fraction of dose escaping gut metabolism. This is readily estimated using parameters obtained from routine IV and oral toxicokinetic analyses. Rearranging the common pharmacokinetic relationship F = Fa × Fg × Fh yields Fa × Fg = F/Fh, where F is the bioavailability at the high dose in the BA-LRA study, and Fh is estimated based on comparison of drug clearance (CL) to hepatic blood flow, or an understanding of the role of hepatic elimination in overall CL. Across a set of 153 internal test compounds, many of which represent retested analogs of compounds with positive BA-LRA scores, the BA-LRA score was plotted as a function of the HAD for 242 unique dose × compound treatment groups. As expected, some drugs with low peripheral exposures still achieve significant liver exposures based on HAD (Figure 6). As a result, low LRA scores for these compounds can be interpreted as resulting from adequate testing, not poor liver exposure to metabolites, and this may not be apparent based on a peripheral parent drug exposure threshold.
Figure 6.
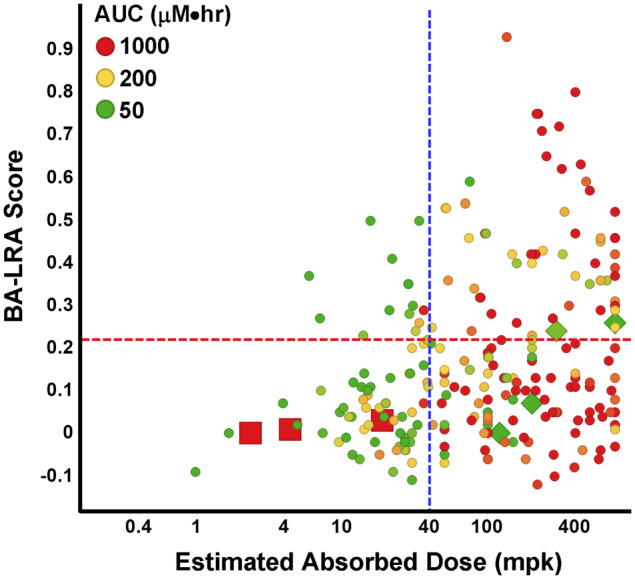
Liver response score versus systemic exposure and hepatic available dose. Plotted is the bioactivation liver response assay (BA-LRA) score versus the estimated hepatic absorbed dose across a set of 150 internal test compounds. Red dashed line indicates BA-LRA threshold of 0.22; green dashed line indicates 40-mpk absorbed dose. Dot color indicates area under the curve (AUC).
The data suggest that a HAD less than approximately 40 mpk in rats does not reliably deliver sufficient parent drug/metabolites to the liver to activate the liver bioactivation transcriptional response under the conditions of this study. Below 40-mpk HAD, just 20% of compounds × dose combinations had BA-LRA scores > 0.22, whereas 38% of those > 40 mpk were above 0.22.
Another key observation is with drugs that have low HAD yet high peripheral parent drug exposures, exceeding 200 µM/h. Examples appear in Figure 6 of drugs that have extremely high plasma protein binding and low clearance with a long half-life, but poor Fa (red squares); for these drugs, it cannot be concluded that CRM formation potential is low, but rather that liver exposure was low and that the drug was inadequately tested in our assay. In contrast to this, examples are shown in Figure 6 of drugs with highly bioavailability (F), yet due to high CL have low peripheral parent drug exposures (green diamonds). Based on a relatively high HAD, these can be considered adequately tested despite low systemic exposure to parent drug. Taken together, these examples illustrate that the HAD is an improved metric, over either administered dose or peripheral exposure, to benchmark adequate liver exposure in this nonclinical study, as well as rationalize the BA-LRA dose-response.
The added value of the HAD estimation approach is clear given the identification of compounds as being adequately tested and negative for BA-LRA, that would otherwise have been deemed inadequately tested. Obviously, this is an important missing element to any approach that would rely solely on administered dose or peripheral parent drug exposure, when it is the tissue metabolite exposure that is deemed more relevant. It is important to distinguish among these various possibilities and properly classify well-absorbed compounds with low peripheral and liver parent exposures due to high CL in rats, because in these cases, parent drug exposure can be low due to conversion to metabolites, which indicates adequate testing.
PXR Activation Provides Insight to an Alternative Source of Oxidative Stress to Elevate BA-LRA Score in Rat Liver
For PXR inducers we reasoned that excessive generation of oxygen free radicals known to result from “leaky microsomes” associated with PXR induction in rat (Dostalek et al., 2007; Mishin et al., 2014) might contribute significantly to BA-LRA due to modification of cellular redox state. We indeed noticed some association of elevated BA-LRA scores with strong PXR activation (Podtelezhnikov et al., 2020). Others have found similar association between NRF2 and CAR activation in mouse (Rooney et al., 2019). Caution therefore must be exercised in not overestimating risk from CRM formation potential in rat when interpreting BA-LRA results when evidence for strong PXR induction is also identified.
A HEK Cell Screen Identifies NRF2 Activation Mechanisms That Are Independent of CRM Formation
Because the liver transcriptional response to CRM is likely mediated, in part, through activation of the NRF2/Keap1 pathway, we sought to develop an in vitro, cell-based assay which could discriminate between the effects induced by CRM from those mediated by drugs which activate NRF2 through a direct mechanism independent of oxidative reactive metabolite formation, such as Bardoxolone and Dimethyl Fumarate (ie, Tecfidera). To this end, we established HEK293-derived cell lines stably transfected with expression constructs for each of the 8 major human cytochrome P450 (CYP450) enzymes carrying a fused Flag tag epitope at the C-terminus under the control of a tetracycline-regulated CMV promoter (see Materials and Methods section). Doxycycline- (Dox, a water-soluble analog of tetracycline) induced expression of the Flag-tagged enzymes in these cells was confirmed by immunoblot analysis with an anti-Flag antibody (Figs. 7A–C). In addition, the expressed enzymes were functionally evaluated and found to be metabolically active toward their respective substrates by LC-MS analysis (data not shown).
Figure 7.

Activation of nuclear factor erythroid 2-related factor 2 (NRF2) by acetaminophen (APAP) and ticlopidine is dependent on CYP450-mediated metabolism and by bardoxolone in a CYP450-independent mechanism. HEK293 cells expressing CYP2E1, CYP2C9, or CYP3A4 were exposed to increasing concentrations of (A) APAP, (B) ticlopidine, and (C) bardoxolone, respectively, for 6 h. Whole cell lysates were prepared, subjected to SDS-PAGE and analyzed by immunoblotting with anti-NRF2, anti-Flag (for CYP450), and anti-GAPDH antibodies. Abbreviations: BDX, bardoxolone; Dox, doxycycline.
Using these CYP450-transfected cell lines, we assessed whether metabolic activation of drugs mediated by CYP450s that form CRM could provoke a cellular response resulting in the activation of the NRF2 pathway. The results of these experiments for 2 compound examples, acetaminophen (APAP) and ticlopidine are shown in Figures 7A and 7B, respectively. When the CYP450-transfected HEK293 cells were exposed to APAP, which has been well characterized to be metabolized by CYP450 to form the reactive intermediate N-Acetyl-p-benzoquinone imine (NAPQI) that is believed to contribute to cellular injury in the liver, we found that cells actively expressing CYP2E1 exhibited a concentration-dependent increase in NRF2 protein level starting at approximately 250 µM. However, such effects were not apparent in cells not expressing CYP2E1 (when no DOX was present) (Figure 7A) or in cells expressing any of the other 7 CYP isoforms (data not shown). Although metabolite analysis was not performed, these results are consistent with APAP being converted to NAPQI by CYP450-2E1 in these cells resulting in activation of the NRF2 pathway. Similarly, ticlopidine was also found to induce activation of NRF2 in a concentration-dependent manner beginning at approximately 25 µM in induced [+Dox] CYP2C9 expressing cells, but not in noninduced cells [−Dox] (Figure 7B). Ticlopidine had no effects on the NRF2 protein level in cells expressing other CYP isoforms (data not shown). Together, these results indicate that formation of CRM following CYP450-mediated drug bioactivation leads to activation of the NRF2 pathway and provide mechanistic support for our in vivo assessment of DILI risk through a CRM-mediated mechanism for these drugs based on their positive BA-LRA scores.
In contrast, we anticipated that drugs which induce NRF2 activation in a metabolism-independent manner such as bardoxolone (BDX), which directly activates NRF2 via modifications to Keap1, would do so regardless of CYP450 expression. To confirm this, we exposed all 8 CYP450-expressing cell lines to BDX in the absence or presence of DOX. As shown in Figure 10C, BDX indeed induced NRF2 activation in a similar fashion under both [− Dox] and [+ Dox] conditions in the HEK-CYP3A4 cell line, consistent with a mechanism independent of CYP-mediated metabolism. Similar results were observed for the other 7 CYP-expressing cell lines (data not shown). Other metabolism-independent NRF2 activators, including sulforaphane, dimethyl fumarate, omeprazole, and oltipraz, gave similar results (data not shown). We have only observed 4 out of hundreds of development candidates that activate NRF2 in a CYP-independent manner and in 1 case this was limited to a series that contained a Michael acceptor substituent. Even though these NRF2 direct activators are rare, it is still prudent to assess any compound that induces a BA-LRA signal without any clear evidence of CRM as to avoid undue termination of drug candidates without CRM-mediated DILI potential.
In Vivo Studies With NRF2 KO Rats Confirm NRF2/KEAP1 Pathway Contribution to the Liver Transcriptional Bioactivation Response Signature and the Involvement of Additional Transcription Factors
A NRF2 knockout rat model was created using custom-designed ZFN targeting a site within exon 1 resulting in approximately1.5 kb deletion of exon 1 and its flanking regions. The NRF2 KO rats showed greater than 80% reduction of NRF2 mRNA using a PCR primer/probe set spanning the junction of exons 2 and 3, and no detectable mRNA using a PCR primer/probe set spanning exons 1 and 2 junction (data not shown). The expression level of a number of NRF2-regulated genes was significantly reduced when compared with that of the WT versus KO (data not shown).
To confirm the contribution of NRF2 to the activation of BA-LRA genes by BDX, NRF2 KO rats, and their WT littermates were treated for 4 days with the direct NRF2 activator BDX or with the CRM generating drug, ticlopidine. When compared with the WT, BDX-induced BA-LRA scores were attenuated 70% in the NRF2 KO rats, whereas ticlopidine-induced BA-LRA scores were not significantly reduced (Figure 8B). Interestingly, even BA-LRA genes shown to be direct targets of NRF2 (Podtelezhnikov et al., 2020; Tamburino et al., unpublished data) continued to respond robustly to ticlopidine in the NRF2 KO (Figure 8A), suggesting the significant contribution of additional mechanisms such as NRF1 in mediating drug-induced BA-LRA responses. This implies that NRF1 may compensate in the absence of NRF2 activation, as a redundant pathway for protective BA-LRA gene activation (Supplementary Table 4). When reagents become available for NRF1 chromatin precipitation experiments this will be important to confirm.
Figure 8.

Liver response in nuclear factor erythroid 2-related factor 2 (NRF2) knockout (KO) rats. Heatmap of Log10 RNA expression ratio values in liver from compound (10-mpk bardoxolone or 400-mpk ticlopidine) treated rats versus respective vehicle-treated wild type (WT) or NRF2 KO rats. Genes are arranged by signature loading coefficients [10] for NRF2 (left side of heatmap from high to low) or NRF1 (right side of heatmap from low to high). Bar plot indicates mean bioactivation liver response assay (BA-LRA) score ± SD for the respective experimental groups in the heatmap; *t test p = .002.
Case Study Examples Illustrating Utility and Principles of Applying a BA-LRA Signature Score for Informing Early DILI Risk Assessment
Case example 1, CGRP receptor antagonist
The CGRP receptor antagonist Telcagepant was dosed for 9 months in NHP at a high dose that achieved AUC exposure multiples exceeding 7-fold the high dose Ph3 clinical trial exposures. In rats and mice AUC exposure margins in studies of 6-month duration exceeded 15-fold and 14-fold, respectively. No significant conventional liver safety signals of concern were noted (Table 1). In Phase 3 studies, for patients receiving doses of up to 280 mg bid for 2 or more consecutive weeks, the incidence of ALT rises > 3-fold ULN was significantly increased, with unconfounded cases of symptomatic hepatitis and ALT rises > 10-fold observed, and development of the drug was discontinued (Hargreaves and Olesen, 2019). Transaminase elevations generally occurred while on drug and resolved rapidly upon discontinuation.
Table 1.
Summary Results of Pivotal In Vivo Toxicology, In Vivo Rat BA-LRA, and In Vitro Rat HepatoPac Studies Conducted for Telcagepant and MK-3207
| |||
|---|---|---|---|
| Species | Duration | MK-0974/Telcagepant Studies Liver Findings | Margins AUCa |
| Rat | 6 months | No liver safety signal at Highest Dose tested (except for < 3X ALT/AST with no histopathology) | 15X |
| NHP | 9 months | No liver safety signal at Highest Dose tested | 7X |
| Mouse | 6 months | No liver safety signal at Highest Dose tested (except for < 2X ALT/AST with no histopathology) | 14X |
| Rat BA-LRA | 4 days | BA-LRA score = 0.30 at 400 mkd × 4 days seen with evidence of transcriptional suppression so maximum daily dose < 600 mg | |
| Rat HepatoPac | 9 days | BA-LRA > 0.2 at 50 µM, with no effect on urea to 100 µM (mito fx); no effect on bile acid transport (at 10.7X est. unbound Liver inlet Cmax) | |
| Species |
Duration | MK-3207 Studies Liver Findings | Margins AUCb |
|---|---|---|---|
| Rat | 6 months | No liver safety signal at Highest Dose tested | 30X |
| NHP | 9 months | No liver safety signal at Highest Dose tested | 4X |
| Mouse | 6 months | No liver safety signal at Highest Dose tested | 14X |
| Dog | 1 month | Slight periportal vacuolation with < 4X ALT/AST elevations also associated with excessive body weight loss | 17X |
| Rat BA-LRA | 4 days | BA-LRA score = 0.34 at 600 mkd × 4-day predicting maximum daily dose boundary of 300 mg | |
| Rat HepatoPac | 9 days | BA-LRA > 0.2 at 50 µM with no effect on urea to 80 µM (mito fx); no effect on bile acid transport (at 4.4X est. unbound Liver inlet Cmax) | |
|
| |||
Telcagepant Phase 3 clinical study exposures based on a mean of 70 µM/h at a dose of 280 mg bid.
MK-3207 Phase 2 clinical study exposures based on a mean of 60 µM/h at a dose of 900 mg daily.
Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; AUC, area under the curve; BA-LRA, bioactivation liver response assay.
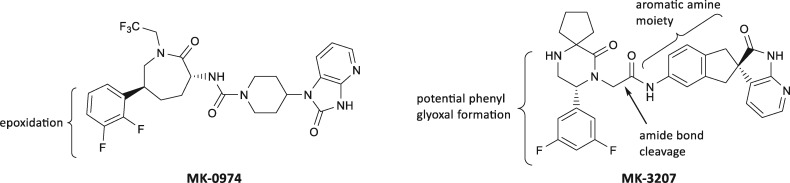
Modeling a Telcagepant dose of 280 mg, an IC50 of 26.3 µM measured in a bile salt export pump (BSEP) membrane vesicle assay, represents an exposure multiple of 17.5-fold above the calculated free unbound liver inlet Cmax (Hafey et al., unpublished data). In human HepatoPac, a Biliary Excretion Index IC50 of 16 µM was measured, which represents an exposure multiple of 10.7-fold above the calculated free unbound liver inlet Cmax (Hafey et al., unpublished data). Furthermore, using an in vivo siRNA BSEP KD rat model no evidence for synergy expected for a BSEP inhibitor was noted for Telcagepant (Li et al., 2019). Taken together, these data led to the conclusion that bile acid perturbation was of low concern as a potential mechanism of DILI. Mitochondrial toxicity was also concluded as being an unlikely mechanism of concern based on results of a 24-h HepG2-based glucose-galactose shift assay (Xu et al., 2019) and no inhibition of urea synthesis in rat or human HepatoPac cultures after 9 days of exposure to Telcagepant. Telcagepant was tested in the HEK Cyp3A4 and parental cell lines up to 90 µM and no effect was seen on NRF2 activation, or on Cyp3A4 degradation (data not shown). In contrast, significant responses were observed in the rat BA-LRA study. Because Telcagepant was poorly tolerated at repeat daily doses of 600 mkd, the standard design was altered to assess livers from animals terminated earlier (day 2/3) at high doses (600 mkd) and after 4–7 days of better-tolerated lower doses (200–400 mkd). A peak LRA score of 0.30 was observed after 4 days dosing with 400 mkd, however clear evidence of innate immune activation and NSAID suppression was observed suggesting underestimation of the full BA-LRA signal to Telcagepant-mediated CRM. Furthermore, Telcagepant elevated an in vitro surrogate of the short-term in vivo rat BA-LRA for response to CRM formation potential using either rat or human HepatoPac (Kang et al., 2020), and dose-dependent increases in the transcriptional response to CRM responsive genes were observed well exceeding the threshold of the assay at 50 µM. Follow-up studies using radiolabeled Telcagepant confirmed high covalent binding in short-term conventional cultured human hepatocytes.
The CGRP receptor antagonist MK-3207 was dosed for 9 months in NHP at a high dose achieving AUC exposures exceeding 4-fold the highest clinical trial target test dose exposures. In rats and mice AUC-based exposure margins in studies of 6-month duration that exceeded 30-fold and 14-fold, respectively. No liver safety signals of concern were noted (Table 1). In a 1-month dog study at high doses exceeding 17-fold target clinical exposures, slight periportal vacuolation was seen along with ALT/AST elevations of approximately 4-fold baseline levels but this dose was also associated with excessive body weight loss, calling into question the significance of the transaminase elevations. During clinical dose escalation in Phase 2, the incidence of subjects experiencing ALT rises of > 3-fold ULN after 2 weeks of dosing was 1% at daily doses of ≤ 100 mg (2/197), and 42% (5/12) at > 500 mg, and development of the drug was discontinued (Hargreaves and Olesen, 2019). Among the 5 patients dosed above 500 mg daily were 3 with > 20-fold ALT rises, 1 symptomatic with Hy’s Law. The ALT rises were generally delayed in onset, for as long as 2 and 3 months, and slow to resolve. Modeling a MK-3207 daily clinical dose of 900 mg, a maximally soluble 50 µM concentration achieving approximately 4.4 times the calculated free unbound liver inlet of the clinical exposure, resulted in no significant impact on biliary excretion of taurocholic acid (Hafey et al., unpublished data) in human HepatoPac cultures. No evidence for mitochondrial toxicity was seen using a 24-h HepG2-based glucose-galactose shift assay (Xu et al., 2019) or based on lack of urea synthesis inhibition of rat and human HepatoPac cultures exposed for 9 days (data not shown). MK-3207 was tested in the HEK Cyp3A4 and parental cell lines up to 90 µM. Although there was no CYP-dependent effect on NRF2 activation, a clear dose-dependent effect on Cyp3A4 degradation was observed that was blocked by ketoconazole, suggesting formation of CRM that could not escape the Cyp3A4 catalytic site in this test system. In the rat BA-LRA study, a median signature score of 0.34 was achieved predicting a lower clinical risk for DILI at projected clinical doses of < 200 mg, but more significant risk at daily doses > 300 mg. Furthermore, MK-3207 (up to 50 µM) was evaluated in an in vitro surrogate of the short-term in vivo rat BA-LRA for response to CRM formation potential using rat HepatoPac (Kang et al., 2020), and dose-dependent increases in the transcriptional response to CRM responsive genes were observed, well exceeding the positive threshold of the assay. MK-3207 is extensively metabolized via multiple biotransformation pathways. One outcome of such metabolism is the cleavage of MK-3207 into difluorophenylglyoxal and a 2-oxo-N-phenylacetamide derivative (Table 1). To track the potential of covalent binding by each of these halves, MK-3207 was strategically tritiated at 2 sites of the parent molecule. The result of this study indicated that both halves of MK-3207 efficiently labeled proteins. Identification of the piperazinone moiety as a primary soft spot, as well as the influencer, for bioactivation of MK-3207 to these electrophilic metabolites led to its removal and subsequent redesign of the molecular scaffold to produce improved backup molecules MK-1602 and MK-8031.
These 2 CGRP case studies provide good examples of how a molecule may present no evidence of liver safety concerns in 3 species of conventional animal toxicology studies, despite achieving good exposures, and still provoke liver signals in the clinic. Evidence of significant formation of CRM corroborated by several independent mechanistic studies was successfully leveraged to develop back-up molecules with reduced CRM formation and DILI potential. Ubrogepant (MK-1602) was designed to maintain efficacy while minimizing potential for CRM formation using the rat BA-LRA to guide successful safety derisking and has demonstrated a superior clinical liver safety profile (Hargreaves and Olesen, 2019) in clinical trials and was recently approved for marketing by the FDA with no liver safety label precautions.
Case example 2, Bruton tyrosine kinase reversible covalent inhibitors
This case is unique in that these drugs are “reversible covalent binders,” drugs which are designed to covalently bind to their targets resulting in sustained target engagement and inhibition, but then be subsequently cleaved to reduce immunogenicity risk expected of irreversible covalent binders. Two promising early candidates, MRK-A and MRK-B demonstrated high in vivo rat BA-LRA scores raising concern that the BA-LRA may not be useful for derisking reversible covalent inhibiting drugs. These findings led to an effort demonstrating alternatively that metabolic activation pathways and formation of CRM, not the on-target reversible covalent binding, were likely responsible for the positive BA-LRA scores. MRK-A and MRK-B were incubated in NADPH-supplemented rat liver microsomes with semicarbazide, glutathione, and cyanide as trapping agents. Although there were minimal adducts of cyanide and glutathione to the compounds, semicarbazide-trapped metabolites were the major products observed for both (Figure 9) (30%–60% by mass spectral ionization efficiency, assuming similar response across metabolites). This finding led to the identification of the piperidine moiety in MRK-A and MRK-B as the sites of bioactivation. The piperidine moiety was replaced with a pyrrolidinone in MRK-C and the semicarbazide-trapping study results indicated that the ring-opened aldehyde formation seen in MRK-A and MRK-B had been minimized by this change in the structural scaffold and MRK-C subsequently tested negative in the rat BA-LRA (Figure 9). This case study showcases how BA-LRA results used in conjunction with metabolite ID studies can guide SAR approaches to dial out metabolic liability and demonstrates that target selective covalent binders can be differentiated on the basis of the BA-LRA score. This approach has been successfully extended to other programs and compound scaffolds.
Figure 9.

Bruton tyrosine kinase case study. A, MRK-A, B, or C (10 µm) was incubated in rat and human liver microsomes with added trapping agents (glutathione 5 mM, semicarbazide 5 mM, potassium cyanide 1 mM) at 37°C for 1 h. The assays were then quenched with acetonitrile, the debris precipitated, and the supernatant concentrated and analyzed by LC-MS. B, The amounts of metabolites represented are calculated based on mass spectral ionization efficiency, assuming similar response across metabolites.
Case example 3, BA-LRA can identify CRM risk that may result in conventional liver signals in higher nonrodent species or in longer duration rat and nonrodent studies
The short-term rat BA-LRA-based DILI derisking approach may also identify compounds that can result in frank liver histopathology in longer-term rodent studies and/or in short- or longer-term nonrodent species (eg, dog) studies. This is exemplified by 2 compounds, MRK-D for an oncology target and MRK-E for an infectious disease target. For MRK-D, a clear risk for CRM was identified in the in vivo BA-LRA in rats (score of 0.45) without liver pathology which suggested a high potential for DILI risk within the clinical dose range. However, given the life-saving indication for advanced oncology patients that are refractory and resistant to current treatment, and given the uncertainties in this new and evolving BA-LRA-based derisking approach at the time, the molecule was progressed. In a 9-day escalating dose limiting toxicity study conducted using 3 dogs where each of 3 different dose levels were administered daily for 3 consecutive days, there was a substantial escalation in the liver toxicity markers ALT (approximately 8000-fold), ALP (1300-fold), and T Bil (1700-fold) detected beginning at the mid dose. Severe liver necrosis was observed histologically at the scheduled necropsy on day 9.
For MRK-E, an elevated LRA score in a 1-week rat toxicity study suggested a potential CRM concern. In addition, increases in ALT were seen with single cell necrosis in the liver and vacuolation ascribed to phospholipidosis was noted in the liver, kidney, and spleen. However, based on the projected clinical dose the overall DILI risk was considered low and the compound was progressed. In 3-month toxicity studies in the rat and dog there was clear evidence of liver findings. Moreover, based on results from Phase 1 clinical studies the safety margin decreased due to an increase in the projected clinical dose. Based on this change in the overall risk profile the compound was discontinued.
Thus, for MRK-D, despite having no evidence of “classical” liver toxicity markers or liver histopathology in the rat (seen in the 4- to 7-day duration studies, data not shown), the BA-LRA transcriptional score flagged the CRM signal and potential for severe liver toxicity that was manifested acutely in a higher species. For MRK-E the in vivo BA-LRA detected a potential DILI risk that surfaced more definitively in both rat and dog using conventional endpoints but only in longer-term toxicity studies demonstrating the utility of short-term in vivo BA-LRA studies to detect CRM signals earlier during development to trigger SAR studies and the synthesis of alternative molecules to reduce later appearing DILI risk potential. The examples also underscore the critical importance of projecting an accurate clinical dose to make the best decisions.
DISCUSSION
Although there is little doubt that drug bioactivation to form CRMs is an important hazard that can lead to either acute or delayed dose dependent or idiosyncratic hepatotoxicities and that paradigms are needed to assess risk of bioactivation potential, numerous lines of evidence including our additional results reported here with CPB in hepatocytes showing a specificity of 54% support the similar conclusion made by others (Bauman et al., 2009; Obach et al., 2008; Usui et al., 2009) that prediction of clinical liver injury potential based solely on chemical measurements of CPB could result in frequent discontinuation of potentially safe, effective drugs due to a poor level of specificity. When considered together with impractical aspects for routine incorporation of radiolabeled drug and CPB measurements during lead optimization and candidate selection, there is presently reduced routine reliance on CPB during drug development although it remains a valuable tool for targeted problem solving.
We hypothesized that a CRM-based molecular initiating event could trigger a transcriptional defense/protective response in hepatocytes and that the magnitude of this response would be associated with greater or lesser DILI risk with improved specificity over CPB-based strategies. Despite the acknowledged imperfections with CPB assays we successfully leveraged a library of internal compounds benchmarked with CPB assay data to identify a conserved transcriptional network induced by CRM forming drugs we have termed the BA-LRA that is dominated by 3 main defense mechanisms in rat liver: the NRF2/Keap1 oxidative stress pathway, the NRF1 ER proteasomal stress pathway, and an additional small subset of coregulated genes representing an undefined electrophilic defense pathway. Others found a NRF2 biomarker set in mouse studies that overlaps with the BA-LRA in 7 genes (Rooney et al., 2018). The NRF2 KO rat model studies demonstrate the specificity of the bardoxolone activation of the NRF2 pathway, in contrast to the CRM-mediated electrophilic response triggered by ticlopidine which was not blunted in the NFR2 KO rat model and appears to comprise these redundant mechanisms to drive upregulation of many of the same protective proteins. Similar to our results with ticlopidine, others have also identified from experiments conducted with NRF2 KO and WT mice, evidence for alternative mechanisms of gene activation among NRF2 network genes (Chanas et al., 2002; Hayes et al., 2000). We conclude that rat liver is quite adept at utilizing these protective defense systems against reactive electrophiles to maintain tissue integrity, with only few compounds triggering evidence of rat liver pathology despite very high drug exposures. Because the rat is also highly immune tolerant, a hepato-immune phenotypic response is rarely seen to CRM-mediated haptenized proteins, in agreement with recent reports (Metushi et al., 2015) deploying strategies to block immune checkpoint pathways in rodents that yield only small and transient transaminase alterations when also dosed with certain human hepatotoxicants. A greater capacity of the rodent to defend against reactive electrophiles and reduced hepato-immune responsiveness likely accounts for most discrepancies between rat and human conventional hepatotoxicity endpoints.
Drug metabolism across species has typically been found to be qualitatively similar, meaning that all human metabolites are generally formed in preclinical species and very infrequently do we observe species-specific metabolites. That said, for 1 of the 3 false positives, clarithromycin, the hepatotoxicity seen in several animal species including rat, may be ascribed to significant metabolism differences from human. Thus any evidence for significant species metabolism differences must always be considered (Aueviriyavit et al., 2010; Abbott Laboratories, 1993; Chanas et al., 2002; Hayes et al., 2000). We found that for a BA-LRA transcriptomic signal to become reliably evident it was important to maximize opportunity for drug metabolic turnover via all available metabolic pathways through administering daily doses greater than 300 mkd and to dose repeatedly for at least 4 days to approach a steady state. We evolved a modeling approach to best assess BA-LRA response as a function of the dose delivered to the liver to ensure study adequacy and to identify substantive SAR improvements when significant differences may be seen across compounds in hepatic absorbed doses versus peripheral exposures. We found this to be superior to a reliance on plasma exposures.
Any successful new approach to inform clinical doses anticipated to be associated with CRM-mediated DILI potential must be prudent and pragmatic. Because we had implemented a routine transcriptomic-based approach for early in vivo tolerability assessment and candidate selection (Glaab et al., 2018), it became operationally expedient to simply add additional sets of transcriptomic biomarkers as represented here by the BA-LRA. To protect specificity and sensitivity of the BA-LRA for identifying liver injury potential via CRM formation we incorporated 3 important strategies. First, we incorporated an in vitro cell line strategy to distinguish specific noncovalent pharmacologic activators of NRF2/Keap1 signaling (eg, bardoxolone, dimethyl fumarate, sulforaphane) from those drugs that require metabolic activation. Second, we developed a strategy to not falsely implicate as BA-LRA positive those compounds that produce oxygen free radicals via liver microsomal enzyme induction. Specifically, we observed a relationship between PXR induction and BA-LRA response that was notably stronger than that of other receptors (Podtelezhnikov et al., 2020). Although these conclusions are consistent with reports of PXR induction contributions to reactive oxygen species (ROS) signal (Dostalek et al., 2007; Mishin et al., 2014), such published data linking induction levels of CYPs to significant activation of NRF1/NRF2 are lacking. The question is difficult if not impossible to experimentally resolve because PXR inducers may themselves often become substrates of CYPs yielding CRM while at the same time increasing background ROS from induced leaky microsomes. The approach taken here using an extensive data-driven experience accumulated over several years, has directed our approach to subtract potential PXR-based contribution from the BA-LRA score (Podtelezhnikov et al., 2020) as a pragmatic guide for internal decision making. Third, we observed convincing evidence that orthogonal TLR activation pathways following innate immune system activation suppress the BA-LRA measurements. This is consistent with reports from others (Nguyen et al., 2015) that cytokine release and TLR activation pathways reduce expression of liver transcripts including numerous metabolism and transporter genes. Fortunately, such suppression is also readily identifiable by the characteristic uncoupling of the otherwise coherent BA-LRA signature gene responses (Figure 3C), and we can rely on this pattern as well as a small set of genes reflective of TLR activation to flag this complexity (Supplementary Table 3 and Podtelezhnikov et al. [2020]). Integrating these study design parameters, we have demonstrated that incorporation of the BA-LRA endpoint into a 4-day rat study design across a set of 116 compounds has ascribed a drug bioactivation mechanism as contributing to the cause of human liver toxicity for 32% of DILI-positive drugs, while maintaining 92% specificity among drugs considered liver safe (Figure 5C).
Reducing DILI risk from presenting in later stages of drug development must incorporate accurate projections of clinical therapeutic doses at the time CRM formation potential is being considered. Ambiguities in projecting human doses so early will continue to make elimination of clinical DILI extremely challenging even with the best liver safety testing toolbox. Study results with acetaminophen exemplify need for continued resolution of this tool. We noted that several DILI-safe drugs present with a high BA-LRA signal burst at 24 h that subsides quickly over 4 days of continued dosing, and several DILI-positive drugs present with a low BA-LRA score at 24 h that escalates to high steady state levels by day 4. This led us early on to adopt a 4-day study dosing paradigm. However, the DILI-positive drug acetaminophen was found to yield a BA-LRA score at 24 h implicating daily doses exceeding 4 g to be a DILI concern (Figs. 2 and 3, BA-LRA = 0.54), but low BA-LRA scores after 4 days dosing (Supplementary Table 1, BA-LRA = 0.12) suggest the drug to be safe at very high daily doses, certainly within 4 g daily. This early and transient BA-LRA signal at 24 h is also evident in the TG-GATEs data for acetaminophen at 600 mkd (Igarashi et al., 2015) which we used to calculate BA-LRA score of 0.32. These values drop at day 3 of daily dosing to a steady-state Affymetrix platform BA-LRA value of 0.10. Such a pattern may be indicative of the DILI risk associated only with an acute overdose of acetaminophen that would not be a concern for adherence to proper therapeutic dosing. Nevertheless, we have indicated that failure of the BA-LRA to identify clinical DILI risk associated with acetaminophen using the 4-day BA-LRA score to represent a false-negative result.
Numerous investigators have demonstrated that the fundamental ability of a drug molecule to undergo bioactivation to form chemically reactive electrophilic metabolites can yield toxicity in many different tissues, and not just liver (Stepan et al., 2011). Our experience suggests that this stage gate screen using liver to assess potential for a chemical to mount a CRM defense response may help reduce rates of toxicity that might otherwise be seen in kidney, skin, blood, bone marrow, etc. where examples of reactive metabolism-mediated toxicities have also been well documented (Stepan et al., 2011), and so beneficial effects on pipeline attrition reduction may be realized more broadly than for just liver. Clozapine eg, presents with a significant BA-LRA signal in rat liver (Supplementary Table 1) and has shown evidence for clinical safety concerns in both bone marrow and liver. Interestingly, metiamide which was discontinued based on CRM-related clinical agranulocytosis did not show a significant rat liver BA-LRA signal (Supplementary Table 1). Work is in progress to investigate the utility of the bioactivation response signature in other tissues of the rat besides liver, and in the liver and other tissues of additional species besides rat. An additional prominent limitation requiring further research to resolve is our finding that drugs which may form reactive CoA-thioesters or unstable acyl-glucuronides generally failed to induce significant BA-LRA responses suggesting that orders in magnitude of differences in the half-lives of unstable acyl-glucuronide metabolites (hours), versus unstable oxidative intermediates (seconds) may underlie the difference in transcriptional response sensitivity.
Both the rat study and the gene expression PCR-based endpoint measurements described here are easily established within a molecular toxicology group or outsourced to several contract companies capable of generating these data. The biggest hurdle for organizational adoption of this assay is the challenge that drug safety organizations face with implementation of any paradigm change that must come to be viewed positively and not skeptically by governance groups, and drug discovery and development team members. For those molecules discovered to have an unacceptably strong BA-LRA signal, pipeline progress will be impeded in the short-term with timelines impacted as selection is guided toward molecules designed with less CRM-associated DILI risk. Such efforts must be embraced as providing greater probability of overall development success in the long-term, in order for the approach to be adopted and used routinely within an organization. Time for prospective evaluation and verification becomes vital for organizational acceptance. We anticipated that compounds targeted to adduct certain proteins with high specificity would not trigger the BA-LRA defense pathways and this was confirmed as demonstrated by the Bruton’s tyrosine kinase case example where early compounds nominated for candidate selection were flagged for an unacceptably high level of bioactivation potential and selection of improved compounds was guided. The CGRP case examples demonstrated how bioactivation-mediated risk for clinical DILI can elude conventional endpoints in routine animal study designs but with careful attention to BA-LRA signals, improved compounds can be selected that eliminate such risk. These case examples described herein together with undisclosed cases provided convincing evidence of significant, long-term benefit of the BA-LRA approach on clinical attrition rates and the successful development of improved novel therapeutics.
SUPPLEMENTARY DATA
Supplementary data are available at Toxicological Sciences online.
Supplementary Material
ACKNOWLEDGMENTS
The authors wish to thank Dr Joseph DeGeorge, Dr Jose Lebron, Dr Deborah Nicoll Griffith, Dr Warren E. Glaab, and Ms Jennifer P. Gara for their contributions to this work.
DECLARATION OF CONFLICTING INTERESTS
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
REFERENCES
- Aueviriyavit S., Kobayashi K., Chiba K. (2010). Species differences in mechanism-based inactivation of CYP3A in humans, rats and mice. Drug Metab. Pharmacokinet. 25, 93–100. [DOI] [PubMed] [Google Scholar]
- Bauman J. N., Kelly J. M., Tripathy S., Zhao S. X., Lam W. W., Kalgutkar A. S., Obach R. S. (2009). Can in vitro metabolism-dependent covalent binding data distinguish hepatotoxic from nonhepatotoxic drugs? An analysis using human hepatocytes and liver S-9 fraction. Chem. Res. Toxicol. 22, 332–340. [DOI] [PubMed] [Google Scholar]
- Abbott Laboratories. (1993). Biaxin [Package Insert]. U.S. Food and Drug Administration. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/050662s058,050698s038,050775s026lbl.pdf. Revised June 2017. Accessed June 29, 2020.
- Blomme E. A., Will Y. (2016). Toxicology strategies for drug discovery: Present and future. Chem. Res. Toxicol. 29, 473–504. [DOI] [PubMed] [Google Scholar]
- Chanas S. A., Jiang Q., McMahon M., McWalter G. K., McLellan L. I., Elcombe C. R., Henderson C. J., Wolf C. R., Moffat G. J., Itoh K., et al. (2002). Loss of the Nrf2 transcription factor causes a marked reduction in constitutive and inducible expression of the glutathione S-transferase Gsta1, Gsta2, Gstm1, Gstm2, Gstm3 and Gstm4 genes in the livers of male and female mice. Biochem. J. 365, 405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M., Borlak J., Tong W. (2016). A model to predict severity of drug-induced liver injury in humans. Hepatology 64, 931–940. [DOI] [PubMed] [Google Scholar]
- Chia A. J., Goldring C. E., Kitteringham N. R., Wong S. Q., Morgan P., Park B. K. (2010). Differential effect of covalent protein modification and glutathione depletion on the transcriptional response of Nrf2 and NF-kappaB. Biochem. Pharmacol. 80, 410–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostalek M., Brooks J. D., Hardy K. D., Milne G. L., Moore M. M., Sharma S., Morrow J. D., Guengerich F. P. (2007). In vivo oxidative damage in rats is associated with barbiturate response but not other cytochrome P450 inducers. Mol. Pharmacol. 72, 1419–1424. [DOI] [PubMed] [Google Scholar]
- Evans D. C., Watt A. P., Nicoll-Griffith D. A., Baillie T. A. (2004). Drug-protein adducts: An industry perspective on minimizing the potential for drug bioactivation in drug discovery and development. Chem. Res. Toxicol. 17, 3–16. [DOI] [PubMed] [Google Scholar]
- Glaab W. E., Holder D., He Y. D., Gerhold D. L., Bailey W. J., Beare C., Erdos Z., Lane P., Michna L., Muniappa N., et al. (2018). Universal toxicity gene signatures for early identification of drug induced tissue injuries in rats. Toxicologist Supplement to Toxicol. Sci. 162,1265. [DOI] [PubMed] [Google Scholar]
- Guengerich F. P. (2011). Mechanisms of drug toxicity and relevance to pharmaceutical development. Drug Metab. Pharmacokinet. 26, 3–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves R., Olesen J. (2019). Calcitonin gene-related peptide modulators—The history and renaissance of a new migraine drug class. Headache 59, 951–970. [DOI] [PubMed] [Google Scholar]
- Hayes J. D., Chanas S. A., Henderson C. J., McMahon M., Sun C., Moffat G. J., Wolf C. R., Yamamoto M. (2000). The Nrf2 transcription factor contributes both to the basal expression of glutathione S-transferases in mouse liver and to their induction by the chemopreventive synthetic antioxidants, butylated hydroxyanisole and ethoxyquin. Biochem. Soc. Trans. 28, 33–41. [DOI] [PubMed] [Google Scholar]
- Hu Z., Lausted C., Yoo H., Yan X., Brightman A., Chen J., Wang W., Bu X., Hood L. (2014). Quantitative liver-specific protein fingerprint in blood: A signature for hepatotoxicity. Theranostics 4, 215–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi Y., Nakatsu N., Yamashita T., Ono A., Ohno Y., Urushidani T., Yamada H. (2015). Open TG-GATEs: A large-scale toxicogenomics database. Nucleic Acids Res. 43, D921–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry R. A., Bolstad B. M., Collin F., Cope L. M., Hobbs B., Speed T. P. (2003). Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 31, e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang W., Podtelezhnikov A. A., Tanis K. Q., Pacchione S., Su M., Bleicher K., Wang Z., Laws G., Griffiths T., Kuhls M., et al. (2020). Development and application of a transcriptomic signature of bioactivation in an advanced in vitro liver model to reduce drug-induced liver injury risk early in the pharmaceutical pipeline. Toxicol Sci. 177, 121–139. [DOI] [PubMed] [Google Scholar]
- Leone A., Nie A., Brandon Parker J., Sawant S., Piechta L. A., Kelley M. F., Mark Kao L., Jim Proctor S., Verheyen G., Johnson M. D., et al. (2014). Oxidative stress/reactive metabolite gene expression signature in rat liver detects idiosyncratic hepatotoxicants. Toxicol. Appl. Pharmacol. 275, 189–197. [DOI] [PubMed] [Google Scholar]
- Li Y., Evers R., Hafey M. J., Cheon K., Duong H., Lynch D., LaFranco-Scheuch L., Pacchione S., Tamburino A. M., Tanis K. Q., et al. (2019). Use of a bile salt export pump knockdown rat susceptibility model to interrogate mechanism of drug-induced liver toxicity. Toxicol. Sci. 170, 180–198. [DOI] [PubMed] [Google Scholar]
- Metushi I. G., Hayes M. A., Uetrecht J. (2015). Treatment of PD-1(−/−) mice with amodiaquine and anti-CTLA4 leads to liver injury similar to idiosyncratic liver injury in patients. Hepatology 61, 1332–1342. [DOI] [PubMed] [Google Scholar]
- Mishin V., Heck D. E., Laskin D. L., Laskin J. D. (2014). Human recombinant cytochrome P450 enzymes display distinct hydrogen peroxide generating activities during substrate independent NADPH oxidase reactions. Toxicol. Sci. 141, 344–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama S., Atsumi R., Takakusa H., Kobayashi Y., Kurihara A., Nagai Y., Nakai D., Okazaki O. (2009). A zone classification system for risk assessment of idiosyncratic drug toxicity using daily dose and covalent binding. Drug Metab. Dispos. 37, 1970–1977. [DOI] [PubMed] [Google Scholar]
- Nguyen T., Sherratt P. J., Pickett C. B. (2003). Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu. Rev. Pharmacol. Toxicol. 43, 233–260. [DOI] [PubMed] [Google Scholar]
- Nguyen T. V., Ukairo O., Khetani S. R., McVay M., Kanchagar C., Seghezzi W., Ayanoglu G., Irrechukwu O., Evers R. (2015). Establishment of a hepatocyte-kupffer cell coculture model for assessment of proinflammatory cytokine effects on metabolizing enzymes and drug transporters. Drug Metab. Dispos. 43, 774–785. [DOI] [PubMed] [Google Scholar]
- Obach R. S., Kalgutkar A. S., Soglia J. R., Zhao S. X. (2008). Can in vitro metabolism-dependent covalent binding data in liver microsomes distinguish hepatotoxic from nonhepatotoxic drugs? An analysis of 18 drugs with consideration of intrinsic clearance and daily dose. Chem. Res. Toxicol. 21, 1814–1822. [DOI] [PubMed] [Google Scholar]
- Olson H., Betton G., Robinson D., Thomas K., Monro A., Kolaja G., Lilly P., Sanders J., Sipes G., Bracken W., et al. (2000). Concordance of the toxicity of pharmaceuticals in humans and in animals. Regul. Toxicol. Pharmacol. 32, 56–67. [DOI] [PubMed] [Google Scholar]
- Park B. K., Boobis A., Clarke S., Goldring C. E., Jones D., Kenna J. G., Lambert C., Laverty H. G., Naisbitt D. J., Nelson S., et al. (2011). Managing the challenge of chemically reactive metabolites in drug development. Nat. Rev. Drug Discov. 10, 292–306. [DOI] [PubMed] [Google Scholar]
- Podtelezhnikov A. A., Monroe J. J., Aslamkhan A. G., Pearson K., Qin C., Tamburino A. M., Loboda A. P., Glaab W. E., Sistare F. D., Tanis K. Q. (2020). Quantitative transcriptional biomarkers of xenobiotic receptor activation in rat liver for the early assessment of drug safety liabilities. Toxicol. Sci. 175, 98–112. [DOI] [PubMed] [Google Scholar]
- Rooney J., Oshida K., Vasani N., Vallanat B., Ryan N., Chorley B. N., Wang X., Bell D. A., Wu K. C., Aleksunes L. M., et al. (2018). Activation of Nrf2 in the liver is associated with stress resistance mediated by suppression of the growth hormone-regulated STAT5b transcription factor. PLoS One 13, e0200004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney J. P., Oshida K., Kumar R., Baldwin W. S., Corton J. C. (2019). Chemical activation of the constitutive androstane receptor leads to activation of oxidant-induced Nrf2. Toxicol. Sci. 167, 172–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sistare F. D., Mattes W. B., LeCluyse E. L. (2016). The promise of new technologies to reduce, refine, or replace animal use while reducing risks of drug induced liver injury in pharmaceutical development. ILAR J. 57, 186–211. [DOI] [PubMed] [Google Scholar]
- Stepan A. F., Walker D. P., Bauman J., Price D. A., Baillie T. A., Kalgutkar A. S., Aleo M. D. (2011). Structural alert/reactive metabolite concept as applied in medicinal chemistry to mitigate the risk of idiosyncratic drug toxicity: A perspective based on the critical examination of trends in the top 200 drugs marketed in the United States. Chem. Res. Toxicol. 24, 1345–1410. [DOI] [PubMed] [Google Scholar]
- Suzuki A., Andrade R. J., Bjornsson E., Lucena M. I., Lee W. M., Yuen N. A., Hunt C. M., Freston J. W. (2010). Drugs associated with hepatotoxicity and their reporting frequency of liver adverse events in VigiBase: Unified list based on international collaborative work. Drug Saf. 33, 503–522. [DOI] [PubMed] [Google Scholar]
- Tugendreich S., Pearson C. I., Sagartz J., Jarnagin K., Kolaja K. (2006). NSAID-induced acute phase response is due to increased intestinal permeability and characterized by early and consistent alterations in hepatic gene expression. Toxicol. Pathol. 34, 168–179. [DOI] [PubMed] [Google Scholar]
- Usui T., Mise M., Hashizume T., Yabuki M., Komuro S. (2009). Evaluation of the potential for drug-induced liver injury based on in vitro covalent binding to human liver proteins. Drug Metab. Dispos. 37, 2383–2392. [DOI] [PubMed] [Google Scholar]
- Xu Q., Liu L., Vu H., Kuhls M., Aslamkhan A. G., Liaw A., Yu Y., Kaczor A., Ruth M., Wei C., et al. (2019). Can galactose be converted to glucose in HepG2 cells? Improving the in vitro mitochondrial toxicity assay for the assessment of drug induced liver injury. Chem. Res. Toxicol. 32, 1528–1544. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.