

Abstract
Background:
22q11.2 deletion syndrome (22qDS) presents with myriad symptoms, including multiple neuropsychiatric disorders. Complications associated with the polygenic haploinsufficiency makes symptoms particularly difficult to manage via traditional therapeutic approaches. However, the varying mechanistic consequences often culminate to generate inappropriate regulation of neuronal circuit activity. We explored if managing this aberrant activity in adults could be a therapeutically beneficial strategy.
Methods:
To assess and dissect hippocampal circuit function, we performed functional imaging in acute slices and targeted eloquent circuits (specific sub-circuits tied to specific behavioral tasks) to provide relevant behavioral outputs. For example, the ventral and dorsal CA1 regions critically support social and spatial discrimination, respectively. We focally introduced chemogenetic constructs in 34 control and 24 22qDS model mice via adeno-associated viral vectors, driven by excitatory neuron-specific promoter elements, to manipulate circuit recruitment in an on-demand fashion.
Results:
22qDS model mice exhibited CA1 excitatory ensemble hyperexcitability and concomitant behavioral deficits in both social and spatial memory. Remarkably, acute chemogenetic inhibition of pyramidal cells successfully corrected the memory deficits, and did so in a regionally-specific manner: ventrally targeted constructs only rescued social behavior, while those expressed dorsally selectively affected spatial memory. Additionally, manipulating activity in control mice could recapitulate the memory deficits in a regionally-specific manner.
Conclusions:
These data suggest that retuning activity dysregulation can rescue function in disease-altered circuits, even in the face of a polygenetic haploinsufficiency with a strong developmental component. Targeting circuit excitability in a focal, modular manner may prove an effective therapeutic for treatment-resistant symptoms of mental illness.
Keywords: 22q11.2 deletion syndrome, circuit-based therapies, hippocampus, social memory, spatial memory, chemogenetics
Introduction
The most common microdeletion syndrome in humans, 22q11.2 Deletion Syndrome (22qDS) results in varying degrees of symptoms in multiple systems, including a high penetrance for developmental neuropsychiatric disorders (1). 22qDS is caused by the hemizygous deletion of a 1.5–3 megabase region of chromosome 22’s q arm, resulting in the loss of one copy of 46 protein coding genes for the 3 megabase and 27 for the 1.5 megabase deletion. In addition to intellectual disability and several anatomic phenotypes, psychiatric diagnoses include depression (2), obsessive-compulsive disorder (3), schizophrenia (4), attentional deficit hyperactivity disorder, and autism spectrum disorder (5). Among their constellation of symptoms, 22qDS patients often display pronounced social cognition impairments (6,7), and performance in the social cognitive domain is one of the best measures of the patients’ overall quality of life (8–10).
Managing neuropsychiatric symptoms proves particularly arduous as 22qDS patients are more vulnerable to side effects associated with pharmacotherapy, in some cases increasing the risk of developing additional psychiatric symptoms or inducing seizures (11,12). Further, the disorder is not readily amenable to genetic interventions given the complexity of the genetic defect and the probability that key incipient causes underlying symptoms are developmental in nature.
Psychiatric diseases often involve hippocampal circuit disruptions, which generate diverse disease-related symptoms. For example, the hippocampus in 22qDS patients has altered morphology, including decreased volume (13–16). Furthermore, studies have revealed alterations in hippocampal and thalamic functional connectivity (17). Such dysregulation, including hippocampal hyperexcitability, can be present in prodromal and idiopathic schizophrenia patients (18–22), suggesting these structural and functional alterations in 22qDS patients may critically contribute to psychiatric symptoms. Germane to this study, the hippocampus critically contributes to social cognition in both humans (23,24) and mice (25–28), with mouse studies demonstrating that murine ventral CA1 outputs can dictate social memory performance (27,28). We made use of this “eloquent circuit,” a specific sub-circuit that can be tied to a specific behavioral task, to provide a relevant behavioral output by which we may assess the circuit’s function in both health and disease.
In this study, we sought to employ a circuit-based therapeutic approach to retune hippocampal CA1 outputs via the circuit’s excitatory projection neurons to restore memory function in a 22q mouse model. This genetically engineered mouse line expresses a hemizygous deletion on a syntenic region of mouse chromosome 16, replicating much of the chromosomal alteration found in the human 22qDS (29). We used focally targeted AAVs with a CamKIIα promoter to express Designer Receptors Exclusively Activated by Designer Drugs (DREADDs) selectively in excitatory projection neurons and in a regionally-specific manner within the hippocampus. We demonstrate that bidirectionally retuning circuit activity in the dorsal or ventral hippocampus is sufficient to dictate behavioral performance in health and disease. We conclude that circuit-based interventions that flexibly modulate principal cell recruitment in a specific, regionally-dependent manner could be a promising therapeutic approach to correct later manifestations of genetic neurodevelopmental disease.
Materials and Methods
Animals
Male and female mice were purchased from Taconic Biosciences (Rensselaer, NY), singly housed, and maintained on a 12-hour light/dark cycle with ad libitum food and water. Mice were 22q (ko/wt C57BL/6-Del(16Dgcr2-Hira)1Tac) (29), wt/wt littermates (wt/wt C57BL/6-Del(16Dgcr2-Hira)1Tac), or C57BL/6. Animals were 3–6 months old when evaluated. Mice were assigned to experimental groups randomly after balancing for age, sex, and genotype. Analysis by sex did not reveal significant differences, thus results were pooled (Ctl: 17 female/50 total, t48=0.2268, p=0.8215; 22q: 12 female/36 total, t34=0.3460, p=0.7315). The control group consisted of a mix of C57BL/6 and wt/wt littermates as analysis did not reveal differences (28 wt/wt/50 total, t48=0.8725, p=0.3873). All procedures were approved by the IACUC of the Children’s Hospital of Philadelphia (CHOP).
Viral Induction
Animals (7–8 w old) were anesthetized with isoflurane and received bilateral viral injections to the ventral CA1 (AP: −3.2 mm, ML: ± 3.1 mm, DV: −4.6 mm) or dorsal CA1 (AP: −2 mm, ML: ±1.5 mm, DV: −1.5 mm). Approximately 6×108 infections particles/virus were infused into each site. Behavioral experiments began 3 w post-injection to allow full expression. See Supplemental for vector information.
Behavioral Tests
Social Memory.
The test involved five stages: habituation, social approach, familiarization, separation, and social discrimination. Stimulus-mice (4–6 week old same sex C57BL/6 mice) and objects were placed in pencil cups on either end of the arena (58 cm × 34 cm × 25.5 cm). For habituation, test-mice explored the arena and empty cups for 6 min before returning to their homecage for a 3 min ITI. For social approach (6 min), test-mice freely explored a novel stimulus-mouse or a novel object to examine sociability. Test-mice were familiarized with the stimulus-mice in their homecage for 2 hours. The stimulus-mouse was returned to its homecage for 30 min of separation. For social discrimination (6 min), test-mice explored the now familiar stimulus-mouse or a novel stimulus-mouse. Mice that remember the familiar conspecific should spend more time exploring the novel conspecific. Sessions were video recorded and manually scored by a blinded experimenter for interactions with the stimuli. Location of the novel stimulus was counterbalanced across trials and subjects. Animals received injections of saline (i.p.), CNO (i.p.), and salvinorin b (s.c.) in a counterbalanced order 1 hour prior to the social discrimination trial. Reversal trials were performed last with no injection. Discrimination Score was calculated as follows: 100*[(Novel mouse exploration)-(Familiar mouse or Novel object exploration)]/(Total exploration).
Novel Object Recognition.
As described previously(30,31), this hippocampal-independent task involves habituating animals to the test arena for 5 min/d for 5 d. On training day, animals explored 2 identical objects (15 min). On test day (24 hours later), 1 object was replaced with a novel object. Novel object exploration calculation: 100*(Novel object exploration)/(Total exploration).
Olfaction.
Animals were placed in a clean homecage with a Q-tip dipped in the olfactory cue hanging from the cage lid. Mice were exposed to the cue for 2 min with 1 min ITI between cues. In order, they explored water, vanilla (1:100 dilution McCormick pure vanilla extract), and opposite sex scent (Q-tips rubbed on the floor of a cage containing opposite sex mice).
Spatial object recognition.
As described previously(30,31), animals explored the arena for 6 min trials: habituation, then 3 training trials with 3 identical objects. On test day (24 hrs later), 1 object was repositioned (displaced object). Discrimination index calculation: Displaced Object Δ%Preference - Avg(Non-displaced Object1 Δ%Preference, Non-displaced Object2 Δ%Preference); Object Δ%Preference = Test(100*Object exploration/Total exploration) - 3rd Training Trial(100*Object exploration/Total exploration).
Voltage Sensitive Dye (VSD) slice preparation and recording
Horizontal slices (400 μm) were prepared and then imaged by a blinded experimenter as described previously (30). Briefly, slices were stained immediately before imaging by pipetting 0.125 mg/mL of di-3-ANEPPDHQ (D36801, Invitrogen, Carlsbad, CA) in aCSF onto the slice for 12 minutes. Slices were imaged in an oxygenated interface chamber (32 ± 1°C) using an 80 × 80 CCD camera recording at a 1-kHz frame rate (NeuroCCD, RedShirt Imaging, Decatur, GA) with a 530 nm Green LED (M530L3-C2, Thorlabs, Newton, NJ). The stimulating electrode was placed in the stratum radiatum to stimulate the Schaeffer collateral (SC) axons, and stimulation strength was determined via an input/output curve to produce 70% of the maximum response in the slice (average stim: 204.9 ± 21.1 uA). Trials lasted 1000 ms each, with a 20 s intertrial interval. Data were analyzed using Igor Pro 6.2 (Wavemetrics, Portland, OR).
Histology
Animals were perfused transcardially with 4% PFA in 100 mM PBS. Brains were submerged in 4% PFA for 12–24 hours, then in 100 mM PBS. Slices (50 μm) were mounted on glass slides and imaged using an Olympus FV1000 confocal microscope. Location of viral expression was visualized by the co-expressing fluorescent reporter. Only animals that had viral fluorescent expression localized to the hippocampus in both hemispheres were included in analyses.
Statistics
All statistical analyses were done with R and Prism 8 (GraphPad). The number of mice used for each experiment was appropriate to detect 25% behavioral differences with 80% power and α was set at 0.05. Normality was tested with the D’Agostino & Pearson test. Statistical tests were used as indicated in the figure legends.
Results
Altering circuit tuning disrupts social discrimination.
To ascertain if net changes in ventral CA1 circuit activity could determine social memory behavioral outcomes, thereby confirming the utility of this eloquent circuit in our hands, we sought to manipulate the ventral hippocampus bidirectionally via CamKIIα-driven DREADDs. Both the muscarinic receptor DREADDs (hM3Dq and hM4Di) and the kappa-opioid receptor DREADD (KORD) are mutated receptors that act through GPCR signaling cascades to affect neuronal activity upon activation by an otherwise inert ligand, traditionally clozapine-n-oxide (CNO) for the muscarinic DREADDs and salvinorin b (SB) for the KORD (32,33).
We examined if decreasing circuit activity could affect performance, co-injecting both inhibitory DREADDs (hM4Di and KORD) into control mice (Fig S1) to provide within-subject controls for our manipulations, thus accounting for potential unintended consequences in downstream GPCR-related mechanisms and off-target ligand effects. When the subjects were tested on a social memory paradigm (Fig 1A), DREADD recruitment significantly impacted the subjects’ ability to discriminate novel from familiar conspecifics (F(1.81,21.72)=7.216, p=0.0049, RM one-way ANOVA with Geisser-Greenhouse’s correction; Fig 1B). The mice preferentially explored the novel stimulus-mouse over the familiar stimulus-mouse when treated with saline, demonstrating a 29.0 ± 2.7% discrimination score, the difference in time spent exploring the novel conspecific compared to the other stimulus (see Methods for calculation). However, recruiting either the hM4Di (1 mg/kg CNO) or the KORD (1 mg/kg SB) prevented the same subjects from discriminating such that they explored the novel and familiar stimulus-mice equally (hM4Di: −5.5 ± 10.2%, pSaline vCNO=0.0188, KORD: −0.5 ± 7.7%, pSaline vSB=0.0140, Dunnett’s multiple comparisons correction). The mice returned to discriminating on the reversal trial (30.4 ± 3.0%, pSaline vReversal=0.9774, Dunnett’s correction), confirming that the DREADD-mediated interventions were responsible for the reversible behavioral change.
Fig 1. Disrupting circuit tuning disrupts behavior.

A. Social memory protocol schematic. B. Social discrimination results. Reducing ventral hippocampal activity with either hM4Di (CNO 1 mg/kg) or KORD (SB 1 mg/kg) compromises performance in control mice (F(1.81,21.72)=7.216, p=0.0049, RM one-way ANOVA with Geisser-Greenhouse’s correction and Dunnett’s correction), as does increasing activity via hM3Dq activation (CNO 1 mg/kg; t9=3.618, p=0.0056, paired t-test). C. Social approach results. Mice consistently show a preference for the novel conspecific (inhibitory DREADDS: F(2.31,27.73)=1.008, p=0.3876, RM one-way ANOVA with Geisser-Greenhouse’s correction; hM3Dq: t9=3.618, p=0.1639, paired t-test). Data are represented as mean ± SEM. Each circle represents an animal (number of animals indicated in parentheses). ns p>0.05, *p<0.05, **p<0.01.
We also examined how increasing circuit excitability affects behavior, bilaterally injecting the excitatory DREADD hM3Dq. With saline treatment, the subjects preferred the novel stimulus-mouse (21.1 ± 4.6%); however, hM3Dq recruitment compromised the subjects’ discrimination (1.2 ± 4.3%; t9=3.618, p=0.0056, paired t-test; Fig 1C). Thus, bidirectional manipulation of circuit activity effected behavioral output changes.
Importantly, these results were not a function of retesting the animals. For one, the mice consistently preferred the novel conspecific over the novel object in the social approach trials (inhibitory DREADD trials: F(2.31,27.73)=1.008, p=0.3876, RM one-way ANOVA with Geisser-Greenhouse’s correction; hM3Dq: t9=3.618, p=0.1639, paired t-test; Fig 1C). Further, the mice explored the various stimuli for consistent amounts of time across the social approach (inhibitory DREADDS: F(3,36)=1.946, p=0.1396, RM one-way ANOVA; hM3Dq: t9=0.3736, p=0.7174, paired t-test; Fig S2A) and discrimination trials (inhibitory DREADDS: F(3,36)=1.943, p=0.1401, RM one-way ANOVA; hM3Dq: t9=0.0560, p=0.9538, paired t-test; Fig S2B). Finally, these results were not due to off-target ligand effects as GFP-expressing control animals consistently preferred the novel conspecific regardless of treatment (Fig S3B).
These data suggest that altering activity in a normal, healthy circuit to be either hyper- or hypoactive can perturb behavioral performance; however, a properly tuned circuit supports successful behavioral outcomes.
22q mice express a social discrimination deficit.
Next, we used the social memory paradigm (Fig 1A) to assess ventral CA1 functionality in the 22q mouse model (29). 22q mice failed to distinguish between novel and familiar stimulus-mice (0.1 ± 5.5%), demonstrating a deficit compared to controls (t26=2.846, p=0.0085, Student’s t-test; Fig 2A). We ran a separate cohort of 22q and control mice at a different behavioral testing core and replicated the deficit (t32=4.040, p=0.0003, Student’s t-test; Fig S4D).
Fig 2. 22q mice express a social discrimination deficit.
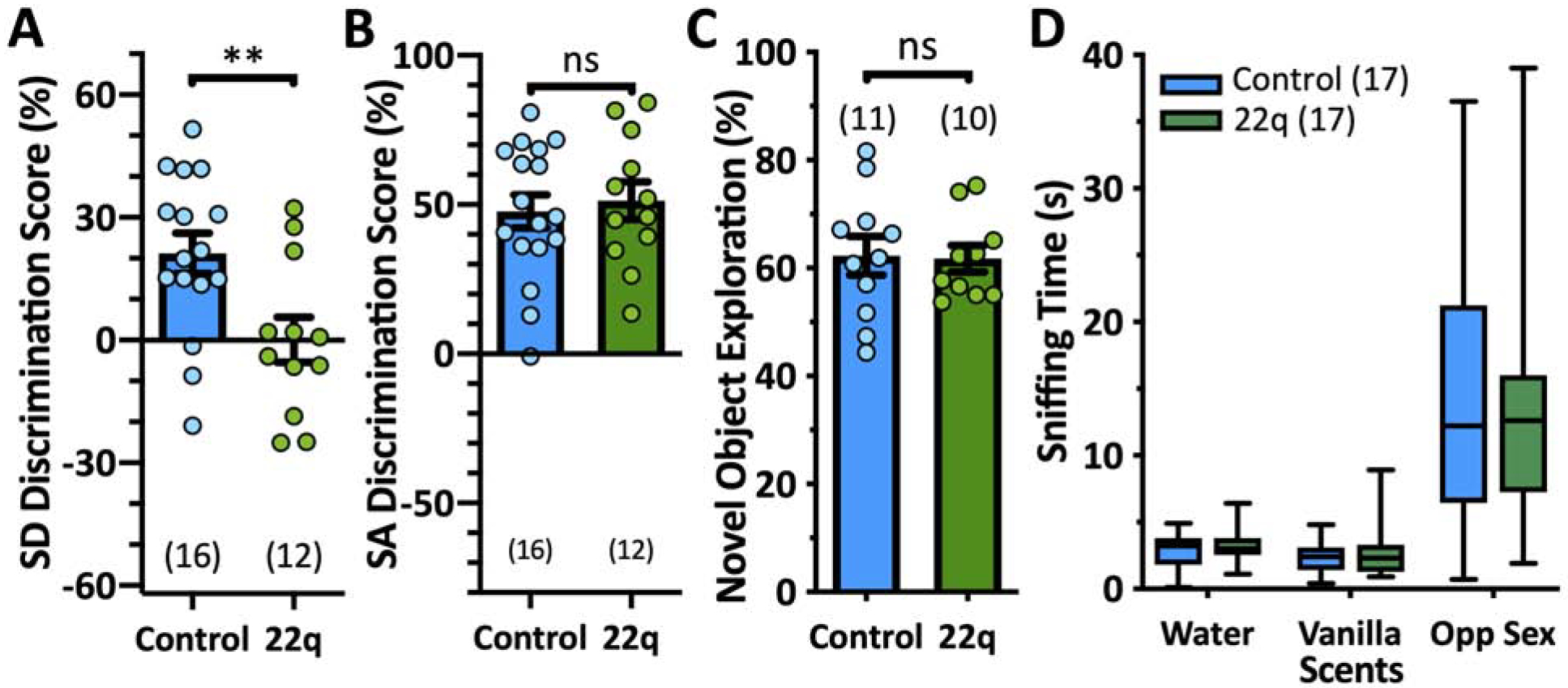
A. Social discrimination results. 22q mice display a deficit (left, t26=2.846, p=0.0085, Student’s t-test). B. Social approach results. 22q mice perform comparably to controls (t26=0.5840, p=0.5643, Student’s t-test), suggesting they do not have sociability issues. C. Novel object recognition results, demonstrating 22q mice do not lack interest in novelty (t19=0.1203, p=0.9055, Student’s t-test). D. 22q mice do not have an olfactory deficit (RM two-way ANOVA results for genotype were as follows: F(1,32)=0.08883, p=0.7676; results for olfactory cue were as follows: F(2,64)=51.74, p=<0.0001; and results for genotype x olfactory cue were as follows: F(2,64)=0.6522, p=0.5243). In A–C, data are represented as mean ± SEM with each circle representing an animal; D is a min-max boxplot (number of animals indicated in parentheses). ns p>0.05, *p<0.05, **p<0.01.
Social approach analysis suggests sociability did not contribute to the discrimination deficit as 22q mice preferred the novel conspecific over the novel object comparably to controls (t26=0.5840, p=0.5643, Student’s t-test; Fig 2B, S3C), a finding consistent with previous reports of normal sociability in 22q mice (34). In both the social approach and discrimination trials, the 22q and control mice explored the stimuli for comparable amounts of time (SA: t26=0.1877, p=0.8525; SD: t26=0.8427, p=0.4074, Student’s t-test; Fig S2), it was merely the distribution of the 22q’s time during the discrimination trial that varied. Furthermore, 22q mice did not lack the ability to recognize nor lack interest in novelty generally, as they preferentially explored a novel object to a familiar one in the novel object recognition task (Fig 2C).
Finally, as olfaction critically influences social behaviors in mice, we confirmed that the 22q mice do not demonstrate an olfactory deficit, particularly to social olfactory cues (RM two-way ANOVA results for genotype were as follows: F(1,32)=0.08883, p=0.7676; results for olfactory cue were as follows: F(2,64)=51.74, p=<0.0001; and results for genotype x olfactory cue were as follows: F(2,64)=0.6522, p=0.5243; Fig 2D). These data collectively suggest that the 22q’s social discrimination deficit could be due to underlying hippocampal circuit dysfunction.
Ventral CA1 is hyperexcitable in 22q mice.
The control mice DREADD manipulations suggest either hyper- or hypoexcitability in the ventral CA1 could generate the 22q-associated behavioral deficit. To assess the circuit directly, we employed in vitro VSD imaging with a CCD camera, which has high spatial and temporal resolution, allowing us to visualize the location and magnitude of synaptic membrane voltage changes in acute slices in real time. We recorded in horizontal slices, using SC stimulation to examine CA1 network recruitment. 22q mice have an increased peak EPSP amplitude compared to controls (t13.96=3.046, p=0.0087, Welch’s t-test; Fig 3A–B). Furthermore, more of the tissue depolarized in response to SC stimulation: we analyzed the number of pixels in which the dye’s fluorescence changed ≥3 standard deviations from baseline over a 250 ms window following stimulation (Fig 3C). This area under the curve calculation demonstrated significantly increased EPSP propagation throughout the CA1 region in 22qs compared to controls (t8.90=3.401, p=0.0080, Welch’s t-test; Fig 3A,C–D). This modest EPSP amplitude increase coupled with the striking EPSP propagation increase may indicate an interneuronopathy, which has been found in multiple hippocampal regions of 22q mouse models (34–36). These data suggest that a hyperexcitable circuit phenotype could be responsible for the social memory behavioral deficit.
Fig 3. VSD reveals ventral CA1 hyperexcitability in 22q mice.

A. Representative micrographs from the CCD camera showing 10 ms bins of the ΔF/F0 (%) response to stimulation in Control (top) and 22q (bottom) slices. B. Percent ΔF/F0 maximal peak response shows a significant increase in 22q tissue (t13.96=3.046, p=0.0034, Welch’s t-test). C. Mean % change in activated pixels (≥3 SD over baseline) vs. time after stimulus plot. D. Area under the curve measures for the data in c quantify the more widely propagating, longer response in 22q tissue (t8.90=3.401, p=0.0080, Welch’s t-test). Data are represented as mean ± SEM. Circles represent a slice per animal (number of slices/animals in parentheses). ns p>0.05, *p<0.05, **p<0.01.
Reducing ventral hippocampal 22q-associated hyperexcitability rescues performance.
The previous DREADD manipulations demonstrated that circuits must be tuned within a permissive window of activity for successful behavioral outcomes to occur. Thus, we explored if this circuit tuning principal could apply to our disease model: could directly imposing constraint on pyramidal cell hyperexcitability be sufficient to restore social discrimination in the 22q mice? We bilaterally co-injected the two inhibitory DREADDs into their ventral hippocampi. This cohort repeated the previously observed social discrimination deficit when treated with saline (−6.7 ± 4.6%; F(3,36)=11.66, p=<0.0001, RM one-way ANOVA; Fig 4). Acutely reducing circuit activity via the muscarinic hM4Di rescued discrimination such that the 22q mice preferentially explored the novel conspecific (23.9 ± 4.1%; pSaline vCNO=0.0001, Dunnett’s correction), and this rescue repeated with KORD recruitment (23.9 ± 5.6%; pSaline vSB=0.0001, Dunnett’s). On the reversal trial, the 22q mice returned to exploring the stimulus-mice indiscriminately (0.3 ± 5.1%; pSaline vReversal=0.5842, Dunnett’s), suggesting the acute circuit retuning was responsible for the behavioral rescue. VSD recordings confirmed that DREADD recruitment successfully reduced circuit excitability in vitro (Fig S5).
Fig 4. Reducing circuit excitability rescues discrimination in 22q mice.

Social discrimination results. Decreasing ventral hippocampal excitability via hM4Di (1 mg/kg CNO) or KORD (1 mg/kg SB) improves performance (F(3, 36)=11.66, p=<0.0001, RM one-way ANOVA with Dunnett’s multiple comparisons correction). Data are represented as mean ± SEM. Each circle represents an animal (number of animals indicated in parentheses). ns p>0.05, * p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
Circuit-based interventions have modular effects.
While promising, these rescue results raised several questions: 1) could we push the circuit modulation to make outputs overly quiescent; 2) are the modulation’s effects localized to the ventral hippocampus; and 3) could this approach apply to other regions and behavioral symptoms? Thus, we bilaterally injected the inhibitory KORD and the inhibitory hM4Di into the ventral and dorsal hippocampi, respectively, in control and 22q mice (Fig 5A, Fig S1).
Fig 5. Modular-specific manipulation rescues distinct symptoms.

A. Schematic of injection sites for the respective DREADD viral vectors. B. Social discrimination results. SB (1 mg/kg) replicates Fig 4 cohort’s results. Robustly recruiting KORDs with 3 mg/kg SB disrupts performance. Dorsal modulation with 1 mg/kg CNO does not affect performance in control or 22q mice (RM one-way ANOVA with Geisser-Greenhouse’s correction and Dunnett’s correction, for Ctl: F(1.64,16.39)=4.740, p=0.0292; for 22q: F(2.11,21.06)=4.627, p=0.0202). C. SOR schematic. D. SOR results. Dorsal hM4Di recruitment (CNO 1 mg/kg) reverses performance while ventral KORD recruitment (SB 1 mg/kg) has no effect (RM one-way ANOVA with Dunnett’s correction, for Ctl: F(2,20)=4.289, p=0.0300, 22q: F(2,20)=6.603, p=0.0071). Data are represented as mean ± SEM. Each circle represents an animal (number of animals indicated in parentheses). ns p>0.05, *p<0.05, **p<0.01.
If the DREADDs were affecting circuit function the way we expected, reducing circuit hyperexcitability to a point would be beneficial, but too much reduction should again prohibit behavioral performance. Therefore, we explored overly recruiting the inhibitory DREADDs with a higher ligand dose. High CNO doses introduce the possibility of off-target clozapine effects (37); we used 3 mg/kg SB, which has not shown off-target effects, to recruit the KORD. 22q animals, in which 1 mg/kg SB repeated the behavioral rescue (25.9 ± 6.7%; RM one-way ANOVA with Geisser-Greenhouse’s correction F(2.11,21.06)=4.627, p=0.0202; pSaline vSB1=0.0132, Dunnett’s correction; Fig 5B), failed to discriminate when treated with the higher SB dose (−5.7 ± 7.1%; pSaline vSB3=0.9736, Dunnett’s). Predictably, the high SB dose also disrupted performance in control mice (1.8 ± 8.2%; RM one-way ANOVA with Geisser-Greenhouse’s correction F(1.64,16.39)=4.740, p=0.0292; Fig 5B). Thus, the intervention was capable of making the circuit overly quiescent, disrupting behavior.
To examine both the DREADDs’ circuit specificity and potential therapeutic effects in other circuits, we made use of the dorsally-expressed hM4Di in the same cohort. Regarding regional specificity, dorsal DREADD recruitment (1 mg/kg CNO) did not affect social discrimination performance in either controls (pSaline vCNO=0.9532, Dunnett’s correction) or 22qs (pSaline vCNO=0.9700, Dunnett’s correction; Fig 5B).
Multiple groups have reported dorsal hippocampal dysregulation in 22q models using varying investigative strategies and have linked this phenotype to hippocampal-dependent cognitive deficits (34–36,38,39). Thus, to examine if chemogenetic circuit modulation could manage other symptoms, we made use of a second eloquent circuit-behavior: spatial object recognition (SOR; Fig 5C), a non-aversive dorsal hippocampal-dependent task (30,31,40). As demonstrated by the control group when treated with saline, mice will preferentially explore the object that was moved during the SOR test trial compared to the objects that were not (32.7 ± 6.6%; RM one-way ANOVA F(2,18)=4.289, p=0.0300; Fig 5D). Recruiting the dorsal hM4Di (1 mg/kg CNO) disrupted SOR performance (1.0 ± 10.3%; pSaline vCNO=0.0225, Dunnett’s correction), but recruiting the ventral KORD (1 mg/kg SB) had no significant effect (25.7 ± 10.1%; pSaline vSB=0.7632, Dunnett’s). Meanwhile, the 22q cohort demonstrated a marked deficit in SOR performance (−7.8 ± 8.5%; RM one-way ANOVA F(2,18)=6.603, p=0.0071; Fig 5D), which is consistent with previous reports of dorsal hippocampal dysfunction, particularly hyperexcitability in CA1, in similar 22q models (35,36,38,39). Recruiting the dorsal hM4Di rescued their ability to discriminate (26.0 ± 9.4%; pSaline vCNO=0.0105, Dunnett’s correction), while ventral KORD recruitment had no effect (−7.8 ± 10.5%; pSaline vSB=>0.9999, Dunnett’s). These results suggest that chemogenetic circuit-based interventions that target the circuit’s excitatory signaling output are sufficient to restore circuit function in multiple regions, but also in a regionally-specific manner.
Discussion
Due to the underlying polygenic haploinsufficiency, 22qDS is a multisystem disorder with highly variable and complex phenotypic expression that can vary over the course of development (7,41). Most 22qDS patients (60–80%) cope with at least one lifelong psychiatric disorder, and many patients display comorbid psychopathologies (41). Unfortunately, traditional pharmacotherapy remains problematic for managing 22qDS patients’ symptoms, as this patient population is more vulnerable to detrimental side effects (11,12). In this set of studies, we sought to explore an alternative therapeutic approach.
The summed consequences of the 22qDS genetic disruption result in neuronal circuit dysregulation, observed as changes in net circuit excitability. As circuit outputs determine behavioural outcomes, circuit excitability provides a potential downstream, mechanistically-independent therapeutic target to manage symptoms. Thus, we made use of eloquent hippocampal circuits to explore if circuit-based interventions targeting principal cell recruitment, but not the underlying mechanisms generating their aberrant activity, could provide therapeutic results in a 22qDS murine model.
Using chemogenetics in control mice, we were able to illustrate a common concept: normally circuits are tuned optimally to support appropriate behavioral outcomes. Mimicking 22q-related ventral hippocampal hyperexcitability in pyramidal cells - without the underlying mechanistic changes that cause the dysregulation in the disease - was sufficient to compromise social discrimination in control mice; reducing activity via inhibitory DREADDs also compromised performance (Fig 1B). These bidirectional results illustrate a bell curve relationship between aggregate circuit activity and behavioral outcomes: appropriate recruitment supports proper informational coding while too much or too little activity masks or degrades the informational signals, respectively.
Using the same circuit-modulatory approach, we sought to reduce 22q-associated ventral hippocampal hyperexcitability (Fig 3) via inhibitory DREADDs to examine if the intervention could ameliorate their social discrimination deficit (Fig 2A). Remarkably, this acute circuit-modulation rescued social memory performance with both inhibitory DREADDs (Fig 4), suggesting that the circuit can regain coding capabilities despite disease-related changes in underlying mechanisms. Furthermore, while moderate KORD recruitment produced salutary effects, overly recruiting the KORD again compromised 22q discrimination performance (Fig 5B). As with the control mice, we were able to illustrate a circuit recruitment/behavioral performance bell curve relationship, with the disease pushing the circuit to a hyperexcitable basal state, moderate inhibitory DREADD recruitment restoring an appropriate level of circuit activity, and more robust DREADD recruitment producing an overly quiescent circuit unable to support behavioral outcomes. These results were not confirmed with in vivo recordings; however, the extensive literature demonstrating DREADDs’ effects on neural circuitry, coupled with our data demonstrating that the constructs can dictate behavioral outcomes in both health and disease, strongly support these conclusions.
We applied the same approach to the dorsal hippocampus to examine if this type of circuit-based intervention can effect improvement in other forms of cognition, in this case spatial memory (Fig 5C). Dorsal hippocampal dysregulation in areas CA2 (34) and CA1 (35,36,38,39) have been characterized previously with multiple lines of evidence showing disease-associated hyperexcitability. Thus, we recruited inhibitory DREADDs in CA1 pyramidal cells, rescuing spatial discrimination performance in the 22q mice (Fig 5D). Intriguingly, DREADD-mediated behavioral effects acted in a modular manner, remaining specific to the region in which the DREADDs were expressed. Dorsally-expressed DREADD recruitment did not affect ventral-dependent behaviors and vice-versa (Fig 5B,D). These behavioral rescue data coupled with our and previous data illustrating hippocampal hyperexcitability suggest an interesting perspective: though the circuit dysregulation extends across the septotemporal axis, symptom management requires more modular intervention. The specificity of the circuit modulation coupled with its success in rescuing function across multiple regions suggest a promising potential to regulate specific circuits and symptoms on an as-needed basis. Further, these data complement previous work that genetically targeted PV interneurons to recalibrate circuit activity and alleviate symptoms (35,36), demonstrating how focal chemogenetic interventions may rescue multiple disease-related alterations via diverse means. Such individualized manipulations could profoundly benefit patients with complex disease states such as 22qDS, where high variability across individuals demands a more nuanced, personalized therapeutic approach.
In addition to its role as a useful investigative tool, chemogenetics is an attractive, potentially translational therapeutic technology that could circumvent many of the complications 22qDS patients face with more traditional pharmacotherapy. Directed and deliberate DREADD expression coupled with the ligand’s specificity for the designer receptor minimize potential side-effects. Additionally, these synthetic ligands possess minimal endogenous biological activity, but are highly bioavailable and can cross the blood brain barrier. CNO back-metabolizes to clozapine, making it a less selective ligand than originally expected (37); however, multiple groups continue to engineer and to test alternative ligands for the muscarinic DREADDs, to date validating perlapine, compound 21 (42,43), JHU37152, and JHU37160 (44). Furthermore, ongoing efforts to identify neuronal cell type selective promoters suggest that future chemogenetic constructs may eventually select not just excitatory versus inhibitory neural populations (45), but specific subpopulations with specific functional roles within a circuit, based on the neurons’ connectivity or spiking characteristics. Chemogenetics has the additional benefit of not needing permanent external machinery, as is the case in optogenetics and deep brain stimulation. Thus, this technology has the flexibility to act in a neuronal subclass-selective manner and in multiple sites across the brain, potentially managing several different circuits and symptoms, and to affect larger tissue regions in a more consistent manner. Finally, chemogenetics could be employed in conjunction with other neuromodulatory techniques, which have also proven effective in manipulating circuit-related psychiatric symptoms (36,46–51), maximizing individualized and varied intervention strategies for patients.
Many complex mechanistic alterations contribute to disease-related circuit disruptions and their corresponding behavioral symptoms. This study demonstrates that the aggregate sum of these disruptions results in net changes in circuit excitability with the disease-altered circuit moving away from a permissive activity point, which normally supports the successful coding of behavioral outputs. Furthermore, our data highlight that restoring the circuit’s principal cell outputs to an appropriate level is a viable therapeutic option, restoring circuit and behavioral functionality irrespective of the mechanisms underlying the disease-related dysfunction. These data demonstrate that restoring circuit tuning with circuit-based therapies in a specific, modular manner can provide promising therapeutic results.
Supplementary Material
Key Resources Table
| Resource Type | Specific Reagent or Resource | Source or Reference | Identifiers | Additional Information |
|---|---|---|---|---|
| Add additional rows as needed for each resource type | Include species and sex when applicable. | Include name of manufacturer, company, repository, individual, or research lab. Include PMID or DOI for references; use “this paper” if new. | Include catalog numbers, stock numbers, database IDs or accession numbers, and/or RRIDs. RRIDs are highly encouraged; search for RRIDs at https://scicrunch.org/resources. | Include any additional information or notes if necessary. |
| Bacterial or Viral Strain | AAV5-CamKIIα-GFP | University of Pennsylvania Vector Core | N/A | |
| Bacterial or Viral Strain | AAV5-CamKIIα-hM3Dq-mcherry | Addgene | 50476 | |
| Bacterial or Viral Strain | AAV5-CamKIIα-hM4Di-mcherry | Addgene | 50477 | |
| Recombinant DNA reagent | AAV5-CamKIIα-HA-KORD-IRES-mCitrine plasmid | Addgene | 65418 | |
| Chemical Compound or Drug | Clozapine n-oxide | Tocris | 4936 | |
| Chemical Compound or Drug | Salvinorin B | Cayman Chemical | 23582 | |
| Chemical Compound or Drug | di-3-ANEPPDHQ | Invitrogen | D36801 | |
| Organism/Strain | Mouse (male & female): ko/wt C57BL/6-Del(16Dgcr2-Hira)1Tac | Taconic Biosciences | 11026-F/M | |
| Organism/Strain | Mouse (male & female): wt/wt C57BL/6-Del(16Dgcr2-Hira)1Tac | Taconic Biosciences | 11026-F/M | |
| Organism/Strain | Mouse (male & female): C57Bl/6 | Taconic Biosciences | B6-F/M MPF | |
| Software; Algorithm | Prism 8 | Graphpad | RRID:SCR_000306 | |
| Software; Algorithm | R | The R Foundation | RRID:SCR_001905 | |
| Software; Algorithm | ANYmaze | Stoelting | 60000 | |
| Software; Algorithm | Igor Pro 6.2 | Wavemetrics | RRID:SCR_000325 | |
| Other | 530 nm Green LED | Thorlabs | M530L3-C2 | |
| Other | 80 × 80 CCD camera | RedShirt Imaging | NeuroCCD |
Acknowledgments
This research was supported by NIH grants from the National Institute for Mental Health (T32MH017168 to J.B.K., T32MH019112 to R.G.P., R01MH110185 to S.A.A.) and the Eunice Kennedy Shriver National Institute of Child Health and Human Development to the Intellectual and Developmental Disabilities Research Center at CHOP/PENN (1U54HD086984-01), and a Foerderer pilot grant from CHOP. We thank Alicia White and the Neurobehavior Testing Core at Penn and the IDDRC at CHOP/Penn for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors report no biomedical financial interests or potential conflicts of interest.
References
- 1.Van L, Boot E, Bassett AS (2017): Update on the 22q11.2 deletion syndrome and its relevance to schizophrenia. Curr Opin Psychiatry 30: 191–196. [DOI] [PubMed] [Google Scholar]
- 2.Green T, Gothelf D, Glaser B, Debbane M, Frisch A, Kotler M, et al. (2009): Psychiatric Disorders and Intellectual Functioning Throughout Development in Velocardiofacial (22q11.2 Deletion) Syndrome. J Am Acad Child Adolesc Psychiatry 48: 1060–1068. [DOI] [PubMed] [Google Scholar]
- 3.Gothelf D, Presburger G, Zohar AH, Burg M, Nahmani A, Frydman M, et al. (2004): Obsessive-compulsive disorder in patients with velocardiofacial (22q11 deletion) syndrome. Am J Med Genet 126B: 99–105. [DOI] [PubMed] [Google Scholar]
- 4.Schneider M, Schaer M, Mutlu AK, Menghetti S, Glaser B, Debbané M, Eliez S (2014): Clinical and cognitive risk factors for psychotic symptoms in 22q11.2 deletion syndrome: A transversal and longitudinal approach. Eur Child Adolesc Psychiatry 23: 425–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Niklasson L, Rasmussen P, Óskarsdóttir S, Gillberg C (2009): Autism, ADHD, mental retardation and behavior problems in 100 individuals with 22q11 deletion syndrome. Res Dev Disabil 30: 763–773. [DOI] [PubMed] [Google Scholar]
- 6.Gur RE, Yi JJ, McDonald-McGinn DM, Tang SX, Calkins ME, Whinna D, et al. (2014): Neurocognitive development in 22q11.2 deletion syndrome: comparison with youth having developmental delay and medical comorbidities. Mol Psychiatry 19: 1205–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Den Heuvel E, Jonkers E, Rombouts E, Manders E, Zink I, Swillen A (2018): Exploratory study on cognitive abilities and social responsiveness in children with 22q11.2 deletion syndrome (22q11DS) and children with idiopathic intellectual disability (IID). Res Dev Disabil 81: 89–102. [DOI] [PubMed] [Google Scholar]
- 8.Shashi V, Veerapandiyan A, Schoch K, Kwapil T, Keshavan M, Ip E, Hooper S (2012): Social skills and associated psychopathology in children with chromosome 22q11.2 deletion syndrome: Implications for interventions. J Intellect Disabil Res 56: 865–878. [DOI] [PubMed] [Google Scholar]
- 9.Campbell LE, McCabe KL, Melville JL, Strutt PA, Schall U (2015): Social cognition dysfunction in adolescents with 22q11.2 deletion syndrome (velo-cardio-facial syndrome): relationship with executive functioning and social competence/functioning. J Intellect Disabil Res 59: 845–859. [DOI] [PubMed] [Google Scholar]
- 10.Lattanzi GM, Buzzanca A, Frascarelli M, Di Fabio F (2018): Genetic and clinical features of social cognition in 22q11.2 deletion syndrome. J Neurosci Res 96: 1631–1640. [DOI] [PubMed] [Google Scholar]
- 11.Mosheva M, Korotkin L, Gur RE, Weizman A, Gothelf D (2019): Effectiveness and side effects of psychopharmacotherapy in individuals with 22q11.2 deletion syndrome with comorbid psychiatric disorders: a systematic review. Eur Child Adolesc Psychiatry 1: 3. [DOI] [PubMed] [Google Scholar]
- 12.Eaton CB, Thomas RH, Hamandi K, Payne GC, Kerr MP, Linden DEJ, et al. (2019): Epilepsy and seizures in young people with 22q11.2 deletion syndrome: Prevalence and links with other neurodevelopmental disorders. Epilepsia 60: 818–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kates WR, Miller AM, Abdulsabur N, Antshel KM, Conchelos J, Fremont W, Roizen N (2006): Temporal Lobe Anatomy and Psychiatric Symptoms in Velocardiofacial Syndrome (22q11.2 Deletion Syndrome). J Am Acad Child Adolesc Psychiatry 45: 587–595. [DOI] [PubMed] [Google Scholar]
- 14.Flahault A, Schaer M, Ottet M-C, Debbané M, Eliez S (2012): Hippocampal volume reduction in chromosome 22q11.2 deletion syndrome (22q11.2DS): A longitudinal study of morphometry and symptomatology. Psychiatry Res Neuroimaging 203: 1–5. [DOI] [PubMed] [Google Scholar]
- 15.Scott JA, Goodrich-Hunsaker N, Kalish K, Lee A, Hunsaker MR, Schumann CM, et al. (2016): The hippocampi of children with chromosome 22q11.2 deletion syndrome have localized anterior alterations that predict severity of anxiety. J Psychiatry Neurosci 41: 203–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mancini V, Sandini C, Padula MC, Zöller D, Schneider M, Schaer M, Eliez S (2019): Positive psychotic symptoms are associated with divergent developmental trajectories of hippocampal volume during late adolescence in patients with 22q11DS. Mol Psychiatry 1. [DOI] [PubMed] [Google Scholar]
- 17.Schleifer C, Lin A, Kushan L, Ji JL, Yang G, Bearden CE, Anticevic A (2019): Dissociable Disruptions in Thalamic and Hippocampal Resting-State Functional Connectivity in Youth with 22q11.2 Deletions. J Neurosci 39: 1301–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shenton ME, Dickey CC, Frumin M, McCarley RW (2001): A review of MRI findings in schizophrenia. Schizophr Res 49: 1–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steen RG, Mull C, Mcclure R, Hamer RM, Lieberman JA (2006): Brain volume in first-episode schizophrenia. Br J Psychiatry 188: 510–518. [DOI] [PubMed] [Google Scholar]
- 20.Samudra N, Ivleva EI, Hubbard NA, Rypma B, Sweeney JA, Clementz BA, et al. (2015): Alterations in hippocampal connectivity across the psychosis dimension. Psychiatry Res Neuroimaging 233: 148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McHugo M, Talati P, Armstrong K, Vandekar SN, Blackford JU, Woodward ND, Heckers S (2019): Hyperactivity and reduced activation of anterior hippocampus in early psychosis. Am J Psychiatry 176: 1030–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Talati P, Rane S, Kose S, Blackford JU, Gore J, Donahue MJ, Heckers S (2014): Increased hippocampal CA1 cerebral blood volume in schizophrenia. NeuroImage Clin 5: 359–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rubin RD, Watson PD, Duff MC, Cohen NJ (2014): The role of the hippocampus in flexible cognition and social behavior. Front Hum Neurosci 8: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Montagrin A, Saiote C, Schiller D (2018): The social hippocampus. Hippocampus 28: 672–679. [DOI] [PubMed] [Google Scholar]
- 25.Meira T, Leroy F, Buss EW, Oliva A, Park J, Siegelbaum SA (2018): A hippocampal circuit linking dorsal CA2 to ventral CA1 critical for social memory dynamics. Nat Commun 9: 4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chiang MC, Huang AJY, Wintzer ME, Ohshima T, McHugh TJ (2018): A role for CA3 in social recognition memory. Behav Brain Res 354: 22–30. [DOI] [PubMed] [Google Scholar]
- 27.Okuyama T, Kitamura T, Roy DS, Itohara S, Tonegawa S (2016): Ventral CA1 neurons store social memory. Science 353: 1536–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Phillips ML, Robinson HA, Pozzo-Miller L (2019): Ventral hippocampal projections to the medial prefrontal cortex regulate social memory. Elife 8: 1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Didriksen M, Fejgin K, Nilsson SRO, Birknow MR, Grayton HM, Larsen PH, et al. (2017): Persistent gating deficit and increased sensitivity to NMDA receptor antagonism after puberty in a new mouse model of the human 22q11.2 microdeletion syndrome: A study in male mice. J Psychiatry Neurosci 42: 48–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kahn JB, Port RG, Yue C, Takano H, Coulter DA (2019): Circuit-based interventions in the dentate gyrus rescue epilepsy-associated cognitive dysfunction. Brain 142: 2705–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oliveira AMM, Hawk JD, Abel T, Havekes R (2010): Post-training reversible inactivation of the hippocampus enhances novel object recognition memory. Learn Mem 17: 155–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL (2007): Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci 104: 5163–5168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vardy E, Robinson JE, Li C, Olsen RHJ, DiBerto JF, Giguere PM, et al. (2015): A New DREADD Facilitates the Multiplexed Chemogenetic Interrogation of Behavior. Neuron 86: 936–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Piskorowski RA, Nasrallah K, Diamantopoulou A, Mukai J, Hassan SI, Siegelbaum SA, et al. (2016): Age-Dependent Specific Changes in Area CA2 of the Hippocampus and Social Memory Deficit in a Mouse Model of the 22q11.2 Deletion Syndrome. Neuron 89: 163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marissal T, Salazar RF, Bertollini C, Mutel S, De Roo M, Rodriguez I, et al. (2018): Restoring wild type-like network dynamics and behaviour during adulthood in a mouse model of schizophrenia. Nat Neurosci 21: 1412–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mukherjee A, Carvalho F, Eliez S, Caroni P (2019): Long-Lasting Rescue of Network and Cognitive Dysfunction in a Genetic Schizophrenia Model. Cell 178: 1387–1402.e14. [DOI] [PubMed] [Google Scholar]
- 37.Gomez JL, Bonaventura J, Lesniak W, Mathews WB, Sysa-Shah P, Rodriguez LA, et al. (2017): Chemogenetics revealed: DREADD occupancy and activation via converted clozapine. Science 357: 503–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zaremba JD, Diamantopoulou A, Danielson NB, Grosmark AD, Kaifosh PW, Bowler JC, et al. (2017): Impaired hippocampal place cell dynamics in a mouse model of the 22q11.2 deletion. Nat Neurosci 20: 1612–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Earls LR, Bayazitov IT, Fricke RG, Berry RB, Illingworth E, Mittleman G, Zakharenko SS (2010): Dysregulation of Presynaptic Calcium and Synaptic Plasticity in a Mouse Model of 22q11 Deletion Syndrome. 10.1523/JNEUROSCI.1425-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lopez AJ, Kramar E, Matheos DP, White AO, Kwapis J, Vogel-Ciernia A, et al. (2016): Promoter-Specific Effects of DREADD Modulation on Hippocampal Synaptic Plasticity and Memory Formation. J Neurosci 36: 3588–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schneider M, Debbané M, Bassett AS, Chow EWC, Fung WLA, van den Bree MBM, et al. (2014): Psychiatric Disorders From Childhood to Adulthood in 22q11.2 Deletion Syndrome: Results From the International Consortium on Brain and Behavior in 22q11.2 Deletion Syndrome. Am J Psychiatry 171: 627–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen X, Choo H, Huang XP, Yang X, Stone O, Roth BL, Jin J (2015): The first structure-activity relationship studies for designer receptors exclusively activated by designer drugs. ACS Chem Neurosci 6: 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thompson KJ, Khajehali E, Bradley SJ, Navarrete JS, Huang XP, Slocum S, et al. (2018): DREADD Agonist 21 Is an Effective Agonist for Muscarinic-Based DREADDs in Vitro and in Vivo. ACS Pharmacol Transl Sci 1: 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bonaventura J, Eldridge MAG, Hu F, Gomez JL, Sanchez-Soto M, Abramyan AM, et al. (2019): High-potency ligands for DREADD imaging and activation in rodents and monkeys. Nat Commun 10: 4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dimidschstein J, Chen Q, Tremblay R, Rogers SL, Saldi G-A, Guo L, et al. (2016): A viral strategy for targeting and manipulating interneurons across vertebrate species. Nat Neurosci 19: 1743–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wolff AR, Bygrave AM, Sanderson DJ, Boyden ES, Bannerman DM, Kullmann DM, Kätzel D (2018): Optogenetic induction of the schizophrenia-related endophenotype of ventral hippocampal hyperactivity causes rodent correlates of positive and cognitive symptoms. Sci Rep 8: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Donegan JJ, Tyson JA, Branch SY, Beckstead MJ, Anderson SA, Lodge DJ (2017): Stem cell-derived interneuron transplants as a treatment for schizophrenia: preclinical validation in a rodent model. Mol Psychiatry 22: 1492–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Donegan JJ, Boley AM, Yamaguchi J, Toney GM, Lodge DJ (2019): Modulation of extrasynaptic GABAA alpha 5 receptors in the ventral hippocampus normalizes physiological and behavioral deficits in a circuit specific manner. Nat Commun 10: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gilani AI, Chohan MO, Inan M, Schobel SA, Chaudhury NH, Samuel P, et al. (2014): Interneuron precursor transplants in adult hippocampus reverse psychosis-relevant features in a mouse model of hippocampal disinhibition. Proc Natl Acad Sci U S A 111: 7450–7455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Neves GA, Grace AA (2018): α7 Nicotinic receptor-modulating agents reverse the hyperdopaminergic tone in the MAM model of schizophrenia. Neuropsychopharmacology 43: 1712–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ewing SG, Grace AA (2013): Deep brain stimulation of the ventral hippocampus restores deficits in processing of auditory evoked potentials in a rodent developmental disruption model of schizophrenia. Schizophr Res 143: 377–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.