

Abstract
Mitochondrial disorders refer to the complex group of conditions affecting energy metabolism. A number of mitochondrial disorders can lead to the development of diabetes mellitus, and mitochondrial diabetes is thought to account for up to 3% of all diabetes mellitus cases. Depending on the degree of preservation of beta cell secretory capacity and peripheral muscle insulin sensitivity, the phenotype of mitochondrial diabetes may resemble that of type 1 or type 2 diabetes. Additionally, mitochondrial diabetes may rarely present with diabetic ketoacidosis, and can be distinguished from other forms of monogenic diabetes including maturity onset diabetes of the young by the presence of multi-organ involvement, particularly pre-senile sensorineural hearing loss, maternal transmission, and later-onset diagnosis, typically affecting adults over 35 years. Various guidelines on diabetes care do not address this important subset of cases, and this diagnosis is easily missed. Additionally, there is paucity of data on tailored diabetes therapies for mitochondrial diabetes, particularly in the era of novel therapies including glucagon-like peptide-1 receptor agonist and sodium glucose co-transporter-2 inhibitors. Here, we report three patients with mitochondrial diabetes who responded well to the addition of these novel agents and propose a new treatment algorithm for this condition.
Keywords: mitochondrial diabetes, mitochondrial disorders, MELAS, MIDD, Glucagon-like peptide receptor agonists, sodium-glucose cotransporter 2 inhibitors
Introduction
Mitochondrial diseases represent a heterogeneous group of genetic conditions that are caused by pathological variants in mitochondrial or nuclear DNA genes encoding mitochondrial proteins which negatively affect energy metabolism.1 The inheritance is vaired with maternally inherited disease-causing variants seen in the mitochondrial DNA (mtDNA), and nuclear DNA genetic alterations presenting mostly with recessive changes; however, dominant, X-linked and de novo disease-causing variants have also been reported.2 The relationship of diabetes mellitus with disease-causing variants in the mtDNA has been extensively researched and will be the primary focus of this article. Mitochondria are foundational organelles found in all mammalian nucleated cells, and are the main site of adenosine triphosphate (ATP) production, the intracellular molecular energy currency for all living organisms. Mitochondria take various reduced co-factor substrates derived from food (e.g. glucose, faty acids, ketone bodies) and convert them to ATP through the process of oxidative phosphorylation.
Mitochondrial disorders of oxidative phosphorylation are among the most common causes of inborn errors of metabolism, and given that mitochondria are essential to basic cellular processes, disruption from genetic alterations have the potential for pervasive and catastrophic impacts. Common final pathways involved in mitochondrial disorders include; lower ATP production, increased oxidative stress from reactive oxygen species, and uncoupling of the membrane potential,3,4 leading to differing clinical phenotypes. Interestingly, individuals with the same genetic defect may present with differing phenotypes, attesting to the complex expression of these proteins, making clinical diagnosis difficult.5 There has been identification of over one thousand of proteins contributing to mitochondrial function (https://www.broadinstitute.org/scientific-community/science/programs/metabolic-disease-program/publications/mitocarta/mitocarta-in-0), explaining the diverse features of mitochondrial disorders including, but not limited to, sensorineural hearing loss, optic neuropathy, encephalopathy, lactic acidosis, stroke-like episodes, cardiomyopathy, ataxia, cognitive impairment, and myopathy.
The endocrinological manifestations of mitochondrial disorders are widespread and include diabetes, dyslipidemia, hypothalamic-pituitary axis dysregulation including adrenal insufficiency, hyponatremia, hypoparathyroidism, hypothyroidism, growth hormone deficiency, hypogonadism, and hypoadrenalism.6–9 Diabetes is the most common endocrinological manifestation of mitochondrial disorders.10 In patients with mitochondrial diabetes, the key abnormality lies in the reduced amount of oxidative phosphorylation due to defects in the tricarboxylic acid cycle and the electron transport chain (Figure 1). This results in the inefficient and suboptimal glucose-stimulated insulin secretion. The American Diabetes Association (ADA) classifies mitochondrial diabetes under the category of “Other; genetic defects of the beta cell” and is thought to account for up to 3% of all diabetes cases.11,12,13,14 Mitochondrial diabetes should be suspected when there is a maternal inheritance pattern of transmission in association with multiorgan dysfunction as mentioned above, but, as other modes of inheritance are also possible, the lack of maternal inheritance does not exclude the diagnosis.15
Figure 1. Mechanism of impaired insulin secretion in mitochondrial diabetes.

1A: Normal beta cell with normally functioning mitochondria. Glucose from the peripheral blood enters the beta cell through GLUT-2 transporter and is converted to glucose-6-phosphate (G-6-P) through the enzyme glucokinase. G-6-P then undergoes glycolysis to form pyruvate which enters the mitochondria to participate in the Krebs cycle. The electron-rich co-factors such as NADH and FADH2 are crucial components of the electron transport chain (ETC) and the resultant oxidative phosphorylation that occurs in the inner membrane of mitochondria, which then leads to the production of adenosine triphosphate (ATP). With increased blood glucose, more glucose molecules enter the beta cells through GLUT-2 transporter and undergo the above reactions, which results in the production of more ATP. The increased ATP concentrations lead to inhibition of the ATP-gated K+ channels. This leads to reduced efflux of K+, which in turn depolarizes the cell membrane, leading to the increased influx of Ca++ into the cell through voltage-dependent Ca++ channels. The increased intracellular Ca++ stimulates the exocytosis of insulin-containing granules, which leads to what is called as ‘glucose-mediated insulin secretion’. 1B: A beta cell with defective mitochondria. The key abnormality lies in the reduced amount of oxidative phosphorylation due to defects in the Krebs cycle and the ETC. This results in the inefficient and suboptimal glucose-mediated insulin secretion. Courtesy of Dr Sriram Gubbi, NIDDK, NIH.
Mitochondrial diabetes due to disease-causing variants in mtDNA can be distinguished from other forms of monogenic diabetes including maturity onset diabetes of the young (MODY), by the presence of multi-organ involvement, particularly sensorineural hearing loss, maternal transmission (if present), low-to-normal body mass index (BMI), and later-onset presentation typically affecting adults over 35 years.6 Confirmation is made by genotyping DNA and, although disease-causing variants in mtDNA may be detected in DNA isolated from blood leucocytes, a negative correlation exists between age of diabetes onset and mutant load.16 Thus, DNA extracted from urine epithelial cells is the preferred source for analysis as it may increase diagnostic yield.
The most common form of mitochondrial diabetes is found in the MT-TL1 (m.3243A>G) genotype, manifesting clinically as maternally inherited diabetes and deafness (MIDD) or mitochondrial encephalopathy with lactic acidosis and stroke-like episodes (MELAS) (Table 1). Other genotypes predispose to mitochondrial diabetes, including m.12258C>A, m.14709T>C, and POLG (Table 1).
Table 1. List of mitochondrial disorders that predispose to mitochondrial diabetes mellitus.
Adapted from Yeung et al.6
| Mitochondrial disorder | Genotype | Inheritance | Major clinical features | Risk of DM |
|---|---|---|---|---|
| Maternally inherited diabetes and deafness (MIDD) | MT-TL1 (m.3243A>G) | Maternal | Late-onset sensorineural hearing loss preceding diabetes by a few years | ↑↑↑ |
| Mitochondrial encephalopathy with lactic acidosis and stroke-like episodes (MELAS) | MT-TL1 (m.32423A>G) in most patients | Maternal | Stroke-like episodes, seizures, migraine, cognitive decline, lactic acidosis, endocrinopathies | ↑↑ |
| Kearns-Sayre syndrome (KSS) | mtDNA deletion | Usually sporadic | Progressive external ophthalmoplegia, retinitis pigmentosa, cardiomyopathy, heart block, endocrinopathies | ↑ |
| Mitochondrial neurogastrointestinal encephalopathy (MNGIE) | TYMP | Autosomal recessive, maternal | Progressive gastrointestinal dysmotility, ophthalmoplegia, leukoencephalopathy, peripheral neuropathy, endocrinopathies | ↑ |
| Pearson syndrome | mtDNA deletion | Usually sporadic | Sideroblastic anaemia, pancytopenia, exocrine pancreatic dysfunction | ↑ |
| Chronic progressive external ophthalmoplegia with myopathy (CPEO+) | mtDNA deletions or mtDNA mutations: POLG RRM2B | Usually sporadic, rare - maternal, autosomal dominant | Progressive external ophthalmoplegia with skeletal myopathy, endocrinopathies | ↑ |
| Myoclonic epilepsy with ragged-red fibres (MERRF) | MT-TK (m.8344A>G) POLG | Maternal, autosomal recessive | Myoclonus, seizures, cerebellar ataxia, myopathy, endocrinopathies | ↑ |
| Leber hereditary optic neuropathy (LHON) | mtDNA point mutations: m.11778G>A m.3460G>A m.14484T>C | Maternal | Optic neuropathy, endocrinopathies | ↑ |
Highest risk
Moderate risk
Very low risk
As our understanding of mitochondria unfolds, increasing evidence points to mitochondrial dysfunction in type 1 diabetes and type 2 diabetes.17 This review will focus on the diabetes associated with primary mitochondrial disorders. The phenotype of mitochondrial diabetes may share features of both type 1 and type 2 diabetes. This heterogenous presentation is often responsible for the misdiagnosis of mitochondrial diabetes by providers.6 A subset of patients with mitochondrial diabetes progress to complete insulin dependency due beta-cell failure, although they rarely manifest with diabetic ketoacidosis, either as a presenting feature or at later stages of disease. Fortunately, most patients with mitochondrial diabetes do not require insulin therapy at diagnosis, but may progress more rapidly to requiring insulin than patients with type 2 diabetes.
There is paucity of evidence on tailored diabetes therapies for mitochondrial diabetes, particularly in the era of novel therapies including glucagon-like peptide-1 receptor agonists (GLP-1 RA, such as liraglutide and semaglutide) and sodium glucose co-transporter-2 inhibitors (SGLT-2i, such as empagiflozin, dapagliflozin, and canagliflozin), and various guidelines on diabetes care do not address this important and growing topic as existing guidelines published in 2013 predate widespread availability of these novel diabetes therapies.3,18 Treatment of mitochondrial diabetes should be individualized based on phenotype, while incorporating the patient’s needs and preferences. Here, we report three patients with mitochondrial diabetes who responded well to the addition of these novel agents and propose a new treatment algorithm for this condition.
Illustrative cases
Case 1.
A 51-year-old man with MELAS (MT-TL1, m.3243A>G, 60% heteroplasmy in peripheral blood leucocyte DNA), manifesting with stroke-like episodes and diabetes, was referred to the endocrine clinic for medical optimization. He was of normal weight and had not sustained weight loss (BMI 21 kg/m2). He had no known family history of MELAS or diabetes. Prior to his MELAS diagnosis, his diabetes was initially managed with metformin 1 gram orally twice daily but metformin therapy was discontinued when he developed lactic acidosis and stroke like episodes. He was transitioned to empagliflozin 25 mg oral daily, gliclazide 30 mg oral daily, and sitagliptin 100 mg oral daily. His glycemic control was suboptimal on this therapy, with fingerstick glucose measurements ranging from 8–10 mmol/L with HbA1C of 9%. He had no evidence of hypoglycemia, neuropathy, cardiac disease, kidney disease or retinopathy. He was transitioned to semaglutide 1 mg subcutaneous weekly injection as monotherapy. Within 6 months of semaglutide therapy, his HbA1c improved to 6.9% and was associated with 2 kg weight loss and no gastrointestinal symptoms. His morning fasting glucose continues to be slightly elevated in the 7–8 mmol/L range.
Case 2.
A 32-year-old woman with a maternal history of diabetes and bilateral sensorineural hearing loss presented to the endocrine clinic for medical optimization. She was of normal weight and had not sustained weight loss (BMI 22 kg/m2). She was diagnosed with MIDD (MTTL m.3243A>G, 30% heteroplasmy in peripheral blood leucocyte DNA). She was started on metformin 1 gram orally twice daily and gliclazide 40 mg orally twice daily. Her plasma glucose measurements were suboptimal on this therapy, ranging from 7–9 mmol/L with HbA1C of 8%. She had no evidence of hypoglycemia, neuropathy, cardiac disease, kidney disease, or retinopathy. She was transitioned to empagliflozin 25 mg orally daily as monotherapy. Within 6 months of empagliflozin therapy, her HbA1c improved to 7% and was associated with 1 kg weight loss. She tolerated the medication well with no urinogenital infection.
Case 3.
A 72-year-old woman with MELAS (MT-TL1, m.3243A>G, 10% heteroplasmy in peripheral blood leucocyte DNA), diagnosed in her 60s following evaluation of a stroke-like episode, had developed diabetes 5 years before her diagnosis of MELAS. She was initially treated with metformin 1 gram orally twice daily and did not develop lactic acidosis. She was transitioned to glyburide 15 mg oral daily and sitagliptin 50 mg oral daily after the diagnosis of MELAS. Low dose (8 units) glargine U-100 therapy was trialed for a few weeks but was associated with recurrent hypoglycemia. She had cataracts, but no evidence of neuropathy, cardiac disease, kidney disease, or retinopathy. She was referred for evaluation of her suboptimal HbA1c of 9.5%. She noted polyuria and was of normal weight (BMI 22 kg/m2). She was transitioned to empagliflozin 25 mg oral daily, sitagliptin 100 mg oral daily, and glyburide was discontinued. Her HbA1c improved to 8% on current therapy, with no hypoglycemia.
Discussion
Mitochondrial diabetes can mimic the clinical manifestations of type 1 or type 2 diabetes, often confusing providers.19 The presentation is often insidious, and the average age of onset is 37 years.20 For mitochondrial diabetes diagnosis in patients with known mitochondrial disease, the oral glucose tolerance test has been traditionally used to evaluate impaired glucose tolerance. The use of HbA1c for the diagnosis of mitochondrial diabetes has not been validated for this purpose, but as demonstrated in our case series, can be used for diagnosis and/or follow up as long as confounding medical factors such as anemia or inflammation leading to increased red cell turnover, which can affect the HbA1c, are absent.21,22 Cutoff values for oral glucose tolerance testing, or elevated random glucose remain the same for mitochondrial diabetes as other forms of diabetes mellitus.
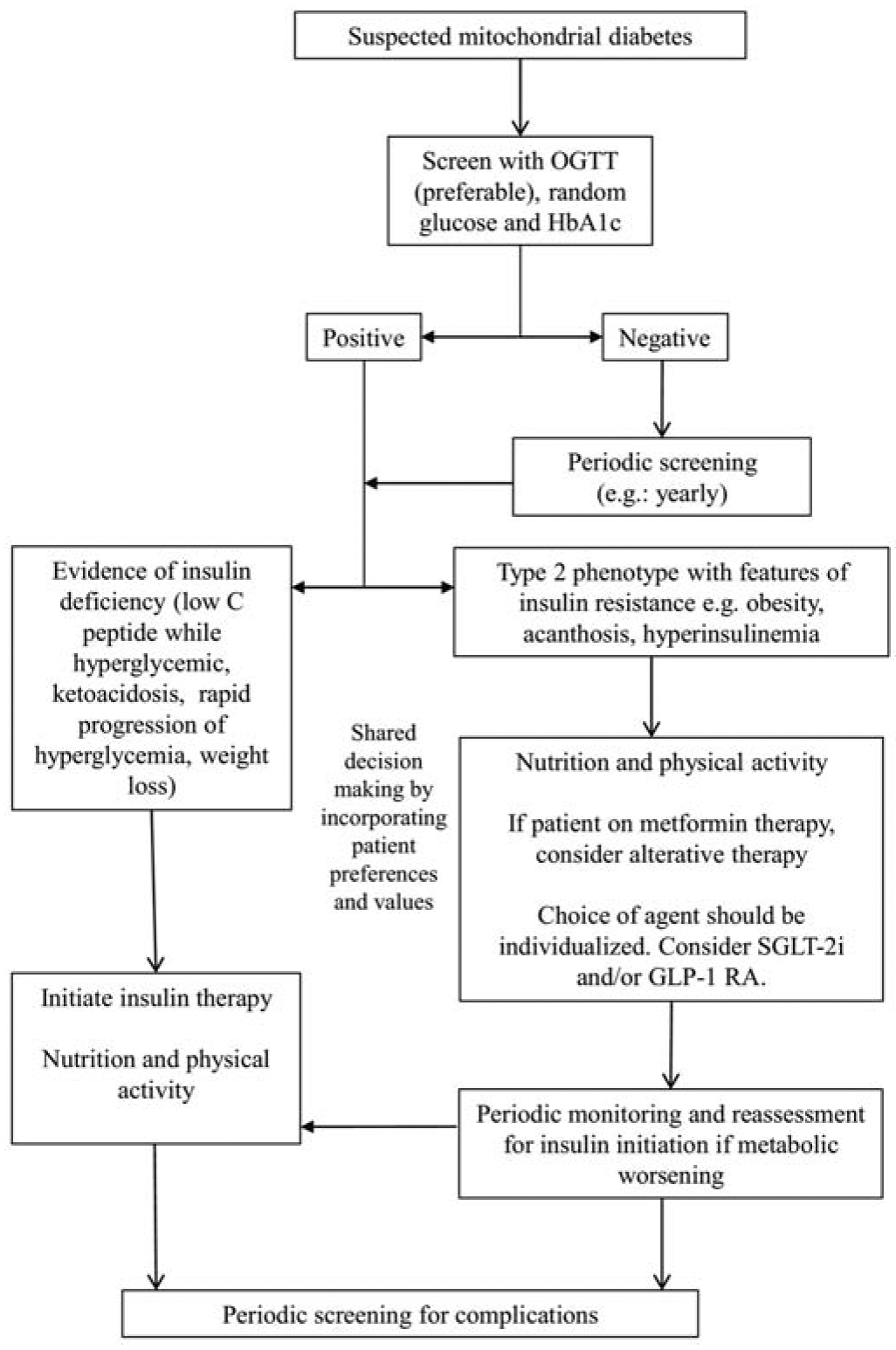
Mechanisms underlying the pathogenesis of hyperglycemia are varied, as differing disease-causing variants in mitochondrial or nuclear genes have shown to have differing effects on both insulin signaling release, as well as peripheral muscle insulin sensitivity.3,23–26 It is important to consider testing for beta cell depletion and concomitant autoimmune diabetes by investigating for evidence of diabetes autoantibodies, ketosis, and/or levels of fasting or stimulated C-peptide in conjunction with concurrent glucose values, especially if patients present with low body mass index, rapid symptomatic progression of weight loss, polydipsia, and/or polyuria. These patients should be started on insulin for immediate metabolic support. For asymptomatic patients who are antibody negative and have existing C-peptide levels, the various type 2 diabetes therapies should be considered (Figure 2).
Figure 2. Treatment algorithm for mitochondrial diabetes. Colour not necessary.
Treatments for mitochondrial disorders
Although there are agents in clinical trials to treat mitochondrial disorders, there are currently very few treatments which have been shown to be effective. Arginine27, citrulline28, and taurine29 have been shown to have some limited efficacy in patients with the common MELAS genotype but the effects of these therapies are not generalizable to patients with other genotypes. Primary mitochondrial disorders are often treated with respiratory chain cofactors (succinate, riboflavin, thiamine, and Coenzyme Q10), antioxidants (Coenzyme Q10, idebenone), and agents that correct the secondary biochemical deficits (creatine, levocarnitine, and folate) but evidence is lacking for most of these therapies.4 The paucity of proven therapies means strong emphasis on supportive care and lifestyle measures is essential to patient care. Aerobic and resistance exercise training have been shown to improve respiratory chain enzyme activity, strength, and mitochondrial biogenesis.30 Patients with mitochondrial diabetes should be encouraged to perform physical activity regularly, according to their individualized capacity. Patients should be well-hydrated and partially fed while doing physical activity, and should avoid exercising while unwell.
Nutrition
While lower carbohydrate and ketogenic diets have been shown to improve glycemic control in type 1 and type 2 diabetes,31 medical nutrition therapy for mitochondrial disorders remains understudied. Ketogenic diets have shown promise in in vitro experiments where ketogenic media shifted the heteroplasmy of diseased mtDNA and selected for the healthy wild-type mtDNA.32 When tested in the transgenic Deletor mouse model, a disease model for progressive late-onset mitochondrial myopathy, the ketogenic diet prevented mitochondrial ultrastructural deterioration in skeletal muscle.33 A pilot study of five patients with mitochondrial myopathy were treated with a high fat, low carbohydrate diet (3–9% of caloric intake) which unfortunately resulted in subacute progressive damage of muscle fibers in all five patients. The authors of the latter study recommended careful monitoring of muscular function and creatine kinase levels in patients with mitochondrial diabetes attempting low carbohydrate diets, and much work remains to be done in this area.34
Pharmacotherapy
There is little published evidence specific to mitochondrial diabetes to guide pharmacologic treatment, with few cases published on mitochondrial diabetes and newer agents,35 so an individualized approach must be taken. Providers are encouraged to engage in shared decision making around which therapy to initiative in a patient with mitochondrial diabetes. Though metformin has been the first line agent for use in type 2 diabetes, treatment of mitochondrial diabetes with metformin therapy is generally avoided due to the high risk of lactic acidosis.36,37 This is likely related to the mechanism of action of metformin, suppressing gluconeogenesis in the liver by inhibiting mitochondrial glycerophosphate dehydrogenase.38 However, the incidence of lactic acidosis with the use of metformin is estimated to be less than 10 in 100 000 patient years which appears to be no higher than the background incidence of lactic acidosis in the general population.39 Lactic acidosis with metformin is most frequently related to the presence of comorbidities such as low cardiac output, anemia, hypoxemia, liver, or kidney injury. Metformin should still be avoided in advanced kidney disease when eGFR is less than 30 mL/min/1.73m2 (Table 2).9 However, in the era of novel therapies that confer cardiovascular protection, metformin should generally be avoided or used with extreme caution in this population.39,40 A proposed recommendation for use of metformin in patients with mitochondrial diabetes is to start metformin at a regular dose when the eGFR is greater than 60 mL/min/1.73m2, half the regular dose when the eGFR is between 30–59 mL/min/1.73m2, and to avoid metformin when the eGFR is below 30 mL/min/1.73m2 (Table 2). Frequent monitoring of lactate level every three to six months is recommended depending on eGFR (Table 2).41
Table 2. Proposed recommendations for use of metformin in patients with mitochondrial diabetes.
Adapted from Yeung et al6.
| eGFR (mL/min per 1.73 m2) | Metformin | Lactate levels |
|---|---|---|
| >60 | Start at regular dose | Every three to six months |
| 59–30 | Start at half the suggested dose, monitor renal function ever three to six months | Every three months |
| <29 | Contraindicated | |
| Risk of acute kidney injury | Cautious use | More frequent monitoring |
Sulfonylureas, meglitinides, and thiazolidinediones were traditionally recommended as priority agents in patients with mitochondrial diabetes that were deemed non-insulin dependent as weight gain is less of an issue in this population; however, a significant risk of hypoglycemia or cardiovascular toxicity is still an issue for these patients.18,39 Therefore, care must be taken in prescribing these agents. Dipeptidyl peptidase-4 inhibitors (DPP4i) are generally considered safe and well tolerated in patients with mitochondrial diabetes.
Given the widespread efficacy of SGLT-2i and GLP-1 RA in type 2 diabetes, in addition to low hypoglycemia risk with use as monotherapy and evidence supporting cardiovascular benefit and decreased progression of chronic kidney disease in type 2 diabetes,42,43 these classes of medication hold potential that is yet to be fully explored in mitochondrial diabetes. Due to the uncontrolled production of reactive oxygen species, mitochondrial dysfunction in the cardiovascular system may lead to hypertension, atherosclerosis, ischemia-reperfusion injury, and cardiomyopathy. Therefore, patients with a mitochondrial disorder are considered high-risk for the development of cardiovascular death. Thus, the decision to treat with a cardioprotective agent, such as GLP-1 RA or SGLT-2i should be considered independently of baseline HbA1c or individualized HbA1c target. To date, there are limited data on their use in patients with mitochondrial diabetes; however, our cases illustrate the potential beneficial metabolic effects for patients.
There is an increasing interest in the mitochondrial contributions to diabetes pathogenesis, and exploration of how SGLT-2i and GLP-1 RAs are modulating these effects. Recent experiments in diabetic rat models showed that the SGLT-2i empagliflozin was able to improve atrial mitochondrial respiratory function and biogenesis while also preventing diabetes-associated atrial electrical remodeling and atrial fibrillation inducibility.44 Dapagliflozin, another SGLT-2i, has also been shown to be superior to insulin in remediating cardiac myocyte mitochondrial dysfunction of membrane potential in rat models of metabolic syndrome.45 There is also evidence to show that empagliflozin remarkably reduces diabetes-associated ultrastructural remodeling of the neurovascular unit and neuroglia in a mouse model, in addition to deleterious changes to mitochondrial structure in the central nervous system.
GLP1 RA also have some intriguing animal data to support their role in mitochondrial-mediated disease; liraglutide was shown to reduce the mitochondrial stress in a rat model of epilepsy subjected to status epilepticus.46 Liraglutide has also been shown to reduce mitochondrial apoptosis in myocardial infarction induced in diabetic rats.47 In an in vitro experiment using retinal ganglion cells originally derived from rats, liraglutide protected mitochondrial biogenesis, decreased mitophagy, and enhanced beneficial mitochondrial function.48 Animals models and pilot data from patients with Freidreich ataxia, an autosomal recessive neurodegenerative disease with high diabetes prevalence, show that exenatide improved mitochondrial function.49 Guidelines on mitochondrial disease suggest avoiding GLP-1 RAs to those with personal or family history of pancreatitis,3 though recent review of GLP-1 RA clinical trial data suggest rates similar to comparator arms with reassuring safety record.50 Given the lack of therapeutic options for the multiorgan failure in mitochondrial disease, these data provide hope that novel pathways ameliorating glucose homeostasis may offer wider systemic benefits to patients, and perhaps should be used more widely in those with mitochondrial disease.
GLP1 RAs have multiple pleiotropic effects, including weight loss, which can help reduce polypharmacy in patients with mitochondrial diabetes,51 as in our cases. However, providers should err on the side of caution in using these therapies for the management of the elderly or frail individuals with mitochondrial diabetes for several reasons. First, GLP-1 RA suppress appetite, leading to weight loss and decreased caloric intake, which may not be desired in certain patients.52 Second, clinically relevant adverse effects, including vomiting, nausea, and diarrhea is prevalent, in up to 50% of users, albeit transient.53–55 These gastrointestinal symptoms may, in theory, mask the gastrointestinal manifestations of a mitochondrial disorder or lead to dehydration and acute kidney injury, an undesired outcome in the general management of mitochondrial disease. Thus, oral hydration may be encouraged in users. Moreover, primary mitochondrial disease patients with a predominant gastrointestinal phenotype, such as mitochondrial neuro-gastrointestinal encephalopathy syndrome (MNGIE, Table 1), should not receive these therapies to avoid worsening or possible unmasking of gastrointestinal symptoms. Third, both GLP-1 RA and SGLT2-i should be avoided in patients with advanced or rapidly progressive kidney disease as they are renally cleared. Fourth, the predictors and pathogenesis of SGLT-2i -associated euglycemic diabetic ketoacidosis remain uncertain, especially in mitochondrial diabetes, so it is prudent to minimize or avoid SGLT-2i use in patients with insulin deficiency, previous diabetic ketoacidosis, established microvascular complications, or those at risk of volume depletion (eg. prescribed diuretics)56–58.
In special populations without robust safety data, more conservative treatment is recommended. The gold standard of care in pregnant females with mitochondrial diabetes remains nutrition, physical activity, and insulin therapy if pharmacotherapy is needed. Insulin therapy should also be used for the management of acute inpatients with mitochondrial diabetes.
Conclusion
Mitochondrial disorders refer to the complex group of conditions affecting energy metabolism, affecting multiple endocrine organs including the pancreas, manifesting with diabetes mellitus. Various guidelines on diabetes care do not address this important subset of cases, and this diagnosis is easily missed. There is paucity of data on tailored diabetes therapies for mitochondrial diabetes, particularly in the era of novel therapies including GLP-1 RA and SGLT-2i. Providers are encouraged to prioritize these therapies in their patients with mitochondrial disorders due to their favorable cardiovascular and renal profiles.
Acknowledgements:
This research was funded in part by the intramural program of the NIH/NIDDK. We thank our patients for allowing us to share their cases.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests: Dr. Yeung reports personal fees from Merck, personal fees from Sanofi, personal fees from Novo Nordisk, grants from Astra Zeneca, outside the submitted work .Dr. Sirrs has participated in research projects funded by Sanofi-Genzyme, Takeda, Idorsia, and Amicus. She has received travel support to attend meetings from Sanofi-Genzyme, Takeda and Amicus. She has not received any funding to support this project. She does not have intellectual property rights related to this project. Dr. Tarnopolsky reports grants and personal fees from Sanofi-Genzyme, other from CEO of Exerkine Corporation, outside the submitted work; In addition, Dr. Tarnopolsky has a patent IL-15 and mitochondrial biogenesis (Canada and USA) issued. All other authors declare no conflict of interest.
References
- 1.McFarland R, Taylor RW, Turnbull DM. A neurological perspective on mitochondrial disease. Lancet Neurol. 2010;9(8):829–840. [DOI] [PubMed] [Google Scholar]
- 2.Gorman GS, Schaefer AM, Ng Y, et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann Neurol. 2015;77(5):753–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schaefer A, McFarland R, Hart Y, Turnbull D. Newcastle Mitochondrial Disease Guidelines. Newcastle Mitochondrial Centre, NHS Specialised Services for Rare Mitochondrial Disorders of Adults and Children. 2010. [Google Scholar]
- 4.Parikh S, Goldstein A, Koenig MK, et al. Diagnosis and management of mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med. 2015;17(9):689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lightowlers RN, Taylor RW, Turnbull DM. Mutations causing mitochondrial disease: What is new and what challenges remain? Science. 2015;349(6255):1494–1499. [DOI] [PubMed] [Google Scholar]
- 6.Yeung RO, Hannah-Shmouni F, Niederhoffer K, Walker MA. Not quite type 1 or type 2, what now? Review of monogenic, mitochondrial, and syndromic diabetes. Rev Endocr Metab Disord. 2018;19(1):35–52. [DOI] [PubMed] [Google Scholar]
- 7.Hannah-Shmouni F, Stratakis CA. An overview of inborn errors of metabolism manifesting with primary adrenal insufficiency. Rev Endocr Metab Disord. 2018;19(1):53–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hannah-Shmouni F, Sirrs S, Mezei MM, Waters PJ, Mattman A. Increased Prevalence of Hypertension in Young Adults with High Heteroplasmy Levels of the MELAS m.3243A>G Mutation. JIMD reports. 2014;12:17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chow J, Rahman J, Achermann JC, Dattani MT, Rahman S. Mitochondrial disease and endocrine dysfunction. Nat Rev Endocrinol. 2017;13(2):92–104. [DOI] [PubMed] [Google Scholar]
- 10.Manwaring N, Jones MM, Wang JJ, et al. Population prevalence of the MELAS A3243G mutation. Mitochondrion. 2007;7(3):230–233. [DOI] [PubMed] [Google Scholar]
- 11.Ohkubo K, Yamano A, Nagashima M, et al. Mitochondrial gene mutations in the tRNA(Leu(UUR)) region and diabetes: prevalence and clinical phenotypes in Japan. Clin Chem. 2001;47(9):1641–1648. [PubMed] [Google Scholar]
- 12.Walker M, Taylor RW, Turnbull DM. Mitochondrial diabetes. Diabet Med. 2005;22 Suppl 4:18–20. [DOI] [PubMed] [Google Scholar]
- 13.Standards of Medical Care in Diabetes—2017: Summary of Revisions. Diabetes Care. 2017;40(Supplement 1):S4–S5. [DOI] [PubMed] [Google Scholar]
- 14.Diagnosis and classification of diabetes mellitus. Diabetes Care. 2009;32 Suppl 1:S62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choo-Kang AT, Lynn S, Taylor GA, et al. Defining the importance of mitochondrial gene defects in maternally inherited diabetes by sequencing the entire mitochondrial genome. Diabetes. 2002;51(7):2317–2320. [DOI] [PubMed] [Google Scholar]
- 16.Whittaker RG, Schaefer AM, McFarland R, Taylor RW, Walker M, Turnbull DM. Prevalence and progression of diabetes in mitochondrial disease. Diabetologia. 2007;50(10):2085–2089. [DOI] [PubMed] [Google Scholar]
- 17.Karaa A, Goldstein A. The spectrum of clinical presentation, diagnosis, and management of mitochondrial forms of diabetes. Pediatr Diabetes. 2015;16(1):1–9. [DOI] [PubMed] [Google Scholar]
- 18.Schaefer AM, Walker M, Turnbull DM, Taylor RW. Endocrine disorders in mitochondrial disease. Mol Cell Endocrinol. 2013;379(1–2):2–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maassen JA, ‘t Hart LM, van Essen E, et al. Mitochondrial Diabetes. Molecular Mechanisms and Clinical Presentation. 2004;53(suppl 1):S103–S109. [DOI] [PubMed] [Google Scholar]
- 20.Karaa A, Goldstein A. The spectrum of clinical presentation, diagnosis, and management of mitochondrial forms of diabetes. Pediatr Diabetes. 2015;16(1):1–9. [DOI] [PubMed] [Google Scholar]
- 21.Frederiksen AL, Jeppesen TD, Vissing J, et al. High Prevalence of Impaired Glucose Homeostasis and Myopathy in Asymptomatic and Oligosymptomatic 3243A>G Mitochondrial DNA Mutation-Positive Subjects. The Journal of Clinical Endocrinology & Metabolism. 2009;94(8):2872–2879. [DOI] [PubMed] [Google Scholar]
- 22.Bonora E, Tuomilehto J. The pros and cons of diagnosing diabetes with A1C. Diabetes Care. 2011;34 Suppl 2:S184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maechler P, Wollheim CB. Mitochondrial signals in glucose-stimulated insulin secretion in the beta cell. The Journal of physiology. 2000;529(1):49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nita II, Hershfinkel M, Kantor C, Rutter GA, Lewis EC, Sekler I. Pancreatic β-cell Na+ channels control global Ca2+ signaling and oxidative metabolism by inducing Na+ and Ca2+ responses that are propagated into mitochondria. The FASEB Journal. 2014;28(8):3301–3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes. 2002;51(10):2944–2950. [DOI] [PubMed] [Google Scholar]
- 26.Anderson EJ, Lustig ME, Boyle KE, et al. Mitochondrial H 2 O 2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. The Journal of clinical investigation. 2009;119(3):573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koga Y, Akita Y, Nishioka J, et al. L-arginine improves the symptoms of strokelike episodes in MELAS. Neurology. 2005;64(4):710–712. [DOI] [PubMed] [Google Scholar]
- 28.El-Hattab AW, Hsu JW, Emrick LT, et al. Restoration of impaired nitric oxide production in MELAS syndrome with citrulline and arginine supplementation. Mol Genet Metab. 2012;105(4):607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohsawa Y, Hagiwara H, Nishimatsu S-i, et al. Taurine supplementation for prevention of stroke-like episodes in MELAS: a multicentre, open-label, 52-week phase III trial. J Neurol Neurosurg Psychiatry. 2019;90(5):529–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tarnopolsky MA. Exercise as a therapeutic strategy for primary mitochondrial cytopathies. J Child Neurol. 2014;29(9):1225–1234. [DOI] [PubMed] [Google Scholar]
- 31.Kirkpatrick CF, Bolick JP, Kris-Etherton PM, et al. Review of current evidence and clinical recommendations on the effects of low-carbohydrate and very-low-carbohydrate (including ketogenic) diets for the management of body weight and other cardiometabolic risk factors: A scientific statement from the National Lipid Association Nutrition and Lifestyle Task Force. J Clin Lipidol. 2019;13(5):689–711.e681. [DOI] [PubMed] [Google Scholar]
- 32.Santra S, Gilkerson RW, Davidson M, Schon EA. Ketogenic treatment reduces deleted mitochondrial DNAs in cultured human cells. Ann Neurol. 2004;56(5):662–669. [DOI] [PubMed] [Google Scholar]
- 33.Ahola-Erkkila S, Carroll CJ, Peltola-Mjosund K, et al. Ketogenic diet slows down mitochondrial myopathy progression in mice. Hum Mol Genet. 2010;19(10):1974–1984. [DOI] [PubMed] [Google Scholar]
- 34.Ahola S, Auranen M, Isohanni P, et al. Modified Atkins diet induces subacute selective ragged-red-fiber lysis in mitochondrial myopathy patients. EMBO Mol Med. 2016;8(11):1234–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murakami T, Ueba Y, Shinoto Y, et al. Successful Glycemic Control Decreases the Elevated Serum FGF21 Level without Affecting Normal Serum GDF15 Levels in a Patient with Mitochondrial Diabetes. Tohoku J Exp Med. 2016;239(2):89–94. [DOI] [PubMed] [Google Scholar]
- 36.Sharma MA, Lee JYJ, Tam A, et al. A mitochondrial DNA D loop insertion detected almost exclusively in non-replicating tissues with maternal inheritance across three generations. Mitochondrion. 2019;46:298–301. [DOI] [PubMed] [Google Scholar]
- 37.Parikh S, Goldstein A, Karaa A, et al. Patient care standards for primary mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med. 2017;19(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Madiraju AK, Erion DM, Rahimi Y, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014;510(7506):542–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schaefer AM, Walker M, Turnbull DM, Taylor RW. Endocrine disorders in mitochondrial disease. Mol Cell Endocrinol. 2013;379(1):2–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hannah-Shmouni F, Sirrs SM, Mattman A. Metformin Therapy and Lactate Levels in Adult Patients with Melas and Diabetes Mellitus. Paper presented at: ENDOCRINE REVIEWS2014. [Google Scholar]
- 41.Yeung RO, Hannah-Shmouni F, Niederhoffer K, Walker MA. Not quite type 1 or type 2, what now? Review of monogenic, mitochondrial, and syndromic diabetes. Reviews in Endocrine and Metabolic Disorders. 2018;19(1):35–52. [DOI] [PubMed] [Google Scholar]
- 42.Zinman B, Wanner C, Lachin JM, et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N Engl J Med. 2015;373(22):2117–2128. [DOI] [PubMed] [Google Scholar]
- 43.Wanner C, Inzucchi SE, Lachin JM, et al. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N Engl J Med. 2016;375(4):323–334. [DOI] [PubMed] [Google Scholar]
- 44.Shao Q, Meng L, Lee S, et al. Empagliflozin, a sodium glucose co-transporter-2 inhibitor, alleviates atrial remodeling and improves mitochondrial function in high-fat diet/streptozotocin-induced diabetic rats. Cardiovasc Diabetol. 2019;18(1):165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Durak A, Olgar Y, Degirmenci S, Akkus E, Tuncay E, Turan B. A SGLT2 inhibitor dapagliflozin suppresses prolonged ventricular-repolarization through augmentation of mitochondrial function in insulin-resistant metabolic syndrome rats. Cardiovasc Diabetol. 2018;17(1):144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang RF, Xue GF, Holscher C, et al. Post-treatment with the GLP-1 analogue liraglutide alleviate chronic inflammation and mitochondrial stress induced by Status epilepticus. Epilepsy Res. 2018;142:45–52. [DOI] [PubMed] [Google Scholar]
- 47.Qiao H, Ren H, Du H, Zhang M, Xiong X, Lv R. Liraglutide repairs the infarcted heart: The role of the SIRT1/Parkin/mitophagy pathway. Mol Med Rep. 2018;17(3):3722–3734. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48.Ma X, Lin W, Lin Z, et al. Liraglutide alleviates H2O2-induced retinal ganglion cells injury by inhibiting autophagy through mitochondrial pathways. Peptides. 2017;92:1–8. [DOI] [PubMed] [Google Scholar]
- 49.Igoillo-Esteve M, Oliveira AF, Cosentino C, et al. Exenatide induces frataxin expression and improves mitochondrial function in Friedreich ataxia. JCI Insight. 2020;5(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Monami M, Nreu B, Scatena A, et al. Safety issues with glucagon-like peptide-1 receptor agonists (pancreatitis, pancreatic cancer and cholelithiasis): Data from randomized controlled trials. Diabetes Obes Metab. 2017;19(9):1233–1241. [DOI] [PubMed] [Google Scholar]
- 51.Onoviran OF, Li D, Toombs Smith S, Raji MA. Effects of glucagon-like peptide 1 receptor agonists on comorbidities in older patients with diabetes mellitus. Ther Adv Chronic Dis. 2019;10:2040622319862691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.O’Neil PM, Birkenfeld AL, McGowan B, et al. Efficacy and safety of semaglutide compared with liraglutide and placebo for weight loss in patients with obesity: a randomised, double-blind, placebo and active controlled, dose-ranging, phase 2 trial. Lancet. 2018;392(10148):637–649. [DOI] [PubMed] [Google Scholar]
- 53.Sun F, Chai S, Yu K, et al. Gastrointestinal adverse events of glucagon-like peptide-1 receptor agonists in patients with type 2 diabetes: a systematic review and network meta-analysis. Diabetes Technol Ther. 2015;17(1):35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Buse JB, Rosenstock J, Sesti G, et al. Liraglutide once a day versus exenatide twice a day for type 2 diabetes: a 26-week randomised, parallel-group, multinational, open-label trial (LEAD-6). The Lancet. 2009;374(9683):39–47. [DOI] [PubMed] [Google Scholar]
- 55.Bettge K, Kahle M, Abd El Aziz MS, Meier JJ, Nauck MA. Occurrence of nausea, vomiting and diarrhoea reported as adverse events in clinical trials studying glucagon-like peptide-1 receptor agonists: A systematic analysis of published clinical trials. Diabetes, Obesity and Metabolism. 2017;19(3):336–347. [DOI] [PubMed] [Google Scholar]
- 56.Goldenberg RM, Berard LD, Cheng AYY, et al. SGLT2 Inhibitor-associated Diabetic Ketoacidosis: Clinical Review and Recommendations for Prevention and Diagnosis. Clin Ther 2016;38(12):2654–2664.e2651. [DOI] [PubMed] [Google Scholar]
- 57.Ogawa W, Sakaguchi K. Euglycemic diabetic ketoacidosis induced by SGLT2 inhibitors: possible mechanism and contributing factors. Journal of diabetes investigation. 2016;7(2):135–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim YG, Jeon JY, Han SJ, Kim DJ, Lee KW, Kim HJ. Sodium-glucose co-transporter-2 inhibitors and the risk of ketoacidosis in patients with type 2 diabetes mellitus: A nationwide population-based cohort study. Diabetes Obes Metab. 2018;20(8):1852–1858. [DOI] [PubMed] [Google Scholar]