

Abstract
Laboratory studies have suggested oncogenic roles of lipids, as well as anticarcinogenic effects of statins. Here we assess the potential effect of statin therapy on cancer risk using evidence from human genetics. We obtained associations of lipid-related genetic variants with the risk of overall and 22 site-specific cancers for 367,703 individuals in the UK Biobank. In total, 75,037 individuals had a cancer event. Variants in the HMGCR gene region, which represent proxies for statin treatment, were associated with overall cancer risk (odds ratio [OR] per one standard deviation decrease in low-density lipoprotein [LDL] cholesterol 0.76, 95% confidence interval [CI] 0.65–0.88, p=0.0003) but variants in gene regions representing alternative lipid-lowering treatment targets (PCSK9, LDLR, NPC1L1, APOC3, LPL) were not. Genetically predicted LDL-cholesterol was not associated with overall cancer risk (OR per standard deviation increase 1.01, 95% CI 0.98–1.05, p=0.50). Our results predict that statins reduce cancer risk but other lipid-lowering treatments do not. This suggests that statins reduce cancer risk through a cholesterol independent pathway.
Research organism: Human
Introduction
Statins are inhibitors of 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMGCR), which is the rate-limiting enzyme in the mevalonate pathway; a pathway producing a range of cell signaling molecules with the potential to regulate oncogenesis. This is supported by strong laboratory evidence that statins induce anticarcinogenic effects on cell proliferation and survival in various cell lines (Sławińska-Brych et al., 2014; Crosbie et al., 2013; Ishikawa et al., 2014; Chang et al., 2013), and reduce tumor growth in a range of in vivo models (Narisawa et al., 1996a; Inano et al., 1997; Narisawa et al., 1996b; Clutterbuck et al., 1998; Kikuchi et al., 1997; Hawk et al., 1996). Furthermore, epidemiological studies of pre-diagnostic use of statins have been associated with reduced risk of specific cancer types (Khurana et al., 2007; Poynter et al., 2005; Pocobelli et al., 2008). However, meta-analyses of cardiovascular-focused randomized controlled trials have shown no effect of statins on cancer (Dale et al., 2006; Farooqi et al., 2018). Conclusions from these trials are limited as they lack adequate power and longitudinal follow-up necessary for assessing the impact on cancer risk. At present, no clinical trials have been designed to assess the role of statins in primary cancer prevention and their role in chemoprevention remains uncertain.
A putative protective effect of statins on cancer development could be through either cholesterol-dependent or independent effects (Goldstein and Brown, 1990; Mullen et al., 2016; Yeganeh et al., 2014; Denoyelle et al., 2001). Cholesterol is a key mediator produced by the mevalonate pathway and is essential to cell signaling and membrane structure, with evidence demonstrating the potential to drive oncogenic processes and tumor growth (Chen and Resh, 2002; Li et al., 2006). However, the epidemiological relationships between circulating cholesterol and cancer risk remain unclear. Individual observational studies have reported positive (Kitahara et al., 2011; Strohmaier et al., 2013), inverse (Kitahara et al., 2011; Strohmaier et al., 2013; Melvin et al., 2012; Katzke et al., 2017), and no association (Salonen, 1982; JPHC Study Group et al., 2009; Van Hemelrijck et al., 2012; Ma et al., 2016) between circulating levels of total cholesterol, low-density lipoprotein (LDL) cholesterol, high-density lipoprotein (HDL) cholesterol, and triglycerides with the risk of overall and site-specific cancers. Different cancer types have distinct underlying pathophysiology, and meta-analyses of observational studies highlight a likely complex relationship which varies according to both lipid fraction (Vílchez et al., 2014; Alsheikh-Ali et al., 2008) and cancer type (Ma et al., 2016; Lin et al., 2017; Passarelli and Newcomb, 2016; Yao and Tian, 2015). Furthermore, cancer can lower cholesterol levels for up to 20 months before diagnosis (Kritchevsky et al., 1991). Thus, the true relationship between lipids and cancer development remains equivocal.
Mendelian randomization is an epidemiological approach that assesses associations between genetically predicted levels of a risk factor and a disease outcome to predict the causal effect of the risk factor on an outcome (Davey Smith and Hemani, 2014). The use of genetic variants minimizes the influence of reverse causality and confounding factors on estimates. Mendelian randomization studies also have the potential to predict the outcomes of trials for specific therapeutic interventions. A limited number of Mendelian randomization studies have investigated the relationship between HMGCR inhibition and cancer (PRACTICAL consortium et al., 2016; Rodriguez-Broadbent et al., 2017; Orho-Melander et al., 2018; Nowak and Ärnlöv, 2018; Yarmolinsky et al., 2020), with protective associations observed for prostate cancer (PRACTICAL consortium et al., 2016), colorectal cancer (Rodriguez-Broadbent et al., 2017), breast cancer (Orho-Melander et al., 2018; Nowak and Ärnlöv, 2018), and ovarian cancer (Yarmolinsky et al., 2020). However, no comprehensive Mendelian randomization investigation has evaluated the predicted impact of HMGCR inhibition or the causal role of specific lipid fractions on the risk of many of the most common site-specific cancers.
Here we investigate the relationship between HMGCR inhibition and the risk of overall cancer and site-specific cancers using genetic variants in the HMGCR gene region. To understand whether statins may influence cancer risk through lipid-related mechanisms, we also assess the relationship between lipids and cancer risk by polygenic Mendelian randomization analyses using common lipid-associated genetic variants. Additionally, to mimic other lipid-lowering pharmaceutical interventions, gene-specific analyses were performed using variants in or near gene regions targeted by these therapies.
Results
Participant characteristics and power calculations
Baseline characteristics of the participants in the UK Biobank and numbers of outcomes are provided in Table 1. In total, 75,037 of the participants had a cancer event, of which 48,674 participants had one of the 22 defined site-specific cancers. Power calculations for the various analyses are presented in Figure 1 (site-specific cancers) and Supplementary file 1 (overall cancer). The number of cases ranged from 324 for liver cancer to 13,666 for breast cancer with an overall median number of 1462 cases across cancer sites. Gene-specific analyses were only well-powered for overall cancer. Polygenic analyses were well-powered to detect moderate effects for overall cancer and common site-specific cancers but less well-powered for less common site-specific cancers.
Table 1. Baseline characteristics of the UK Biobank participants included in this study and the numbers of outcome events.
| Characteristic or cancer site/type | Mean (SD) or N (%)† |
|---|---|
| Sample size | 367,703 (100) |
| Female | 198,904 (54.1) |
| Age at baseline | 57.2 (8.1) |
| Body mass index | 27.3 (4.8) |
| Systolic blood pressure | 137.6 (18.6) |
| Diastolic blood pressure | 82.0 (10.1) |
| Smoking status (current/ex/ never)* | 37,866 (10.3)/185,704 (50.5)/143,777 (39.1) |
| Alcohol status (current/ex/ never)* | 342,797 (93.2)/12,732 (3.5)/11,646 (3.2) |
| History of type 2 diabetes | 15,834 (4.3) |
| Overall cancer | 75,037 (20.4) |
| Breast | 13,666 (6.9) |
| Prostate | 7872 (4.7) |
| Lung | 2838 (0.8) |
| Bowel | 5486 (1.5) |
| Melanoma | 4869 (1.3) |
| Non-Hodgkin’s lymphoma | 2296 (0.6) |
| Kidney | 1310 (0.4) |
| Head/neck | 1615 (0.4) |
| Brain | 810 (0.2) |
| Bladder | 2588 (0.7) |
| Pancreas | 1264 (0.3) |
| Uterus | 1931 (1.0) |
| Leukaemia | 1403 (0.4) |
| Esophagus | 843 (0.2) |
| Ovaries | 1520 (0.8) |
| Gastric | 736 (0.2) |
| Liver | 324 (0.1) |
| Myeloma | 656 (0.2) |
| Thyroid | 375 (0.1) |
| Biliary | 387 (0.1) |
| Cervix | 1928 (1.0) |
| Testes | 735 (0.4) |
*Excluding 356 participants with smoking status absent and 528 participants with alcohol consumption status absent.
†For sex-specific cancers, this is the percentage of individuals of the relevant sex.
Figure 1. Power calculations for polygenic and gene-specific analyses, displaying the Mendelian randomization estimate that can be detected with 80% power assuming a sample size of 367,703 individuals for site-specific cancers.

Gene-specific analyses for HMGCR and other drug proxy variants
Associations for specific gene regions representing targets of lipid-lowering drugs are displayed in Figure 2 and Figure 2—figure supplements 1–6. For overall cancer, there was evidence of association for variants in the HMGCR gene region (odds ratio [OR] 1.32, 95% confidence interval [CI] 1.13–1.53, p=0.0003) but not for other gene regions: for PCSK9 (OR 1.03, 95% CI 0.92–1.14, p=0.66), for LDLR (OR 0.99, 95% CI 0.92–1.07, p=0.86), for NPC1L1 (OR 0.87, 95% CI 0.73–1.04, p=0.13), for APOC3 (OR 1.08, 95% CI 0.98–1.19, p=0.15), or for LPL (OR 1.03, 95% CI 0.95–1.13, p=0.45). The association of variants in the HMGCR gene region with overall cancer remained broadly similar when restricting outcomes to the 48,674 individuals who had one of the 22 site-specific cancers (OR 1.29, 95% CI 1.08–1.54, p=0.005) and when excluding outcomes that were self-reported only (70,734 remaining cases, OR 1.30, 95% CI 1.12–1.52, p=0.0007).
Figure 2. Gene-specific Mendelian randomization estimates (odds ratio with 95% confidence interval per one standard deviation increase in lipid fraction) for variants in gene regions representing targets of lipid-lowering treatments.
Estimates are scaled to a one standard deviation increase in LDL-cholesterol for the HMGCR, PCSK9, LDLR, and NPC1L1 regions, and a one standard deviation increase in triglycerides for the APOC3 and LPL regions. A: associations with overall cancer for each gene region in turn. B: associations with site-specific cancers for variants in the HMGCR gene region.

Figure 2—figure supplement 1. Gene-specific Mendelian randomization estimates (odds ratio with 95% confidence interval per one standard deviation increase in LDL-cholesterol) for variants in the PCSK9 gene region.

Figure 2—figure supplement 2. Gene-specific Mendelian randomization estimates (odds ratio with95%confidence interval per one standard deviation increase in LDL-cholesterol) for variants in theLDLRgene region.

Figure 2—figure supplement 3. Gene-specific Mendelian randomization estimates (odds ratio with 95% confidence interval per one standard deviation increase in LDL-cholesterol) for variants in theNPC1L1gene region.

Figure 2—figure supplement 4. Gene-specific Mendelian randomization estimates (odds ratio with 95% confidence interval per one standard deviation increase in LDL-cholesterol) for variants in theAPOC3gene region.

Figure 2—figure supplement 5. Gene-specific Mendelian randomization estimates (odds ratio with 95% confidence interval per one standard deviation increase in LDL-cholesterol) for variants in theLPLgene region.

Figure 2—figure supplement 6. Genetic associations with LDL-cholesterol (standard deviation units) plotted against genetic associations with overall cancer (log odds ratios) for six variants in the HMGCR gene region.
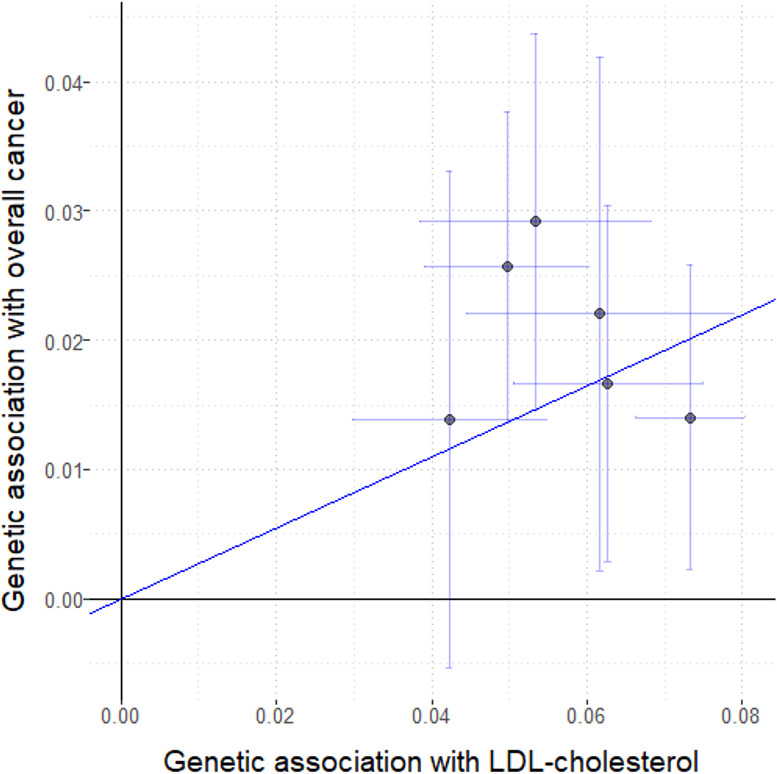
Figure 2—figure supplement 7. Gene-specific Mendelian randomization estimates (odds ratio with 95% confidence interval per one standard deviation increase in LDL-cholesterol) for variants in the HMGCR gene region excluding self-reported outcomes.

Figure 2—figure supplement 8. Gene-specific Mendelian randomization estimates (odds ratio with 95% confidence interval per one standard deviation increase in LDL-cholesterol) for variants in the HMGCR gene region excluding those with a cancer diagnosis other than site-specific cancer under analysis.

For site-specific cancers, the HMGCR gene region showed positive associations for five of the six most common cancer sites (breast, prostate, melanoma, lung, and bladder; not for bowel), although none of these results individually reached a conventional level of statistical significance. Similar results were observed for analyses of site-specific cancers when excluding individuals with solely self-reported outcomes from the analysis (Figure 2—figure supplement 7) and when individuals with a cancer diagnosis other than the site-specific cancer under analysis were omitted from the analysis rather than treated as a control (Figure 2—figure supplement 8); estimates were generally slightly higher, but findings were unchanged. There was little evidence for associations in site-specific analyses for other lipid-lowering drug targets.
Polygenic analyses for all lipid-related variants
Polygenic Mendelian randomization estimates are displayed in Figure 3 for HDL-cholesterol, LDL-cholesterol, and triglycerides (see also Supplementary file 1), and Figure 4 for total cholesterol. For overall cancer, the OR per one standard deviation increase in genetically-predicted levels of the risk factor was 1.01 (95% CI 0.98–1.05, p=0.50) for LDL-cholesterol, 0.99 (95% CI 0.95–1.03, p=0.54) for HDL-cholesterol, 1.00 (95% CI 0.95–1.05, p=0.85) for triglycerides, and 1.01 (95% CI 0.98–1.05; p=0.57) for total cholesterol. Results for the lipid subfractions were similar using the multivariable MR-Egger method (Supplementary file 1). Similar results were observed when omitting self-reported outcomes from the analysis (Supplementary file 1).
Figure 3. Multivariable Mendelian randomization estimates for HDL-cholesterol, LDL-cholesterol, and triglycerides (odds ratio with 95% confidence interval per one standard deviation increase in lipid fraction) from polygenic analyses including all lipid-associated variants.

Figure 3—figure supplement 1. Multivariable Mendelian randomization estimates for HDL-cholesterol, LDL-cholesterol, and triglycerides (odds ratio with 95% confidence interval per one standard deviation increase in lipid fraction) from polygenic analyses including all lipid-associated variants excluding self-reported outcomes.

Figure 3—figure supplement 2. Multivariable Mendelian randomization estimates for HDL-cholesterol, LDL-cholesterol, and triglycerides (odds ratio with 95% confidence interval per one standard deviation increase in lipid fraction) from polygenic analyses including all lipid-associated variants excluding those with a cancer diagnosis other than the site-specific cancer under analysis.

Figure 4. Univariable Mendelian randomization estimates for total cholesterol (odds ratio with 95% confidence interval per one standard deviation increase in lipid fraction) from polygenic analyses including all lipid-associated variants.

Figure 4—figure supplement 1. Univariable Mendelian randomization estimates for total cholesterol (odds ratio with 95% confidence interval per one standard deviation increase in lipid fraction) from polygenic analyses including all lipid-associated variants excluding self-reported outcomes.

Figure 4—figure supplement 2. Univariable Mendelian randomization estimates for total cholesterol (odds ratio with 95% confidence interval per one standard deviation increase in lipid fraction) from polygenic analyses including all lipid-associated variants excluding those with a cancer diagnosis other than the site-specific cancer under analysis.

Figure 4—figure supplement 3. Scatterplot to assess heterogeneity of genetic associations with total cholesterol (horizontal axis, standard deviation units) against genetic associations with overall cancer (vertical axis, log odds ratios).

For site-specific cancers, there were positive associations between risk of bowel cancer and genetically predicted levels of total cholesterol (OR 1.18, 95% CI 1.06–1.32, p=0.002) and LDL-cholesterol (OR 1.16, 95% CI 1.04–1.29, p=0.006). Compared to primary analyses, results were attenuated in robust methods (Supplementary file 1). While these robust methods are univariable analyses, evidence for a causal effect is most reliable when it is supported by multiple methods that make different assumptions, which was not the case here. No other associations were statistically significant at p<0.01. Again, similar results were observed when omitting self-reported outcomes from the analysis (Figure 3—figure supplement 1 and Figure 4—figure supplement 1), and when omitting individuals with a different cancer diagnosis from the analysis (Figure 3—figure supplement 2 and Figure 4—figure supplement 2). The exception was for liver cancer, where estimates for LDL-cholesterol (OR 0.52, 95% CI 0.30–0.0.88, p=0.016) and total cholesterol (OR 0.54, 95% CI 0.31–0.94, p=0.028) became stronger on the omission or exclusion of individuals with solely self-reported outcomes. The numbers of events that were self-reported only for each outcome are reported in Supplementary file 1. Heterogeneity I (Crosbie et al., 2013) statistics were around 40% for the analysis of overall cancer, and generally lower for site-specific cancers (Supplementary file 1). The burden of heterogeneity was shared amongst several genetic variants; there were no striking outliers and hence no specific variants strongly driving heterogeneity (see Figure 4—figure supplement 3 for scatterplot of genetic associations with total cholesterol and overall cancer).
To address the possibility of null results arising due to low power, we combined data on gastrointestinal cancers (liver, stomach, bowel, esophagus, biliary tract, and pancreas). Estimates were somewhat stronger for LDL-cholesterol and total cholesterol compared to the analysis for overall cancer, but did not reach a conventional level of statistical significance: LDL-cholesterol 1.05 (95% CI 0.97–1.14, p=0.23), HDL-cholesterol 1.07 (95% CI 0.97–1.18, p=0.17), triglycerides 1.02 (95% CI 0.91–1.15, p=0.70), and total cholesterol 1.07 (95% CI 0.99–1.16, p=0.10).
Discussion
Our comprehensive Mendelian randomization investigation shows a positive association between overall cancer and variants in the HMGCR gene region which can be considered as proxies for statin therapy. However, gene regions which can be considered as proxies for alternative lipid-lowering therapies were not associated with cancer risk. Furthermore, there was little consistent evidence of an association between genetically-predicted lipid fractions and cancer outcomes in polygenic analyses either for overall cancer or for any site-specific cancer. Taken together, our findings predict that statins lower the risk of cancer, and provide important evidence that this occurs through mechanisms other than lipid lowering.
We found that genetic variants in the HMGCR region, serving as proxies for targets of statin therapy, were associated with a 26% decrease in risk of overall cancer per standard deviation (around 39 mg/dL or 1.0 mmol/L) reduction in genetically-predicted LDL-cholesterol. Our result replicates protective associations previously observed for prostate cancer (PRACTICAL consortium et al., 2016), colorectal cancer (Rodriguez-Broadbent et al., 2017), breast cancer (Orho-Melander et al., 2018; Nowak and Ärnlöv, 2018), and ovarian cancer (Yarmolinsky et al., 2020), although with a stronger weight of statistical evidence due to the additional number of cases analyzed here. For coronary artery disease, the short-term impact of statins in trials is around one-third of the genetic estimate, which represents the impact of lifelong reduced levels of LDL-cholesterol (Ference et al., 2012). Under the assumption that even if LDL-cholesterol may not be the relevant causal risk factor, it is a relevant prognostic factor for assessing the degree of HMGCR inhibition, this suggests that any reduction in cancer risk from statins in practice is likely to be modest. Mechanistically, cardiovascular risk reduction by statins is predominantly due to cholesterol lowering (Liao and Laufs, 2005), whereas we give evidence this is not the case for cancer.
While our results should be seen as tentative until trials have demonstrated benefit, associations of HMGCR variants show broad concordance with statin therapy for many continuous phenotypes (Würtz et al., 2016), and suggest that statins reduce the risk of coronary artery disease (Ference et al., 2016), increase risk of type 2 diabetes (Lotta et al., 2016), and increase risk of intracerebral hemorrhage (Sun et al., 2019; Allara et al., 2019a), as confirmed in clinical trials (ASCOT investigators et al., 2003; DIAGRAM Consortium et al., 2015; Collins et al., 2016). Genetic evidence pertaining to HMGCR has been proven to be a reliable guide for the performance of statins in trials. Clinical trials are required to confirm our promising findings for primary prevention of cancer risk.
The notion that statins could be used for chemoprevention is longstanding. Nobel Prize winners Goldstein and Brown proposed that this occurs through non-lipid lowering mechanisms (Goldstein and Brown, 1990). We provide evidence from human genetics to support this theory. Our results suggest that with respect to genetically predicted HMGCR inhibition and cancer risk, LDL-cholesterol is simply a biomarker of HMGCR inhibition that is accessible, but the true causal pathway is likely via another molecule whose levels are correlated with its LDL-cholesterol lowering effect. HMGCR catalyzes the rate-limiting step of the mevalonate pathway; a pathway with an arm leading to the endpoint of cholesterol synthesis and another arm leading to isoprenoid synthesis. Measuring levels of intra-cellular isoprenoids is challenging but these molecules are implicated in cancer via their role as major post-translational modifiers of key oncogenic proteins (Mullen et al., 2016). In particular, mevalonate and other isoprenoid metabolites are required for the prenylation and functioning of the Ras and Rho GTPases, which are oncoproteins, and involved in important cellular processes including apoptosis, phagocytosis, vascular trafficking, cell proliferation, transmigration, cytoskeleton organization, and recruitment of inflammatory cells. Statin inhibition of these metabolites has demonstrated anti-oncological effects in vivo and in vitro (Yeganeh et al., 2014) including the promotion of tumor cell death and apoptosis (Spampanato et al., 2012; Ghosh-Choudhury et al., 2010; Parikh et al., 2010; Misirkic et al., 2012), inhibition of angiogenesis (Park et al., 2008), and reduction of tumor cell invasion and metastasis (Wang et al., 2000; Yang et al., 2013). Other potential statin-mediated mechanisms of tumor suppression include the reduction of systemic inflammatory mediators like interleukin 1-beta and tumor necrosis factor (Park et al., 2008; Bruegel et al., 2006), and epigenetic regulation through inhibiting HMGCR-mediated deacetylation (Lin et al., 2008), which contributes to colorectal cancer in mouse models (Pisanti et al., 2014). Thus, our findings based on large-scale human genetic data are consistent with pre-clinical studies on statins in cancer which have repeatedly argued for a cholesterol independent mechanism for statin effects on cancer.
We have here presented the first wide-angled Mendelian randomization analysis of lipid subtypes for a range of site-specific cancers. We corroborate previous Mendelian randomization studies suggesting no causal role of lipids in the development of pancreatic cancer (Carreras-Torres et al., 2017a) and prostate cancer (PRACTICAL consortium et al., 2016). We also extend these findings to show little convincing evidence for total cholesterol levels or lipid fractions as a risk factor for any cancer type studied. However, it is plausible that lack of power or heterogeneity between cancer types could have contributed to these null results as previous large Mendelian randomization studies of the most common cancers (colorectal, lung, and breast) have shown significant associations with LDL-cholesterol, including results in both directions. Although our associations did not achieve a conventional level of statistical significance, we mirror the positive associations of genetically-predicted LDL-cholesterol and HDL-cholesterol with breast cancer (Nowak and Ärnlöv, 2018; Beeghly-Fadiel et al., 2019) observed in the Breast Cancer Association Consortium which studied over 60,000 cases of cancer. However, we did not observe the negative association observed between LDL-cholesterol and lung cancer found in a Mendelian randomization analysis of 29,266 cases, though in the same study rare LDL-cholesterol variants showed the opposite association (Carreras-Torres et al., 2017b). Of all the site-specific cancers studied, the present study only implicated dyslipidemia in driving bowel (i.e. colorectal) cancer with positive associations demonstrated for total and LDL-cholesterol levels. These findings corroborate a previous Mendelian randomization study of 26,397 colorectal cancer patients (Cornish et al., 2020). Furthermore, in a smaller Mendelian randomization study, genetically-predicted total cholesterol levels, but not LDL-cholesterol, were associated with colorectal cancer risk (Rodriguez-Broadbent et al., 2017), and several previous meta-analyses of observational studies have associated dyslipidemia with increased risk of colorectal adenoma (Passarelli and Newcomb, 2016) and cancer (Yao and Tian, 2015; Tian et al., 2015). However, the associations we found for colorectal cancer were attenuated in sensitivity analyses and must therefore be interpreted with caution. Overall, there was little consistent evidence for total cholesterol levels or lipid fractions as a risk factor for cancer, although this must be interpreted with caution.
Our headline finding relates to overall cancer, which is a combination of different malignancies. While they may have different underlying aetiologies, all cancers are known to share common underlying ‘hallmark’ molecular and cellular aberrations and there may thus be pathophysiologic relevance in combining outcomes (Hanahan and Weinberg, 2011). Furthermore, analyses for overall cancer are highly relevant from a public health perspective. In addition to the cardiovascular benefits of statins, any individual patient decision regarding whether to take them for primary cancer prevention is likely to reflect the risk of all cancer types, not one particular subtype. Overall, clinical trials are needed to confirm the protective effect of statins in the primary prevention of cancer and should characterize the adverse risks of statins before they are advocated in clinical guidance. In particular, any potential adverse effect of LDL-cholesterol lowering on lung cancer should be monitored, despite the lack of replication for this finding in the present analysis.
Our investigation has many strengths, but also limitations. The large sample size of over 360,000 participants and the broad set of outcomes analyzed render this the most comprehensive Mendelian randomization analysis of lipids and cancer outcomes conducted to date. However, the investigation has a number of limitations. For many site-specific cancers, there were not enough outcome events to obtain adequate power to rule out the possibility of moderate causal effects. This is particularly relevant to analyses of gene-specific target regions for LDL-cholesterol, which were not adequately powered to detect small effect sizes. Conversely, the study of a large number of outcomes across multiple risk factors as we have done is prone to type one errors, meaning that results are falsely found to be significant. However, correction for multiple testing may lead to type two errors, meaning that results are falsely judged to be inconclusive. We encourage readers to weigh the evidence presented carefully rather than to reduce findings to a binary designation of ‘significant’ or ‘not significant’. While there is evidence to support our assumption that genetic variants in relevant gene regions can be used as proxies for pharmacological interventions, our findings should be considered with caution until they have been replicated in clinical trials. Our investigation was able to compare subgroups of the population with different lifelong average levels of lipid fractions, but the impact of lowering a particular lipid fraction in practice is likely to differ from the genetic association, particularly quantitatively (Burgess et al., 2012). Combining cancer types to study overall cancer risk has the aforementioned benefits and also results in the largest number of cases and so the greatest power to detect a causal effect. However, this assumes a consistent effect between cancer types, so there is potential for directional heterogeneity and a consequent reduction in power. Furthermore, this combined endpoint is dependent on the characteristics of the analytic sample and the relative prevalence of different cancer types. In particular, cancers with greater survival chances will be overrepresented in the case sample. Finally, analyses were conducted in UK-based participants of European ancestries. While it is recommended to have a well-mixed study population for Mendelian randomization to ensure that genetic associations are not influenced by population stratification, it means that results may not be generalizable to other ethnicities or nationalities.
In conclusion, our findings suggest that HMGCR inhibition may have a chemopreventive role in cancer through non-lipid lowering properties and that this role may apply across cancer sites. The efficacy of statins for cancer prevention must be urgently evaluated.
Materials and methods
Study design and data sources
We performed two-sample Mendelian randomization analyses, taking genetic associations with risk factors (i.e. serum lipid levels) from one dataset, and genetic associations with cancer outcomes from an independent dataset, as performed previously for cardiovascular diseases (Allara et al., 2019b).
We obtained genetic associations with serum lipid concentrations (total cholesterol, LDL-cholesterol, HDL-cholesterol, and triglycerides) from the Global Lipids Genetic Consortium (GLGC) on up to 188,577 individuals of European ancestry (Global Lipids Genetics Consortium et al., 2013). Genetic associations were estimated with adjustment for age, sex, and genomic principal components within each participating study after inverse rank quantile normalization of lipid concentrations, and then meta-analyzed across studies.
We estimated genetic associations with cancer outcomes on 367,703 unrelated individuals of European ancestry from the UK Biobank, a population-based cohort recruited between 2006 and 2010 at 22 assessment centers throughout the UK and followed-up until 31st March 2017 or their date of death (recorded until 14th February 2018; Sudlow et al., 2015). We defined cancer outcomes for overall cancer and for the 22 most common site-specific cancers in the UK (Supplementary file 1). Outcomes were based on electronic health records, hospital episode statistics data, national cancer registry data, and death certification data, which were all coded according to ICD-9 and ICD-10 diagnoses. Further cancer outcomes were captured by self-reported information validated by an interview with a trained nurse and from cancer histology data in the national cancer registry. To obtain genetic association estimates for each outcome, we conducted logistic regression with adjustment for age, sex, and 10 genomic principal components using the snptest software program. For sex-specific cancers (breast, uterus, and cervix for women; prostate and testes for men), analyses were restricted to individuals of the relevant sex. For overall cancer, each individual could contribute to the analysis as a case once. For site-specific cancers, an individual could contribute to the analysis of multiple cancers. Controls were defined as individuals without the disease outcome under consideration. Hence an individual with one cancer could be a control for analyses of another cancer. We also performed sensitivity analyses excluding individuals with solely self-reported cancer outcomes from the analyses, and for site-specific cancers, excluding individuals with a cancer diagnosis other than the site-specific cancer under analysis (so that controls were only those without any cancer diagnosis).
Gene-specific analyses for HMGCR and other drug proxy variants
We performed targeted analyses for variants in the HMGCR gene region that can be considered as proxies for statin therapy. Additionally, we conducted separate analyses for the PCSK9, LDLR, NPC1L1, APOC3, and LPL gene regions, mimicking other lipid-altering therapies (Supplementary file 1). These regions were chosen as they contain variants that explain enough variance in lipids to perform adequately powered analyses. Variants in each gene region explained 0.4% (HMGCR), 1.2% (PCSK9), 1.0% (LDLR), 0.2% (NPC1L1), 0.1% (APOC3), and <0.1% (LPL) of the variance in LDL-cholesterol. The APOC3 and LPL variants also explained 1.0% and 0.9% of the variance in triglycerides, respectively. Variants were chosen based on their associations with the relevant lipid trait from a conditional analysis in the GLGC (Supplementary methods). We performed the inverse-variance weighted method accounting for correlations between the variants using generalized weighted linear regression (Burgess et al., 2016). This was implemented using the ‘correl’ option in the MendelianRandomization package (Yavorska and Burgess, 2017). Estimates for the HMGCR, PCSK9, LDLR, and NPC1L1 gene regions are scaled to a one standard deviation increase in LDL-cholesterol, whereas estimates for the APOC3 and LPL gene regions are scaled to a one standard deviation increase in triglycerides.
Selection of variants for gene-specific analyses
Variants for the gene-specific analyses were selected to match the choice in a parallel analysis of cardiovascular diseases (Allara et al., 2019a). Variants in the HMGCR and PCSK9 regions were originally selected by Ference et al., 2016. Variants in the LPL region were originally selected by Lotta et al., 2016. Variants in the NPC1L1 region were originally selected by Ference et al., 2015. Variants in the LDLR and APOC3 regions were selected by Do et al., 2013. All variants were chosen based on their associations with lipid levels in conditional analyses using data from the GLGC. Variants are all conditionally associated with the relevant lipid trait (either LDL-cholesterol or triglycerides) and not strongly correlated (r2 <0.4). The variants are listed in Supplementary file 1.
Polygenic analyses for all lipid-related variants
We carried out polygenic analyses based on 184 genetic variants previously demonstrated to be associated with at least one of total cholesterol, LDL-cholesterol, HDL-cholesterol, or triglycerides at a genome-wide level of significance (p < 5 × 10−8) in the GLGC (Do et al., 2013). These variants explained 15.0% of the variance in total cholesterol, 14.6% in LDL-cholesterol, 13.7% in HDL-cholesterol, and 11.7% in triglycerides in the GLGC. Variants were reported as uncorrelated in the original publication by the GLGC, but some pairs of correlated variants remained in the analysis.
To obtain the associations of genetically-predicted values of LDL-cholesterol, HDL-cholesterol, and triglycerides with each cancer outcome while accounting for measured genetic pleiotropy via each other, we performed multivariable Mendelian randomization analyses using the inverse-variance weighted method (Sanderson et al., 2019). For total cholesterol, we performed univariable Mendelian randomization analyses using the inverse-variance weighted method (Burgess et al., 2013). The analysis for total cholesterol was conducted both because cancer risk may be influenced by total cholesterol rather than any particular lipid subfraction and to mitigate against the loss of power from adjustment for the various lipid subfractions in the multivariable analysis. To account for between-variant heterogeneity, we used random-effects models in all analyses. Heterogeneity between the estimates for different variants was quantified by Cochran’s Q statistic and Higgins’ I (Crosbie et al., 2013) statistic. For polygenic analyses that provided evidence of a causal effect, we additionally performed robust methods for Mendelian randomization, in particular the MR-Egger (Bowden et al., 2015) and weighted median methods (Bowden et al., 2016). All estimates are expressed per one standard deviation increase in the corresponding lipid fraction (in the GLGC, one standard deviation was 45.6 mg/dL for total cholesterol, 39.0 mg/dL for LDL-cholesterol, 15.8 mg/dL for HDL-cholesterol, and 90.5 mg/dL for triglycerides). Correlation between variants was accounted for in the inverse-variance weighted and MR-Egger methods; for the weighted median analysis, one of each pair of correlated variants was dropped from the analysis.
As power calculators have not been developed for multivariable Mendelian randomization analyses, we performed power calculations for polygenic analyses based on univariable Mendelian randomization for each lipid fraction in turn, and for gene-specific analyses for each gene region in turn (Burgess, 2014). We carried out all analyses using R (version 3.4.4) unless otherwise stated. All statistical tests and p-values presented are two sided.
Acknowledgements
Stephen Burgess is supported by Sir Henry Dale Fellowship jointly funded by the Wellcome Trust and the Royal Society (grant number 204623/Z/16/Z). Stephen Burgess and Amy Mason are supported by the UK National Institute for Health Research Cambridge Biomedical Research Centre.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Contributor Information
Stephen Burgess, Email: sb452@medschl.cam.ac.uk.
Andrew P Morris, University of Liverpool, United Kingdom.
Eduardo Franco, McGill University, Canada.
Funding Information
This paper was supported by the following grants:
Wellcome Trust and Royal Society 204623/Z/16/Z to Stephen Burgess.
National Institute for Health Research to Amy M Mason, Stephen Burgess.
Additional information
Competing interests
No competing interests declared.
Author contributions
Investigation, Writing - original draft.
Conceptualization, Writing - original draft.
Investigation, Writing - review and editing.
Investigation, Writing - review and editing.
Methodology, Writing - review and editing.
Investigation, Writing - review and editing.
Conceptualization, Formal analysis, Methodology, Writing - review and editing.
Additional files
Data availability
All data generated or analysed during this study are publicly-available and/or provided in the supporting files. The UK Biobank can be accessed online at http://biobank.ctsu.ox.ac.uk/crystal/. The Global Lipids Genetics Consortium data can be accessed at http://csg.sph.umich.edu/willer/public/lipids2013/.
The following datasets were generated:
References
- Allara E, Morani G, Carter P. Genetic determinants of lipids and cardiovascular disease outcomes: a Wide-Angled mendelian randomization investigation. Circ Genomic Precis Med. 2019a;12:e002711. doi: 10.1161/CIRCGEN.119.002711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allara E, Morani G, Carter P. Genetic determinants of lipids and cardiovascular disease outcomes: a wide-angled mendelian randomization investigation. bioRxiv. 2019b doi: 10.1101/668970. [DOI] [PMC free article] [PubMed]
- Alsheikh-Ali AA, Trikalinos TA, Kent DM, Karas RH. Statins, low-density lipoprotein cholesterol, and risk of Cancer. Journal of the American College of Cardiology. 2008;52:1141–1147. doi: 10.1016/j.jacc.2008.06.037. [DOI] [PubMed] [Google Scholar]
- ASCOT investigators. Sever PS, Dahlöf B, Poulter NR, Wedel H, Beevers G, Caulfield M, Collins R, Kjeldsen SE, Kristinsson A, McInnes GT, Mehlsen J, Nieminen M, O'Brien E, Ostergren J. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian cardiac outcomes trial--lipid lowering arm (ASCOT-LLA): a multicentre randomised controlled trial. The Lancet. 2003;361:1149–1158. doi: 10.1016/S0140-6736(03)12948-0. [DOI] [PubMed] [Google Scholar]
- Beeghly-Fadiel A, Khankari NK, Delahanty RJ, Shu X-O, Lu Y, Schmidt MK, Bolla MK, Michailidou K, Wang Q, Dennis J, Yannoukakos D, Dunning AM, Pharoah PDP, Chenevix-Trench G, Milne RL, Hunter DJ, Per H, Kraft P, Simard J, Easton DF, Zheng W. A mendelian randomization analysis of circulating lipid traits and breast Cancer risk. International Journal of Epidemiology. 2019;68:dyz242. doi: 10.1093/ije/dyz242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and Bias detection through egger regression. International Journal of Epidemiology. 2015;44:512–525. doi: 10.1093/ije/dyv080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genetic Epidemiology. 2016;40:304–314. doi: 10.1002/gepi.21965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruegel M, Teupser D, Haffner I, Mueller M, Thiery J. Statins reduce macrophage inflammatory protein-1alpha expression in human activated monocytes. Clinical and Experimental Pharmacology and Physiology. 2006;33:1144–1149. doi: 10.1111/j.1440-1681.2006.04493.x. [DOI] [PubMed] [Google Scholar]
- Burgess S, Butterworth A, Malarstig A, Thompson SG. Use of mendelian randomisation to assess potential benefit of clinical intervention. BMJ. 2012;345:e7325. doi: 10.1136/bmj.e7325. [DOI] [PubMed] [Google Scholar]
- Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genetic Epidemiology. 2013;37:658–665. doi: 10.1002/gepi.21758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess S. Sample size and power calculations in mendelian randomization with a single instrumental variable and a binary outcome. International Journal of Epidemiology. 2014;43:922–929. doi: 10.1093/ije/dyu005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess S, Dudbridge F, Thompson SG. Combining information on multiple instrumental variables in mendelian randomization: comparison of allele score and summarized data methods. Statistics in Medicine. 2016;35:1880–1906. doi: 10.1002/sim.6835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreras-Torres R, Johansson M, Gaborieau V, Haycock PC, Wade KH, Relton CL, Martin RM, Davey Smith G, Brennan P. The role of obesity, type 2 diabetes, and metabolic factors in pancreatic Cancer: a mendelian randomization study. JNCI: Journal of the National Cancer Institute. 2017a;109:djx012. doi: 10.1093/jnci/djx012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreras-Torres R, Johansson M, Haycock PC, Wade KH, Relton CL, Martin RM, Davey Smith G, Albanes D, Aldrich MC, Andrew A, Arnold SM, Bickeböller H, Bojesen SE, Brunnström H, Manjer J, Brüske I, Caporaso NE, Chen C, Christiani DC, Christian WJ, Doherty JA, Duell EJ, Field JK, Davies MPA, Marcus MW, Goodman GE, Grankvist K, Haugen A, Hong YC, Kiemeney LA, van der Heijden E, Kraft P, Johansson MB, Lam S, Landi MT, Lazarus P, Le Marchand L, Liu G, Melander O, Park SL, Rennert G, Risch A, Haura EB, Scelo G, Zaridze D, Mukeriya A, Savić M, Lissowska J, Swiatkowska B, Janout V, Holcatova I, Mates D, Schabath MB, Shen H, Tardon A, Teare MD, Woll P, Tsao MS, Wu X, Yuan JM, Hung RJ, Amos CI, McKay J, Brennan P. Obesity, metabolic factors and risk of different histological types of lung Cancer: a mendelian randomization study. PLOS ONE. 2017b;12:e0177875. doi: 10.1371/journal.pone.0177875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H-L, Chen C-Y, Hsu Y-F, Kuo W-S, Ou G, Chiu P-T, Huang Y-H, Hsu M-J. Simvastatin induced HCT116 colorectal Cancer cell apoptosis through p38MAPK-p53-survivin signaling cascade. Biochimica Et Biophysica Acta (BBA) - General Subjects. 2013;1830:4053–4064. doi: 10.1016/j.bbagen.2013.04.011. [DOI] [PubMed] [Google Scholar]
- Chen X, Resh MD. Cholesterol depletion from the plasma membrane triggers Ligand-independent activation of the epidermal growth factor receptor. Journal of Biological Chemistry. 2002;277:49631–49637. doi: 10.1074/jbc.M208327200. [DOI] [PubMed] [Google Scholar]
- Clutterbuck RD, Millar BC, Powles RL, Newman A, Catovsky D, Jarman M, Millar JL. Inhibitory effect of simvastatin on the proliferation of human myeloid leukaemia cells in severe combined immunodeficient (SCID) mice. British Journal of Haematology. 1998;102:522–527. doi: 10.1046/j.1365-2141.1998.00783.x. [DOI] [PubMed] [Google Scholar]
- Collins R, Reith C, Emberson J, Armitage J, Baigent C, Blackwell L, Blumenthal R, Danesh J, Smith GD, DeMets D, Evans S, Law M, MacMahon S, Martin S, Neal B, Poulter N, Preiss D, Ridker P, Roberts I, Rodgers A, Sandercock P, Schulz K, Sever P, Simes J, Smeeth L, Wald N, Yusuf S, Peto R. Interpretation of the evidence for the efficacy and safety of statin therapy. The Lancet. 2016;388:2532–2561. doi: 10.1016/S0140-6736(16)31357-5. [DOI] [PubMed] [Google Scholar]
- Cornish AJ, Law PJ, Timofeeva M, Palin K, Farrington SM, Palles C, Jenkins MA, Casey G, Brenner H, Chang-Claude J, Hoffmeister M, Kirac I, Maughan T, Brezina S, Gsur A, Cheadle JP, Aaltonen LA, Tomlinson I, Dunlop MG, Houlston RS. Modifiable pathways for colorectal Cancer: a mendelian randomisation analysis. The Lancet Gastroenterology & Hepatology. 2020;5:55–62. doi: 10.1016/S2468-1253(19)30294-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosbie J, Magnussen M, Dornbier R, Iannone A, Steele TA. Statins inhibit proliferation and cytotoxicity of a human leukemic natural killer cell line. Biomarker Research. 2013;1:33. doi: 10.1186/2050-7771-1-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale KM, Coleman CI, Henyan NN, Kluger J, White CM. Statins and Cancer risk: a meta-analysis. Journal of the American Medical Association. 2006;295:74–80. doi: 10.1001/jama.295.1.74. [DOI] [PubMed] [Google Scholar]
- Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Human Molecular Genetics. 2014;23:R89–R98. doi: 10.1093/hmg/ddu328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denoyelle C, Vasse M, Körner M, Mishal Z, Ganné F, Vannier JP, Soria J, Soria C. Cerivastatin, an inhibitor of HMG-CoA reductase, inhibits the signaling pathways involved in the invasiveness and metastatic properties of highly invasive breast Cancer cell lines: an in vitro study. Carcinogenesis. 2001;22:1139–1148. doi: 10.1093/carcin/22.8.1139. [DOI] [PubMed] [Google Scholar]
- DIAGRAM Consortium. MAGIC Consortium. InterAct Consortium. Swerdlow DI, Preiss D, Kuchenbaecker KB, Holmes MV, Engmann JE, Shah T, Sofat R, Stender S, Johnson PC, Scott RA, Leusink M, Verweij N, Sharp SJ, Guo Y, Giambartolomei C, Chung C, Peasey A, Amuzu A, Li K, Palmen J, Howard P, Cooper JA, Drenos F, Li YR, Lowe G, Gallacher J, Stewart MC, Tzoulaki I, Buxbaum SG, van der A DL, Forouhi NG, Onland-Moret NC, van der Schouw YT, Schnabel RB, Hubacek JA, Kubinova R, Baceviciene M, Tamosiunas A, Pajak A, Topor-Madry R, Stepaniak U, Malyutina S, Baldassarre D, Sennblad B, Tremoli E, de Faire U, Veglia F, Ford I, Jukema JW, Westendorp RG, de Borst GJ, de Jong PA, Algra A, Spiering W, Maitland-van der Zee AH, Klungel OH, de Boer A, Doevendans PA, Eaton CB, Robinson JG, Duggan D, Kjekshus J, Downs JR, Gotto AM, Keech AC, Marchioli R, Tognoni G, Sever PS, Poulter NR, Waters DD, Pedersen TR, Amarenco P, Nakamura H, McMurray JJ, Lewsey JD, Chasman DI, Ridker PM, Maggioni AP, Tavazzi L, Ray KK, Seshasai SR, Manson JE, Price JF, Whincup PH, Morris RW, Lawlor DA, Smith GD, Ben-Shlomo Y, Schreiner PJ, Fornage M, Siscovick DS, Cushman M, Kumari M, Wareham NJ, Verschuren WM, Redline S, Patel SR, Whittaker JC, Hamsten A, Delaney JA, Dale C, Gaunt TR, Wong A, Kuh D, Hardy R, Kathiresan S, Castillo BA, van der Harst P, Brunner EJ, Tybjaerg-Hansen A, Marmot MG, Krauss RM, Tsai M, Coresh J, Hoogeveen RC, Psaty BM, Lange LA, Hakonarson H, Dudbridge F, Humphries SE, Talmud PJ, Kivimäki M, Timpson NJ, Langenberg C, Asselbergs FW, Voevoda M, Bobak M, Pikhart H, Wilson JG, Reiner AP, Keating BJ, Hingorani AD, Sattar N. HMG-coenzyme A reductase inhibition, type 2 diabetes, and bodyweight: evidence from genetic analysis and randomised trials. The Lancet. 2015;385:351–361. doi: 10.1016/S0140-6736(14)61183-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do R, Willer CJ, Schmidt EM, Sengupta S, Gao C, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, Mora S, Beckmann JS, Bragg-Gresham JL, Chang H-Y, Demirkan A, Den Hertog HM, Donnelly LA, Ehret GB, Esko T, Feitosa MF, Ferreira T, Fischer K, Fontanillas P, Fraser RM, Freitag DF, Gurdasani D, Heikkilä K, Hyppönen E, Isaacs A, Jackson AU, Johansson Åsa, Johnson T, Kaakinen M, Kettunen J, Kleber ME, Li X, Luan J, Lyytikäinen L-P, Magnusson PKE, Mangino M, Mihailov E, Montasser ME, Müller-Nurasyid M, Nolte IM, O'Connell JR, Palmer CD, Perola M, Petersen A-K, Sanna S, Saxena R, Service SK, Shah S, Shungin D, Sidore C, Song C, Strawbridge RJ, Surakka I, Tanaka T, Teslovich TM, Thorleifsson G, Van den Herik EG, Voight BF, Volcik KA, Waite LL, Wong A, Wu Y, Zhang W, Absher D, Asiki G, Barroso I, Been LF, Bolton JL, Bonnycastle LL, Brambilla P, Burnett MS, Cesana G, Dimitriou M, Doney ASF, Döring A, Elliott P, Epstein SE, Eyjolfsson GI, Gigante B, Goodarzi MO, Grallert H, Gravito ML, Groves CJ, Hallmans G, Hartikainen A-L, Hayward C, Hernandez D, Hicks AA, Holm H, Hung Y-J, Illig T, Jones MR, Kaleebu P, Kastelein JJP, Khaw K-T, Kim E, Klopp N, Komulainen P, Kumari M, Langenberg C, Lehtimäki T, Lin S-Y, Lindström J, Loos RJF, Mach F, McArdle WL, Meisinger C, Mitchell BD, Müller G, Nagaraja R, Narisu N, Nieminen TVM, Nsubuga RN, Olafsson I, Ong KK, Palotie A, Papamarkou T, Pomilla C, Pouta A, Rader DJ, Reilly MP, Ridker PM, Rivadeneira F, Rudan I, Ruokonen A, Samani N, Scharnagl H, Seeley J, Silander K, Stančáková A, Stirrups K, Swift AJ, Tiret L, Uitterlinden AG, van Pelt LJ, Vedantam S, Wainwright N, Wijmenga C, Wild SH, Willemsen G, Wilsgaard T, Wilson JF, Young EH, Zhao JH, Adair LS, Arveiler D, Assimes TL, Bandinelli S, Bennett F, Bochud M, Boehm BO, Boomsma DI, Borecki IB, Bornstein SR, Bovet P, Burnier M, Campbell H, Chakravarti A, Chambers JC, Chen Y-DI, Collins FS, Cooper RS, Danesh J, Dedoussis G, de Faire U, Feranil AB, Ferrières J, Ferrucci L, Freimer NB, Gieger C, Groop LC, Gudnason V, Gyllensten U, Hamsten A, Harris TB, Hingorani A, Hirschhorn JN, Hofman A, Hovingh GK, Hsiung CA, Humphries SE, Hunt SC, Hveem K, Iribarren C, Järvelin M-R, Jula A, Kähönen M, Kaprio J, Kesäniemi A, Kivimaki M, Kooner JS, Koudstaal PJ, Krauss RM, Kuh D, Kuusisto J, Kyvik KO, Laakso M, Lakka TA, Lind L, Lindgren CM, Martin NG, März W, McCarthy MI, McKenzie CA, Meneton P, Metspalu A, Moilanen L, Morris AD, Munroe PB, Njølstad I, Pedersen NL, Power C, Pramstaller PP, Price JF, Psaty BM, Quertermous T, Rauramaa R, Saleheen D, Salomaa V, Sanghera DK, Saramies J, Schwarz PEH, Sheu WH-H, Shuldiner AR, Siegbahn A, Spector TD, Stefansson K, Strachan DP, Tayo BO, Tremoli E, Tuomilehto J, Uusitupa M, van Duijn CM, Vollenweider P, Wallentin L, Wareham NJ, Whitfield JB, Wolffenbuttel BHR, Altshuler D, Ordovas JM, Boerwinkle E, Palmer CNA, Thorsteinsdottir U, Chasman DI, Rotter JI, Franks PW, Ripatti S, Cupples LA, Sandhu MS, Rich SS, Boehnke M, Deloukas P, Mohlke KL, Ingelsson E, Abecasis GR, Daly MJ, Neale BM, Kathiresan S. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nature Genetics. 2013;45:1345–1352. doi: 10.1038/ng.2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooqi MAM, Malhotra N, Mukherjee SD, Sanger S, Dhesy-Thind SK, Ellis P, Leong DP. Statin therapy in the treatment of active Cancer: a systematic review and meta-analysis of randomized controlled trials. PLOS ONE. 2018;13:e0209486. doi: 10.1371/journal.pone.0209486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ference BA, Yoo W, Alesh I, Mahajan N, Mirowska KK, Mewada A, Kahn J, Afonso L, Williams KA, Flack JM. Effect of Long-Term exposure to lower Low-Density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease. Journal of the American College of Cardiology. 2012;60:2631–2639. doi: 10.1016/j.jacc.2012.09.017. [DOI] [PubMed] [Google Scholar]
- Ference BA, Majeed F, Penumetcha R, Flack JM, Brook RD. Effect of naturally random allocation to lower low-density lipoprotein cholesterol on the risk of coronary heart disease mediated by polymorphisms in NPC1L1, HMGCR, or both: a 2 × 2 factorial mendelian randomization study. Journal of the American College of Cardiology. 2015;65:1552–1561. doi: 10.1016/j.jacc.2015.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ference BA, Robinson JG, Brook RD, Catapano AL, Chapman MJ, Neff DR, Voros S, Giugliano RP, Davey Smith G, Fazio S, Sabatine MS. Variation in PCSK9 and HMGCR and risk of cardiovascular disease and diabetes. The New England Journal of Medicine. 2016;375:2144–2153. doi: 10.1056/NEJMoa1604304. [DOI] [PubMed] [Google Scholar]
- Ghosh-Choudhury N, Mandal CC, Ghosh-Choudhury N, Ghosh Choudhury G. Simvastatin induces derepression of PTEN expression via NFkappaB to inhibit breast Cancer cell growth. Cellular Signalling. 2010;22:749–758. doi: 10.1016/j.cellsig.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Global Lipids Genetics Consortium. Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, Mora S, Beckmann JS, Bragg-Gresham JL, Chang HY, Demirkan A, Den Hertog HM, Do R, Donnelly LA, Ehret GB, Esko T, Feitosa MF, Ferreira T, Fischer K, Fontanillas P, Fraser RM, Freitag DF, Gurdasani D, Heikkilä K, Hyppönen E, Isaacs A, Jackson AU, Johansson Å, Johnson T, Kaakinen M, Kettunen J, Kleber ME, Li X, Luan J, Lyytikäinen LP, Magnusson PKE, Mangino M, Mihailov E, Montasser ME, Müller-Nurasyid M, Nolte IM, O'Connell JR, Palmer CD, Perola M, Petersen AK, Sanna S, Saxena R, Service SK, Shah S, Shungin D, Sidore C, Song C, Strawbridge RJ, Surakka I, Tanaka T, Teslovich TM, Thorleifsson G, Van den Herik EG, Voight BF, Volcik KA, Waite LL, Wong A, Wu Y, Zhang W, Absher D, Asiki G, Barroso I, Been LF, Bolton JL, Bonnycastle LL, Brambilla P, Burnett MS, Cesana G, Dimitriou M, Doney ASF, Döring A, Elliott P, Epstein SE, Ingi Eyjolfsson G, Gigante B, Goodarzi MO, Grallert H, Gravito ML, Groves CJ, Hallmans G, Hartikainen AL, Hayward C, Hernandez D, Hicks AA, Holm H, Hung YJ, Illig T, Jones MR, Kaleebu P, Kastelein JJP, Khaw KT, Kim E, Klopp N, Komulainen P, Kumari M, Langenberg C, Lehtimäki T, Lin SY, Lindström J, Loos RJF, Mach F, McArdle WL, Meisinger C, Mitchell BD, Müller G, Nagaraja R, Narisu N, Nieminen TVM, Nsubuga RN, Olafsson I, Ong KK, Palotie A, Papamarkou T, Pomilla C, Pouta A, Rader DJ, Reilly MP, Ridker PM, Rivadeneira F, Rudan I, Ruokonen A, Samani N, Scharnagl H, Seeley J, Silander K, Stančáková A, Stirrups K, Swift AJ, Tiret L, Uitterlinden AG, van Pelt LJ, Vedantam S, Wainwright N, Wijmenga C, Wild SH, Willemsen G, Wilsgaard T, Wilson JF, Young EH, Zhao JH, Adair LS, Arveiler D, Assimes TL, Bandinelli S, Bennett F, Bochud M, Boehm BO, Boomsma DI, Borecki IB, Bornstein SR, Bovet P, Burnier M, Campbell H, Chakravarti A, Chambers JC, Chen YI, Collins FS, Cooper RS, Danesh J, Dedoussis G, de Faire U, Feranil AB, Ferrières J, Ferrucci L, Freimer NB, Gieger C, Groop LC, Gudnason V, Gyllensten U, Hamsten A, Harris TB, Hingorani A, Hirschhorn JN, Hofman A, Hovingh GK, Hsiung CA, Humphries SE, Hunt SC, Hveem K, Iribarren C, Järvelin MR, Jula A, Kähönen M, Kaprio J, Kesäniemi A, Kivimaki M, Kooner JS, Koudstaal PJ, Krauss RM, Kuh D, Kuusisto J, Kyvik KO, Laakso M, Lakka TA, Lind L, Lindgren CM, Martin NG, März W, McCarthy MI, McKenzie CA, Meneton P, Metspalu A, Moilanen L, Morris AD, Munroe PB, Njølstad I, Pedersen NL, Power C, Pramstaller PP, Price JF, Psaty BM, Quertermous T, Rauramaa R, Saleheen D, Salomaa V, Sanghera DK, Saramies J, Schwarz PEH, Sheu WH, Shuldiner AR, Siegbahn A, Spector TD, Stefansson K, Strachan DP, Tayo BO, Tremoli E, Tuomilehto J, Uusitupa M, van Duijn CM, Vollenweider P, Wallentin L, Wareham NJ, Whitfield JB, Wolffenbuttel BHR, Ordovas JM, Boerwinkle E, Palmer CNA, Thorsteinsdottir U, Chasman DI, Rotter JI, Franks PW, Ripatti S, Cupples LA, Sandhu MS, Rich SS, Boehnke M, Deloukas P, Kathiresan S, Mohlke KL, Ingelsson E, Abecasis GR. Discovery and refinement of loci associated with lipid levels. Nature Genetics. 2013;45:1274–1283. doi: 10.1038/ng.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of Cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hawk MA, Cesen KT, Siglin JC, Stoner GD, Ruch RJ. Inhibition of lung tumor cell growth in vitro and mouse lung tumor formation by lovastatin. Cancer Letters. 1996;109:217–222. doi: 10.1016/S0304-3835(96)04465-5. [DOI] [PubMed] [Google Scholar]
- Inano H, Suzuki K, Onoda M, Wakabayashi K. Anti-carcinogenic activity of simvastatin during the promotion phase of radiation-induced mammary tumorigenesis of rats. Carcinogenesis. 1997;18:1723–1727. doi: 10.1093/carcin/18.9.1723. [DOI] [PubMed] [Google Scholar]
- Ishikawa S, Hayashi H, Kinoshita K, Abe M, Kuroki H, Tokunaga R, Tomiyasu S, Tanaka H, Sugita H, Arita T, Yagi Y, Watanabe M, Hirota M, Baba H. Statins inhibit tumor progression via an enhancer of zeste homolog 2-mediated epigenetic alteration in colorectal Cancer. International Journal of Cancer. 2014;135:2528–2536. doi: 10.1002/ijc.28672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JPHC Study Group. Iso H, Ikeda A, Inoue M, Sato S, Tsugane S. Serum cholesterol levels in relation to the incidence of Cancer: the JPHC study cohorts. International Journal of Cancer. 2009;125:2679–2686. doi: 10.1002/ijc.24668. [DOI] [PubMed] [Google Scholar]
- Katzke VA, Sookthai D, Johnson T, Kühn T, Kaaks R. Blood lipids and lipoproteins in relation to incidence and mortality risks for CVD and Cancer in the prospective EPIC-Heidelberg cohort. BMC Medicine. 2017;15:218. doi: 10.1186/s12916-017-0976-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana V, Bejjanki HR, Caldito G, Owens MW. Statins reduce the risk of lung Cancer in humans: a large case-control study of US veterans. Chest. 2007;131:1282–1288. doi: 10.1378/chest.06-0931. [DOI] [PubMed] [Google Scholar]
- Kikuchi T, Nagata Y, Abe T. In vitro and in vivo antiproliferative effects of simvastatin, an HMG-CoA reductase inhibitor, on human glioma cells. Journal of Neuro-Oncology. 1997;34:233–239. doi: 10.1023/a:1005753523949. [DOI] [PubMed] [Google Scholar]
- Kitahara CM, Berrington de González A, Freedman ND, Huxley R, Mok Y, Jee SH, Samet JM. Total cholesterol and Cancer risk in a large prospective study in Korea. Journal of Clinical Oncology. 2011;29:1592–1598. doi: 10.1200/JCO.2010.31.5200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kritchevsky SB, Wilcosky TC, Morris DL, Truong KN, Tyroler HA. Changes in plasma lipid and lipoprotein cholesterol and weight prior to the diagnosis of Cancer. Cancer Research. 1991;51:3198–3203. [PubMed] [Google Scholar]
- Li YC, Park MJ, Ye SK, Kim CW, Kim YN. Elevated levels of cholesterol-rich lipid rafts in Cancer cells are correlated with apoptosis sensitivity induced by cholesterol-depleting agents. The American Journal of Pathology. 2006;168:1107–1118. doi: 10.2353/ajpath.2006.050959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao JK, Laufs U. Pleiotropic effects of statins. Annual Review of Pharmacology and Toxicology. 2005;45:89–118. doi: 10.1146/annurev.pharmtox.45.120403.095748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YC, Lin JH, Chou CW, Chang YF, Yeh SH, Chen CC. Statins increase p21 through inhibition of histone deacetylase activity and release of promoter-associated HDAC1/2. Cancer Research. 2008;68:2375–2383. doi: 10.1158/0008-5472.CAN-07-5807. [DOI] [PubMed] [Google Scholar]
- Lin X, Lu L, Liu L, Wei S, He Y, Chang J, Lian X. Blood lipids profile and lung Cancer risk in a meta-analysis of prospective cohort studies. Journal of Clinical Lipidology. 2017;11:1073–1081. doi: 10.1016/j.jacl.2017.05.004. [DOI] [PubMed] [Google Scholar]
- Lotta LA, Sharp SJ, Burgess S, Perry JRB, Stewart ID, Willems SM, Luan J, Ardanaz E, Arriola L, Balkau B, Boeing H, Deloukas P, Forouhi NG, Franks PW, Grioni S, Kaaks R, Key TJ, Navarro C, Nilsson PM, Overvad K, Palli D, Panico S, Quirós J-R, Riboli E, Rolandsson O, Sacerdote C, Salamanca-Fernandez E, Slimani N, Spijkerman AMW, Tjonneland A, Tumino R, van der A DL, van der Schouw YT, McCarthy MI, Barroso I, O’Rahilly S, Savage DB, Sattar N, Langenberg C, Scott RA, Wareham NJ. Association between Low-Density lipoprotein Cholesterol–Lowering Genetic Variants and Risk of Type 2 Diabetes. Jama. 2016;316:1383–1391. doi: 10.1001/jama.2016.14568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma HQ, Cui LH, Li CC, Yu Z, Piao JM. Effects of serum triglycerides on prostate Cancer and breast Cancer risk: a Meta-Analysis of prospective studies. Nutrition and Cancer. 2016;68:1073–1082. doi: 10.1080/01635581.2016.1206582. [DOI] [PubMed] [Google Scholar]
- Melvin JC, Seth D, Holmberg L, Garmo H, Hammar N, Jungner I, Walldius G, Lambe M, Wigertz A, Van Hemelrijck M. Lipid profiles and risk of breast and ovarian Cancer in the swedish AMORIS study. Cancer Epidemiology Biomarkers & Prevention. 2012;21:1381–1384. doi: 10.1158/1055-9965.EPI-12-0188. [DOI] [PubMed] [Google Scholar]
- Misirkic M, Janjetovic K, Vucicevic L, Tovilovic G, Ristic B, Vilimanovich U, Harhaji-Trajkovic L, Sumarac-Dumanovic M, Micic D, Bumbasirevic V, Trajkovic V. Inhibition of AMPK-dependent autophagy enhances in vitro antiglioma effect of simvastatin. Pharmacological Research. 2012;65:111–119. doi: 10.1016/j.phrs.2011.08.003. [DOI] [PubMed] [Google Scholar]
- Mullen PJ, Yu R, Longo J, Archer MC, Penn LZ. The interplay between cell signalling and the mevalonate pathway in Cancer. Nature Reviews Cancer. 2016;16:718–731. doi: 10.1038/nrc.2016.76. [DOI] [PubMed] [Google Scholar]
- Narisawa T, Morotomi M, Fukaura Y, Hasebe M, Ito M, Aizawa R. Chemoprevention by Pravastatin, a 3-Hydroxy-3-methylglutaryl-coenzyme A Reductase Inhibitor, of N -Methyl- N -nitrosourea-induced Colon Carcinogenesis in F344 Rats. Japanese Journal of Cancer Research. 1996a;87:798–804. doi: 10.1111/j.1349-7006.1996.tb02103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narisawa T, Fukaura Y, Tanida N, Hasebe M, Ito M, Aizawa R. Chemopreventive efficacy of low dose of pravastatin, an HMG-CoA reductase inhibitor, on 1,2-dimethylhydrazine-induced Colon carcinogenesis in ICR mice. The Tohoku Journal of Experimental Medicine. 1996b;180:131–138. doi: 10.1620/tjem.180.131. [DOI] [PubMed] [Google Scholar]
- Nowak C, Ärnlöv J. A mendelian randomization study of the effects of blood lipids on breast Cancer risk. Nature Communications. 2018;9:3957. doi: 10.1038/s41467-018-06467-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orho-Melander M, Hindy G, Borgquist S, Schulz CA, Manjer J, Melander O, Stocks T. Blood lipid genetic scores, the HMGCR gene and Cancer risk: a mendelian randomization study. International Journal of Epidemiology. 2018;47:495–505. doi: 10.1093/ije/dyx237. [DOI] [PubMed] [Google Scholar]
- Parikh A, Childress C, Deitrick K, Lin Q, Rukstalis D, Yang W. Statin-induced autophagy by inhibition of geranylgeranyl biosynthesis in prostate Cancer PC3 cells. The Prostate. 2010;70:971–981. doi: 10.1002/pros.21131. [DOI] [PubMed] [Google Scholar]
- Park KW, Hwang KK, Cho HJ, Hur J, Yang HM, Yoon CH, Kang HJ, Oh BH, Park YB, Kim HS. Simvastatin enhances endothelial differentiation of peripheral blood mononuclear cells in hypercholesterolemic patients and induces pro-angiogenic cytokine IL-8 secretion from monocytes. Clinica Chimica Acta. 2008;388:156–166. doi: 10.1016/j.cca.2007.10.027. [DOI] [PubMed] [Google Scholar]
- Passarelli MN, Newcomb PA. Blood lipid concentrations and colorectal adenomas: a systematic review and Meta-Analysis of colonoscopy studies in Asia, 2000-2014. American Journal of Epidemiology. 2016;183:691–700. doi: 10.1093/aje/kwv294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisanti S, Picardi P, Ciaglia E, D'Alessandro A, Bifulco M. Novel prospects of statins as therapeutic agents in Cancer. Pharmacological Research. 2014;88:84–98. doi: 10.1016/j.phrs.2014.06.013. [DOI] [PubMed] [Google Scholar]
- Pocobelli G, Newcomb PA, Trentham-Dietz A, Titus-Ernstoff L, Hampton JM, Egan KM. Statin use and risk of breast Cancer. Cancer. 2008;112:27–33. doi: 10.1002/cncr.23129. [DOI] [PubMed] [Google Scholar]
- Poynter JN, Gruber SB, Higgins PD, Almog R, Bonner JD, Rennert HS, Low M, Greenson JK, Rennert G. Statins and the risk of colorectal Cancer. New England Journal of Medicine. 2005;352:2184–2192. doi: 10.1056/NEJMoa043792. [DOI] [PubMed] [Google Scholar]
- PRACTICAL consortium. Bull CJ, Bonilla C, Holly JM, Perks CM, Davies N, Haycock P, Yu OH, Richards JB, Eeles R, Easton D, Kote-Jarai Z, Amin Al Olama A, Benlloch S, Muir K, Giles GG, MacInnis RJ, Wiklund F, Gronberg H, Haiman CA, Schleutker J, Nordestgaard BG, Travis RC, Neal D, Pashayan N, Khaw KT, Stanford JL, Blot WJ, Thibodeau S, Maier C, Kibel AS, Cybulski C, Cannon-Albright L, Brenner H, Park J, Kaneva R, Batra J, Teixeira MR, Micheal A, Pandha H, Smith GD, Lewis SJ, Martin RM. Blood lipids and prostate Cancer: a mendelian randomization analysis. Cancer Medicine. 2016;5:1125–1136. doi: 10.1002/cam4.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Broadbent H, Law PJ, Sud A, Palin K, Tuupanen S, Gylfe A, Hänninen UA, Cajuso T, Tanskanen T, Kondelin J, Kaasinen E, Sarin AP, Ripatti S, Eriksson JG, Rissanen H, Knekt P, Pukkala E, Jousilahti P, Salomaa V, Palotie A, Renkonen-Sinisalo L, Lepistö A, Böhm J, Mecklin JP, Al-Tassan NA, Palles C, Martin L, Barclay E, Farrington SM, Timofeeva MN, Meyer BF, Wakil SM, Campbell H, Smith CG, Idziaszczyk S, Maughan TS, Kaplan R, Kerr R, Kerr D, Passarelli MN, Figueiredo JC, Buchanan DD, Win AK, Hopper JL, Jenkins MA, Lindor NM, Newcomb PA, Gallinger S, Conti D, Schumacher F, Casey G, Aaltonen LA, Cheadle JP, Tomlinson IP, Dunlop MG, Houlston RS. Mendelian randomisation implicates hyperlipidaemia as a risk factor for colorectal Cancer. International Journal of Cancer. 2017;140:2701–2708. doi: 10.1002/ijc.30709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salonen JT. Risk of Cancer and death in relation to serum cholesterol A longitudinal study in an eastern finnish population with high overall cholesterol level. American Journal of Epidemiology. 1982;116:622–630. doi: 10.1093/oxfordjournals.aje.a113445. [DOI] [PubMed] [Google Scholar]
- Sanderson E, Davey Smith G, Windmeijer F, Bowden J. An examination of multivariable mendelian randomization in the single-sample and two-sample summary data settings. International Journal of Epidemiology. 2019;48:713–727. doi: 10.1093/ije/dyy262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sławińska-Brych A, Zdzisińska B, Kandefer-Szerszeń M. Fluvastatin inhibits growth and alters the malignant phenotype of the C6 glioma cell line. Pharmacological Reports. 2014;66:121–129. doi: 10.1016/j.pharep.2014.01.002. [DOI] [PubMed] [Google Scholar]
- Spampanato C, De Maria S, Sarnataro M, Giordano E, Zanfardino M, Baiano S, Cartenì M, Morelli F. Simvastatin inhibits Cancer cell growth by inducing apoptosis correlated to activation of bax and down-regulation of BCL-2 gene expression. International Journal of Oncology. 2012;40:935–941. doi: 10.3892/ijo.2011.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohmaier S, Edlinger M, Manjer J, Stocks T, Bjørge T, Borena W, Häggström C, Engeland A, Nagel G, Almquist M, Selmer R, Tretli S, Concin H, Hallmans G, Jonsson H, Stattin P, Ulmer H. Total serum cholesterol and Cancer incidence in the metabolic syndrome and Cancer project (Me-Can) PLOS ONE. 2013;8:e54242. doi: 10.1371/journal.pone.0054242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudlow C, Gallacher J, Allen N. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLOS Medicine. 2015;12:e1001779. doi: 10.1371/journal.pmed.1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Clarke R, Bennett D, Guo Y, Walters RG, Hill M, Parish S, Millwood IY, Bian Z, Chen Y, Yu C, Lv J, Collins R, Chen J, Peto R, Li L, Chen Z. Causal associations of blood lipids with risk of ischemic stroke and intracerebral hemorrhage in chinese adults. Nature Medicine. 2019;25:569–574. doi: 10.1038/s41591-019-0366-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Wang K, Li J, Wang J, Wang Z, Fan Y, Ye Y, Ji G, Li Y. The association between serum lipids and colorectal neoplasm: a systemic review and meta-analysis. Public Health Nutrition. 2015;18:3355–3370. doi: 10.1017/S1368980015000646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Hemelrijck M, Garmo H, Hammar N, Jungner I, Walldius G, Lambe M, Holmberg L. The interplay between lipid profiles, glucose, BMI and risk of kidney Cancer in the swedish AMORIS study. International Journal of Cancer. 2012;130:2118–2128. doi: 10.1002/ijc.26212. [DOI] [PubMed] [Google Scholar]
- Vílchez JA, Martínez-Ruiz A, Sancho-Rodríguez N, Martínez-Hernández P, Noguera-Velasco JA. The real role of prediagnostic high-density lipoprotein cholesterol and the Cancer risk: a concise review. European Journal of Clinical Investigation. 2014;44:103–114. doi: 10.1111/eci.12185. [DOI] [PubMed] [Google Scholar]
- Wang IK, Lin-Shiau SY, Lin JK. Suppression of invasion and MMP-9 expression in NIH 3t3 and v-H-Ras 3t3 fibroblasts by lovastatin through inhibition of ras isoprenylation. Oncology. 2000;59:245–254. doi: 10.1159/000012168. [DOI] [PubMed] [Google Scholar]
- Würtz P, Wang Q, Soininen P, Kangas AJ, Fatemifar G, Tynkkynen T, Tiainen M, Perola M, Tillin T, Hughes AD, Mäntyselkä P, Kähönen M, Lehtimäki T, Sattar N, Hingorani AD, Casas JP, Salomaa V, Kivimäki M, Järvelin MR, Davey Smith G, Vanhala M, Lawlor DA, Raitakari OT, Chaturvedi N, Kettunen J, Ala-Korpela M. Metabolomic profiling of statin use and genetic inhibition of HMG-CoA reductase. Journal of the American College of Cardiology. 2016;67:1200–1210. doi: 10.1016/j.jacc.2015.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Chen M, Sun T. Simvastatin attenuates TGF-β1-induced epithelial-mesenchymal transition in human alveolar epithelial cells. Cellular Physiology and Biochemistry. 2013;31:863–874. doi: 10.1159/000350104. [DOI] [PubMed] [Google Scholar]
- Yao X, Tian Z. Dyslipidemia and colorectal Cancer risk: a meta-analysis of prospective studies. Cancer Causes & Control. 2015;26:257–268. doi: 10.1007/s10552-014-0507-y. [DOI] [PubMed] [Google Scholar]
- Yarmolinsky J, Bull CJ, Vincent EE, Robinson J, Walther A, Smith GD, Lewis SJ, Relton CL, Martin RM. Association between genetically proxied inhibition of HMG-CoA reductase and epithelial ovarian Cancer. Jama. 2020;323:646–655. doi: 10.1001/jama.2020.0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yavorska OO, Burgess S. MendelianRandomization: an R package for performing mendelian randomization analyses using summarized data. International Journal of Epidemiology. 2017;46:1734–1739. doi: 10.1093/ije/dyx034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeganeh B, Wiechec E, Ande SR, Sharma P, Moghadam AR, Post M, Freed DH, Hashemi M, Shojaei S, Zeki AA, Ghavami S. Targeting the mevalonate cascade as a new therapeutic approach in heart disease, Cancer and pulmonary disease. Pharmacology & Therapeutics. 2014;143:87–110. doi: 10.1016/j.pharmthera.2014.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]