

Abstract
Feline McDonough Sarcoma-like tyrosine kinase 3 (FLT3), a tyrosine-protein kinase involved in hematopoiesis, is detectable on the cell surface of approximately 80% of leukemia isolates from adult patients with acute myeloid leukemia (AML). AMG 553 is an investigational chimeric antigen receptor (CAR) T-cell immunotherapy for the treatment of AML. FLT3 expression analysis and in vitro and in vivo studies were leveraged to evaluate the nonclinical safety of AMG 553. Cynomolgus monkeys administered autologous anti-FLT3 CAR T cells demonstrated no evidence of CAR T-cell-mediated toxicity, expansion, or persistence, likely due to restricted cell surface FLT3 protein expression in healthy animals. This highlights the limited value of such in vivo studies for safety assessment of the CAR T-cell modality when directed against a target with restricted expression. To complement these studies and directly evaluate the potential toxicities of eliciting T-cell-mediated cytotoxicity against cells with surface expression of FLT3 protein in vivo, data from cynomolgus monkey toxicology studies with 2 bispecific T-cell engager molecules targeting FLT3 were leveraged; findings were consistent with the targeted killing of bone marrow cells expressing cell surface FLT3. Potential AMG 553-induced cytotoxicity was assessed against a wide range of normal human primary cells and cell lines; cytotoxicity was observed against FLT3-positive AML cell lines and a percentage of primary bone marrow CD34+ cells. In conclusion, the nonclinical safety data suggest that AMG 553 can target FLT3 protein on AML cells, whereas only affecting a percentage of normal hematopoietic stem and progenitor cells, supporting clinical development.
Keywords: chimeric antigen receptor T cells, acute myeloid leukemia, safety
Acute myeloid leukemia (AML) is a hematological disorder characterized by the accumulation of malignant myeloid progenitor cells in the bone marrow and blood. Acute myeloid leukemia is the most common form of acute leukemia in adults (Wiese and Daver, 2018). In 2018, an estimated 19 520 new cases of AML were expected in the United States with approximately 10 670 deaths from this disease (Howlader et al., 2018). Recent approvals of new therapies come after approximately 30 years of minimal changes to the standard therapeutic regimen (Bohl et al., 2019; Tiong and Wei, 2019). However, many of these new therapies are targeted toward patients expressing specific genetic mutations therefore limiting the number of patients receiving benefit. T-cell therapeutics such as bispecific T-cell engager (BiTE) molecules and chimeric antigen receptor (CAR) T cells redirect a patient’s own T cells to eliminate a tumor expressing a specific tumor-associated antigen through T-cell-mediated cytotoxicity. Such therapeutics have shown promise in hematologic malignancies such as CD19-positive acute lymphoblastic leukemia and non-Hodgkin’s lymphoma. CD19-binding blinatumomab is approved for various types of acute lymphoblastic leukemia and has been shown to induce complete responses in B-cell lymphomas (Goebeler and Bargou, 2016) and the CD19-directed CAR T-cell therapies tisagenlecleucel (Kymriah; Novartis, East Hanover, New Jersey) (Maude et al., 2018) and axicabtagene ciloleucel (Yescarta; Kite Pharma, Santa Monica, California) (Neelapu et al., 2017) were approved by the Food and Drug Administration (FDA) in 2017. Similar approaches may be possible for treating AML.
Feline McDonough Sarcoma (FMS)-like tyrosine kinase 3 (FLT3), also referred to as CD135, is a tyrosine kinase receptor expressed on the surface of a subset of hematopoietic stem and progenitor cells that plays a key role in differentiation. This receptor is detected on the cell surface of approximately 80% of leukemia isolates from adult and pediatric patients with AML (Brauchle et al., 2020; Meshinchi and Appelbaum, 2009; Rosnet et al., 1996; Tarlock et al., 2017), with no significant difference in expression level at the time of initial diagnosis versus at the time of relapse (Lindl et al., 2017). FLT3 is one of the most commonly mutated genes in AML disease samples (Ley et al., 2013), suggesting that FLT3 has an important role in AML pathogenesis. FLT3 mutations, detected in approximately 30% of AML patients, occur in the intracellular signal transduction region. Internal tandem duplication of base pairs within the juxtamembrane region or point mutations in the second kinase domain of FLT3 results in ligand-independent constitutive activation (Gilliland and Griffin, 2002; Nakao et al., 1996). The discovery of these mutations led to the development of small molecule inhibitors of mutant FLT3, including midostaurin (Fischer et al., 2010; Schlenk et al., 2019; Stone et al., 2017), crenolanib (Zimmerman et al., 2013), and gilteritinib (Mori et al., 2014; Perl et al., 2017) which have been approved by the FDA for AML patients whose disease harbors mutant FLT3. Although FLT3 inhibitors are expected to benefit a subset of patients with disease expressing FLT3 mutations, therapies eliciting T-cell-mediated cytotoxicity by targeting the extracellular domain of FLT3, such as a CAR T-cell or BiTE molecule therapy, would provide benefit irrespective of FLT3 mutation status.
AMG 553 is an investigational, adoptive cellular immunotherapy for the treatment of relapsed/refractory AML, consisting of autologous T cells that have been genetically modified ex vivo to express a transmembrane CAR to target FLT3 protein on the surface of AML cells. The AMG 553 CAR construct consists of single chain variable fragment (scFv) that binds an epitope in the extracellular domain of FLT3, a CD28 costimulatory domain, and a CD3 zeta chain subunit-activation domain. A combination of studies was leveraged to extensively evaluate the nonclinical safety of AMG 553. First, FLT3 transcript, protein expression, and cellular localization were thoroughly assessed in a wide range of normal human tissues to determine potential off-tumor, on-target liabilities (Brauchle et al., 2020). Exploratory toxicology studies were then performed in cynomolgus monkeys administered autologous anti-FLT3 CAR T cells. Additionally, data from cynomolgus monkey toxicology studies with 2 separate BiTE molecules targeting FLT3 protein were leveraged to complement the CAR T-cell exploratory cynomolgus monkey studies and to directly evaluate the potential toxicities of eliciting T-cell-mediated cytotoxicity against cells with surface expression of FLT3 protein in vivo. Finally, in vitro AMG 553-mediated cytotoxicity was assessed in cells from a variety of normal human tissues reported to express FLT3 transcript and/or FLT3 protein expression as well as from major organs such as the liver.
MATERIALS AND METHODS
FLT3 mRNA and FLT3 Protein Assessment in Normal Cynomolgus Monkey Tissues
In situ hybridization, RNA-sequencing (RNA-seq), and immunohistochemistry (IHC) were conducted to assess FLT3 target expression in normal cynomolgus monkey tissues using standard techniques as detailed in the Supplementary Methods.
In Vivo Study Animal Care
Cynomolgus monkeys were cared for in accordance to National Research Council (2011) and individually housed at an indoor American Association for the Accreditation of Laboratory Animal Care international accredited facility in species-specific housing. All research protocols were approved by the Institutional Animal Care and Use Committee. All animals were negative for simian retrovirus and tuberculosis. Cynomolgus monkeys were fed a certified pelleted primate diet daily in amounts appropriate for the age and size of the animals and had ad libitum access to municipal tap water processed through a reverse osmosis filter and UV light treatment, via automatic watering device. Animals were maintained on a 12-h light:12-h dark cycle in rooms at 64°F–84°F and 30%–70% humidity and had access to enrichment opportunities (device, food treat, and/or socialization).
In Vivo Assessment of Autologous Anti-FLT3 CAR T Cells in Cynomolgus Monkeys
Preparation of Autologous Anti-FLT3 CAR T Cells
Peripheral blood mononuclear cells (PBMCs) were isolated from cynomolgus monkeys, transduced with the AMG 553 anti-FLT3 CAR construct, and expanded as detailed in the Supplementary Methods.
Study Design
In the first study, cynomolgus monkeys (1 male/dose group) received a single intravenous (IV) dose of 1.28 × 106, 1.84 × 107, or 2.74 × 107 CAR+ cells/kg or 5.75 × 107 total untransduced T cells (negative control) on day 1 and were necropsied on day 29. In a second study, a single group of 3 male cynomolgus monkeys were pretreated on days −5, −4, and −3 with cyclophosphamide and fludarabine as a nonmyeloablative lymphodepleting and preconditioning treatment. On day 1, the animals received a single IV dose of autologous anti-FLT3 CAR T cells at approximately 1 × 108 cells/kg. Scheduled necropsies were conducted at approximately 24 h postdose for 1 animal and on day 15 for the other 2 animals. Doses in both studies were the maximum feasible dose based off of the PBMCs isolated from serial blood collections from the animals, the transduction efficiency, and successful expansion of the cells. Anti-FLT3 CAR T-cell persistence after infusion, pharmacodynamic (PD; FLT3 transcript and soluble FLT3 ligand [sFLT3L] levels), and safety endpoints (clinical observations, food consumption, clinical pathology, serum cytokine levels, and macro- and microscopic observations) were assessed.
Monitoring of Anti-FLT3 CAR T-Cell Persistence in PBMC by ddPCR
Postinfusion, approximately 200 ng of DNA isolated from PBMC was added to each well of a 96-well PCR plate along with droplet digital polymerase chain reaction (ddPCR) Supermix, and primers/probes for detection of the FLT3-CAR and a single copy endogenous gene (RPP30). Droplets were generated using an automated droplet generator (Bio-Rad, Hercules, CA). The plate was run on a thermocycler using standard conditions and read using the QX200 Droplet Reader (Bio-Rad, Hercules, CA).
Monitoring of FLT3 mRNA Levels by ddPCR
For blood specimens and bone marrow aspirates, the template cDNA for ddPCR applications was generated from 250 to 400 ng of DNase I-treated total RNA using SuperScript III First-Strand Synthesis SuperMix for qRT-PCR (Thermo Fisher Scientific, Waltham, Massachusetts) following the manufacturer’s protocol. PrimeTime quantitative polymerase chain reaction assays (Integrated DNA Technologies, Coralville, Iowa) were designed to exon-exon junction regions in the gene encoding FLT3 based on the cynomolgus reference genome sequence.
Monitoring Cytokine Levels in Cynomolgus Monkey Serum
Serum or plasma samples from cynomolgus monkeys were analyzed using Millipore Milliplex MAP Non-Human Primate Cytokine/Chemokine Kits (Millipore Sigma, Burlington, Massachusetts) on a Luminex platform according to the manufacturer’s protocol or a good laboratory practice (GLP)-validated protocol.
Monitoring sFLT3L Levels in Cynomolgus Monkey Serum
Serum sFLT3L was measured with an R&D System’s Quantikine Enzyme-Linked Immunosorbent Assay Human FLT3 Ligand Immunoassay (R&D Systems, Inc, a biotechne brand, Minneapolis, Minnesota), optimized for use with nonhuman primate samples according to the manufacturer’s protocol.
In Vivo Assessment of 2 Anti-FLT3 BiTE Molecules in Cynomolgus Monkeys
Study Designs
In an initial exploratory study, 3 female cynomolgus monkeys were administered escalating doses of the test article, an anti-FLT3 BiTE molecule (“canonical BiTE molecule”) which does not contain a half-life extending (HLE) Fc moiety, via IV infusion for 16 days as described (Brauchle et al., 2020). The low dose was administered days 1–3, middose 1 (3× low dose) days 4–6, middose 2 (9× low dose) days 7–9, and high dose (20× low dose) days 10–16. In the second study, cynomolgus monkeys were administered an anti-FLT3 BiTE molecule containing an HLE Fc moiety (“HLE anti-FLT3 BiTE molecule”), or the control article via IV slow bolus approximately once weekly for 28 days. Animals received either vehicle control (n = 5 animals/sex), low dose (n = 3 animals/sex), middose (5× low dose; n = 5 animals/sex), or a step-dose regimen (low dose/10× low dose/50× low dose; n = 3 animals/sex). The study was performed in accordance with the U.S. Department of Health and Human Services, FDA, United States Code of Federal Regulations, Title 21, Part 58: GLP for Nonclinical Laboratory Studies and as accepted by Regulatory Authorities throughout the European Union (OECD Principles of Good Laboratory Practice) and Japan (MHL W). Toxicokinetic (TK), PD (FLT3 transcript and sFLT3L levels), and safety endpoints (clinical observations, food consumption, ophthalmic and neurologic examinations, cardiovascular examination by jacketed external telemetry, respiratory rate, clinical pathology, serum or plasma cytokine levels, and macro- and microscopic observations) were assessed.
TK Analysis
Toxicokinetic analysis of the canonical BiTE molecule was performed as described elsewhere (Brauchle et al., 2020). For the HLE anti-FLT3 BiTE molecule studies, TK samples were quantified using a validated enzyme-linked immunosorbent assay with an lower limit of quantitation (LLOQ) of 20 ng/ml as outlined in the Supplementary material. Samples that were below the LLOQ were reanalyzed using a qualified, but not validated assay, with an LLOQ of 0.200 ng/ml. Assay reagents were the same as in the validated assay. The modifications to the qualified assay to achieve the 0.200 ng/ml for the LLOQ were decreasing the minimum required dilution in sample pretreatment and increasing the concentration of the streptavidin-poly horseradish peroxidase. Serum samples were also analyzed for the presence of antidrug antibodies (ADAs). Antidrug antibody samples were analyzed for binding antibodies against the HLE anti-FLT3 BiTE molecule using a validated Universal Indirect Species-specific Assay.
Monitoring Serum Cytokine Levels, FLT3 Transcript, and sFLT3L Levels in Cynomolgus Monkey
Cytokine, FLT3 transcript in bone marrow and peripheral blood, and sFLT3L levels were monitored as described above in the study of FLT3 CAR T cells in cynomolgus monkeys. In the 28-day study, FLT3 transcript levels were compared across time points within each group and with time-matched vehicle treated controls. A t test was performed based on the mean and degrees of freedom for each time point using a web-based calculator (http://www.socscistatistics.com/pvalues/Default.aspx; last accessed July 8, 2020). Statistical significance was defined as a p value ≤ .05.
In Vitro Assessment of On-Target, Off-Tumor, and Off-Target Toxicity
Human Primary Cells, Cell Lines, and Human Samples
All human specimens were collected under Institutional Review Board approval with appropriate informed consent. In all cases, materials obtained were surplus to standard clinical practice. Patient identity and protected health information (PHI/identifying information were redacted from tissues and clinical data).
Primary cells and cell lines were selected from tissues reported to express FLT3 transcript or FLT3 protein expression as well as from major organs. Human primary cells (epithelial cells from the renal proximal tubule, pancreas, and prostate; cortical neurons, plasmacytoid dendritic cells (pDCs); osteoclasts differentiated from primary osteoclast precursors; cardiac myocytes; and hepatocytes) and human cell lines (CAL62 thyroid carcinoma, NU-GC-4 stomach adenocarcinoma, NCI-H1975 lung adenocarcinoma, and HuH-7 hepatic carcinoma) were cultured as described in Supplementary Table S1. Chinese hamster ovary (CHO) cells and a B-cell leukemia cell line (Namalwa) served as negative controls, whereas CHO cells overexpressing human FLT3 (CHO-FLT3) and AML cell lines (EOL-1, HL-60, MOLM-13, and MV4-11) served as positive controls and are also described in Supplementary Table S1.
Characterization of Target Expression in Cells and Cell Lines for In Vitro Cytotoxicity Assessment
FLT3 transcript expression was characterized by RNA-seq using standard techniques as detailed in the Supplementary Methods. FLT3 protein expression was characterized with the Sally Sue automated capillary electrophoresis immunoassay system (Protein Simple, San Jose, California) as described in the Supplementary Methods. This single platform assay has more quantitative, reproducible results and a wider dynamic range than a traditional Western analysis (Chen et al., 2013; Xu et al., 2015).
Generation of AMG 553 for In Vitro Cytotoxicity Assessment
Chimeric antigen receptor T-cell transduction
AMG 553 CAR constructs were synthesized by Integrated DNA Technologies (IDT; San Diego, California) and subcloned downstream of the MSCV promoter in the pGAR MSCV eGFP mPGK Puro vector. In total, 293 T cells were transiently transfected with 10 µg of CAR and 120 µg total of packaging plasmids pLP1, pLP2, and pLP/VSVG (Thermo Fisher Scientific) using polethyleneimine. After 72 h, lentiviral particles were collected, filtered, and stored at 4°C for ≤1 week. Commercially obtained normal human PBMCs from 2 donors (PBMCs; Hemacare, Los Angeles, California) were thawed, washed twice in RPMI 1640 supplemented with 10% fetal bovine serum and 1× penicillin streptomycin l-glutamine media, and expanded in cell culture. Cells were seeded at 106 cells/ml and stimulated with 300 IU/ml OKT3 and 50 ng/ml IL-2 for 48 h after which cells were counted, washed, and resuspended at 106 cells/ml. Lentiviral particles were added to cell cultures; cell cultures were normalized to 5 × 105 cells/ml every other day for 12 days.
T-cell-dependent cellular cytotoxicity assay
Chimeric antigen receptor T cells were plated in 1:1 effector:target (E:T) ratio with target cell lines. For flow cytometry-based assays, cells were targeted in round-bottom 96-well plates, and target cell viability was assessed by flow cytometry enumeration of live target cells (CD3−, ToPro3−; BV421 mouse-anti human CD3 Clone SP34-2; BD Biosciences, San Jose, California) and To-Pro-3 (Thermo Fisher Scientific). For imaging-based T-cell-dependent cellular cytotoxicity (TDCC) assays, target cells were plated in black flat bottom 96-well plates and cultured in an incubator equipped with an IncuCyte Zoom (Essen Bioscience, Ann Arbor, Michigan); phase contrast images (4 fields of view/well) were collected every 2 h for 48 h. Next, Hoechst (1:5000 dilution) and To-Pro-3 (1:5000 dilution) were added to each well, plates were incubated 15 min at 37°C then imaged on an Opera Phenix (Perkin Elmer, Waltham, Massachusetts) with a 20× objective (49 fields of view/well). Live cells were enumerated, and percent confluence was analyzed with IncuCyte Zoom 2016B software.
RESULTS
FLT3 Protein Expression in Cynomolgus Monkey Tissues Is Similar to Expression in Human Tissues
The mechanism of action (MOA) of AMG 553 is binding and subsequent T-cell mediated lysis of cells expressing FLT3 protein on the cell surface. Therefore, to understand potential off-tumor, on-target liabilities, FLT3 mRNA and FLT3 protein expression were thoroughly assessed in normal human tissues using numerous complementary methods. Full results are presented elsewhere (Brauchle et al., 2020) but were consistent with published results (Çakmak-Görür et al., 2019; Kikushige et al., 2008). Briefly, cell surface FLT3 protein in human tissues was only detected in a portion of bone marrow hematopoietic stem and progenitor cells in addition to rare, scattered cells in the tonsil; FLT3 protein in other cell types was cytoplasmic.
Cynomolgus monkey FLT3 transcript and FLT3 protein expression was evaluated in a variety of normal tissues to determine if the cynomolgus monkey was a representative nonclinical species for assessment of FLT3 target toxicity. Full results of FLT3 protein expression on cynomolgus monkey hematopoietic cells are presented elsewhere (Brauchle et al., 2020). Briefly, cell surface FLT3 protein detection on cynomolgus monkey hematopoietic cells was similar to human and consistent with previously reported human expression (Kikushige et al., 2008); detectable cell surface FLT3 protein was restricted to a portion of cynomolgus monkey CD34+ bone marrow cells with no detection on the surface of other peripheral blood cell types (Brauchle et al., 2020).
Results for cynomolgus monkey nonhematopoietic tissues with a positive FLT3 expression signal by at least one method, along with comparison to the previously published human results (Brauchle et al., 2020), are provided in Table 1. FLT3 transcript was detected in the gastrointestinal, respiratory, nervous, urinary, musculoskeletal, male and female reproductive systems, eye, tongue, and thyroid of cynomolgus monkey. Because AMG 553 is only anticipated to target FLT3 protein on the cell surface, IHC was used to elucidate the cellular localization of FLT3 protein on FLT3 transcript-expressing tissue. Positive, cytoplasmic FLT3 protein staining was observed by IHC in cynomolgus monkey pancreas, stomach, skeletal muscle, brain (cerebrum, cerebellum, and brain stem), pituitary, testis, and kidney. Representative images of FLT3 IHC staining of cynomolgus monkey tonsil, pancreas, and cerebellum are presented in Figure 1. FLT3 protein staining in the cerebrum, cerebellum, and the ganglion cell layer of the retina consisted of cytoplasmic staining of multifocal neurons. This staining pattern was consistent in multiple sections of brain and peripheral nerve with no membranous staining observed in neurons (Figure 1). These data indicate that FLT3 expression and cellular localization in normal cynomolgus tissues are similar to human and the cynomolgus monkey is a representative nonclinical species for assessment of FLT3 target toxicity.
Table 1.
Summary of Human and Cynomolgus Monkey Nonimmune Tissues Positive for FLT3 mRNA and/or FLT3 Protein Expression in at Least One Platform
| Species |
Cynomolgus Monkey |
Humana |
||||||
|---|---|---|---|---|---|---|---|---|
| Biomolecule |
mRNA |
Proteinb |
mRNA |
Proteinb |
||||
| Organ Systemc/Platform | ISH | RNA-seqd | IHCcyto | IHCmem | qPCRe | RNA-seqd | IHCcyto | IHCmem |
| Endocrine | + | + | − | − | − | + | − | |
| Female reproductive | + | − | − | − | − | − | − | |
| GI | + | + | + | − | − | + | + | − |
| Male reproductive | + | + | + | − | + | − | + | − |
| Musculoskeletal | − | + | + | − | − | + | + | − |
| Nervous | + | + | + | − | + | + | + | − |
| Sense (eye, tongue) | + | + | + | − | − | − | − | |
| Respiratory | − | + | − | − | + | + | − | − |
| Urinary | + | + | + | − | − | + | + | − |
+, positive; −, negative.
Abbreviations: GI, gastrointestinal; IHC, immunohistochemistry; ISH, in situ hybridization; qPCR, quantitative polymerase chain reaction; RNA-seq, RNA-sequencing.
Full results previously published in Brauchle et al. (2020).
Tissues are negative if staining is in resident leukocytes only. IHCcyto, cytoplasmic staining; IHCmem, plasma membrane staining.
Positive when at least one sample of a tissue within the organ system showed a signal defined as + or greater.
Positive when median FPKM ≥ 0.75.
Positive when > 0.75 transcripts/cell.
Figure 1.

Feline McDonough Sarcoma-like tyrosine kinase 3 (FLT3) expression in by immunohistochemistry (IHC) in cynomolgus monkey. FLT3 expression by IHC demonstrates variable expression patterns in cynomolgus monkey tissues. FLT3 expression by IHC was membranous and cytoplasmic in cynomolgus monkey tonsillar mononuclear cells (A) and cytoplasmic only in cynomolgus monkey pancreas (B). FLT3 IHC staining in cerebellum (C) was limited to punctate cytoplasmic staining in neurons (D, higher magnification of C).
No CAR T-Cell-Mediated Toxicity, Persistence, or Expansion After Administration to Cynomolgus Monkeys
Two separate single dose exploratory studies with autologous CAR T cells were conducted in cynomolgus monkey, one with and one without preconditioning lymphodepletion. T cells from cynomolgus monkeys were transduced with the same CAR construct as AMG 553. The transduction efficiency was 24%–74% and in vitro expansion after transduction was up to 9.7-fold. After 40 h of in vitro culture at an effector:target (E:T) ratio of 1:1, transduced T cells demonstrated FLT3 specific in vitro cytotoxicity with 36–57 or 32–57 percent dead target cells for cell lines expressing human FLT3 or nonhuman primate FLT3, respectively, whereas percent dead FLT3-negative cells was 15–19. In addition to demonstrating FLT3-dependent target cell killing, human and cynomolgus monkey FLT3-dependent increases in IFN-γ levels in culture were demonstrated with an increase of 2.6- to 34.9-fold higher than obtained with target cells not expressing FLT3. Autologous CAR T cells were administered to cynomolgus monkeys, with or without preconditioning lymphodepletion. Cell persistence after infusion, PD, and safety endpoints were assessed.
FLT3 CAR T cells were not detectable at any time point in isolated PBMCs in those monkeys who were not lymphodepleted. In the study with lymphodepletion, measurable levels of FLT3 CAR T cells in isolated PBMC (1% of total PBMC) were only detected by ddPCR at the end of the 30-min infusion of the cells in the 1 animal that was assessed at that time point; levels were not detectable above baseline levels at other time points for any animal using flow cytometry or ddPCR (Figure 2). Multiple tissues collected at the time of necropsy (day 2 or 15) including, but not limited to, brain, bone marrow, cerebral spinal fluid (CSF), and spinal cord were negative for FLT3 CAR T cells using ddPCR.
Figure 2.
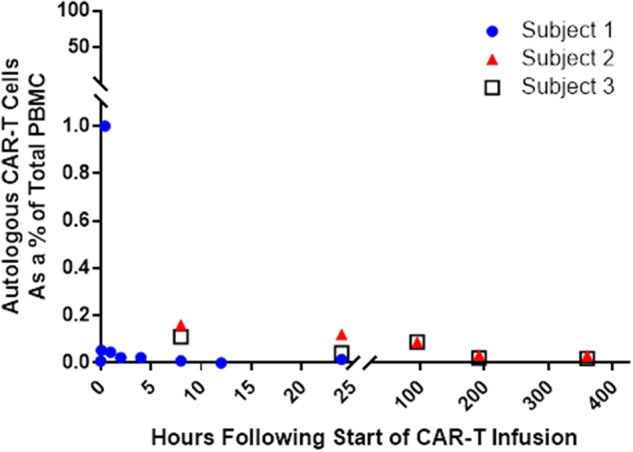
Autologous anti-Feline McDonough Sarcoma-like tyrosine kinase 3 (anti-FLT3) chimeric antigen receptor (CAR) T cells do not persist in cynomolgus monkeys in vivo. Three male cynomolgus monkeys were administered autologous anti-FLT3 CAR T cells. ddPCR was used to quantify the level of FLT3+ CAR T cells in peripheral blood mononuclear cell samples.
FLT3 transcript was measured in whole blood and bone marrow samples as a marker of the number of cells expressing FLT3 transcript to understand the PD effect of the FLT3 CAR T cells. sFLT3L was measured in the serum as an exploratory indicator of the loss of FLT3-expressing cells (Bertho et al., 2001). There were no anti-FLT3 CAR T-cell related changes in blood or bone marrow FLT3 transcript levels, nor serum sFLT3L throughout either study.
In the study without lymphodepletion, clinical signs were limited to a transient increase in body temperature on day 2 in one animal and clinical pathology changes were limited to increased C-reactive protein (CRP) at 8 and/or 24 h after dosing in all animals that received anti-FLT3 CAR T cells; both parameters recovered by day 4. All clinical pathology, flow cytometry, and microscopic changes observed in the study with lymphodepletion were considered to be related to the lymphodepletion regimen (cyclophosphamide and fludarabine) itself and/or handling treatment and not exacerbated by administration of anti-FLT3 CAR T cells. Changes were of similar incidence and/or severity as a previously conducted cyclophosphamide/fludarabine study (data not shown) and/or consistent with reports in the literature (Malik et al., 1996; Philips et al., 1961) and not further affected by anti-FLT3 CAR T-cell administration. There were no anti-FLT3 CAR T-cell-related changes in body weight, food consumption, hematology or coagulation, urinalysis, cytokines, bone marrow or CSF cytology, and flow cytometry or light microscopic changes in any animals in either study.
FLT3 Targeting BiTE Molecules Informed Potential On-Target Toxicities in Cynomolgus Monkeys
Data from cynomolgus monkey toxicology studies with 2 separate BiTE molecules targeting FLT3 were leveraged. The canonical FLT3 BiTE molecule is an exploratory molecule which contains the same anti-FLT3 scFv as AMG 553 and cross reacts with cynomolgus monkey (Brauchle et al., 2020). The HLE anti-FLT3 BiTE molecule cross reacts with cynomolgus monkey and the anti-FLT3 scFv binds the same FLT3 epitope as AMG 553. The molecules have similar binding affinities for cynomolgus monkey FLT3 and CD3 (Brauchle et al., 2020).
In a 16-day exploratory toxicology study, an anti-FLT3 canonical BiTE molecule was administered via continuous IV infusion in a step-dose regimen, resulting in approximately dose-proportionally increased exposures (Brauchle et al., 2020). Signs of FLT3 BiTE molecule-related PD activity (Brauchle et al., 2020) were observed, including dose-independent increases in serum cytokine concentration (Figure 3), decreased circulating leukocytes, and clinical pathology changes, such as increased CRP, consistent with an inflammatory response; many of these parameters were attenuated with continued dosing and/or approached predose values at the end of infusion. At the end of the study, FLT3 BiTE molecule-related changes were observed in bone marrow cytology which predominately affected cells of the myeloid lineage; these changes included decreased neutrophil and/or eosinophil promyelocytes, metamyelocytes, and/or myelocytes and were consistent with T-cell redirected lysis of FLT3-positive bone marrow hematopoietic stem and progenitor cells. No FLT3 BiTE molecule-related changes in body temperature, body weight, food consumption, or light microscopic changes were observed.
Figure 3.

Feline McDonough Sarcoma-like tyrosine kinase 3 (FLT3) bispecific T-cell engager (BiTE) molecule-mediated increases in cytokines in cynomolgus monkeys. Three female cynomolgus monkeys were administered a step dose of the experimental FLT3 BiTE molecule via intravenous infusion for 16 days as described (Brauchle et al., 2020); doses were escalated every 3 days. Serum was collected at the indicated times and kept frozen until analysis. All samples within a dose group were analyzed concurrently. IFN-γ (A), TNF-α (B), MCP-1 (C), and IL-6 (D) serum concentrations were quantified using the MILLIPLEX MAP for Nonhuman Primate Cytokine Magnetic Bead Kit (Millipore Sigma) according to the manufacturer’s protocol and within assay standard curve limits.
The HLE anti-FLT3 BiTE molecule was administered to cynomolgus monkeys approximately once weekly at 2 flat doses (referred to as low- or middose) or using a step-dosing regimen where the last dose was greater than the flat doses in a 1-month GLP compliant toxicology study. Serum exposure levels were approximately dose-proportional at the beginning of the study. Binding ADAs developed during the course of the study and were associated with decreased serum exposure in all animals. Toward the end of the study, BiTE molecule levels in some animals were close to or below the limit of quantification (0.2 ng/ml); there was no quantifiable HLE anti-FLT3 BiTE molecule in the CSF at the end of the study. Evidence of PD was indicated by statistically significant decreases in FLT3 transcript in blood and increased serum levels of sFLT3L at all doses (Figure 4). sFLT3L was measured in the serum as an indicator of the amount of its receptor, FLT3, with an inverse relationship to the amount of FLT3-expressing cells expected because the FLT3 receptor is less available (Bertho et al., 2001). At the end of the study (day 29), FLT3 transcript levels in blood and sFLT3L levels in serum reached or were approaching pretreatment levels which is consistent with the loss of anti-FLT3 BiTE molecule exposure. There were no changes in bone marrow FLT3 transcript levels.
Figure 4.

Half-life extending (HLE) anti-Feline McDonough Sarcoma-like tyrosine kinase 3 (anti-FLT3) bispecific T-cell engager (BiTE) molecule-mediated alterations in FLT3 transcript and soluble FLT3 ligand (sFLT3L) levels. Cynomolgus monkeys were administered the HLE anti-FLT3 BiTE molecule or the vehicle article via intravenous slow bolus approximately once weekly for 28 days. Animals received either vehicle control (n = 5 animals/sex), low dose (n = 3 animals/sex), middose (n = 5 animals/sex), or step dose (n = 3 animals/sex). Serum or peripheral blood mononuclear cell samples were collected pretreatment (0), day 12 predose and day 29, and kept frozen until analysis. All samples within a dose group were analyzed concurrently. FLT3 mRNA (A) was quantified using ddPCR and sFLT3L levels (B) were measured using Quantikine Enzyme-Linked Immunosorbent Assay Human Flt-3 Ligand Immunoassay (R&D Systems, Inc). Note that 5 animals were excluded from the middose analyses due to early mortality. In (A), each line demarks how the FLT3 mRNA changes over time for each cynomolgus monkey.
Administration of the middose resulted in early mortality in 5 out of 10 animals. The cause of death/moribundity was multifactorial including decreased bone marrow cellularity, bacteremia, and/or ADA-mediated thrombocytopenia. Decreased bone marrow cellularity was consistent with T-cell-redirected lysis of FLT3 expressing bone marrow hematopoietic stem and progenitor cells; bacteremia was secondary to the decrease in myeloid cells and subsequent decreased innate immunity. All 5 animals were positive for ADAs. The decreases in platelet counts were consistent with ADA-mediated destruction of platelets and not a direct effect of the anti-FLT3 HLE BiTE molecule because megakaryocyte numbers were deemed adequate to support platelet production and platelet counts were not decreased in animals receiving higher doses. ADA-mediated effects on platelet counts, and specifically, shortened platelet life span due to platelet destruction by drug induced antibodies, and thrombocytopenia are known possible sequelae to the administration of therapeutic proteins (Aster and Bougie, 2007; Everds and Tarrant, 2013; Koren et al., 2008; Mease et al., 2017). Development of ADAs in nonhuman primates is not predictive of immunogenicity in humans (Avery et al., 2005; Ponce et al., 2009; van Meer et al., 2013).
For animals that survived to the scheduled day 29 necropsy, there were no anti-FLT3 HLE BiTE molecule-related changes in body weight, qualitative food consumption, ophthalmic examination, body temperature, neurological examination, respiration rate, urinalysis, CSF parameters, or macroscopic observations. Electrocardiograms were collected during the study via jacketed external telemetry and the only anti-FLT3 HLE BiTE molecule-related effect was a dose-responsive elevation in heart rate after the initial low- or middose administration which was attenuated with repeated dosing.
Consistent with anti-FLT3 HLE BiTE molecule-induced T-cell activation, transient, HLE anti-FLT3 BiTE molecule-related increases in plasma cytokine levels (MCP-1, IFNγ, and IL-6) were observed at all doses 4 h after the first administration. Cytokine levels approached prestudy or vehicle control levels within 24 h in most animals (Figure 5). Following the step dose on days 5 and 8, increased serum MCP-1 concentration was observed 4 h after dose administration, but the increases were attenuated; relatively minor increases were observed in 2 animals on day 26, 4 h after dose administration with return to pretreatment by 24 h.
Figure 5.

Half-life extending (HLE) anti-Feline McDonough Sarcoma-like tyrosine kinase 3 (anti-FLT3) bispecific T-cell engager (BiTE) molecule-mediated increases in cytokine in cynomolgus monkey. Cynomolgus monkeys were administered the HLE anti-FLT3 BiTE molecule or the vehicle control via intravenous slow bolus approximately once weekly for 28 days. Animals received either vehicle control (n = 5 animals/sex), low dose (n = 3 animals/sex), middose (n = 5 animals/sex), or step dose (n = 3 animals/sex). Plasma samples were collected pretreatment (0) and at the indicated timepoints and kept frozen until analysis. All samples within a dose group were analyzed concurrently. IFN-γ (A), IL-6 (B), and MCP-1 (C) concentration as quantified using the MILLIPLEX MAP for Nonhuman Primate Cytokine Magnetic Bead Kit (Millipore Sigma) according to the manufacturer’s protocol and within assay standard curve limits. The average cytokine concentration per dose cohort was plotted versus time post treatment initiation. Note that 5 animals were excluded from the middose analyses due to early mortality. One day 2 sample from the low dose group and one day 27 sample from the Untreated group were processed to serum instead of plasma.
HLE anti-FLT3 BiTE molecule-related hematology changes included decreased circulating leukocytes at all doses at the beginning of the study which remained decreased in some animals at the end of the study. Changes in clinical chemistry parameters, such as an increase in CRP, early in the study were consistent with an acute phase response at all dose levels (Cabana et al., 1996; Cray et al., 2009); changes were attenuated with repeated dosing and most reached or were approaching vehicle treated control levels at the end of the study. At the end of the study, light microscopic changes at all dose levels included decreased bone marrow cellularity which was characterized by decreased numbers of myeloid cells in most animals and decreased erythroid cells in 2 animals. In addition, increased cellularity was observed in the bone marrow, lymph nodes, and/or spleen at all dose levels which was consistent with recovery due to ADA-mediated loss of HLE anti-FLT3 BiTE molecule exposure.
AMG 553 Was Selectively Cytotoxic Against a Portion of Human Primary CD34+ Bone Marrow Cells In Vitro
AMG 553 recognizes an epitope in the extracellular domain of FLT3 and is therefore expected to target only cells that express cell surface FLT3. FLT3 target expression analysis revealed expression of FLT3 transcript and cytoplasmic FLT3 protein in a small number of normal tissues. Because of the potency of the CAR T modality and the number of CARs expressed on a given T-cell, AMG 553-mediated cytotoxicity was assessed in a panel of normal human cell types from various tissues. FLT3 expression in these primary cells and cell lines was assessed by RNA-seq and Western capillary electrophoresis immunoassay (Table 2). FLT3 mRNA and FLT3 protein were detected in primary CD34+ bone marrow cells and primary pDCs (Goldstein, Frank, Dunn, Kim, Wang, Karbowski, Balazs, Coxon, Koppikar, Lebrec, and Arvedson, in preparation). Neither FLT3 transcript nor FLT3 protein (Figure 6) was detected in any other cell type, including those from tissues previously reported to have FLT3 expression (Brauchle et al., 2020). In a TDCC assay comprising AMG 553 and normal primary human CD34+ bone marrow cells, AMG 553-mediated cytotoxicity was observed against a portion of primary CD34+ bone marrow cells (Goldstein, Frank, Dunn, Kim, Wang, Karbowski, Balazs, Coxon, Koppikar, Lebrec, and Arvedson, in preparation) (Table 2), consistent with reported cell surface FLT3 expression on a subset of these cells. AMG 553-mediated cytotoxicity was not observed in TDCC assays against other cell types including primary cortical neurons obtained from 2 donors (Table 2 and Figure 7).
Table 2.
In Vitro AMG 553-Mediated Cytotoxicity Is Restricted to CD34+ Bone Marrow Cells
| Test System Name (Cell Type) |
|||
|---|---|---|---|
| Positive control cells | FLT3 Expressiona | Cytotoxicityb | |
| EOL-1 (AML) | + | + | |
| HL-60 (AML) | + | + | |
| MOLM-13 (AML) | + | + | |
| MV4-11 (AML) | + | + | |
| CHO-FLT3 (human FLT3 overexpressing CHO cell line) | + | + | |
| Negative control cells | |||
| Namalwa (B-cell leukemia) | − | − | |
| CHO | − | − | |
| Cells from tissues with previous evidence of FLT3 cell surface protein expression | |||
| CD34+ bone marrow cells (primary) | + | + | |
| Cells from tissues with previous evidence of FLT3 transcript and/or FLT3 intracellular protein but no detectable FLT3 cell surface protein expression | |||
| Renal proximal tubule epithelial cells (primary) | − | − | |
| CAL-62 (thyroid carcinoma) | − | − | |
| Pancreatic epithelial cells (primary) | − | − | |
| NU-GC-4 (stomach adenocarcinoma) | − | − | |
| Prostate epithelial cells (primary) | − | − | |
| NCI-H1975 (lung adenocarcinoma) | − | − | |
| Cortical neurons (primary) | − | − | |
| Plasmacytoid dendritic cells (primary) | + | − | |
| Osteoclasts (differentiated from primary osteoclast precursors) | − | − | |
| Cells from tissues with no previous evidence of FLT3 protein and/or FLT3 transcript expression | |||
| Cardiac myocytes (primary) | − | − | |
| HuH-7 (hepatic carcinoma) | − | − | |
| Hepatocytes (primary) | − | − | |
FLT3 mRNA was assessed by RNA-sequencing and FLT3 protein was assessed by Western Capillary Electrophoresis Immunoassay. +, positive for both mRNA and protein; −, negative for both mRNA and protein.
Cytotoxicity was assessed as mean percent decrease in live cells. +, positive; −, negative.
Figure 6.

Feline McDonough Sarcoma-like tyrosine kinase 3 (FLT3) protein detection in primary cells and cell lines. FLT3 protein expression was assessed in primary cells and cell lines (A, B) using Western blot capillary electrophoresis immunoassays. FLT3 positive controls included lysates from Chinese hamster ovary (CHO) cells transfected with human FLT3 (CHO-FLT3) and human acute myeloid/monocytic leukemia cancer cell lines EOL-1, HL-60, MOLM-13, and MV4-11. These FLT3 positive cell lines expressed abundant FLT3 protein at the expected molecular weight (black box) and highly glycosylated FLT3 which shows as a smear around 160 kDa. FLT3 negative controls included lysates from CHO parental cells and human B-cell leukemia cancer cell line Namalwa. Beta-actin (42 kDa) is shown for each sample as a loading control. MW, molecular weight.
Figure 7.

No in vitro AMG 553-mediated cytotoxicity against primary cortical neurons. Cellular imaging was used to identify and enumerate live target primary cortical neuron cell nuclei (size-selected, Hoechst-positive, ToPro3-negative) after 44 h of coculture at 1:1 ratio with AMG 553 or untransduced cells from 2 different donors, 3277 and 6527, or no T cells. Primary cortical neurons from 2 different donors (A and B) served as target cells. Mean ± standard deviation shown for 4 technical replicates.
DISCUSSION
Therapies that elicit T-cell-mediated cytotoxicity by targeting cell surface FLT3 could provide benefit to patients with AML, irrespective of FLT3 mutation status. AMG 553 is an investigational, CAR T-cell therapy targeting cell surface-expressed FLT3 protein for the treatment of relapsed/refractory AML. The nonclinical safety evaluation of AMG 553 included assessment of FLT3 expression in normal human tissues (Brauchle et al., 2020), in vitro assessment of AMG 553-mediated cytotoxicity against human cells from a variety of tissues, and toxicology studies in cynomolgus monkeys administered anti-FLT3 CAR T cells or anti-FLT3 BiTE molecules. The data supported the initiation of the clinical development of AMG 553.
In normal human tissues, cell surface FLT3 protein was restricted to a portion of bone marrow hematopoietic stem and progenitor cells and rare, scattered cells in the tonsil. FLT3 protein in other cell types was cytoplasmic (Brauchle et al., 2020). Because AMG 553 recognizes an epitope in the extracellular portion of FLT3, it is unlikely that AMG 553 would exhibit pharmacologic activity against cells other than AML cells and a subpopulation of hematopoietic bone marrow stem and progenitor cells. To determine if FLT3 expression in cynomolgus monkey was representative of human tissue expression, FLT3 mRNA and FLT3 protein expression were assessed in a variety of normal cynomolgus monkey tissues. Overall, FLT3 expression distribution across tissues and cellular localization was similar between human (Brauchle et al., 2020; Çakmak-Görür et al., 2019; Kikushige et al., 2008) and cynomolgus monkey. Cell surface FLT3 protein expression in cynomolgus monkey was restricted to bone marrow hematopoietic stem and progenitor cells (Brauchle et al., 2020); in other tissues, FLT3 protein was cytoplasmic and, therefore not expected to be accessible to AMG 553. Comparable FLT3 expression and cellular localization across tissues between human and cynomolgus monkey indicate that cynomolgus monkey is a representative species for nonclinical safety assessment of a FLT3-targeting molecule with a T-cell-mediated cytotoxicity MOA.
To determine the feasibility of using the cynomolgus monkey as a nonclinical animal model to assess the safety profile of AMG 553, 2 separate single dose exploratory studies were conducted using autologous cynomolgus monkey T cells transduced with the human anti-FLT3 CAR of AMG 553. Anti-FLT3 CAR T-cell doses were the maximum feasible dose based off the PBMCs isolated from serial blood collections from the animals, the transduction efficiency, and successful expansion of the cells. Anti-FLT3 CAR T-cell related changes were limited to a transient increase in body temperature in a single animal and a transient increase in CRP in 1 of 2 studies. Other effects were consistent with the preconditioning regimen and administration of CAR T cells did not exacerbate any effects. Despite in vitro evidence of cytotoxicity against FLT3-positive cells using cynomolgus monkey T cells transduced with anti-FLT3 CAR, no in vivo PD activity was observed. Measurable levels of anti-FLT3 CAR T cells were only detected using ddPCR immediately at the end of infusion in 1 of 3 animals. Anti-FLT3 CAR T cells did not persist in PBMC and were not detected in multiple tissues assessed at the end of the study. The lack of CAR T-cell persistence was likely because of restricted expression of the FLT3 antigen and highlights the limited use of the cynomolgus monkey as an animal model for safety assessment of CAR T cells directed toward FLT3 or other targets with restricted cell surface protein expression.
To complement the cynomolgus monkey studies with autologous CAR T cells and to directly evaluate the potential toxicities of eliciting T-cell-mediated cytotoxicity against cells with surface expression of FLT3 protein in vivo, data from cynomolgus monkey toxicology studies with 2 separate BiTE molecules targeting FLT3 were leveraged. BiTE molecules form a link between T cells (by binding to the CD3 complex of the T-cell receptor) and tumor cells (by binding to a tumor antigen), resulting in T-cell mediated cytotoxicity against cells expressing tumor antigen on the cell surface, in this case, FLT3 protein (Figure 8). Therefore, the 2 BiTE molecules and AMG 553 have a similar MOA, but with a different modality.
Figure 8.

Bispecific T-cell engager (BiTE) molecule- and chimeric antigen receptor (CAR)-mediated recognition of tumor cells by T lymphocytes. Illustration of anti-Feline McDonough Sarcoma-like tyrosine kinase 3 (anti-FLT3) BiTE molecule (left panel) and anti-FLT3 CAR T-cell (right panel) antigen recognition mechanisms to activate tumor-associated antigen specific T cells.
BiTE molecule-related changes in both studies were consistent with the expected pharmacology of a molecule mediating T-cell redirected lysis of bone marrow hematopoietic stem and progenitor cells expressing cell surface FLT3 protein. Early changes were consistent with BiTE molecule-mediated T-cell activation and included transient increases in heart rate and cytokine levels, decreased circulating leukocytes, and clinical chemistry changes consistent with an acute phase response. Many of these changes were attenuated with repeated dosing. Changes in FLT3 transcript and sFLT3L in both studies were consistent with BiTE molecule-mediated redirected T-cell lysis of FLT3 expressing bone marrow hematopoietic stem and progenitor cells. The recovery of these parameters at the end of the HLE anti-FLT3 BiTE molecule study was consistent with the ADA-mediated loss of exposure. At the end of the studies, decreases in circulating leukocytes along with changes in bone marrow cytology or decreased bone marrow cellularity were observed and were consistent with BiTE molecule-mediated redirected T-cell lysis of FLT3 expressing bone marrow hematopoietic stem and progenitor cells. Increased cellularities in bone marrow, lymph nodes, and/or spleen in the HLE anti-FLT3 BiTE molecule study were adaptive responses secondary to decreased hematology parameters and/or decreased bone marrow cellularity earlier in the study (as noted above) and consistent with recovery following ADA-mediated decreases in exposure. The lack of toxicity in tissues other than the bone marrow that had detectable FLT3 mRNA and/or FLT3 protein expression, such as the stomach and kidney, is consistent with analyses indicating that the subcellular localization of FLT3 protein in these tissues is cytoplasmic and is not anticipated to be accessible to a molecule targeting cell surface FLT3 protein (BiTE molecule or CAR T cells).
The cause of death/moribund condition in the HLE anti-FLT3 BiTE molecule treated middose group was multifactorial including ADA-mediated thrombocytopenia. ADA-mediated effects on platelet counts, and specifically, shortened platelet life span due to platelet destruction by drug induced antibodies, and thrombocytopenia are known possible sequela to the administration of therapeutic proteins (Aster and Bougie, 2007; Everds and Tarrant, 2013; Koren et al., 2008; Mease et al., 2017). Development of ADAs in nonhuman primates is not predictive of immunogenicity in humans (Avery et al., 2005; Ponce et al., 2009; van Meer et al., 2013). Therefore, the ADA-mediated thrombocytopenia is not considered to be predictive of effects in humans. Observations that are considered to be translatable to human such as cytokine release, effects on bone marrow, and potential secondary infections are all considered to be manageable risks in the clinic. These risks are also present with current standard of care therapies, thus maintaining a favorable benefit/risk ratio.
The lack of persistence of anti-FLT3 CAR T cells in a healthy cynomolgus monkey in vivo without a tumor burden was consistent with the lack of antigenic stimulation due to the restricted expression of FLT3. Other nonhuman primate models have overcome this through administration of autologous cells transfected with the target antigen in order to provide antigenic stimulation (Berger et al., 2015). In this case, BiTE molecules were used because they are less reliant on T-cell expansion and the frequent dosing was able to provide persistent FLT3 target engagement (Slaney et al., 2018). Fundamental differences between CAR T cells and BiTE molecules make direct comparisons of doses challenging (Slaney et al., 2018). The anti-FLT3 CAR T-cell doses used in the presented cynomolgus monkey studies were maximum feasible doses based on PBMC availability and were not used to determine first-in-human doses. The BiTE molecule doses provide exposure multiples over first-in-human doses for clinical anti-FLT3 BiTE molecule studies. Additionally, anti-FLT3 BiTE molecule-mediated findings were consistent with the anticipated pharmacology of a molecule mediating T-cell redirected lysis of cells expressing cell surface FLT3 protein which in turn provides in vivo data informing potential “off-tumor, on-target” toxicity for AMG 553.
Due to the specificity of AMG 553 for FLT3 (Goldstein, Frank, Dunn, Kim, Wang, Karbowski, Balazs, Coxon, Koppikar, Lebrec, and Arvedson, in preparation), AMG 553 is not anticipated to exhibit off-target cytotoxicity against cells that do not express FLT3 protein. To further assess the activity of AMG 553, in vitro cytotoxicity experiments were performed to assess potential “off-tumor, on-target” effects of AMG 553 on human cells from tissue types with previous evidence of FLT3 transcript and/or FLT3 intracellular protein but no detectable cell surface FLT3 protein expression (Brauchle et al., 2020; Çakmak-Görür et al., 2019). Cells from additional tissues representing major organs with no previous evidence of FLT3 transcript or FLT3 protein were also evaluated to assess potential “off-target” activity. In vitro AMG 553-mediated cytotoxicity was observed for a subset of CD34+ bone marrow cells (Goldstein, Frank, Dunn, Kim, Wang, Karbowski, Balazs, Coxon, Koppikar, Lebrec, and Arvedson, in preparation) and was not observed against other cells, including cortical neurons. Despite some tissues that were previously reported to have detectable FLT3 transcript or FLT3 intracellular protein (Brauchle et al., 2020; Çakmak-Görür et al., 2019), many of the cells obtained for this study did not have detectable FLT3 transcript or FLT3 protein. Donor variability, heterogeneity of cell types in a tissue, and/or resident immune cells may explain the lack of FLT3 transcript and FLT3 protein detection in the cultured cells in this study. For example, although lung had detectable FLT3 transcript, the protein was shown to be in the cytoplasm of alveolar macrophages indicating that the mRNA signal was likely from resident immune cells (Brauchle et al., 2020) and FLT3 expression would not be expected in the cells in this study. Although FLT3 transcript and FLT3 protein were detected in pDCs in this study, cell surface FLT3 protein was undetectable by flow cytometry in a previous study (Brauchle et al., 2020), which is consistent with the lack of AMG 553-mediated cytotoxicity in pDCs in this study. Thus, these data indicate that AMG 553 is anticipated to have minimal effects on normal tissues.
In conclusion, the robust nonclinical safety assessment of AMG 553 consisted of thorough assessment of FLT3 expression in normal human (Brauchle et al., 2020) and cynomolgus monkey tissues, in vitro assessment of cytotoxicity against a wide range of human cells, and toxicology studies in the cynomolgus monkey administered either autologous T cells transduced with the AMG 553 CAR or BiTE molecules. Collectively, these data suggest that AMG 553 is anticipated to elicit T-cell-mediated cytotoxicity against only FLT3 cell surface positive tumor cells with “off-tumor, on-target” effects limited to a subpopulation of bone marrow hematopoietic stem and progenitor cells.
SUPPLEMENTARY DATA
Supplementary data are available at Toxicological Sciences online.
DECLARATION OF CONFLICTING INTERESTS
C.K., R.G., B.F., K.K., C.-M.L., O.H., K.H., B.B., X.W., Q.Y., R.H., M.B., T.A., and H.L. are employed by Amgen Inc. and report Amgen stock.
FUNDING
Amgen Inc.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge Elizabeth Leight of Leight Medical Communications, LLC (St Charles, Missouri), whose work was supported by Amgen Inc., for providing medical writing support.
REFERENCES
- Aster R. H., Bougie D. W. (2007). Drug-induced immune thrombocytopenia. N. Engl. J. Med. 357, 580–587. [DOI] [PubMed] [Google Scholar]
- Avery D. T., Ellyard J. I., Mackay F., Corcoran L. M., Hodgkin P. D., Tangye S. G. (2005). Increased expression of CD27 on activated human memory B cells correlates with their commitment to the plasma cell lineage. J. Immunol. 174, 4034–4042. [DOI] [PubMed] [Google Scholar]
- Berger C., Sommermeyer D., Hudecek M., Berger M., Balakrishnan A., Paszkiewicz P. J., Kosasih P. L., Rader C., Riddell S. R. (2015). Safety of targeting ROR1 in primates with chimeric antigen receptor-modified T cells. Cancer Immunol. Res. 3, 206–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertho J. M., Demarquay C., Frick J., Joubert C., Arenales S., Jacquet N., Sorokine-Durm I., Chau Q., Lopez M., Aigueperse J., et al. (2001). Level of Flt3-ligand in plasma: A possible new bio-indicator for radiation-induced aplasia. Int. J. Radiat. Biol. 77, 703–712. [DOI] [PubMed] [Google Scholar]
- Bohl S. R., Bullinger L., Rucker F. G. (2019). New targeted agents in acute myeloid leukemia: New hope on the rise. Int. J. Mol. Sci. 20, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauchle B., Goldstein R., Karbowski C., Henn A., Li C.-M., Bücklein V., Krupka C., Boyle M., Koppikar P., Haubner S., et al. (2020). Characterization of a novel Flt3 BiTE® antibody construct for the treatment of acute myeloid leukemia. Mol. Cancer Ther. doi: 10.1158/1535-7163.MCT-19-1093. [DOI] [PubMed] [Google Scholar]
- Cabana V. G., Lukens J. R., Rice K. S., Hawkins T. J., Getz G. S. (1996). HDL content and composition in acute phase response in three species: Triglyceride enrichment of HDL a factor in its decrease. J. Lipid Res. 37, 2662–2674. [PubMed] [Google Scholar]
- Çakmak-Görür N., Radke J., Rhein S., Schumann E., Willimsky G., Heppner F. L., Blankenstein T., Pezzutto A. (2019). Intracellular expression of Flt3 in Purkinje cells: Implications for adoptive T-cell therapies. Leukemia 33, 1039–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. Q., Heldman M. R., Herrmann M. A., Kedei N., Woo W., Blumberg P. M., Goldsmith P. K. (2013). Absolute quantitation of endogenous proteins with precision and accuracy using a capillary western system. Anal. Biochem. 442, 97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cray C., Zaias J., Altman N. H. (2009). Acute phase response in animals: A review. Comp. Med. 59, 517–526. [PMC free article] [PubMed] [Google Scholar]
- Everds N. E., Tarrant J. M. (2013). Unexpected hematologic effects of biotherapeutics in nonclinical species and in humans. Toxicol. Pathol. 41, 280–302. [DOI] [PubMed] [Google Scholar]
- Fischer T., Stone R. M., Deangelo D. J., Galinsky I., Estey E., Lanza C., Fox E., Ehninger G., Feldman E. J., Schiller G. J., et al. (2010). Phase IIB trial of oral midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J. Clin. Oncol. 28, 4339–4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilliland D. G., Griffin J. D. (2002). The roles of FLT3 in hematopoiesis and leukemia. Blood 100, 1532–1542. [DOI] [PubMed] [Google Scholar]
- Goebeler M. E., Bargou R. (2016). Blinatumomab: A CD19/CD3 bispecific T cell engager (BiTE) with unique anti-tumor efficacy. Leuk. Lymphoma 57, 1021–1032. [DOI] [PubMed] [Google Scholar]
- Howlader N, Noone A. M., Krapcho M., Miller D., Brest A., Yu M., Ruhl J., Tatalovich Z., Mariotto A., Lewis D. R., et al. , eds. 2018. SEER Cancer Statistics Review, 1975–2015 National Cancer Institute, Bethesda, MD. Available at: https://seercancergov/csr/1975_2015/, based on November 2017 SEER data submission, posted to the SEER web site, April 2018.
- Kikushige Y., Yoshimoto G., Miyamoto T., Iino T., Mori Y., Iwasaki H., Niiro H., Takenaka K., Nagafuji K., Harada M., et al. (2008). Human Flt3 is expressed at the hematopoietic stem cell and the granulocyte/macrophage progenitor stages to maintain cell survival. J. Immunol. 180, 7358–7367. [DOI] [PubMed] [Google Scholar]
- Koren E., Smith H. W., Shores E., Shankar G., Finco-Kent D., Rup B., Barrett Y. C., Devanarayan V., Gorovits B., Gupta S., et al. (2008). Recommendations on risk-based strategies for detection and characterization of antibodies against biotechnology products. J. Immunol. Methods 333, 1–9. [DOI] [PubMed] [Google Scholar]
- Ley T. J., Miller C., Ding L., Raphael B. J., Mungall A. J., Robertson A., Hoadley K., Triche T. J. Jr, Laird P. W., Baty J. D., et al. (2013). Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 368, 2059–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindl B., Krupka C., Chapman-Arvedson T., Kischel R., Kufer P., Haubner S., Lichtenegger F., Koehnke T., Metzeler K., Konstandin N., et al. (2017). Midostaurin reduces BiTE® mediated cytotoxicity against acute myeloid leukemia. Blood 130, 2656. [Google Scholar]
- Malik S. W., Myers J. L., DeRemee R. A., Specks U. (1996). Lung toxicity associated with cyclophosphamide use. Two distinct patterns. Am. J. Respir. Crit. Care Med. 154(6 Pt 1), 1851–1856. [DOI] [PubMed] [Google Scholar]
- Maude S. L., Laetsch T. W., Buechner J., Rives S., Boyer M., Bittencourt H., Bader P., Verneris M. R., Stefanski H. E., Myers G. D., et al. (2018). Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 378, 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mease K. M., Kimzey A. L., Lansita J. A. (2017). Biomarkers for nonclinical infusion reactions in marketed biotherapeutics and considerations for study design. Curr. Opin. Toxicol. 4, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meshinchi S., Appelbaum F. R. (2009). Structural and functional alterations of FLT3 in acute myeloid leukemia. Clin. Cancer Res. 15, 4263–4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M., Kaneko N., Ueno Y., Tanaka R., Cho K., Saito R., Kondoh Y., Shimada I., Kuromitsu S. (2014). ASP2215, a novel FLT3/AXL inhibitor: Preclinical evaluation in acute myeloid leukemia (AML). J. Clin. Oncol. 32, 7070. [Google Scholar]
- Nakao M., Yokota S., Iwai T., Kaneko H., Horiike S., Kashima K., Sonoda Y., Fujimoto T., Misawa S. (1996). Internal tandem duplication of the FLT3 gene found in acute myeloid leukemia. Leukemia 10, 1911–1918. [PubMed] [Google Scholar]
- National Research Council. (2011). Guide for the Care and Use of Laboratory Animals, 8th ed The National Academies Press, Washington, DC: Available at: https://grants.nih.gov/grants/olaw/guide-for-the-care-and-use-of-laboratory-animals.pdf. Accessed July 8, 2020. [Google Scholar]
- Neelapu S. S., Locke F. L., Bartlett N. L., Lekakis L. J., Miklos D. B., Jacobson C. A., Braunschweig I., Oluwole O. O., Siddiqi T., Lin Y., et al. (2017). Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 377, 2531–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perl A. E., Altman J. K., Cortes J., Smith C., Litzow M., Baer M. R., Claxton D., Erba H. P., Gill S., Goldberg S., et al. (2017). Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: A multicentre, first-in-human, open-label, phase 1–2 study. Lancet Oncol. 18, 1061–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips F. S., Sternberg S. S., Cronin A. P., Vidal P. M. (1961). Cyclophosphamide and urinary bladder toxicity. Cancer Res. 21, 1577–1589. [PubMed] [Google Scholar]
- Ponce R., Abad L., Amaravadi L., Gelzleichter T., Gore E., Green J., Gupta S., Herzyk D., Hurst C., Ivens I. A., et al. (2009). Immunogenicity of biologically-derived therapeutics: assessment and interpretation of nonclinical safety studies. Regul. Toxicol. Pharmacol. 54, 164–182. [DOI] [PubMed] [Google Scholar]
- Rosnet O., Buhring H. J., Marchetto S., Rappold I., Lavagna C., Sainty D., Arnoulet C., Chabannon C., Kanz L., Hannum C., et al. (1996). Human FLT3/FLK2 receptor tyrosine kinase is expressed at the surface of normal and malignant hematopoietic cells. Leukemia 10, 238–248. [PubMed] [Google Scholar]
- Schlenk R. F., Weber D., Fiedler W., Salih H. R., Wulf G., Salwender H., Schroeder T., Kindler T., Lubbert M., Wolf D., et al. (2019). Midostaurin added to chemotherapy and continued single-agent maintenance therapy in acute myeloid leukemia with FLT3-ITD. Blood 133, 840–851. [DOI] [PubMed] [Google Scholar]
- Slaney C. Y., Wang P., Darcy P. K., Kershaw M. H. (2018). Cars versus bites: A comparison between T cell-redirection strategies for cancer treatment. Cancer Discov. 8, 924–934. [DOI] [PubMed] [Google Scholar]
- Stone R. M., Mandrekar S. J., Sanford B. L., Laumann K., Geyer S., Bloomfield C. D., Thiede C., Prior T. W., Dohner K., Marcucci G., et al. (2017). Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N. Engl. J. Med. 377, 454–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarlock K., Alonzo T. A., Loken M. R., Gerbing R. B., Ries R. E., Aplenc R., Sung L., Raimondi S. C., Hirsch B. A., Kahwash S. B., et al. (2017). Disease characteristics and prognostic implications of cell-surface FLT3 receptor (CD135) expression in pediatric acute myeloid leukemia: A report from the children’s oncology group. Clin. Cancer Res. 23, 3649–3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiong I. S., Wei A. H. (2019). New drugs creating new challenges in acute myeloid leukemia. Genes Chromosomes Cancer 58, 903–914. [DOI] [PubMed] [Google Scholar]
- van Meer P. J., Kooijman M., Brinks V., Gispen-de Wied C. C., Silva-Lima B., Moors E. H., Schellekens H. (2013). Immunogenicity of mAbs in non-human primates during nonclinical safety assessment. mAbs 5, 810–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiese M., Daver N. (2018). Unmet clinical needs and economic burden of disease in the treatment landscape of acute myeloid leukemia. Am. J. Manag. Care 24(16 Suppl), S347–S355. [PubMed] [Google Scholar]
- Xu D., Mane S., Sosic Z. (2015). Characterization of a biopharmaceutical protein and evaluation of its purification process using automated capillary Western blot. Electrophoresis 36, 363–370. [DOI] [PubMed] [Google Scholar]
- Zimmerman E. I., Turner D. C., Buaboonnam J., Hu S., Orwick S., Roberts M. S., Janke L. J., Ramachandran A., Stewart C. F., Inaba H., et al. (2013). Crenolanib is active against models of drug-resistant FLT3-ITD-positive acute myeloid leukemia. Blood 122, 3607–3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.