

Abstract
DNA polymerase ζ (Polζ) belongs to the same B-family as high-fidelity replicative polymerases, and yet is specialized for the extension reaction in translesion DNA synthesis (TLS). Despite its importance in TLS, the structure of Polζ is unknown. We present cryo-EM structures of S.cerevisiae Polζ holoenzyme in the act of DNA synthesis (3.1Å) and without DNA (4.1Å). Polζ displays a pentameric ring-like architecture, with catalytic Rev3 and accessory Pol31, Pol32 and two Rev7 subunits forming an uninterrupted daisy chain of protein-protein interactions. We also uncover the features that impose high fidelity during the nucleotide incorporation step and those that accommodate mismatches and lesions during the extension reaction. Collectively, we decrypt the molecular underpinnings of Polζ’s role in TLS and provide a framework for new cancer therapeutics.
Introduction
The survival of all organisms depends critically on the ability to faithfully replicate DNA. However, cellular DNA is susceptible to damage by normal metabolic activities and environmental factors such as UV light, ionizing radiation, and industrial carcinogens that can cause lesions that evade DNA repair and stall the replication machinery. To avoid compromising genomic integrity, both prokaryotes and eukaryotes possess specialized translesion synthesis (TLS) DNA polymerases (Pols) that can replicate through these lesions. Most of the TLS polymerases belong to the Y-family, which includes the single subunit Polη, Polι, Polκ and Rev1 in humans1,2. In contrast, Polζ is a multi-subunit TLS polymerase containing catalytic Rev3 and accessory Rev7, Pol31 and Pol32 subunits. Rev3 belongs to the same B-family as Pol1, Pol2, and Pol3, the catalytic subunits of the high-fidelity eukaryotic replicative polymerases α, ε, and δ, respectively2–4. However, unlike the replicative polymerases, Polζ is specialized for the extension step of lesion bypass, whereby it is recruited to add nucleotides once another TLS polymerase has added a nucleotide opposite the lesion4,5. The ability of Polζ to carry out synthesis downstream of mismatched termini and diverse DNA lesions is important in maintaining genome integrity and preventing cancer6,7. At the same time, human Polζ has emerged as an important determinant for tumor resistance to chemotherapeutic agents in various cancers6,8,9.
The Rev3 sequence differs from that of Pol1, Pol2 and Pol3 in containing a large insert which comprises the Rev7 binding sites (Fig. 1a). Rev7 (also known as MAD2B) increases the activity of Rev310, and a mutation in mouse Rev7 that disrupts its association with Rev3 leads to defects in development and to the accumulation of DNA damage11. Rev7 is a member of the HORMA (Hop1, Rev7 and Mad2) family of proteins12, and has roles outside of Polζ, including association with the spindle assembly checkpoint protein Mad2 during chromosome segregation13. The Pol31–Pol32 sub-complex associates with Polζ via interactions between the Rev3 C-terminal domain (CTD) and Pol3114–16. The Rev3 CTD contains two cysteine-rich metal-binding modules, CysAD and CysBD (Fig. 1a), analogous to the modules at the C-termini of Pol1, Pol2, and Pol3.
Fig. 1.
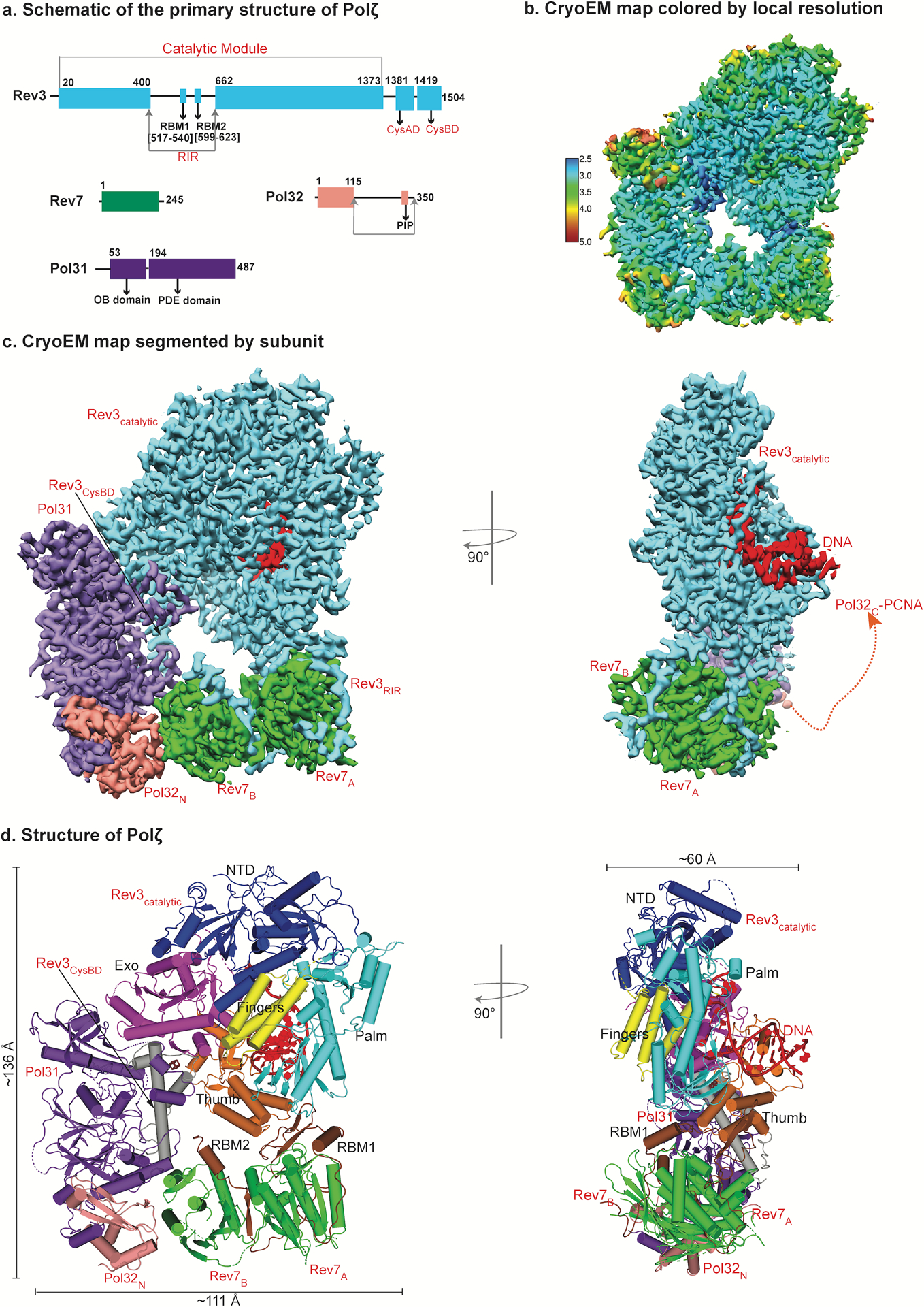
The architecture of DNA bound Polζ holoenzyme. a, Schematic of the primary structure of S. Cerevisiae Polζ subunits. Different colors denote each subunit. b, Near atomic resolution cryo-EM density map of DNA bound Polζ holoenzyme colored by local resolution. c, The three-dimensional reconstruction of Polζ holoenzyme viewed (left) perpendicular and parallel (right) to the DNA axis. A red dashed connector represents disordered Pol32c and the arrowhead marks the putative interaction location with PCNA. d, Cryo-EM structure of DNA bound Polζ colored by domain, and viewed from the same orientations as in (c).
Despite its importance in protecting eukaryotic cells from DNA damage, the structural basis of the ability of Polζ to function as the “master” extender in TLS remains unknown. Available structural information is limited to structures of human Rev7 in complex with short human Rev3 peptides17,18, and to structures of Pol31 and Pol32N and their human counterparts (p50 and p66N)19,20. There is no structural information on the catalytic subunit Rev3, which due to its large size (1,505 amino acids in yeast Rev3 and 3,130 amino acids in human Rev3L)21 and tendency to aggregate has resisted crystallization. A low-resolution (~22 Å) model of apo Polζ based on negatively stained electron microscopy provided some information on the overall shape of the holoenzyme, but provided no information on subunit architecture or protein-protein and protein-DNA interactions22.
We present here cryo-EM structures of the complete yeast Polζ holoenzyme on (3.1 Å resolution) and off (4.1 Å resolution) DNA (Table 1 and Extended Data Figs. 1–3). The structures unveil the mechanism by which Polζ synthesizes DNA and resolve the longstanding conundrum of how it differs from replicative polymerases to perform TLS.
Table 1:
Cryo-EM data collection, refinement and validation statistics
| Polζ-DNA-dCTP (EMD-21115, PDB 6V93) |
Polζ Apo State (EMD-21108, PDB 6V8P) |
|
|---|---|---|
| Data collection and processing | ||
| Magnification | 22500 | 105,000 |
| Voltage (kV) | 300 | 300 |
| Electron exposure (e–/Å2) | 71.63 | 87.62 |
| Defocus range (μm) | −0.5 to −2.5 | −1.5 to −2.5 |
| Pixel size (Å) | 0.537 | 1.1 |
| Symmetry imposed | C1 | C1 |
| Initial particle images (no.) | 205,914 | 1,784,587 |
| Final particle images (no.) | 156,067 | 311,800 |
| Map resolution (Å). | 3.1 | 4.1 |
| FSC threshold | 0.143 | 0.143 |
| Map resolution range (Å) | 2.5 – 5.0 | 4.0.- 6.0 |
| Refinement | ||
| Initial model used | PDB: 3IAY, 3EOJ, 3ABD | PDB: 6V93 |
| Model resolution (Å) | 3.2 | 4.3 |
| FSC threshold | 0.5 | 0.5 |
| Map sharpening B factor (Å2) | −100 | −224 |
| Model composition | ||
| Non-hydrogen atoms | 18,105 | 14,552 |
| Protein residues | 2198 | 2117 |
| DNA/other | 27/3 | 0/1 |
| Water | 220 | 0 |
| B factors (Å2) | ||
| Protein. | 57.06 | 173.97 |
| DNA/other | 76.01/147.63 | −132.86 |
| Water | 58.22 | - |
| R.M.S. deviations | ||
| Bond lengths (Å) | 0.002 | 0.002 |
| Bond angles (°) | 0.519 | 0.483 |
| Validation | ||
| MolProbity score. | 1.76 | 1.60 |
| Clashscore | 5.76 | 3.96 |
| Poor rotamers (%) | 0 | 0 |
| Ramachandran plot | ||
| Favored (%) | 93.00 | 93.64 |
| Allowed (%) | 6.96 | 6.31 |
| Disallowed (%) | 0.05 | 0.05 |
Results
Ring-like architecture of Polζ
The holoenzyme consists of one catalytic Rev3, and two Rev7 (Rev7A and Rev7B), one Pol31, and one Pol32 accessory subunits assembled in a pentameric ring-like architecture with approximate dimensions of 136 Å × 111 Å × 60 Å (Fig. 1b–d). This ring-like architecture is unique among multi-subunit DNA polymerases and resembles an uninterrupted daisy chain of protein-protein interactions, where Pol32 is the only subunit that does not make direct contact with Rev3.
The catalytic subunit Rev3 alone makes all of the contacts to the DNA (Figs. 1c,d and 2 and Supplementary Fig. 1). The duplex portion of the template-primer has a B-DNA like conformation with average helical twist and rise values of 29.7° and 3.11 Å, respectively. For convenience, we refer to positions of nucleotides by TN-PN, where T and P refer to the template and primer strands, respectively, and the subscript N refers to the number of base pairs from the templating position (Fig. 2a). At the replicative end, the templating base G (position T0) establishes Watson-Crick (W-C) base pairing with incoming dCTP (position P0).
Fig. 2.

Structure and cryo-EM density details of Rev3. a, Close-up view of the active site of Rev3 depicting key residues forming the T0-P0 binding site, including ligands and metal ions. Highlighted on the right is the well-resolved density for the T0 and T1 positions (red) and the sequence of the palindromic DNA employed to form the ternary complex. The region of the template-primer duplex enclosed in the box was built into the final model. b, Structure of Rev3 colored by domain. Dark blue, brown, magenta, cyan, yellow, orange and grey denote, respectively, the N-terminal, RIR, exonuclease, palm, fingers, thumb, and C-terminal domains of Rev3. Cryo-EM density for selected regions of Rev3 that highlight the differences in sequence with Pol3 (PDB ID: 3IAY), including residues in the inactive exonuclease domain, residues in close proximity to the CysBD, and the near absence of the β-hairpin region in Rev3. Also shown are close-up views of the coordination around the 4Fe-4S cluster, and the interfaces between the RIR and the palm and thumb domains.
Unexpectedly, two Rev7 subunits bind in a unique head-to-tail arrangement, unexampled in the HORMA family13. Pol31 and Pol32N are held rigidly in a radial arrangement with respect to the DNA duplex. Pol32C (residues 116–350) is disordered but a PCNA interaction (PIP) motif at its C-terminus can potentially extend to the downstream portion of the DNA duplex for putative interactions with PCNA (Fig. 1a,c).
Catalytic Rev3 structure
Rev3 embraces the template-primer with its palm (residues 329–373; 941–1043; 1098–1215), fingers (residues 302–328; 1044–1097), thumb (residues 1216–1372), exonuclease (residues 662–894), and N-terminal (NTD; residues 1–301; 374–400; 895–940) domains (Figs. 1c and 2b and Supplementary Fig. 1). The palm interacts with the replicative end of the template-primer and carries the active site residues (D975 and D1144) (Figs. 2a and 3a). The fingers domain drapes over the nascent G:dCTP base pair in a closed conformation (described below) (Fig. 2). The thumb grips the duplex portion of the primer-template, making contacts through the minor groove. The inactive exonuclease domain lies on the opposite side of the DNA as the thumb, extending towards the major groove (Fig. 2b). The NTD bridges the exonuclease and fingers domains and makes numerous contacts with the unpaired portion of the template strand (Figs. 1d and 2b).
Fig. 3.
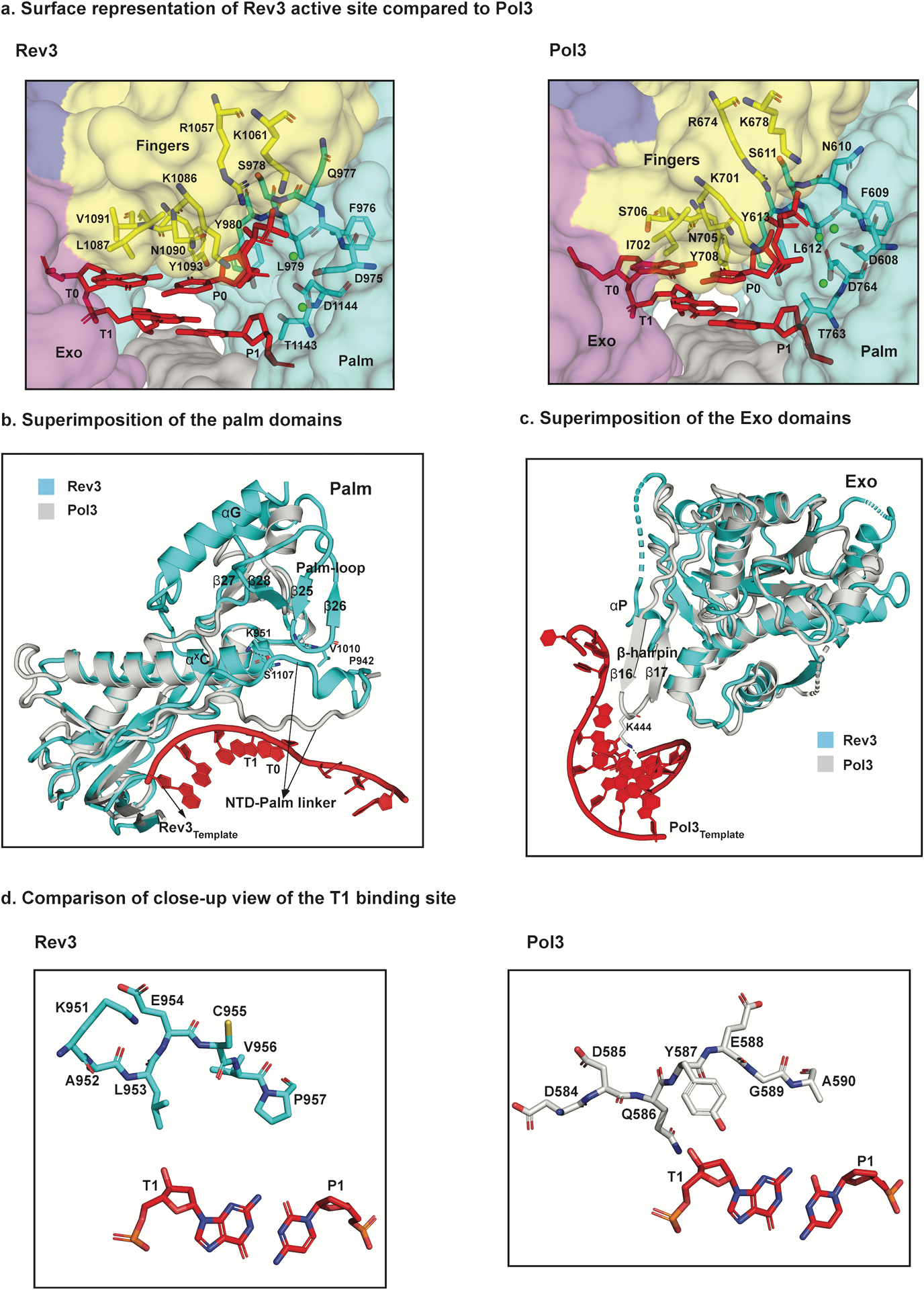
Structural basis for fidelity and mismatch extension. a, Surface representation of a close-up view of the active sites of Rev3 and Pol3 depicting conserved residues interacting with the incoming nucleotide (dCTP) as well as the templating base (G). b, Superimposition of the palm domain of Rev3 and Pol3. The Rev3 template DNA strand is shown in red and the T1 base is highlighted. In comparison to Pol3, the palm-loop (β25 and β26) is a unique Rev3 structural element that interacts with the NTD-palm linker. Key residues in the NTD-palm linker are shown as sticks interacting with the palm-loop residues as well as the αXC palm helix. Another unique structural element, αG, which interacts with β27 and β28 is also highlighted. c, Superimposition of the exonuclease domains of Rev3 and Pol3. Overlay of the exonuclease domains of Rev3 and Pol3 shows a much shorter and disordered helical loop in Rev3 in comparison to a well-defined β-hairpin in Pol3. The Pol3 DNA is highlighted in red. d, Comparison of the T1 binding site in Rev3 and Pol3, showing details of the interaction between the NTD-palm linker region and the T1 base. The residue E954 in Rev3 points away from the T1 base, whereas residue Y587 stacks against the sugar in Pol3. L953 in Rev3 is the only residue close to the T1 base, resulting in a less constrained pocket in comparison to Pol3.
The Rev3 palm is dominated by a mixed six-stranded β-sheet (β23, β29, β30, β32–β34) flanked by two long α-helices (αXC and αXD) from one side and short helix (αX) from the other (Figs. 2b and 3b and Supplementary Fig. 1). Compared to Pol319,23, the palm domain differs by an extra helix (αG) and a long loop (Palm-loop) that droops over the connecting linker between the NTD and the palm (NTD-palm linker) (Fig. 3b). The fingers domain contains three α-helices (αF, αXA and αXB) (Fig. 2b). αXA and αXB are analogous to the two long α-helices in B-family polymerases that drape over the nascent base pair24, whereas αF is unique to Polζ and is involved in contacts with the NTD. The NTD is much more extended than in Pol3 and contains additional secondary structural elements and loops that make contacts with the other domains of Rev3 (Extended Data Fig. 4).
The large insert in Rev3 between the NTD and the exonuclease domain comprises the Rev7 interaction region (RIR; residues 401–661). The RIR is disordered except for the region (513–624) that contains the two Rev7 binding motifs (RBM1, residues 517–540; RBM2, residues 599–623) that interact with the Rev7A and Rev7B subunits, respectively (Figs. 1d and 2b and Supplementary Fig. 1). Of the two cysteine-rich metal-binding modules at the C-terminal end of Rev3, CysAD (residues 1381–1418) is disordered, whereas CysBD (residues 1419–1504) is ordered and serves to recruit Pol31 (Fig. 1c). CysBD contains a well-resolved 4Fe-4S (Fig. 2b) cluster, which, as in the case of Polδ may modulate DNA synthesis in response to oxidative stress19,25.
Polζ lacks 3’−5’ exonucleolytic proofreading activity observed in most B-family polymerases1,26. The Rev3 exonuclease domain (Fig. 2b) is rendered inactive by the absence of two of the carboxylates normally associated with exonuclease activity (E323 and D407 in Pol3 is substituted by H675 and S773 in Rev3, for example). In most B-family polymerases, the exonuclease domain is further defined by a so-called “β-hairpin”, postulated to facilitate the transition of the primer between the polymerase and exonuclease active sites27,28. Strikingly, the β-hairpin is almost non-existent in Rev3 (Fig. 3c), concordant with the lack of proofreading activity in Polζ.
Overall, Rev3 is well poised for catalysis. The triphosphate moiety of incoming dCTP weaves a path between the fingers and palm domains and draws the two domains together (Fig. 2a). Two calcium ions (A and B) are located between the triphosphate tail and the primer terminus, analogous to metals “A” and ‘B” in other DNA polymerases29. Although calcium inhibits DNA polymerase activity, the positions of the catalytic residues (D975 and D1144) and the metal ions are appropriate for a two-metal mechanism of catalysis29 (Fig. 2a).
Fidelity and mismatch extension
The structure provides insights into why Polζ is unable to incorporate nucleotides opposite DNA lesions1, but is able to extend synthesis from mismatches and DNA lesions1,5. From the structure, Rev3’s inability to insert nucleotides opposite DNA lesions is due to residues L1087, N1090, V1091, Y1093 and G1094 from the fingers domain, and Y980 from the palm domain (Figs. 2a and 3a). L1087 and N1090 fit snuggly atop the templating base, while Y980, Y1093 and G1094 impinge on the nascent base-pair from the minor groove side (Figs. 2a and 3a). The contacts are primarily van der Waals in nature and together they impose a strong preference for a W-C base pair at T0-P0. These contacts are remarkably similar to those observed with Pol3 (Fig. 3a), suggesting that Polζ operates in much the same way as a high-fidelity replicative polymerase during the nucleotide incorporation step, sharing (in a steric sense) the same intolerance for mismatches and DNA lesions at the incipient T0-P0 position.
The ability of Polζ to tolerate mismatches and lesions at the T1-P1 position sets it apart from all other eukaryotic B-family polymerases1,26. We trace the ability of Polζ to tolerate DNA distortions at the T1-P1 position and to extend synthesis from the aberrant junction to divergence in the path of the NTD-palm linker and its sequestration by the Palm-loop.
In Pol3, the NTD-palm linker spans the width of the template-primer, with Y587 lying flush against the sugar of T1 nucleotide23 (Fig. 3d and Extended Data Fig. 5). Y587 is the only amino acid that makes direct contacts with the T1 nucleotide, whereas all of the other contacts are water-mediated. In Rev3, Y587 is replaced by a glutamate (E954), but most importantly, this segment of the NTD-palm linker traces a different path than in Pol3, particularly in the region that abuts against the T1 nucleotide in Pol3 (Fig. 3b). The linker is fixed in this alternative position in Rev3 by interaction with the Palm-loop, as well as helix αxC of the palm domain. Notably, the Palm-loop is a unique structural feature of the Rev3 palm domain. The net result of this movement in the NTD-palm linker is the creation of additional space around the T1 nucleotide that, in principle, can more easily contain DNA distortion and deviations from W-C geometry at the T1-P1 position (Fig. 3b,d and Extended Data Fig. 5).
Polζ incorporates two Rev7 subunits in a novel head-to-tail arrangement
The presence of Rev7 in Polζ is the main difference with Polδ in terms of subunit composition1,26. Rev7 is a member of the HORMA family of proteins that can exist in topologically distinct open (O) to the closed (C) states13,30,31. Until recently, Polζ was thought to incorporate only a single copy of Rev74. We show here that Polζ contains in fact two copies of Rev7 (Fig. 1c,d), with the holoenzyme assembled as a five-subunit (Rev3–Rev7A–Rev7B–Pol31–Pol32) complex. The Rev7 monomers adopt a novel head-to-tail arrangement unlike anything seen previously with other HORMA proteins13.
The N-terminal approximate two-thirds of Rev7 (residues 1–148) comprises the “core”, dominated by a three-stranded β-sheet (β4, β5 and β6) flanked by three α-helices (αA, αB and αC) from one side (Fig. 4a). The C-terminal region (residues 149–245; ensuing β6) is the “seatbelt” that can adopt different topological conformations: packing (as strands β7 and β8) against β6 side of the core in the open state, but rearranging (as strands β8’ and β8”) to the β5 side of the core in the closed state and encircling the bound peptide13,30,31 (Fig. 4a).
Fig. 4:
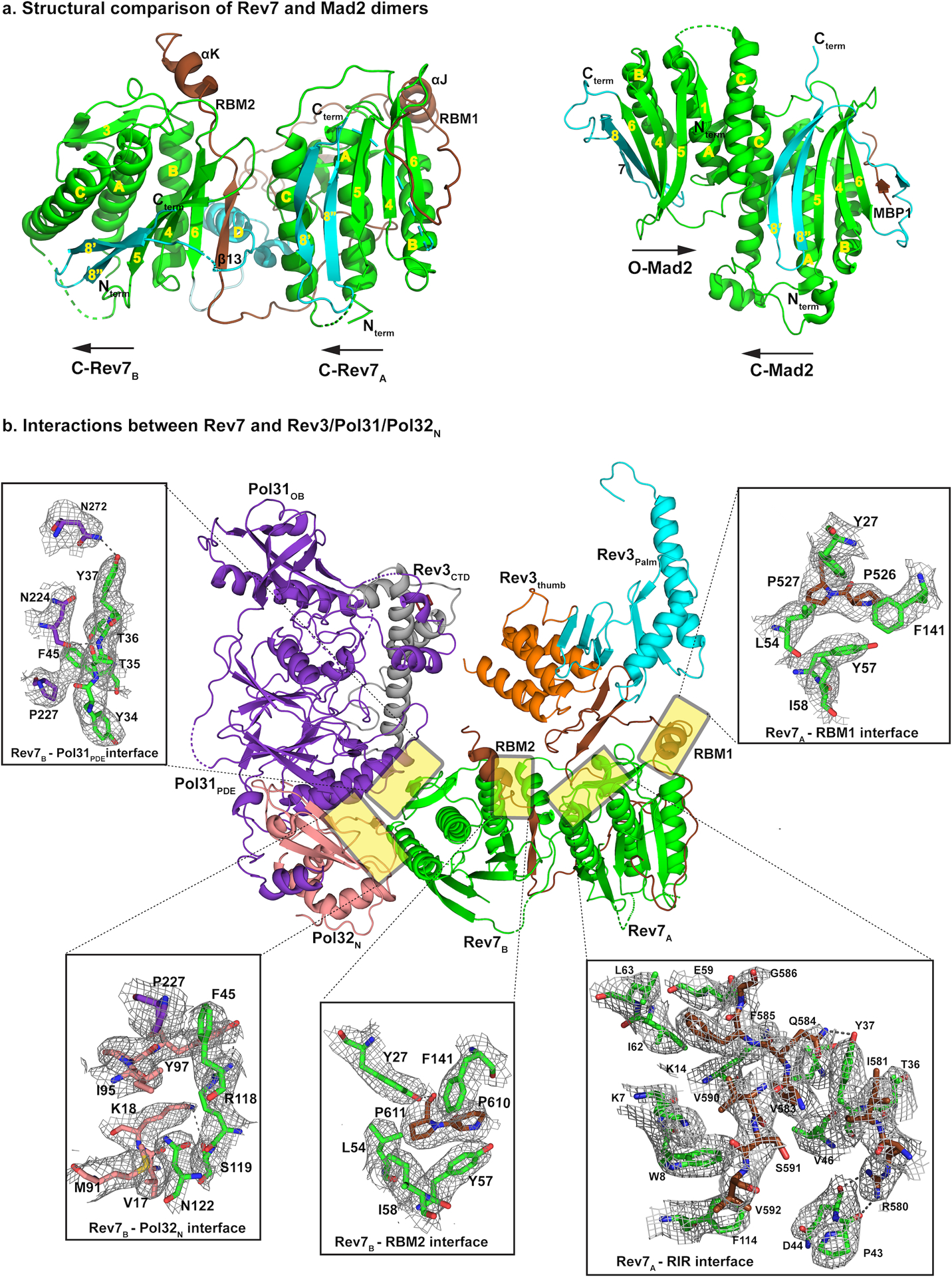
Rev7 dimer is the organizing center of Polζ holoenzyme. a, Rev7 dimer presents a novel head-to-tail arrangement (left) in comparison to other HORMA family members (right, PDB ID: 2V64). The C-terminal seatbelt region is highlighted in cyan. Both Rev7 subunits are in the closed state with Rev7A binding RBM1 and Rev7B binding RBM2. b, Both Rev7 subunits are required for assembly of the Polζ holoenzyme. Structure and cryo-EM density details of the Rev7 dimer interface with Rev3, Pol31 and Pol32N. RBM1 and RBM2 share a consecutive proline-proline motif that interacts with similar set of residues (Y27, F141, L54, Y57, for example) from both monomers. Interactions at the Rev7A and RIR interface that further stabilize the Rev7A:Rev7B dimer are shown. In addition, interactions of Rev7B with Pol31PDE–Pol32N depicted here aid in restricting the movement of Pol31–Pol32N subcomplex relative to Rev3.
In Polζ, both Rev7 subunits are in the closed state with Rev7A binding RBM1 and Rev7B binding RBM2. However, the arrangement is fundamentally different from that observed with other HORMA proteins (Fig. 4a). Mad2, for example, has been resolved as both a symmetric (C-Mad2:C-Mad2)32 and an asymmetric (C-Mad2:O-Mad2) dimer (Fig. 4a)33, but the arrangement in both cases is anti-parallel or head-to-head with the dimer interface dominated by helix αC from each monomer (the “head” here is defined as the side of HORMA protein containing helix αC). By contrast, Rev7A and Rev7B adopt a parallel or head-to-tail arrangement, resulting in helices αC (one from each monomer) being far apart (Fig. 4a). As such, the Rev7A-Rev7B interface is entirely asymmetric with helix αC and the β2-β3 pseudo hairpin of Rev7A making hydrogen bonds and van der Waals contacts with the seatbelt region of Rev7B. The interface is rather sparse, burying ~ 524 Å2 of solvent accessible surface area as compared to 1960 Å2 in C-Mad2:O-Mad2, with the Rev7 dimeric arrangement stabilized additionally by contacts with Rev3 as well as Pol31 and Pol32N (Fig. 4). As such, the Rev7A and Rev7B subunits act as a bridge between the catalytic Rev3 and the accessory Pol31 and Pol32 subunits (Figs. 1c,d and 4b).
Rev3-Rev7 interactions
Rev7A and Rev7B bind simultaneously to RBM1 and RBM2, marking the first time that a HORMA dimeric arrangement has been captured with a peptide bound to each monomer (Fig. 4a). RBM2 folds into a β-strand, linker, and an α-helix, whereas RBM1 consists of only the linker and α-helix (the β-strand in RBM1 and a portion of the seatbelt of Rev7A have weak density and are mostly disordered). The two RBMs contain consecutive prolines (P526 and P527 in RBM1 and P610 and P611 in RBM2) that are central in interactions with Rev7A and Rev7B (Fig. 4b). The aliphatic rings of P526 and P610 make hydrophobic contacts with the aromatic rings of Y57 and F141 of Rev7, whereas the aliphatic ring of P527 and P611 make hydrophobic contacts with the side chain of L54 (Fig. 4b and Extended Data Fig. 6). These hydrophobic contacts are augmented by a hydrogen bond between the main chain carbonyl of P611 and the hydroxyl of Y27 of Rev7B. Overall, these interactions are similar to those observed in the structure of human Rev7 with a human Rev3 RBM1 or RBM2 peptide17,18 and are likely to extend to Polζ from other eukaryotic species.
Strikingly, the entire segment of RIR connecting RBM1 and RBM2 (residues 513–624) is well-defined in our structure, weaving a path between Rev7A and Rev7B, as well as making contacts with the palm and thumb domains (Fig. 2b). Contacts with Rev7A are extensive, with amino acids such as V583, V590, V592 and F585 making hydrophobic contacts with residues from helices αA and αB of Rev7A (Fig. 4b and Extended Data Fig. 6). Overall, these contacts supplement (and even exceed) those made by RBM1 and RBM2 and appear to be critical in stabilizing the Rev7A:Rev7B homodimer in the non-canonical head-to-tail arrangement within Polζ.
Pol31 and Pol32
Considering their different roles in DNA replication and repair it is quite remarkable that Polζ and Polδ share the same accessory Pol31 and Pol32 subunits. Pol31 and Pol32N do not engage the DNA in either polymerase (Fig. 5a), despite containing domains (an OB fold and an inactive phosphodiesterase (PDE) domain in Pol31 and a winged helix-turn-helix domain in Pol32N) that are potentially capable of binding DNA (Figs. 1c,d and 5a). In both polymerases, CysBD at the C-terminus of the catalytic subunit interacts with both the Pol31OB and Pol31PDE domains of Pol31 (Fig. 5a), though the contacts vary between Polζ and Polδ19 (Fig. 5b). The size and structure of CysBD is, however, similar in Rev3 and Pol3, composed of two long antiparallel α-helices and a 4Fe-4S cluster (Extended Data Fig. 7). By contrast, the equivalent domains in the catalytic subunits of Polα and Polε are larger and observed to bind to a single divalent Zn2+ ion34–36.
Fig. 5:
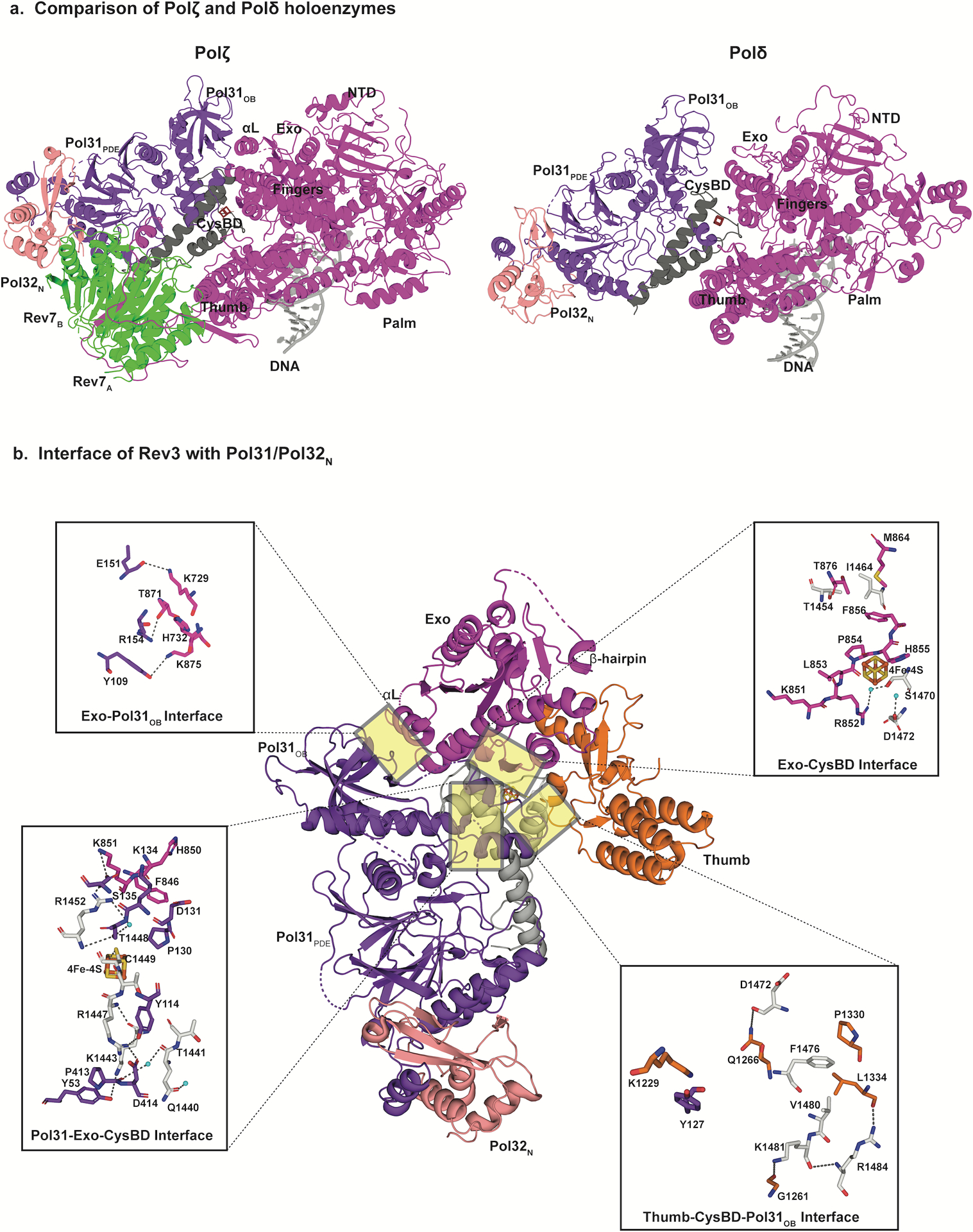
Rigidity of Polζ holoenzyme. a, Comparison of the cryo-EM structures of DN bound Polζ (left) and Polδ (right, PDB ID: 6P1H) holoenzymes viewed parallel to the DNA axis. Compared to their positioning relative to the catalytic subunit in Polδ, the Pol31–Pol32N subcomplex pivots away from Rev3 through a combination of rotation and translation. This movement helps accommodate the Rev7 dimer. In addition, in Polζ the interface between the exonuclease domain and Pol31OB includes helix αL, and is more compact than in Polδ. b, Detailed views of selected interaction sites between Rev3 and Pol31 and Pol32N interface. Mobility of the Pol31-Pol32N subcomplex is further restricted due to extensive contacts at the interface between exonuclease, CysBD and Pol31. The 4Fe-4S cluster is shown as a box with orange and yellow sticks.
Although Pol31 and Pol32N are situated radially with respect to the DNA in Polζ and Polδ, their exact position and orientation differs in the two enzymes (Fig. 5a). In particular, to make room for Rev7 in Polζ, Pol31 and Pol32N rotate by ~18 and translate by ~ 10Å relative to their positions in Polδ (Fig. 5a). This motion is along a direction roughly perpendicular to the DNA axis and it positions Pol31OB closer to the exonuclease domain in Polζ compared to Polδ (Fig. 5a). Thus, whereas there is a sizeable gap at the interface between Pol31OB and the exonuclease domain in Polδ, the interface is more tightly packed in Polζ and includes an α-helix (αL) from Rev3 not present in Polδ (Fig. 5a).
Overall, Pol31 and Pol32N are held much more rigidly than in Polδ19. Besides contacts with CysBD and the exonuclease domain of Rev3 (Fig. 5b), Pol31 and Pol32N also make numerous contacts with Rev7B (Fig. 4b), with ~695 Å2 of surface area is buried at this interface (Fig. 5b). This rigidity is highlighted by the resolution of Pol31 and Pol32N in the Polζ cryo-EM map, which is comparable to that of Rev3 and Rev7 (Fig. 1b). By contrast, in the Polδ cryo-EM structure19, the resolution of Pol31 and Pol32N was relatively low and only improved after multibody refinement (reflecting flexibility).
Conformational changes on DNA binding
A hallmark of replicative DNA polymerases is a conformational change in the fingers domain, from an “open” to a “closed” state on correct dNTP binding37,38. The apo Polζ structure reveals the fingers domain in the open conformation, with helices αF, αxA and αxB rotated outwards by 15° compared to the ternary complex (Fig. 6). Thus, analogous to replicative polymerases, Polζ’s fidelity during the nucleotide incorporation step appears to be augmented by selection based on opening and closing of the fingers domain. Also, in the apo structure, parts of the thumb domain that contact the DNA minor groove in the ternary complex are disordered or located away from the DNA (Fig. 6). Thus, many of the positively charged residues that interact with the DNA sugar-phosphate backbone in the ternary complex are either completely disordered (such as K1280 and K1283) or are far away from the DNA and partially disordered (such as K1272, K1273, R1309 and R1357) (Fig. 6).
Fig. 6:
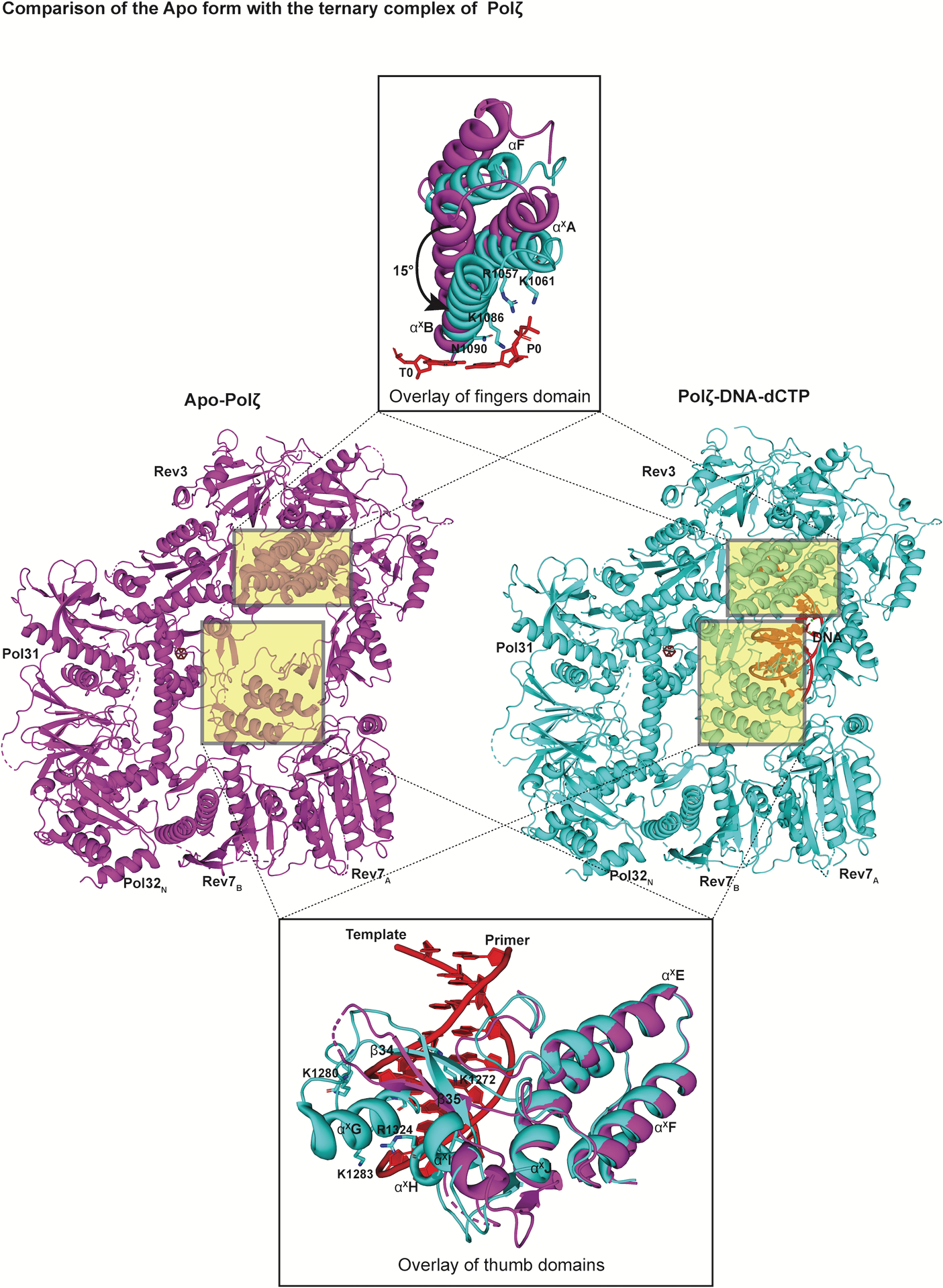
Conformational changes upon DNA binding. Structures of the ‘open’ apo state of Polζ (left, colored in magenta) in comparison to its ‘closed’ ternary complex (right, colored in cyan) viewed perpendicular to the DNA axis. An overlay of the fingers and the thumb domain of both states highlight significant conformational changes. In addition to the inward rotation of the fingers domain, various structural elements and loops (including αxG highlighted here) in the thumb domain become ordered upon DNA binding.
Discussion
Polζ stands out as the central DNA polymerase for the extension step in the bypass of the vast majority of DNA lesions formed in eukaryotic cells. For decades, structural studies of Polζ holoenzyme, or even just its large catalytic subunit Rev3, have been hampered by low yields and unattainability of well-diffracting crystals. Here, we employ cryo-EM to present the near atomic resolution structures of the complete yeast Polζ holoenzyme, with and without bound DNA. The structures reveal a pentameric ring-like architecture for Polζ, whereby the subunits form an uninterrupted daisy chain of protein-protein interactions.
The inability of Polζ to insert nucleotides opposite DNA lesions is readily understood from the DNA bound structure. The juxtaposition between the fingers helices and the nascent base-pair (position T0-P0) is remarkably similar to that observed with Pol319,23 and the sheer density of these contacts is incompatible with DNA distortion or deviations from W-C geometry at the T0-P0 position. The ability of Polζ to tolerate mismatches and lesions at the T1-P1 position and to extend synthesis from the aberrant junction appears to be due primarily to the divergence in the path of the NTD-palm linker in Rev3 and its sequestration by the Palm-loop. Specifically, the NTD-palm linker in Rev3 traces a different path than in Pol3, particularly in the region that abuts against the T1 nucleotide in Pol3 (Fig. 3b). The linker is fixed in this alternative position in Rev3 by interaction with the Palm-loop and αxC where the Palm-loop is unique to the Rev3 palm domain. The net result of this movement in the NTD-palm linker is the creation of extra space around the T1 nucleotide that, in principle, can more easily contain DNA distortion and deviations from W-C geometry at the T1-P1 position (Fig. 3b,d and Extended Data Fig. 5).
Interestingly in DNA Pol II structures39, a B-family polymerase that perform TLS in E.coli, the NTD-palm linker (referred to as the N-palm linker) tracks a similar path as in Rev3, but there is no equivalent of a Palm-loop to draw the linker away from the T1 nucleotide. Instead, shortening of the distance between the NTD and the palm domain in DNA Pol II has been suggested to relax the linker for lesion bypass39. Also, in the structure of DNA Pol II with an abasic lesion at the T1 position, both the lesion and the adjoining 5’ nucleotide are looped out in the space (or cavity) adjacent to the NTD-palm linker39. However, when we model the two looped out nucleotides in Rev3, there is a severe steric clash with the main chain carbonyl of E954 emanating from the NTD-palm linker (Extended Data Fig. 8). The NTD-palm linker in Rev3 appears to be held more rigidly than in DNA Pol II (due to contacts with the Palm-loop and αxC, described above), but could adjust its position to contain the looped out, nucleotides. Overall, Polζ may employ a dual strategy, where DNA lesions and mismatches resulting in mild distortion at T1P1 are extended without looping out of the nucleotides, whereas lesions and mismatches that cause severe distortion are looped out but may incur an energetic penalty for the required rearrangement of the NTD-palm linker.
Another salient feature of Rev3 is the near absence of the β-hairpin in its inactive exonuclease domain. This is important because the β-hairpin in B-family polymerases is postulated to hold the template strand in place while the mismatched primer strand separates and migrates to the exonuclease active site27,28. The near absence of the β-hairpin in Rev3 is concordant with the lack of proofreading activity in Rev3 and thus the need to transfer a mismatched primer strand from the polymerase to the exonuclease active site. Also, because the template strand makes far fewer contacts with the β-hairpin it may augment the accommodation of DNA distortion resulting from mismatches and lesions at the T1-P1 position. In contrast to Rev3, DNA Pol II has proofreading activity and a β-hairpin39, but an alteration in the position of the β-hairpin has been suggested to increase the dwell time of the DNA substrate in the Pol II polymerase active site for TLS to occur39. The near absence of the β-hairpin in Rev3 should, in principle, increase the dwell or residence time of a DNA substrate in the Rev3 polymerase active site, which coupled to fewer overall contacts to the template strand may increase the opportunity for TLS to occur. In future studies, it will be interesting to probe the consequences on TLS of altering the lengths and sequences of the NTD-palm linker and the β-hairpin in Rev3.
Rev7 is the only subunit of Polζ that does not have a counterpart in other B-family polymerases such as Pols α, ε, and δ. Until recently, the identification of a single RBM in human Rev3 (residues 1877–1887) pointed to human Polζ incorporating only one copy of Rev74. In 2015, a second RBM was characterized in human Rev3 (residues 1993–2003)40, leading to the suggestion that Polζ might actually incorporate two copies of Rev718,40. The equivalent RBM motifs had been difficult to discern in yeast Rev3, but from the structure we can now identify them as residues 517–540 (RBM1) and 599–623 (RBM2). The two RBMs bind simultaneously to the two Rev7 subunits, which for the first time captures a HORMA dimeric arrangement with a peptide bound to each monomer. Rev7A and Rev7B arrange in a non-canonical head-to-tail configuration, lending to an interface that is fundamentally different from that observed in C-Mad2:O-apoMad2 or C-apoMad2:C-apoMad232,33 (Fig. 4a and Extended Data Fig. 6). Notably, the solvent accessible surface area buried at the Rev7A:Rev7B interface is significantly less than that typically observed in oligomeric proteins41, implying that the Rev7A:Rev7B homodimer is stabilized in the head-to-tail arrangement (within Polζ) by additional contacts that Rev7A establishes with the RIR region connecting RBM1 and RBM2, and that Rev7B establishes with Pol31 and Pol32N (Figs. 1d and 4b). Interestingly, Rev7 has been proposed to homo-dimerize independently of Polζ42, although the dimer interaction appears to be weak compared to Mad243. Rev7 has also a been suggested from biophysical studies to dimerize in a canonical head-to-head arrangement when complexed to RBM1 and RMB2 of human Rev318. An intriguing question for future work is whether Rev7 dimerizes in a head-to-tail or a head-to-head configuration independently of Polζ. An interesting possibility is that Rev7 forms weak head-to-head dimers independently of Polζ, but switches to a non-canonical head-to-tail arrangement in the context of Polζ, following interactions with RBM1 and RBM2, the RIR between RBM1 and RBM2, and Pol31 and Pol32N.
What is the role of Rev7A and Rev7B in Polζ activity? The two subunits emerge from our structure as the organizing center of Polζ. Most importantly, the incorporation of two copies of Rev7 in Polζ increases the potential surface area available for interactions with other components of the TLS machinery, including DNA polymerase Rev1. The C-terminal domain of Rev1 (Rev1-CTD) can interact simultaneously with Rev7 and Y-family polymerases and serves to link the master extender Polζ to inserters Polη, Polι or Polκ for lesion bypass44–47. Intriguingly, when we model Rev1-CTD on Rev7A and Rev7B, only Rev7B is capable of accommodating Rev1 (Extended Data Fig. 9). This stoichiometry is consistent with a biophysical study showing that only a single Rev1-CTD is capable of binding human Rev7 when tethered to RBM1 and RBM218. We find potentially stabilizing interactions between Rev1-CTD and Pol32N (Extended Data Fig. 9), complementing evidence that Rev1-CTD can interact with a portion of Pol32C48. In all, the ability of Rev7 to assemble as a head-to-tail homodimer has major implications for how Polζ interacts with other components of the TLS machinery.
Polζ has emerged as an important determinant for tumor resistance to chemotherapeutic agents and radiotherapy in several types of cancers6,8,9. Indeed, in mouse cancer models, depletion of Rev3 sensitizes non-small cell lung and prostate cancers to conventional chemotherapy49,50. Similarly, depletion of Rev7 sensitizes ovarian cancer cells to chemotherapy51, and enhances the sensitivity of glioma cells to ionizing radiation52. However, the development of inhibitors of Polζ has been hampered by the lack of structural information on the polymerase. As such, most of the effort to date has been directed at the Rev7:RBM1 and Rev7:Rev1 interfaces for which structural information is available53,54. The structure of Polζ presented here will spur new efforts to develop inhibitors of this unique polymerase. We identify several new protein-protein interfaces that can potentially serve as models for targeting by small molecules or stapled peptides, including the Rev7B:Pol31, Rev7B:Pol32, Rev7A:RIR and Pol31OB:exonuclease interfaces. The structure of Rev3 itself offers new opportunities to directly target the catalytic activity of Polζ. The Rev3 fingers and palm domains and the NTD are significantly different from those in other polymerases and may provide a basis for the selective binding of small molecules to Rev3. Although human Rev3 (containing 3,130 amino acids) is about twice the length of yeast Rev3, this difference is due almost entirely to increase in the length of the RIR, which, except for RBM1 and RBM2, is predicted to be disordered. Indeed, deletion of ~ 1000 residues (from amino acids 526 to 1588) of human Rev3’s RIR has no significant effect on the biochemical activities of human Polζ21. Importantly, all of the domains and protein-protein interaction modalities identified here are also present in human Polζ.
Altogether, we unveil here for the first time the pentameric ring-like architecture of Polζ and identify structural elements that allow this unique polymerase to synthesize DNA and perform TLS. The structures provide an unprecedented new framework for genetic and biochemical studies aimed at understanding the role of Polζ in protecting organisms from environmental and cellular genotoxic stresses, and a framework for the discovery of therapeutics in the treatment of chemotherapy resistant tumors.
Methods
Protein expression and purification
S. cerevisiae Polζ holoenzyme, comprised of the full length Rev3 (residues 1–1,504), Rev7 (residues 1–245), Pol31 (residues 1–487), and Pol32 (residues 1–350) subunits, was expressed in yeast from plasmids pBJ1462 and pBJ1524 as described15. Rev3 and Pol31 subunits harbor PreScission protease cleavable Flag-MAT and GST tags, respectively. Protein was purified as described15 with several modifications. In brief, yeast cells were lysed with a mechanical bead beater using 0.5 mm pre-cooled zirconia beads. Lysis was done in the presence of β-mercaptoethanol and the protease inhibitors benzamidin, leupeptin, aprotinin and pepstatin A. After lysis, polyethylenimine was added to a final concentration of 0.04%. Proteins in the clarified cell extract were precipitated with 48% ammonium sulfate55. Pellets containing the Polζ holoenzyme were then solubilized and purified over manually packed glutathione Sepharose (GST) (GE healthcare) column and the bound protein was eluted with 40 mM Glutathione. The eluate was then passed over an anti-FLAG agarose (Sigma) column. The GST and FLAG tags were cleaved with GST-PreScission Protease and free GST derived from the Pol31 subunit and PreScission Protease were removed by further incubation with GST beads. The protein was concentrated and purified (Supplementary Fig. 2) over a Superose-6 Gel Filtration (GE healthcare) column. The complex with DNA was prepared by incubating the Polζ holoenzyme in CaCl2 supplemented buffer with a HPLC purified (Integrated DNA technologies) palindromic DNA (5´ TAATGGTAGGGGAGGGAATCCCTCCCCTACdd 3´) added in 1.5 molar excess, yielding G as the templating base. The addition of incoming dCTP (1 mM) allowed the capture of Polζ in the act of DNA synthesis. Final buffer for the ternary complex is 25 mM Bis-Tris, pH 6.8, 120 mM NaCl, 5 mM CaCl2, 2.5 % glycerol, 2 mM TCEP. The sample of Polζ without DNA was obtained fortuitously. The Polζ holoenzyme was prepared with a short primer–template (11-nt–16-nt; 5´-TAACCGCGTTCdd-3´–5’-CTCTTGAACGCGGTTA-3’) in the absence of incoming nucleotide but a subsequent cryo-EM map, at a nominal resolution of 4.1 Å, revealed it as structure of the apo holoenzyme (described below). The switch from a short primer–template) to the longer palindromic DNA (described above) was motivated by the success of this longer DNA in capturing the Polδ ternary complex.
Cryo-EM specimen preparation
Initial specimen preparation of Polζ was done using conventional blotting and vitrification techniques on 300 mesh carbon lacey grids. The data collected from these grids resulted in preferred particle orientation and the subsequent undersampling of Fourier components. This led to a loss of resolution along the preferred orientation axis, strong anisotropy of the 3D reconstruction and a ‘smearing effect’ on the map (Extended Data Fig. 1). The use of quantifoil, c-flat or continuous carbon grids, in combination with different glow discharging protocols, cryo-plunging instruments and additives, including detergents, did not improve the preferred orientation issue. Finally, better sampling of orientations for the DNA bound complex was achieved using a Chameleon EP-2 robotic plunge-freezer. Based on Spotiton56, Chameleon is a blot-free system that requires pico-liter sample volumes for vitrification (https://www.sptlabtech.com/products/sample-preparation/chameleon). 300-mesh carbon self-blotting nanowire grids of 0.8 μm hole size and 1.3 μm spacing were made at New York Structural Biology center (NYSBC)57. Following glow discharging for 20 s at 12 mA using a PELCO EasiGlow (Ted Pella), the Chameleon piezo tip aspirated a 5 μl aliquot of protein and spotted 2 nl per grid. The grids were then plunged into liquid ethane at 134 ms spot-to-plunge speed.
3 μl aliquots of the apo Polζ were adsorbed onto 300 mesh gold-coated lacey grids that were plasma cleaned with O2 and H2 for 30 s using a Solarus plasma cleaner (Gatan), manually one-side blotted from the back of the grid and flash frozen in liquid ethane using a manual homemade cryoplunger.
Cryo-EM data collection
The ternary complex was imaged with a Titan Krios microscope (Thermo Fisher Scientific) operated at 300 keV and equipped with a K2 direct electron detector (Gatan) operating in super-resolution mode at a calibrated pixel size of 0.537 Å. The data was subsequently binned by two during frame alignment to yield a pixel size of 1.07 Å. Movies were recorded at a frame rate of 200 ms for 10 s yielding a total accumulated dose of 71.63 e−/Å2. A nominal defocus range of 0.5 to 2.5 μm was employed and movies were recorded automatically acquired using Leginon to control both the microscope and the K2 camera58,59. Frames were aligned using MotionCor2 with dose weighting60 and the contrast transfer function (CTF) estimation was performed with CTFFIND461. A total of 5297 micrographs were collected from two 48-hr sessions, out of which 1916 micrographs were selected using image rejector in Appion by applying a cutoff of 4.5 Å at the 0.5 confidence level for the CTF fit. This unusual attrition in the number of selected micrographs was the result of significant variation in hole quality across our Chameleon vitrified grids
All the images for the apo state of Polζ were also acquired using a Titan Krios microscope (Thermo Fisher Scientific) operated at 300 keV and equipped with a K2 direct electron detector (Gatan), but this time operated in counting mode at calibrated pixel size of 1.1 Å. Images were automatically recorded using Leginon. Due to preferred orientation of the particles (Extended Data Fig. 1), the stage was tilted to an angle of 40° and data were collected following a recently reported strategy62. As a part of this strategy, we compensated for the loss of signal to noise ratio, resulting from the increase at high tilt angles in the path of electrons through vitreous ice, by recording movies at a frame rate of 200 ms for 12 s (yielding a total accumulated dose of 87.62 e−/Å2), at a nominal defocus of 1.5 to 2.5 μm, and with the microscope operating an energy filter. Another consequence of the high tilt was, in general, a significant increase in drift between frames compared to the untilted data. Frame alignment at a single pixel level with MotionCor2 improved data quality. CTF estimation was done on a per particle basis using GCTF63 to account for particles at different Z-heights due to the specimen tilt.
Image processing and 3D reconstruction
Particle picking for the ternary complex was initially done using template-based picking (FindEM) in Appion64 using re-projections from the negative stain reconstruction of Polζ22. Initial processing of micrographs from the first session in cryoSPARC65 resulted in a 3D model with a sphericity of 0.93 out of 1 (Extended Data Fig. 2). Final particles from processing of this session (Session 1: 48,559 particles) and another session (Session 2: 30,961 particles) were reextracted with RELION366,67 and used as input for training Topaz, a neural network-based particle picker68. The micrographs were binned by 4 and used with resnet8 neural network architecture. Pi, the expected fraction of positive pixels was set to 0.035 and the radius parameter, which sets the number of pixels around a labeled particle coordinate, was set to 3. A total of 25 iterations were used to train Topaz using 1/3 of the particles as an input. The best model was then applied to all the images using a binned radius of 20, followed by thresholding that resulted in particle picks that were reextracted in RELION3 for further processing.
Particle picking for the apo-state of Polζ was done with FindEM using templates from the negative-stain Polζ reconstruction. Particles were picked based on a relaxed threshold in an attempt to include all the possible particles on the micrographs. Particles were then extracted using RELION2 for further processing.
Re-extracted particles for the ternary complex were subjected to multiple rounds of 2D-classification in cryoSPARC2. A final set of 181,726 particles were subjected to an ab-initio clean-up, which allowed for removal of particles corresponding to a low-resolution model with preferred orientation. The resulting particles were refined in cryoSPARC2 to give a 3D reconstruction at nominal resolution of 3.2 Å based on the Fourier shell correlation (FSC) value of 0.143 between independently refined half-sets69,70. The cryo-EM map was checked for directional anisotropy (https://3dfsc.salk.edu) and gave a value of 0.96 out of 1. Processing in RELION3 with the Topaz picked particles also gave similar results and map quality. In order to improve the resolution further, separate masks for Rev3 and Rev7A–Rev7B–Pol31–Pol32N were generated using Segger, an extension of UCSF chimera71. Masks were applied to the volume and 3D refinement without particle extraction was done for each region. The resulting consensus maps were at a FSC0.143 of 3.02 Å and 3.08 Å for Rev3 and Rev7A–Rev7B–Pol31–Pol32N region, respectively (Extended Data Fig. 2). The maps were then combined in PHENIX72 to generate a composite map which was used for model building. Local resolution estimates shown (Fig. 1b) were calculated using ResMap73.
In the case of apo Polζ, particles extracted with RELION2 were subjected to iterative rounds of 2D classification in cryoSPARC (Extended Data Fig. 3). A set of 457,987 particles were selected from the 2D class averages, which were then used to do an ab-initio clean-up to generate an initial model. This model from 357,328 particles was subjected to 3D refinement resulting in a 3D reconstruction with a FSC0.143 of 4.1 Å with and a sphericity of 0.92 over 1. A final set of 311,800 particles were exported from cryoSPARC using pyem script (https://github.com/asarnow/pyem). Per-particle CTF refinement was done in cisTEM74 resulting in the cryo-EM map with a sphericity of 0.95 that used for model building (Extended Data Fig. 3).
Model building, refinement and analysis
The structure of the Pol domain of Rev3 (excluding residues 303–400) was built manually using the cryo-EM map of the ternary complex. Model building was done in COOT75 using a structure based sequence alignment of Rev3 with Pol3 (PDB ID: 3IAY) as a guide. In comparison to Pol3, the well-resolved side chain densities guide the building of various insertions in Rev3 NTD, exonuclease, palm and the thumb domains. Unambiguous density for 22 bases of the DNA, incoming nucleotide and metal ions helped in building them accurately. The density for the 4Fe-4S cluster and the CTD of Rev3 was also well-defined, especially for the region closest to the interface of exonuclease domain of Rev3 with Pol31, Pol32N. Pol31 and Pol32N were built based on homology models from their human homologs, p50 and p66N (PDB ID: 3E0J) as well as the complex of Pol1 CTD with the B-subunit in yeast Polα (PDB ID: 3FLO). We built Rev7B by using a homology model of human Rev7 (PDB ID: 3ABD) and placing it in the density for the domain next to Pol32N. Rev7A had weaker density in comparison to Rev7B, but was identified as a Mad2 fold using BALBES76. A careful analysis of the side chain density confirmed it to be a second Rev7 molecule in the Polζ holoenzyme.
The refined cryo-EM structure of the ternary complex displays clear density (Fig. 1b–d) for almost all of the secondary structures and ~85% of the side chains, 22 of the 26 bases of the DNA duplex, templating base G, dCTP, 220 solvent molecules, as well as the active site metal ions. The model for the apo structure was built by fitting the coordinates of the ternary complex into the cryo-EM map using COOT and Chimera. Manual adjustments and deletions of disordered regions were done in COOT.
Both models were refined using real-space refinement in PHENIX and validated using Molprobity77 and EMringer78. Figures were prepared using Chimera and PyMOL79. Superimposition of structures was performed in COOT. Sequence alignments were done in Clustal Omega80, PHYRE281 and PROMALS3D82. Buried surface area was calculated in Chimera.
Extended Data
Extended Data Fig. 1. Preferred specimen orientation.

a, Data collected at 0° stage angle resulted in disproportionally low number of classes for side-views of the ternary complex of Polζ depicted in the 2D class averages. This resulted in a ‘smeared 3D model’ as shown by the anisotropic 3DFSC plot. Scale bar = 123 Å. b, Data collected at a stage angle of 0° for the apo-state of Polζ also had preferred set of views as shown in the 2D class averages. The final construction was anisotropic as depicted by the directional FSC plot. Scale bar = 123 Å..
Extended Data Fig. 2. Cryo-EM data collection and processing of Polζ-DNA-dCTP complex.

a, Data were collected on Chameleon grids and particles from one session were picked with template based picker (FindEM) and processed in cryoSPARC to give a consensus map with a FSC0.143 of 3.57 Å. Major stages of processing are shown schematically and particles involved at each stage are highlighted in green. Scale bar = 137 Å. b, Final particles from two sessions were merged and used to train Topaz. Data processing from Topaz picked particles in cryoSPARC2 improved the sphericity. A schematic representation of the improved consensus map displaying a FSC0.143 of 3.2 Å is shown. Scale bar = 137 Å. c, Focused refinement of the final volume was done in cryoSPARC2. Masks were created (along the blue dashed line) for 3D refinement of Rev3 and accessory subunits separately to give consensus maps at 3.02 Å and 3.08 Å, respectively..
Extended Data Fig. 3. Cryo-EM data collection and processing for Polζ apo state.
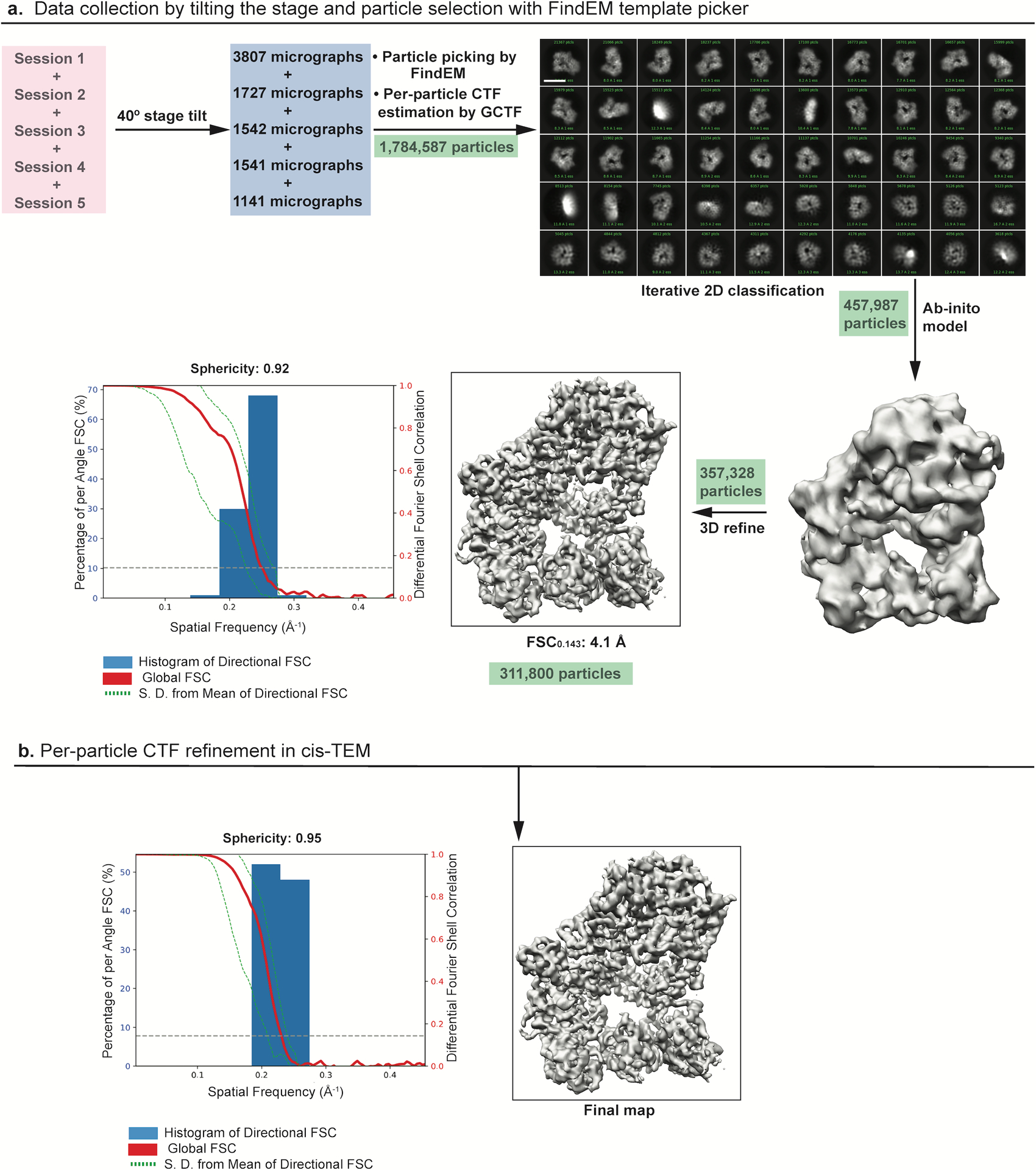
a, Data were collected at a 40° tilt angle and processed in cryoSPARC to give a good distribution of particles (green) with different views depicted in the 2D class averages. The final 3D reconstruction displaying a FSC0.143 of 4.1 Å showed an isotropic map amenable for model building. Scale bar = 123 Å. b, Per- particle CTF refinement of the map improved the sphericity further as shown by the 3DFSC plot..
Extended Data Fig. 4. Comparison of the NTD of Rev3 and Pol3.
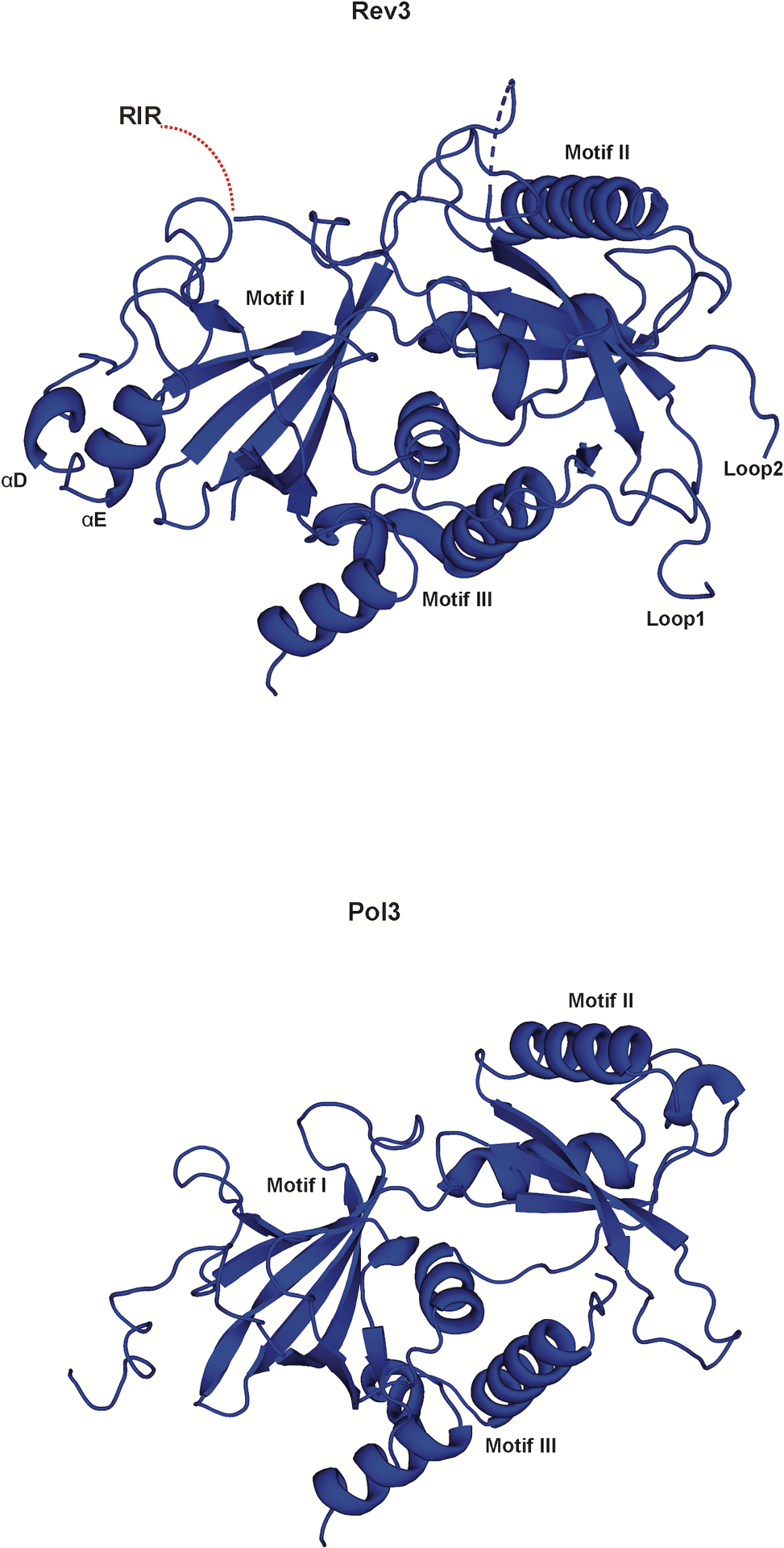
The NTD in Rev3 and Pol3, is composed of three motifs (I, II, III) but is much more elaborate and extended in Rev3. Loop 1 and Loop 2 contact all three motifs and connect the NTD to the fingers and palm domains, respectively..
Extended Data Fig. 5. Surface representation of the T1 binding site.
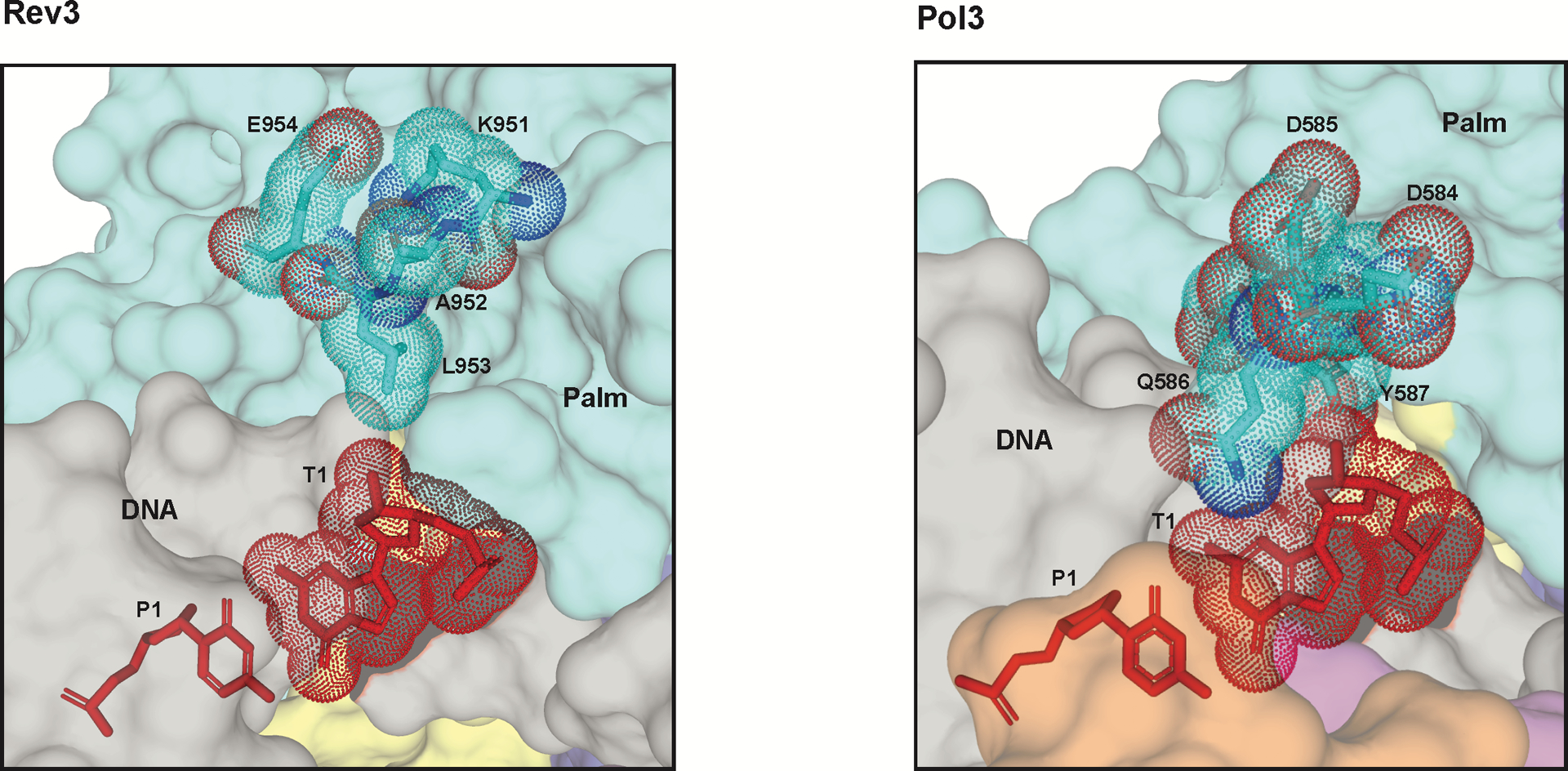
Residues around the T1 site are shown (sticks) for Rev3 (left) and Pol3 (right). Surface for the palm domains and DNA are shown in cyan and grey, respectively. The T1 base (red) and the key residues are highlighted in dots..
Extended Data Fig. 6. Comparison of yeast and human Rev7-RIR complexes.
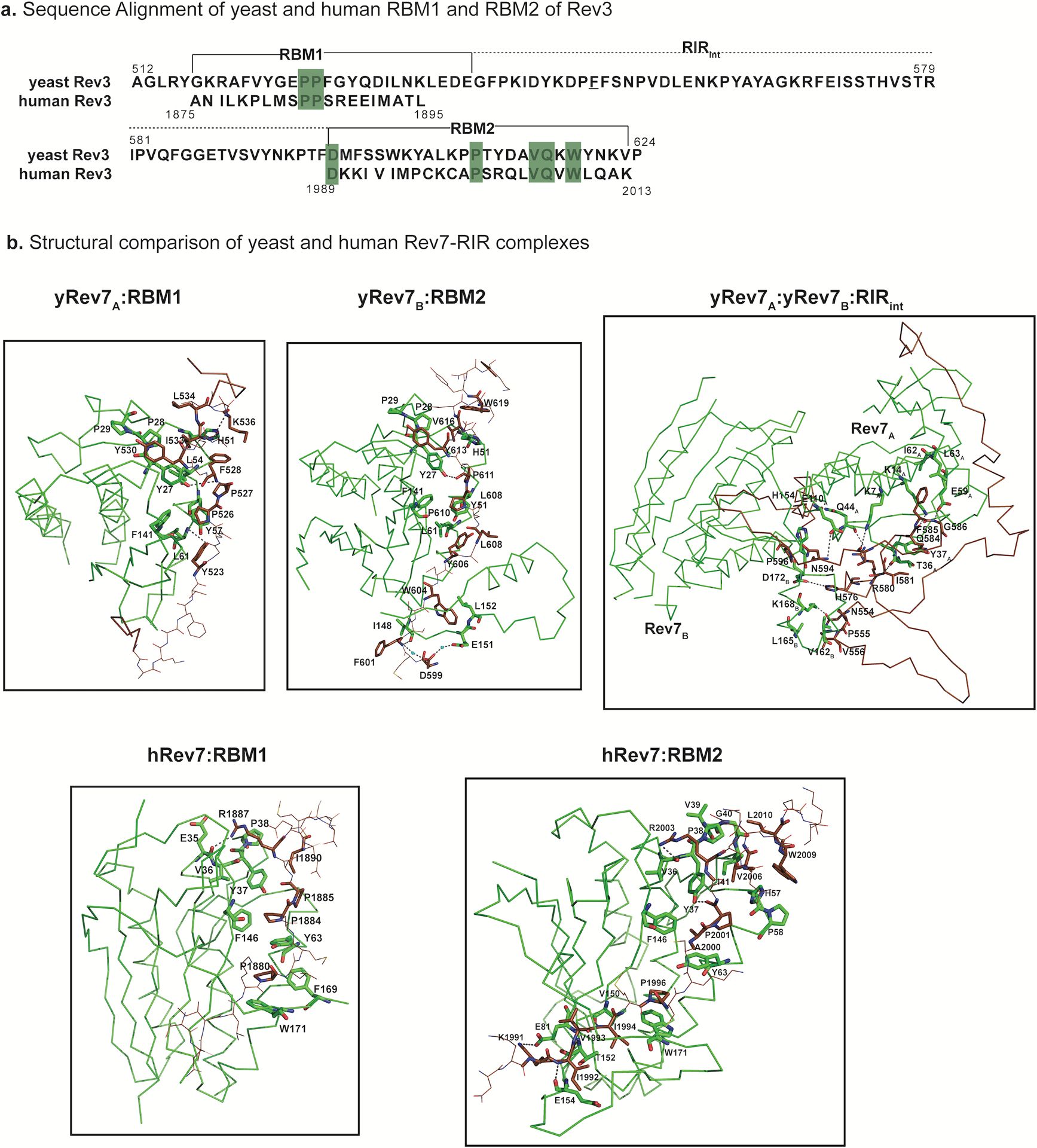
a, Sequence alignment of yeast and human RBM1 and RBM2 regions of Rev3. Conserved prolines within RBM1 and RBM2 are highlighted in green. Also, highlighted are the conserved residues among the yeast and human homologs within the RIR region. b, Structural comparison of the yeast and human RBM1 and RBM2. Individual structures of human Rev7 with RBM1 peptide (hRev7:RBM1; PDB ID: 3ABD) and RBM2 peptide (hRev7:RBM2; PDB ID: 6BC8) are compared to the corresponding sub-regions (yRev7A:RBM1; yRev7B:RBM2) in the yeast Polζ holoenzyme. The protein residues involved in the interactions are highlighted in green and the RIR is shown in brown. The interactions of Rev7A and Rev7B with the RIR segment connecting RBM1 and RBM2 (yRev7A:yRev7B:RIRint) is also depicted..
Extended Data Fig. 7. Comparison between the CysBD of Polζ and Polδ.
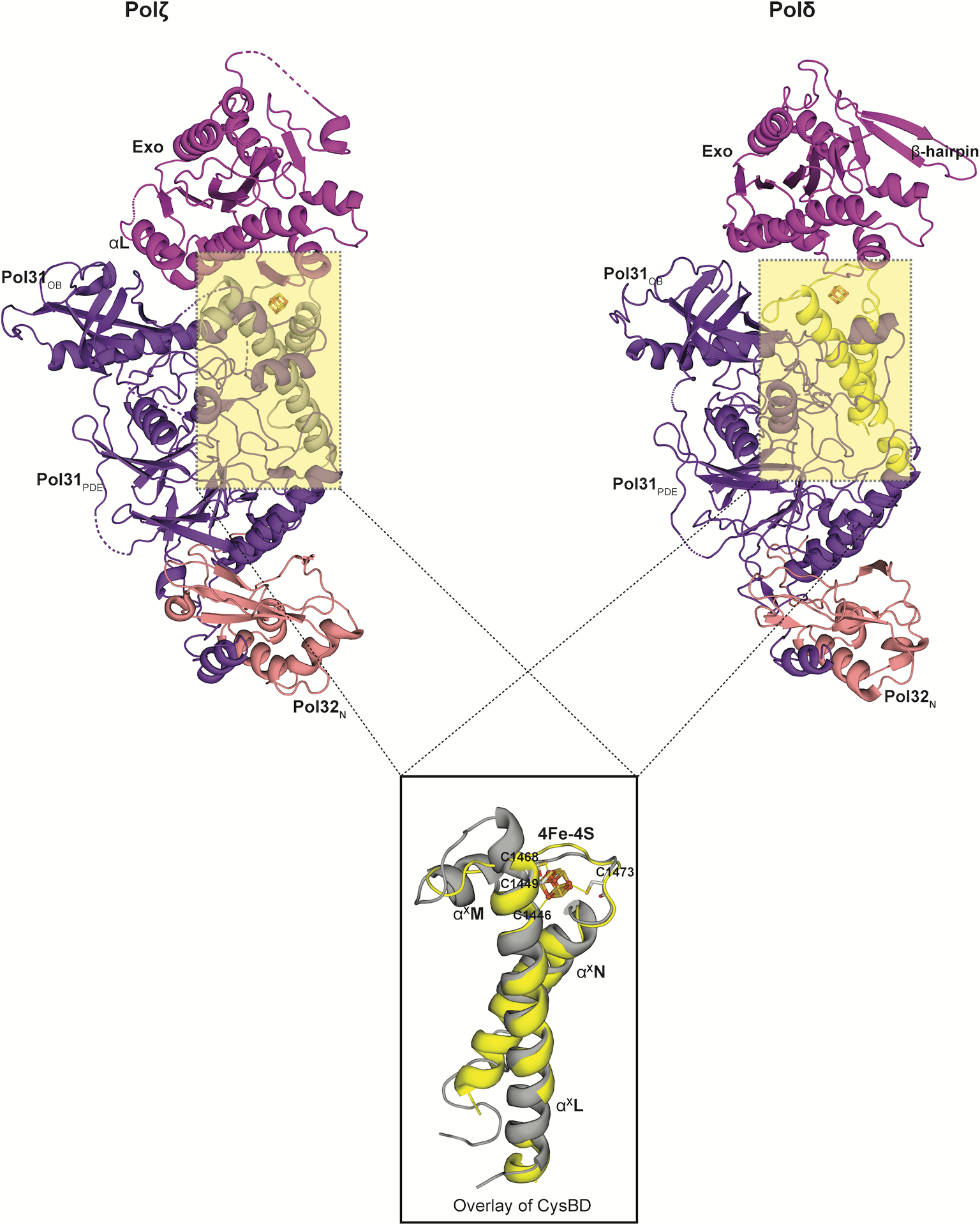
A superimposition of the CysBD of the Polζ (left; grey in color) and Polδ (right; yellow in color; PDB ID: 6P1H) shows conservation in its overall topology. Notably, helix αXM in Polζ CysBD has been substituted by a loop in Polδ (PDB ID: 6P1H). All the four cysteines interacting with the 4Fe-4S cluster in Rev3 are also highlighted..
Extended Data Fig. 8. Comparison of Rev3 and Pol II.
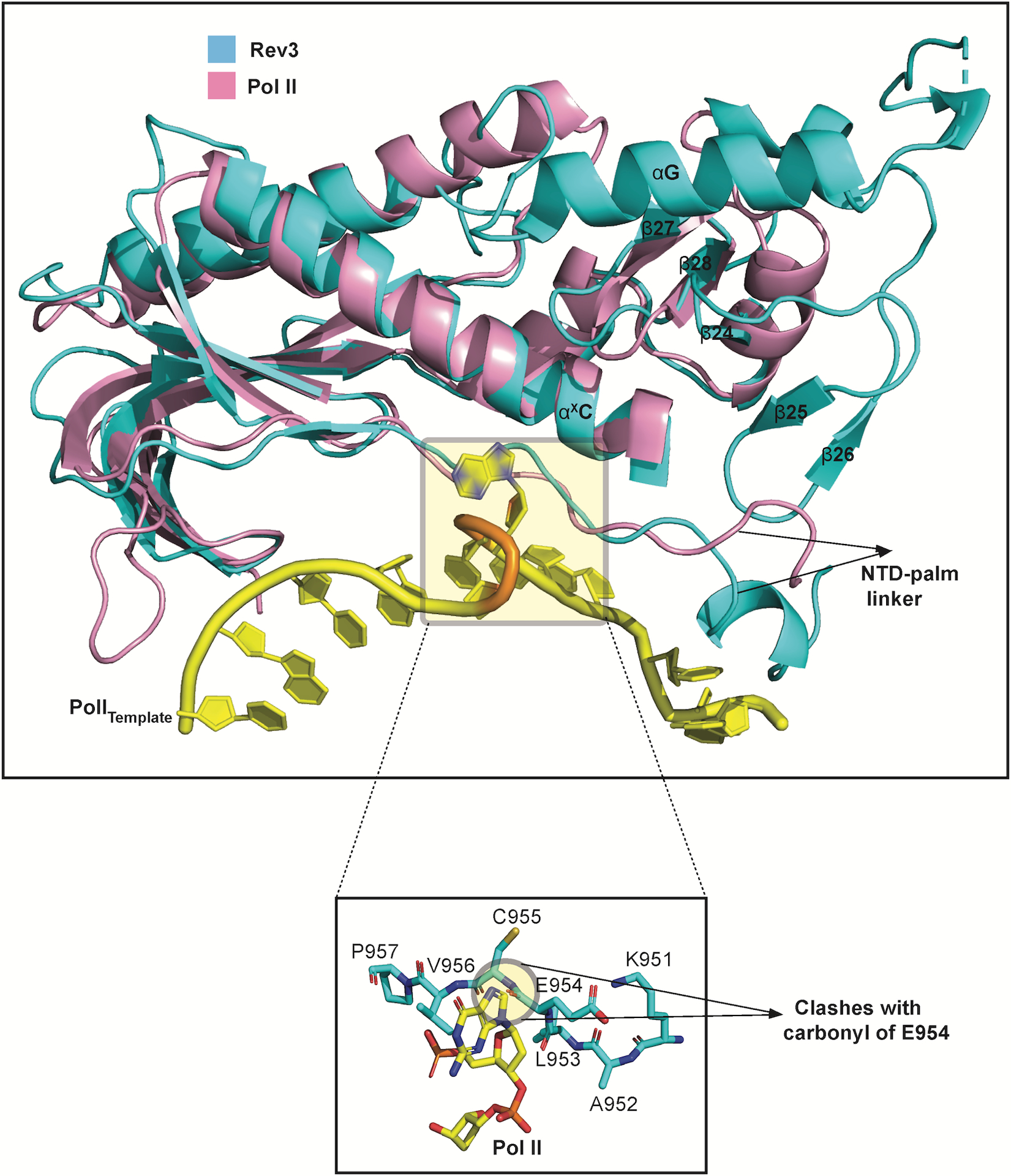
Overlay of the palm domains of Rev3 and Pol II show a similar trajectory for the NTD-palm linker. In Rev3, this trajectory is coupled to interactions with the Palm-loop. The Pol II template DNA strand is shown in yellow (PDB ID: 3K5M). A close-up view of the looped-out abasic lesion and the adjoining 5’ guanine nucleotide. Notably, the guanine base clashes with the backbone carbonyl of E954 in Rev3..
Extended Data Fig. 9. Docking of Rev1 CTD on the Polζ holoenzyme.
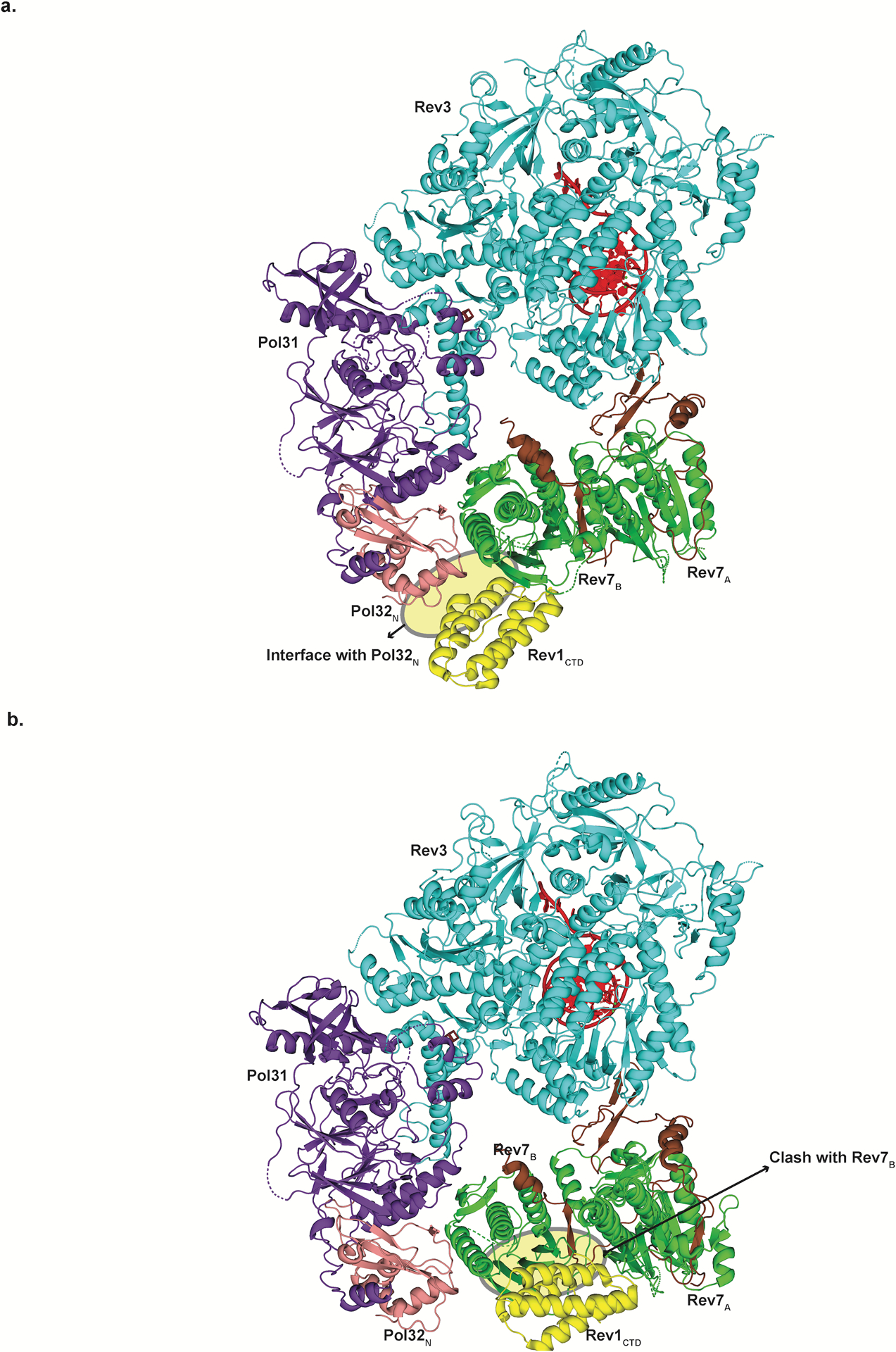
a, Superimposition of human Rev7-RBM1-Rev1CTD (PDB ID: 4EXT) on Rev7B shows close proximity to Pol32N (shown in yellow), highlighting the importance of Pol32N in stabilizing this interaction. b, Superimposition of the human Rev7-RBM1-Rev1CTD on Rev7A shows clashes of Rev1CTD with various secondary structure elements of Rev7B (shown in yellow)..
Supplementary Material
Acknowledgements
We thank Drs. Bridget Carragher, Clint Potter and Edward Eng for helpful advice and discussion throughout the project. We also thank Zhening Zhang and Daija Bobe for help in grid preparation; Yong Zi Tan for help in collecting tilted cryo-EM data, Alan Brown and Tom Terwilliger for help in implementing software, and Deepak Nair for help in model building. This work was primarily funded by grant R01-GM124047 from the National Institutes of Health (NIH). I. U.-B. was supported by a grant PID2019-;104423GB-I00/AEI/10.13039/501100011033 from the Spanish State Research Agency and by the Basque Excellence Research Centre program. Initial EM screening was performed at the Icahn School of Medicine microscope facility supported by a shared instrumentation grant from the NIH (1S10RR026473). Some of this work was performed at the Simons Electron Microscopy Center and National Resource for Automated Molecular Microscopy located at the New York Structural Biology Center, supported by grants from the Simons Foundation (SF349247), NYSTAR, and the NIH National Institute of General Medical Sciences (GM103310) with additional support from Agouron Institute (F00316), NIH (OD019994), and NIH (RR029300). Computing resources needed for this work were provided in part by the High Performance Computing facility of the Icahn School of Medicine at Mount Sinai. Molecular graphics and analyses performed with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311.
Footnotes
Competing interests
The authors declare no competing interests.
Reporting Summary Statement
Further information on experimental design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Accession numbers of the Polζ–DNA–dNTP model and map are Protein Data Bank (PDB)-6V93 and EMD-21115, respectively. Accession numbers of the Polζ (apo form) model and map are PDB-6V8P and EMD-21108, respectively.
References
- 1.Prakash S, Johnson RE & Prakash L Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu Rev Biochem 74, 317–353 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Jain R, Aggarwal AK & Rechkoblit O Eukaryotic DNA polymerases. Curr Opin Struct Biol 53, 77–87 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Makarova AV & Burgers PM Eukaryotic DNA polymerase zeta. DNA Repair (Amst) 29, 47–55 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martin SK & Wood RD DNA polymerase zeta in DNA replication and repair. Nucleic Acids Res 47, 8348–8361 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnson RE, Washington MT, Haracska L, Prakash S & Prakash L Eukaryotic polymerases iota and zeta act sequentially to bypass DNA lesions. Nature 406, 1015–1019 (2000). [DOI] [PubMed] [Google Scholar]
- 6.Lange SS, Takata K & Wood RD DNA polymerases and cancer. Nat Rev Cancer 11, 96–110 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharma S, Helchowski CM & Canman CE The roles of DNA polymerase zeta and the Y family DNA polymerases in promoting or preventing genome instability. Mutat Res 743–744, 97–110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamanaka K, Chatterjee N, Hemann MT & Walker GC Inhibition of mutagenic translesion synthesis: A possible strategy for improving chemotherapy? PLoS Genet 13, e1006842 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sharma S, Shah NA, Joiner AM, Roberts KH & Canman CE DNA polymerase zeta is a major determinant of resistance to platinum-based chemotherapeutic agents. Mol Pharmacol 81, 778–787 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nelson JR, Lawrence CW & Hinkle DC Thymine-thymine dimer bypass by yeast DNA polymerase zeta. Science 272, 1646–1649 (1996). [DOI] [PubMed] [Google Scholar]
- 11.Khalaj M et al. A missense mutation in Rev7 disrupts formation of Polζ, impairing mouse development and repair of genotoxic agent-induced DNA lesions. J Biol Chem 289, 3811–3824 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aravind L & Koonin EV The HORMA domain: a common structural denominator in mitotic checkpoints, chromosome synapsis and DNA repair. Trends Biochem Sci 23, 284–286 (1998). [DOI] [PubMed] [Google Scholar]
- 13.Rosenberg SC & Corbett KD The multifaceted roles of the HORMA domain in cellular signaling. J Cell Biol 211, 745–755 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baranovskiy AG et al. DNA polymerase delta and zeta switch by sharing accessory subunits of DNA polymerase delta. J Biol Chem 287, 17281–17287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson RE, Prakash L & Prakash S Pol31 and Pol32 subunits of yeast DNA polymerase delta are also essential subunits of DNA polymerase zeta. Proc Natl Acad Sci U S A 109, 12455–12460 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Makarova AV, Stodola JL & Burgers PM A four-subunit DNA polymerase zeta complex containing Pol delta accessory subunits is essential for PCNA-mediated mutagenesis. Nucleic Acids Res 40, 11618–11626 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hara K et al. Crystal structure of human REV7 in complex with a human REV3 fragment and structural implication of the interaction between DNA polymerase zeta and REV1. J Biol Chem 285, 12299–12307 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rizzo AA et al. Rev7 dimerization is important for assembly and function of the Rev1/Pol zeta translesion synthesis complex. Proc Natl Acad Sci U S A 115, Eb191–Eb200 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jain R et al. Cryo-EM structure and dynamics of eukaryotic DNA polymerase delta holoenzyme. Nat Struct Mol Biol 26, 955–962 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baranovskiy AG et al. X-ray structure of the complex of regulatory subunits of human DNA polymerase delta. Cell Cycle 7, 3026–3036 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee YS, Gregory MT & Yang W Human Pol zeta purified with accessory subunits is active in translesion DNA synthesis and complements Pol eta in cisplatin bypass. Proc Natl Acad Sci U S A 111, 2954–2959 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gomez-Llorente Y et al. The Architecture of Yeast DNA Polymerase zeta. Cell Reports 5, 79–86 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swan MK, Johnson RE, Prakash L, Prakash S & Aggarwal AK Structural basis of high-fidelity DNA synthesis by yeast DNA polymerase delta. Nat Struct Mol Biol 16, 979–986 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doublie S & Zahn KE Structural insights into eukaryotic DNA replication. Front Microbiol 5, 444 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bartels PL, Stodola JL, Burgers PMJ & Barton JK A Redox Role for the [4Fe4S] Cluster of Yeast DNA Polymerase delta. J Am Chem Soc 139, 18339–18348 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burgers PMJ & Kunkel TA Eukaryotic DNA Replication Fork. Annu Rev Biochem 86, 417–438 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hogg M, Aller P, Konigsberg W, Wallace SS & Doublie S Structural and biochemical investigation of the role in proofreading of a beta hairpin loop found in the exonuclease domain of a replicative DNA polymerase of the B family. J Biol Chem 282, 1432–1444 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Stocki SA, Nonay RL & Reha-Krantz LJ Dynamics of bacteriophage T4 DNA polymerase function: identification of amino acid residues that affect switching between polymerase and 3’ → 5’ exonuclease activities. J Mol Biol 254, 15–28 (1995). [DOI] [PubMed] [Google Scholar]
- 29.Steitz TA DNA polymerases: structural diversity and common mechanisms. J Biol Chem 274, 17395–17398 (1999). [DOI] [PubMed] [Google Scholar]
- 30.Mapelli M & Musacchio A MAD contortions: conformational dimerization boosts spindle checkpoint signaling. Curr Opin Struct Biol 17, 716–725 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Luo X & Yu H Protein metamorphosis: the two-state behavior of Mad2. Structure 16, 1616–1625 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang M et al. Insights into mad2 regulation in the spindle checkpoint revealed by the crystal structure of the symmetric mad2 dimer. PLoS Biol 6, e50 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mapelli M, Massimiliano L, Santaguida S & Musacchio A The Mad2 conformational dimer: structure and implications for the spindle assembly checkpoint. Cell 131, 730–743 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Baranovskiy AG et al. Crystal structure of the human Pol B-subunit in complex with the C-terminal domain of the catalytic subunit. J Biol Chem 292, 15717–15730 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klinge S, Nunez-Ramirez R, Llorca O & Pellegrini L 3D architecture of DNA Pol alpha reveals the functional core of multi-subunit replicative polymerases. EMBO J 28, 1978–1987 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suwa Y et al. Crystal Structure of the Human Pol alpha B Subunit in Complex with the C-terminal Domain of the Catalytic Subunit. J Biol Chem 290, 14328–14337 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Joyce CM & Benkovic SJ DNA polymerase fidelity: kinetics, structure, and checkpoints. Biochemistry 43, 14317–14324 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Doublie S, Sawaya MR & Ellenberger T An open and closed case for all polymerases. Structure 7, R31–35 (1999). [DOI] [PubMed] [Google Scholar]
- 39.Wang F & Yang W Structural insight into translesion synthesis by DNA Pol II. Cell 139, 1279–1289 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tomida J et al. REV7 is essential for DNA damage tolerance via two REV3L binding sites in mammalian DNA polymerase zeta. Nucleic Acids Res 43, 1000–1011 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Janin J, Miller S & Chothia C Surface, subunit interfaces and interior of oligomeric proteins. J Mol Biol 204, 155–164 (1988). [DOI] [PubMed] [Google Scholar]
- 42.Hara K et al. Purification, crystallization and initial X-ray diffraction study of human REV7 in complex with a REV3 fragment. Acta Crystallogr Sect F Struct Biol Cryst Commun 65, 1302–1305 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hanafusa T et al. Overlapping in short motif sequences for binding to human REV7 and MAD2 proteins. Genes Cells 15, 281–296 (2010). [DOI] [PubMed] [Google Scholar]
- 44.Wojtaszek J et al. Structural basis of Rev1-mediated assembly of a quaternary vertebrate translesion polymerase complex consisting of Rev1, heterodimeric polymerase (Pol) zeta, and Pol kappa. J Biol Chem 287, 33836–33846 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pustovalova Y, Bezsonova I & Korzhnev DM The C-terminal domain of human Rev1 contains independent binding sites for DNA polymerase eta and Rev7 subunit of polymerase zeta. FEBS Lett 586, 3051–3056 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pozhidaeva A et al. NMR structure and dynamics of the C-terminal domain from human Rev1 and its complex with Rev1 interacting region of DNA polymerase eta. Biochemistry 51, 5506–5520 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ohashi E et al. Identification of a novel REV1-interacting motif necessary for DNA polymerase kappa function. Genes Cells 14, 101–111 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pustovalova Y et al. Interaction between the Rev1 C-Terminal Domain and the PolD3 Subunit of Polζ Suggests a Mechanism of Polymerase Exchange upon Rev1/Polζ-Dependent Translesion Synthesis. Biochemistry 55, 2043–2053 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Doles J et al. Suppression of Rev3, the catalytic subunit of Polζ, sensitizes drug-resistant lung tumors to chemotherapy. Proc Natl Acad Sci U S A 107, 20786–20791 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu X et al. Enhancing tumor cell response to chemotherapy through nanoparticle-mediated codelivery of siRNA and cisplatin prodrug. Proc Natl Acad Sci U S A 110, 18638–18643 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Niimi K et al. Suppression of REV7 enhances cisplatin sensitivity in ovarian clear cell carcinoma cells. Cancer Sci 105, 545–552 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhao J et al. Mitotic arrest deficient protein MAD2B is overexpressed in human glioma, with depletion enhancing sensitivity to ionizing radiation. J Clin Neurosci 18, 827–833 (2011). [DOI] [PubMed] [Google Scholar]
- 53.Wojtaszek JL et al. A Small Molecule Targeting Mutagenic Translesion Synthesis Improves Chemotherapy. Cell 178, 152–159 e11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Actis ML et al. Identification of the first small-molecule inhibitor of the REV7 DNA repair protein interaction. Bioorg Med Chem 24, 4339–4346 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Green AA & Hughes WL Protein Fractionation on the Basis of Solubility in Aqueous Solutions of Salts and Organic Solvents. Methods in Enzymology 1, 67–90 (1955). [Google Scholar]
- 56.Jain T, Sheehan P, Crum J, Carragher B & Potter CS Spotiton: a prototype for an integrated inkjet dispense and vitrification system for cryo-TEM. J Struct Biol 179, 68–75 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wei H et al. Optimizing “self-wicking” nanowire grids. J Struct Biol 202, 170–174 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lander GC et al. Appion: an integrated, database-driven pipeline to facilitate EM image processing. J Struct Biol 166, 95–102 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Suloway C et al. Automated molecular microscopy: the new Leginon system. J Struct Biol 151, 41–60 (2005). [DOI] [PubMed] [Google Scholar]
- 60.Zheng SQ et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nature Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rohou A & Grigorieff N CTFFIND4: Fast and accurate defocus estimation from electron micrographs. Journal of Structural Biology 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tan YZ et al. Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Nature Methods 14, 793–796 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang K Gctf: Real-time CTF determination and correction. Journal of Structural Biology 193, 1–12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Roseman AM FindEM - a fast, efficient program for automatic selection of particles from electron micrographs. Journal of Structural Biology 145, 91–99 (2004). [DOI] [PubMed] [Google Scholar]
- 65.Punjani A, Rubinstein JL, Fleet DJ & Brubaker MA cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nature Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 66.Scheres SHW RELION: Implementation of a Bayesian approach to cryo-EM structure determination. Journal of Structural Biology 180, 519–530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zivanov J et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 7, e42166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bepler T et al. Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat Methods (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Scheres SHW & Chen SX Prevention of overfitting in cryo-EM structure determination. Nature Methods 9, 853–854 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rosenthal PB & Henderson R Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. Journal of Molecular Biology 333, 721–745 (2003). [DOI] [PubMed] [Google Scholar]
- 71.Pettersen EF et al. UCSF chimera - A visualization system for exploratory research and analysis. Journal of Computational Chemistry 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 72.Klaholz BP Deriving and refining atomic models in crystallography and cryo-EM: the latest Phenix tools to facilitate structure analysis. Acta Crystallogr D Struct Biol 75, 878–881 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kucukelbir A, Sigworth FJ & Tagare HD Quantifying the local resolution of cryo-EMEM density maps. Nature Methods 11, 63–65 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Grant T, Rohou A & Grigorieff N cisTEM, user friendly software for single-particle image processing. Elife 7, e35383 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallographica Section D-Structural Biology 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 76.Brown A et al. Tools for macromolecular model building and refinement into electron cryo-microscopy reconstructions. Acta Crystallogr D Biol Crystallogr 71, 136–153 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen VB et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallographica Section D-Structural Biology 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Barad BA et al. EMRinger: side chain directed model and map validation for 3D cryo-electron microscopy. Nature Methods 12, 943–946 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.DeLano WL & Lam JW PyMOL: A communications tool for computational models. Abstracts of Papers of the American Chemical Society 230, U1371–U1372 (2005). [Google Scholar]
- 80.Sievers F et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7, 539 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kelley LA, Mezulis S, Yates CM, Wass MN & Sternberg MJ The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10, 845–858 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pei J & Grishin NV PROMALS: towards accurate multiple sequence alignments of distantly related proteins. Bioinformatics 23, 802–808 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.