

Abstract
The HIV/AIDS epidemic is one of the world’s most serious health challenges. Although combination antiretroviral therapy provides effective viral suppression, current medicines used against HIV cannot completely eradicate the infectious disease and often have associated toxicities and severe side effects in addition to causing drug resistance. Therefore, the continued development of new antiviral agents with diverse structures and novel mechanisms of action remains a vital need for the management of HIV/AIDS. Natural products are an important source of drug discovery, and certain triterpenes and their analogues have demonstrated potential as pharmaceutical precursors for the treatment of HIV. Over the past decade, natural triterpenoids and analogues have been extensively studied to find new anti-HIV drugs. This review discusses the anti-HIV triterpenoids and analogues reported during the period of 2009 to 2019. The article includes not only a comprehensive review of the recent anti-HIV agent development from the perspective of medicinal chemistry, but also discusses structure–activity relationship analyses of the described triterpenoids.
Keywords: anti-HIV activity, triterpenes, natural products, medicinal plants, structural modification
1 |. INTRODUCTION
As of 2017, about 36.9 million people worldwide were estimated to be living with HIV-1 infection.1,2 Although combination antiretroviral therapy (cART) provides effective viral suppression, current medicines cannot cure HIV-1 infection and often have associated toxicities and severe side effects in addition to causing drug resistance. For example, although access to highly active antiretroviral therapy (HAART) has increased in recent years, rising to nearly 22 million people (59% of people living with HIV) in 2017, this therapy cannot achieve the ultimate goal of complete eradication of HIV and patients have to undergo long-term treatment, which can result in permanent damage to tissues or organs. Besides, the virus rapidly develops resistance to the drugs. Both problems underline the need to develop new therapeutic strategies against HIV/AIDS to complement the existing ones.3,4 During the last decade, much effort has been made to find effective anti-HIV agents from natural products.5,6 Many classes of natural product derivatives have been evaluated with varying success.7,8 For example, triterpenoids and their analogues have been extensively studied for their diverse structures and anti-HIV activities.9 Most recently, triterpenoids have been found to influence a broad range of virus-host fusion by wrapping the HR2 domain widespread in viral envelopes.10 As early as 1994, betulinic acid (1) (Figure 1) was shown to inhibit the infectivity of HIV-1 in vitro.11 Subsequently, numerous betulinic acid derivatives were synthesized with the goal to develop anti-HIV drugs.12,13 Systematic structural modifications ultimately resulted in the identification of 3-O-(3′,3′-dimethylsuccinyl) betulinic acid (DSB), known as bevirimat (BVM, 2) (Figure 1). BVM plays significant anti-HIV activity in H9 lymphocytic cell lines with an EC50 value of less than 0.35 nM and a selectivity index of 20,000.14 BVM is defined as a novel strategy against HIV and termed a maturation inhibitor. BVM inhibits HIV-1 protease, which causes the last cleavage of the Gag polyprotein, leading to the accumulation of the p25 capsid-small peptide 1 (SP1) intermediate and thus resulting in noninfectious HIV-1 virions. BVM dimeglumine (MPC-4326, formerly PA-457), a potent HIV-1 maturation inhibitor, reached the most advanced stage of drug development.15 Although BVM exhibited promising pharmacokinetic (PK) profiles in clinical trials and was shown to be safe and effective in reducing viral loads in HIV-1-infected patients, its effectiveness was compromised by the high baseline drug resistance of HIV-1 variants with polymorphism in the putative drug binding site.16 Naturally occurring polymorphisms in the SP1 region of Gag (e.g., SP1-V7A) (Figure 2) led to a variable response in some BVM-treated patients.17–19 The reduced susceptibility of SP1-polymorphic HIV-1 to BVM led to the discontinuation of its clinical development.20,21 In addition, disadvantages of BVM include its poor solubility in aqueous and biologically pertinent organic media as well as high plasma protein binding. Eventually, the development of BVM was halted in 2010 after reaching Phase IIb clinical trials. Thereafter further investigations of BVM derivatives are being carried out to improve polymorphic virus coverage, reduce protein binding and obtain better pharmaceutical properties.
FIGURE 1.
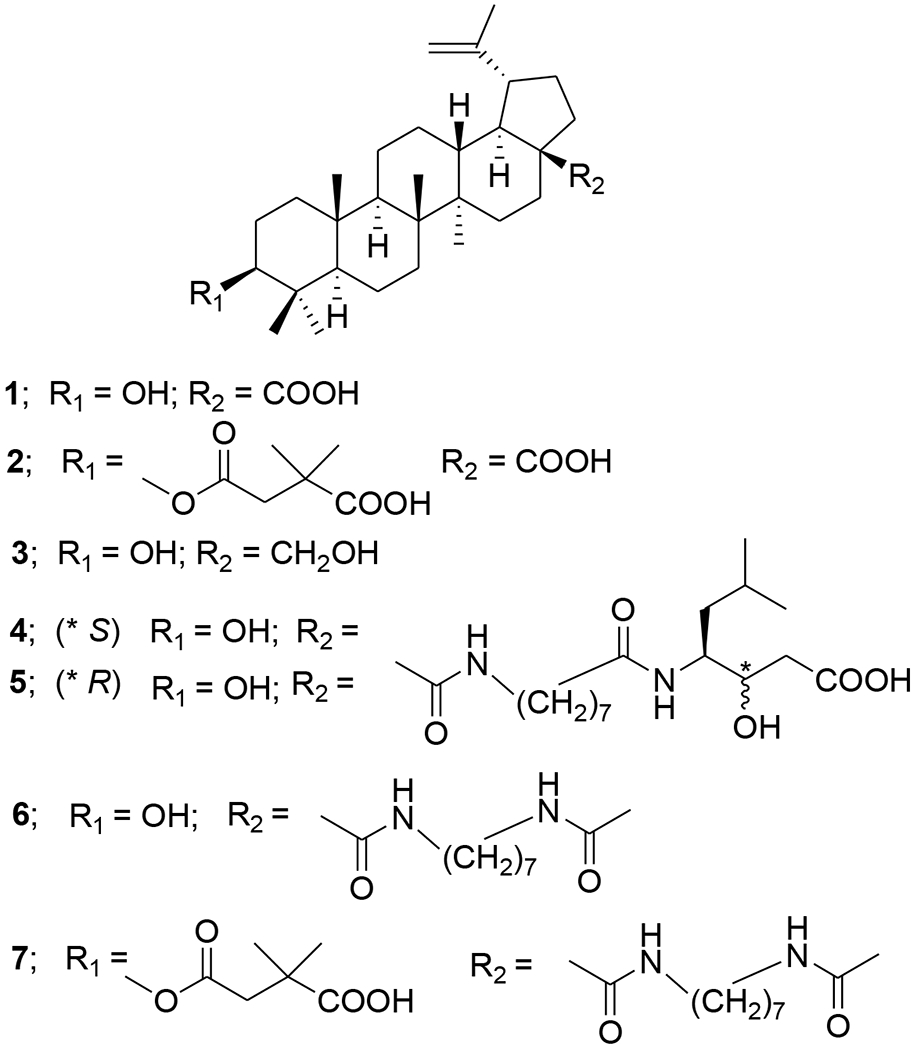
Chemical structures of compounds 1–7
FIGURE 2.
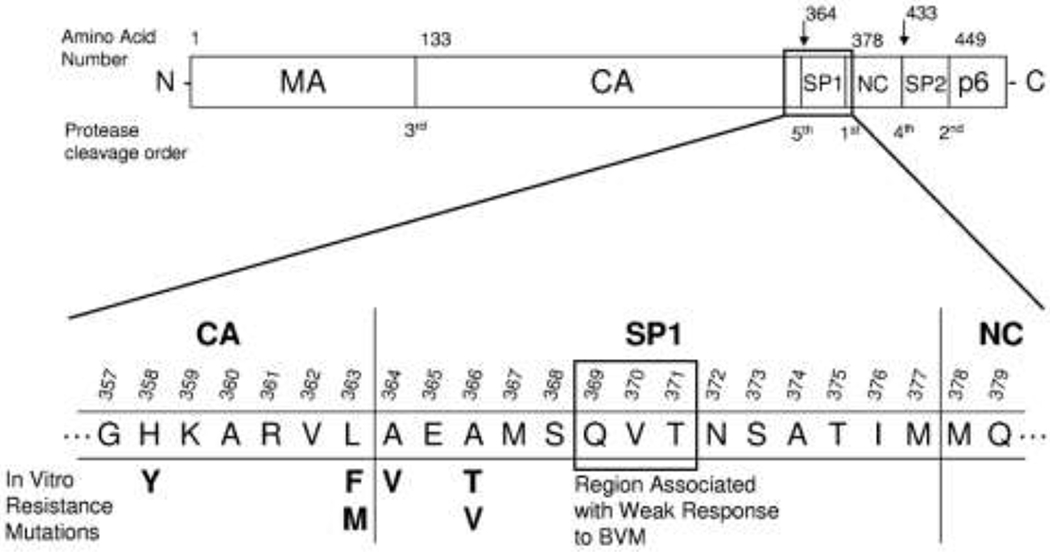
Schematic of HIV-1 Gag polyprotein showing amino acid residues associated with bevirimat susceptibility. MA, matrix; CA, capsid; SP1, small peptide 1; NC, nucleocapsid; SP2, small peptide 2. Reproduced with permission from [19].
Two important reviews by Kuo et al.22 and Singh and Bodiwala23 were published just before the period covered in this article, the former more specifically concentrating on anti-HIV triterpenoids and analogues. Two additional extensive reviews of the literatures reported on anti-HIV triterpenoids and derivatives were also published.24,25 Acyl derivatives of triterpenic oximes exhibit important pharmacological, including anti-HIV, activity.26 Related chemistry and pharmacology of synthetic triterpenoid dimers obtained from natural compounds were reviewed recently.27 The structures and anti-HIV activities of protostane and fusidane triterpenoids covering the literature until 2013 were briefly reviewed (together with several other activities).28 Verma discussed a raft-targeting method for prevention and therapy of AIDS using natural products, especially euphane-type triterpenes.29 In another review, natural products including lignans, triterpenes, chlorogenic acid derivatives and farnesyl hydroquinones, in vivo metabolites and synthesized derivatives as potential anti-viral agents were described.30 A review covering the literature from 1991 to 2012 presented microbial transformation of diterpenoids and some triterpenoids with anti-HIV activities.31 In 2014, the antitumor and antiviral activities of pentacyclic triterpenes were discussed.32 Herein, this review describes the recent advances on anti-HIV triterpenoids and synthetic analogues covering the period from 2009 till 2019, with more than 300 anti-HIV triterpenes discussed. The article includes not only a comprehensive review of the recent anti-HIV drug development from the perspective of medicinal chemistry but also discusses structure–activity relationship analyses of the described triterpenoids.
2 |. PENTACYCLIC TRITERPENOID
2.1 |. The lupane group
Plant-derived pentacyclic triterpenoids of lupane families provide a versatile structural platform for the discovery of new biologically active compounds. Even small structural alterations in these triterpenoid derivatives can cause remarkable differences in their activity, making a convincing case for a systematic study of structure–activity relationships in this compound class. The multi-activity profile of lupane-type terpenoids can be a benefit for overcoming HIV-1 resistance by simultaneously affecting multiple targets. Both betulinic acid (1) and betulin (3) (Figure 1) are important lupane triterpenoids that exhibit many favorable biological effects, including anti-HIV, anti-cancer, anti-malarial and anti-inflammatory properties, but cause minimal toxicity to unaffected cells.33 Betulinic acid is commonly found in some species of the family Betulaceae, while 3 is a major constituent in the bark of white-barked birch trees with isolated yields up to 22% (dry weight) and can be easily converted synthetically to 1 in high yields.34 Many studies have been devoted to the investigation of betulinic acid, betulin and their derived compounds. The last related review was published in 2014.35 It has been pointed out that structural changes at different positions of betulinic acid/betulin can result in significant differences in the anti-HIV mechanism of action. For example, betulinic acid derivatives altered at the C-28 position are HIV-1 entry inhibitors such as compounds RPR103611 (4), IC9564 (5) and A43D (6) (Figure 1), among which A43D was effective against multiple HIV subtypes and displayed the strongest inhibition toward the clade C HIV-1 strains.36 On the other hand, betulinic acid analogues modified at the C-3 rather than C-28 position, like BVM, are HIV-1 maturation inhibitors. Furthermore, simultaneous modifications at C-3 and C-28 led to the identification of A12–2 (7) (Figure 1), a bifunctional HIV inhibitor with an EC50 value of 0.0026 μM at least 20 times more potent than either 2 or 5.37 Over the past ten years, extensive work has been directed towards the optimization of the C-3 and C-28 side chains of betulinic acid and betulin derivatives and analogues (Table 1). C-3 modifications comprise oxidation, acylation, conjugation and heterocycle substitution, while C-28 alterations include amination, esterification and conjugations. In addition, chemical modifications at other positions like C-30 were also reported. Sousa et al. provided insight into the functionalization of betulinic acid and its analogues in the latest review.38
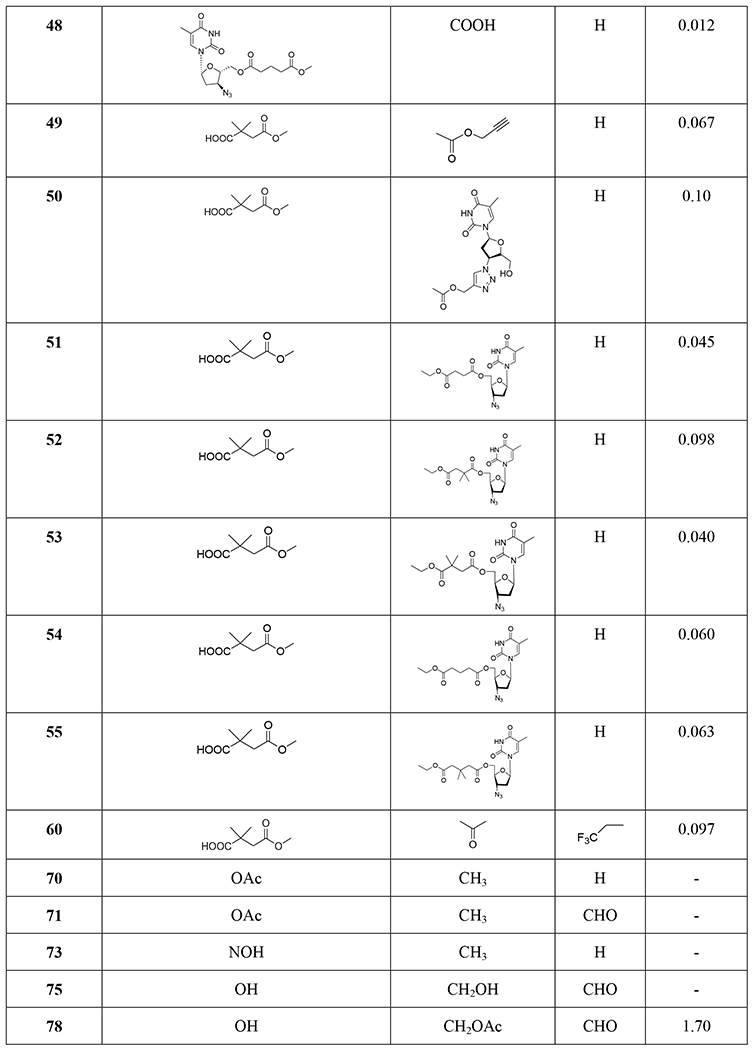
TABLE 1.
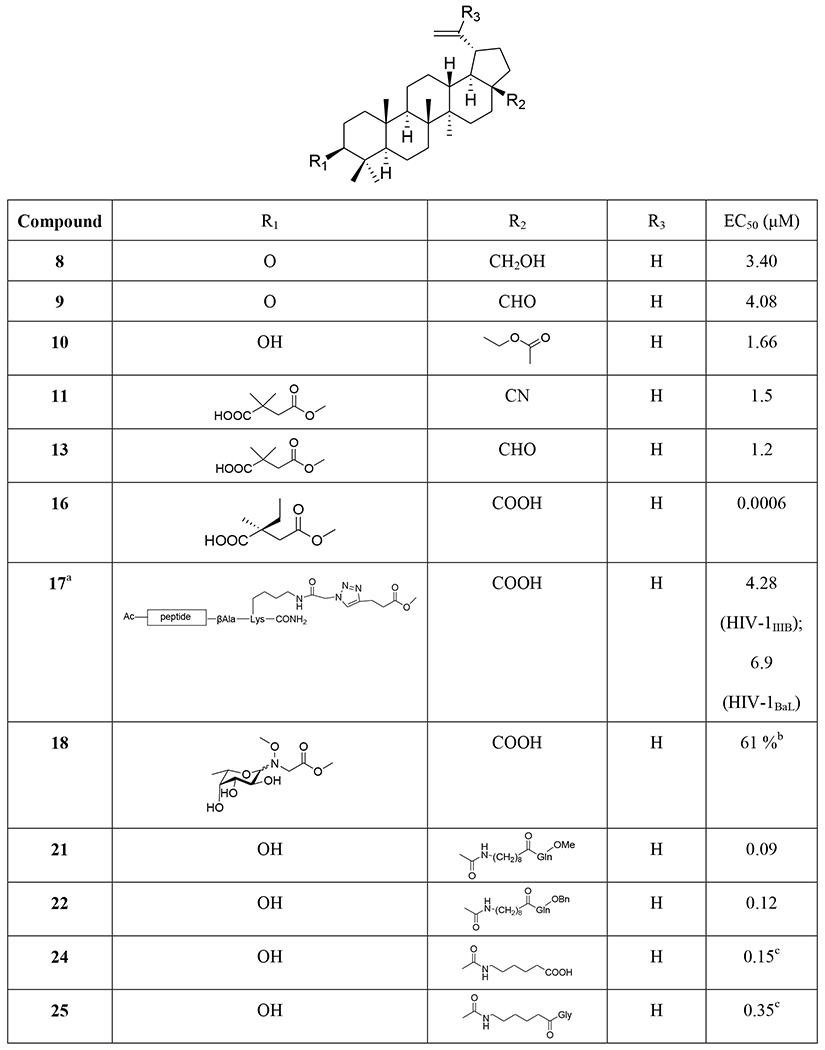
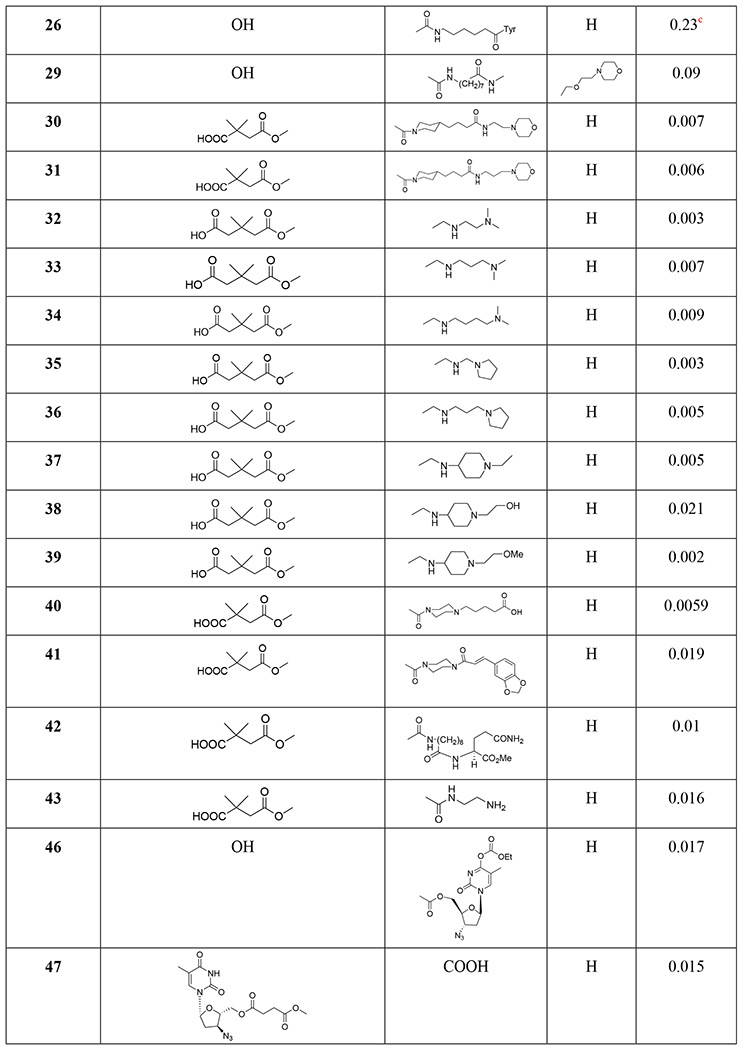
Structures and anti-HIV activity of betulinic acid and betulin derivatives and analogues
 |
 |
 |
Peptide: NNYTSLIHSLIEESQNQQEKNEQELL;
Antiviral activity was assessed by the percent increase of cytoprotective effect over untreated HIV-1-infected cells
anti-HIV-2 activity.
Three betulin analogues (8~10) (Table 1) were found to inhibit HIV-1 infection by acting on multiple steps in the HIV-1 replication cycle in both MT-2 cells and primary human T cells. Betulone (8) targets reverse transcription, integration, viral transcription, Gag production and maturation. 3-Oxo-lup-20(29)-en-28-al (9) inhibits HIV-1 infection by targeting integration, viral transcription and Gag production. 28-Acetoxy-3β-hydroxy-lup-20(29)-en-30-al (10) targets reverse transcription, viral transcription and Gag production. Compounds 8–10 inhibited HIV-1 infection of human primary lymphocytes and infections with protease inhibitor- and BVM-resistant HIV-1 variants with similar IC50 values.39 Betulinic aldehyde and nitrile derivatives were identified to show anti-HIV-1 activity using a single-cycle replication assay, which can differentiate whether a derivative targets the late or early steps in the viral life cycle. The betulinic nitrile derivative (11) and aldehyde derivatives (12–15) (Tables 1 and 2 and Figure 3) inhibit a late step in virus replication in HEK 293T cells.40
TABLE 2.
Structures and anti-HIV activity of dihydrobetulinic acid derivatives and analogues
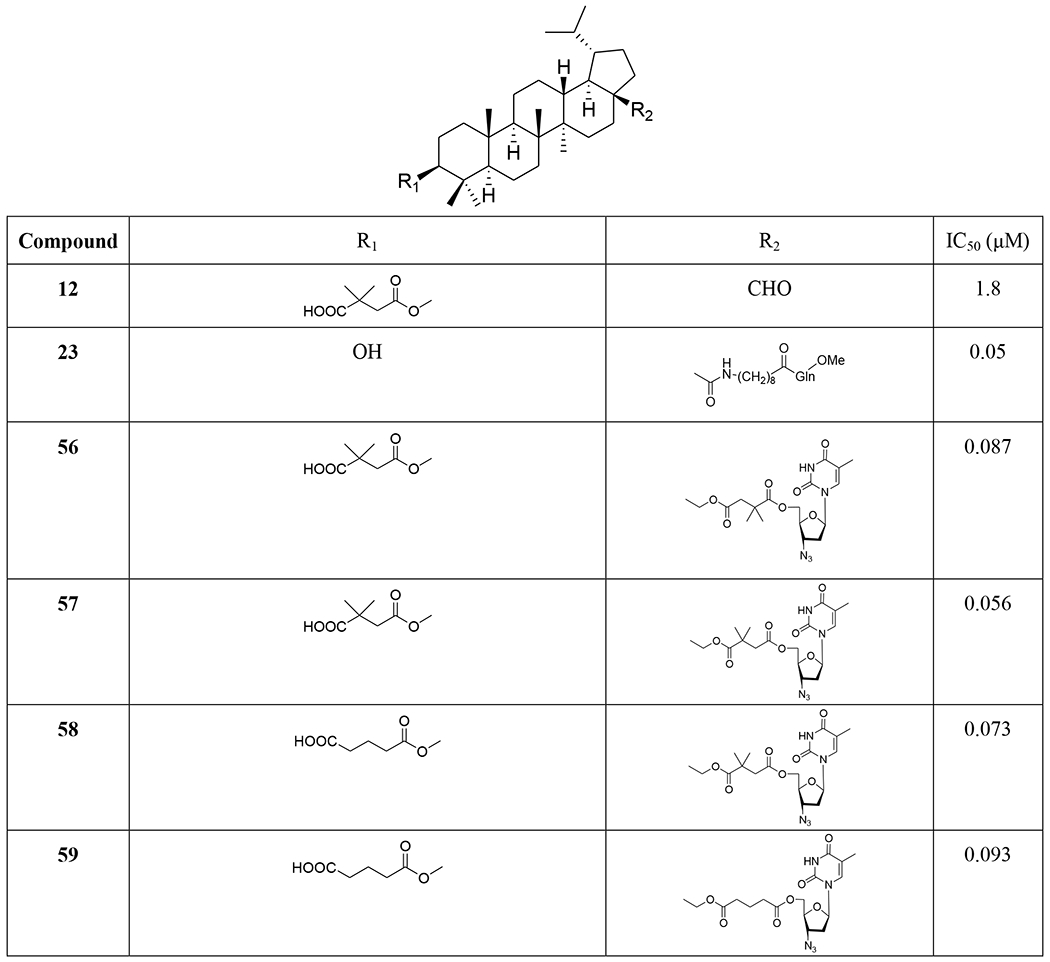 |
FIGURE 3.
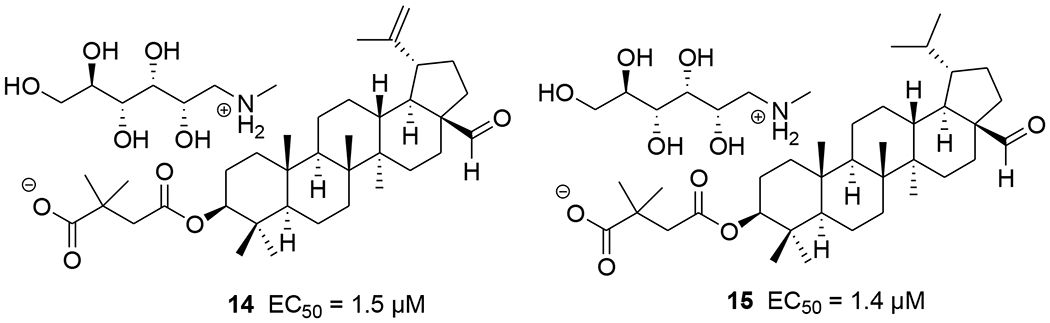
Chemical structures of compounds 14 and 15
Different C-3 conformationally restricted 3-O-acyl derivatives were synthesized to explore the conformational space of the C-3 pharmacophore of betulinic acid. The 3'S isomer of compound 16 (Table 1) displayed slightly better activity than 2 in acutely infected MT-2 cell lines.41 Click chemistry was used to synthesize hybrid molecules of triterpene sapogenins and HBD (helix zone-binding domain)-containing peptides. P26–BApc (17) (Table 1) exhibited anti-HIV-1 activity against both T20-sensitive and -resistant HIV-1 strains and improved pharmacokinetic properties.42 The sugars attached to pharmaceutically important natural products could lead to improved key pharmacological properties and/or molecular mechanisms of action. A 37-member library of betulinic acid C3-neoglycosides was synthesized and each glycosylated derivative was tested for anti-HIV activity in CEM-SS cells infected with HIV-1IIIB. Nineteen of 32 ester-linked compounds displayed at least a twofold improvement over betulinic acid. Among these compounds, L-Fuc derivative (α:β/1:2) 18 (Table 1) was the most active.43 In 2016, Liu et al.44 synthesized a series of C-3 phenyl- and heterocycle-substituted derivatives of C-3 deoxybetulinic acid and deoxybetulin. When compared with BVM, betulinic acid-derived analogue 19 containing a 4-subsituted benzoic acid moiety exhibited comparable anti-HIV activity against wild-type virus in MT-2 cells using variants of the NLRepRlucP373S virus in a multiple cycle assay. Furthermore, compared with 2, the potency of 19 was less affected by the presence of human serum displaying a similar pharmacokinetic profile in rats. In addition, 4-benzoic acid deoxybetulin analogue 20 exhibited comparable antiviral potency toward wild-type and V370A viruses compared with 19, while in the presence of human serum albumin (HAS), 20 was threefold more potent than 19 (Figure 4). Compounds 19 and 20 demonstrated potency towards the polymorphic V370A with EC50 values of 0.23 and 0.21 μM, respectively. 44
FIGURE 4.
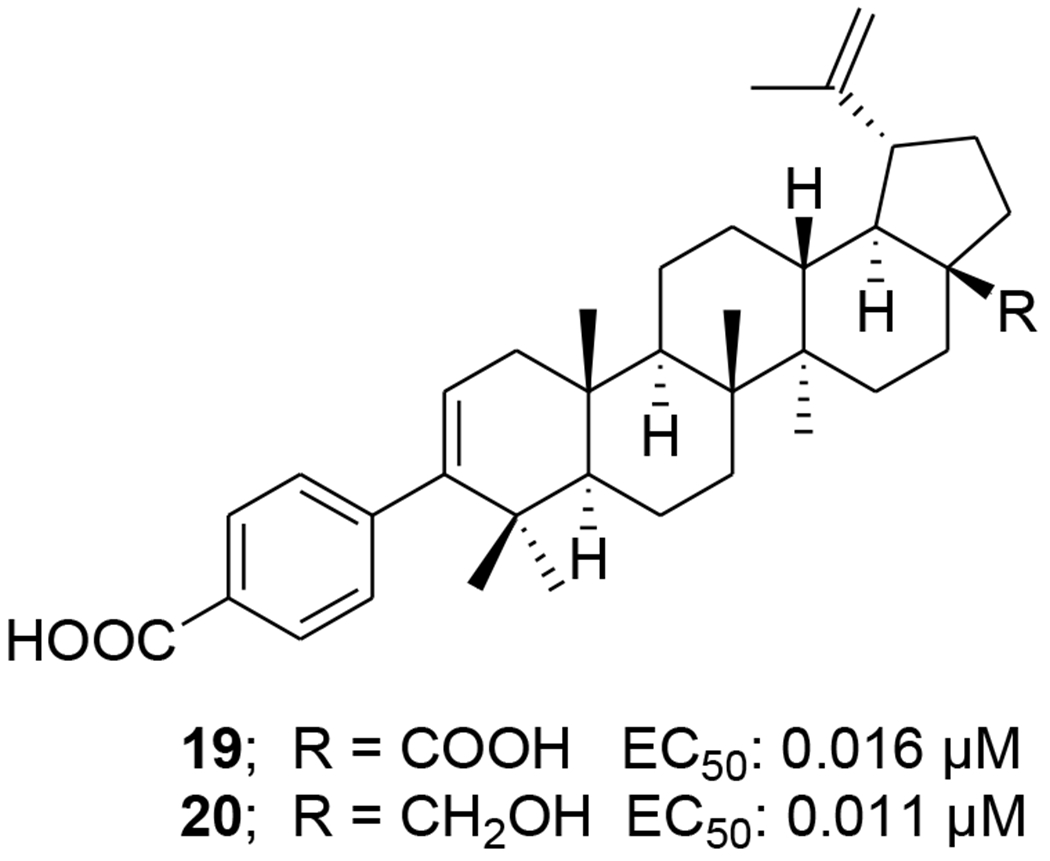
Chemical structures of compounds 19 and 20
Three C-28 glutamine ester derivatives of betulinic acid (21–23) (Tables 1 and 2) were more potent against BVM-resistant viruses in an HIV-1 multi-cycle replication assay (Table 3). In addition, compounds 21–23 showed markedly improved microsomal stability compared with 6.45 Betulinic acid C-28 derivatives (24–26) (Table 1) displayed selective anti-HIV-2 activity at nanomolar concentrations. A shorter C-28 side chain was required for optimal anti-HIV-2 activity.46 Different ionic derivatives of betulinic acid were prepared by straightforward coupling chemistry; they displayed significantly improved water solubility without disrupting the structurally related bioactivity. These compounds had lower IC50 values towards HIV-1 protease than 1; particularly, 27 and 28 (Figure 5) showed IC50 values roughly two and three times lower, respectively, than that of 1.47
TABLE 3.
Antiviral activity of compounds 21–23 toward BVM-resistant virus
| Compound | 21 | 22 | 23 |
|---|---|---|---|
| V370A/EC50 (μM) | 0.02 | 0.05 | 0.02 |
FIGURE 5.
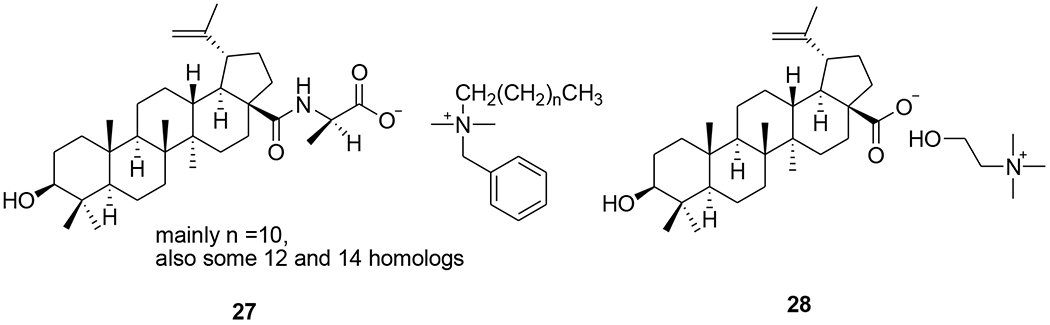
Chemical structures of compounds 27 and 28
Various 28,30-disubstituted and 3,28-disubstituted betulinic acid derivatives were also synthesized. Compound 29 (Table 1) showed improved solubility and anti-HIV potency. Using a cyclic secondary amine to form the C-28 amide bond significantly increased the metabolic stability of the derivatives in pooled human liver microsomes. Compounds 30 and 31 (Table 1) displayed potent anti-HIV activity.48 A collection of C-28 alkyl amine derivatives of BVM was synthesized and tested for their ability to block CA-SP1 processing and virus replication by using wild-type and a V7A variant of NL4–3. Some derivatives (32–39) (Tables 1 and 4) showed markedly greater potency than BVM against an HIV-1 clade B clone (NL4–3) and robust antiviral activity against a variant of NL4–3 containing the V7A polymorphism in SP1. One of the most potent compounds (37) also strongly inhibited a multi-clade panel of primary HIV-1 isolates.49 Among a series of synthetic betulinic acid derivatives with C-28 substitution, all active compounds showed only anti-maturation effects, as confirmed by TZM-bl assay, in blocking the HIV replication. Compound 40 (Table 1) exhibited the best anti-HIV activity and good in vitro metabolic stability in pooled human liver microsomes.50
TABLE 4.
Antiviral activity of compounds 32–39 toward BVM-resistant virus
| Compound | 32 | 33 | 34 | 35 | 36 | 37 | 38 | 39 |
|---|---|---|---|---|---|---|---|---|
| V7A/EC50 (μM) | 0.009 | 0.014 | 0.022 | 0.026 | 0.022 | 0.010 | 0.008 | 0.019 |
The integration of privileged motifs into promising natural product skeletons is an effective strategy for discovering potent derivatives. Two “privileged fragments”, caffeic acid and piperazine were integrated into BVM. Compound 41 (Table 1) is a maturation inhibitor with improved metabolic stability and its activity was increased by threefold against NL4–3 and 51-fold against NL4–3/V370A (EC50: 0.15 μM).51 Compound 42 (Tables 1 and 5) exhibited much improved activity against several HIV-1 strains carrying BVM-R polymorphisms and was at least 20-fold more potent than BVM against the replication of NL4–3/V370A.52 To improve the water solubility of BVM, different hydrophilic substituents were added at the C-28 position. Compound 43 (Table 1) showed higher hydrophilicity associated with a 2.5-fold increase in activity, a higher selectivity index and a better antiviral profile. Also, NMR indicated a direct interaction between 43 and the domain CA-SP1eNC.53 In 2017, a series of betulin-derived a-keto amides was identified as inhibiting HIV-1 maturation. When tested in a panel of 62 HIV-1 isolates covering a diversity of CA-SP1 genotypes including A, AE, B, C, and G using a PBMC based assay, GSK8999 (44) (Figure 6) was potent against 57 of 62 isolates. Particularly, compound 44 showed potency towards the polymorphic V370A and T371A with IC50 values of 0.025 and 0.008 μM, respectively.54 Furthermore, GSK2838232 (45), the only α-keto amide betulin derivative investigated in a Phase I clinical study, had low to moderate relative bioavailability (6%–40%) and was metabolized through hepatic Phase I oxidation, Phase II glucuronidation and biliary excretion, according to these preclinical studies. In clinical studies, GSK2838232 (100 and 200 mg) with 100 mg ritonavir for 11 days exhibited good safety but was significantly influenced by CYP3A4 and P-gp inhibitors.55
TABLE 5.
Antiviral activity of compound 42 toward BVM-resistant viruses
| Compound | EC50 (μM) | |||
|---|---|---|---|---|
| V370A | V370Δ | T371Δ | V362I | |
| 42 | 0.16 | 0.32 | 0.067 | 0.016 |
FIGURE 6.
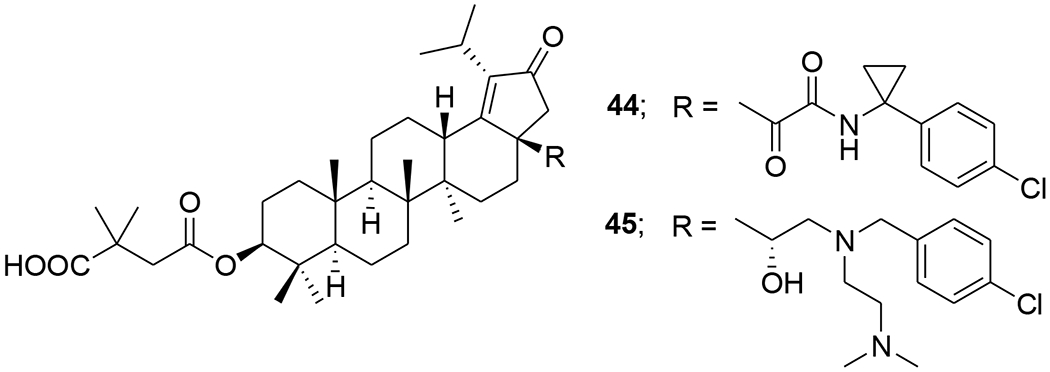
Chemical structure of compounds 44 and 45
Recently, conjugations between betulinic acid/betulin and AZT have been reported. A one-pot synthesis of ester-linked conjugates of betulinic acid with AZT and its derivatives or with 3TC was accomplished. This direct synthesis provided a scalable procedure for preparing anti-HIV hybrid conjugates. Compounds 46–48 (Table 1) exhibited very good anti-HIV activity against HIV-1IIIB infected MT-4 cells.56 Alternatively, betulin/betulinic acid conjugation with AZT was achieved via a triazole linkage by click chemistry. Compounds 49 and 50 (Table 1) showed potent anti-HIV activity.57 Various trisubstituted betulinic acid derivatives were prepared containing 3-O-acyl and 28-amide side chains and a propynyl group at the С-2 position of ring A of the lupane core. Then, a series of C-2 triazole-linked bioconjugates of lupane triterpenoids with AZT were synthesized based on a CuI-catalyzed 1,3-cycloaddition between alkynes and azides (Figure 7). The proposed strategy concerning AZT–betulinic acid hybrid molecules as potential anti-HIV agents makes it possible to vary the C-3 and C-28 pharmacophores in the triterpene moieties.58 Fourteen conjugates of 3,28-di-O-acylbetulins with AZT were prepared and nine conjugates (51–59) (Tables 1 and 2) exhibited potent anti-HIV activity with EC50 values ranging from 0.040 to 0.098 μM in HIV-1NL4–3 infected MT-4 cells.59
FIGURE 7.
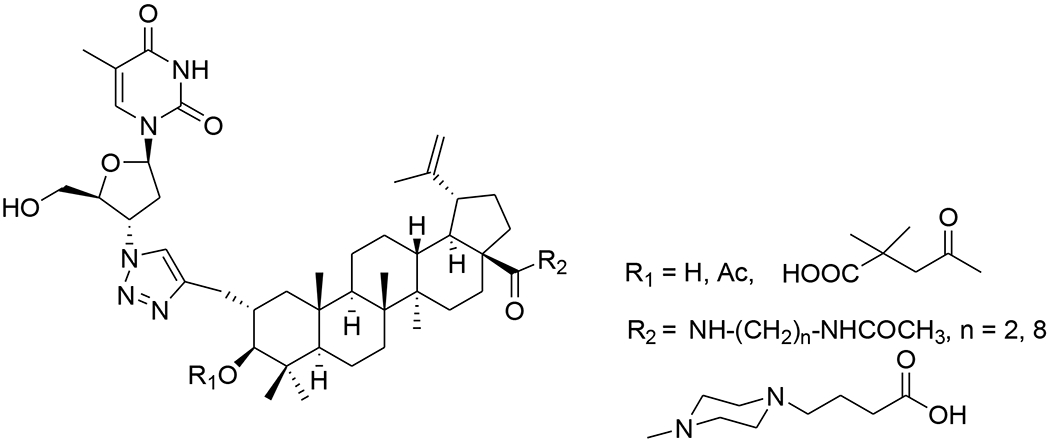
Chemical structure of AZT–betulinic acid conjugates
Several fluorinated derivatives of BVM were synthesized. Compound 60 (Table 1), which has a trifluoromethyl group added to C-30 of its isopropenyl group, exhibited similar potency as BVM against HIV-1NL4–3.60
At the C-3 position, benzoic acid can be a suitable replacement for the dimethyl succinate side chain of BVM. SAR studies showed that the benzoic acid unit conferred topographical constraint on the pharmacophore and was associated with a lower shift in potency in the presence of human serum albumin. To possibly improve the polymorphic coverage of betulinic acid derivatives, a series of C-3 benzoic acid-substituted betulinic acid derivatives was synthesized through modifications at the C-28 position. The dimethylaminoethyl amides 61 and 62 (Figure 8) exhibited improved potency toward BVM-resistant viruses and increased C24 values in rat oral PK studies (Table 6).61 In the continued efforts, C28 amine derivatives were designed and synthesized. Compared with the C-28 amide series, the C-28 amine derivatives exhibited further improvements in HIV-1 inhibitory activity toward polymorphisms in the Gag polyprotein as well as improved activity in the presence of human serum. Compared to the C-28 amide 61, the C-28 amine compound 63 (Figure 8) containing a thiomorpholine dioxide showed two- to four-fold improved potency towards the screened viruses, such as wild type, V370A and ΔV370, exhibited low shifts in the EC50 values toward V370A and ΔV370 viruses in the presence of human serum or human serum albumin, and demonstrated improved potency towards the polymorphic T371A and V362I virus variants as well as low plasma exposure following oral administration to rats (Table 7).62
FIGURE 8.
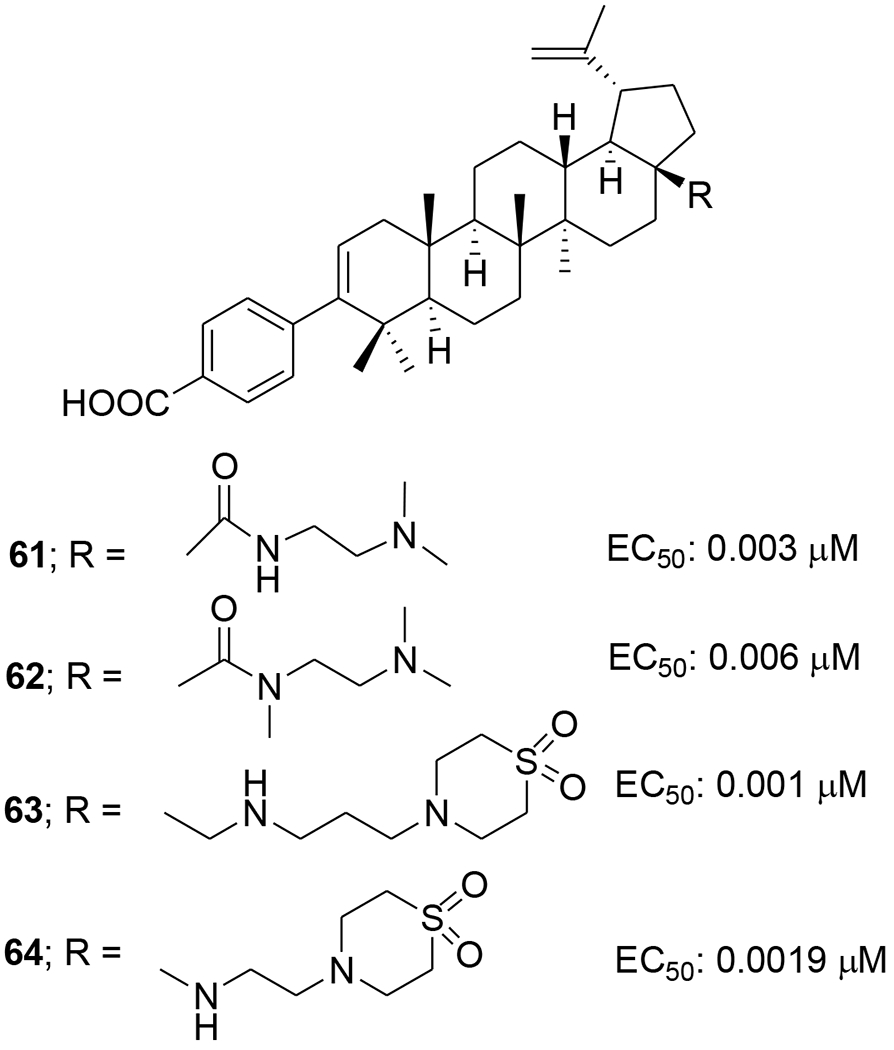
Chemical structures of compounds 61–64
TABLE 6.
Antiviral activity of compounds 61 and 62 toward BVM-resistant viruses
| Compound | EC50 (μM) | |
|---|---|---|
| V370A | ΔV370 | |
| 61 | 0.008 | 0.031 |
| 62 | 0.038 | 0.025 |
TABLE 7.
Antiviral activity of compounds 63 and 64 toward BVM-resistant viruses
| Compound | EC50 (μM) | |||||||
|---|---|---|---|---|---|---|---|---|
| V370A | ΔV370 | Q369H | T371A | V362I | V370M | V370A/ΔT371 | ΔV371 | |
| 63 | 0.0025 | 0.014 | - | 0.0017 | 0.00094 | - | - | - |
| 64 | 0.0027 | 0.013 | 0.0019 | 0.002 | 0.0045 | 0.0028 | 0.0036 | 0.0073 |
-: Not determined
Structure–activity relationships (SARs) led to the design of specific C-17 amine moieties and ultimately enabled the discovery of BMS-955176, also known as GSK3532795 (64) (Figure 8), as a second-generation maturation inhibitor that combines broad coverage of polymorphic viruses (EC50 <15 nM toward a panel of common polymorphisms representative of 96.5% HIV-1 subtype B virus) with a favorable pharmacokinetic profile in preclinical species.63 Compound 64 exhibited potent activity (EC50: 3.9 nM) toward a library (n = 87) of gag/pr recombinant viruses representing 96.5% of subtype B polymorphic Gag diversity near the CA/SP1 cleavage site and a median EC50 of 21 nM toward a library of subtype B clinical isolates assayed in peripheral blood mononuclear cells (PBMCs) (Table 7). Potent activity was maintained against a panel of reverse transcriptase, protease, and integrase inhibitor-resistant viruses, with EC50 values like those for the wild-type virus. A 5.4-fold reduction in EC50 occurred in the presence of 40% human serum plus 27 mg/mL of human serum albumin, which corresponded well to an in vitro measurement of 86% human serum binding. Time-of-addition and pseudotype reporter virus studies confirmed that the compound acts late in the virus replication cycle. Compound 64 inhibits HIV-1 protease cleavage at the CA/SP1 junction within Gag in virus-like particles (VLPs) and HIV-1-infected cells, and it binds reversibly and with high affinity to assembled Gag in purified HIV-1 VLPs. Finally, in in vitro combination studies, the compound showed no antagonistic interactions with representative antiretrovirals of other mechanistic classes.64 A concise and scalable synthesis of 64 starting from betulin in seven steps and 47% overall yield has been described. The synthesis is framed by an oxidation strategy highlighted by a CuI mediated aerobic oxidation of betulin, a highly selective PIF mediated dehydrogenation of an oxime, and a subsequent Lossen rearrangement, which occurs through a unique reaction mechanism for the installation of the C17 amino functionality.65 Compound 64 demonstrated better anti-HIV potency than BVM with no significant safety issues in its Phase IIa studies. However, Phase IIb studies with 64 were terminated due to gastrointestinal intolerance.66
In addition, 3D-QSAR and molecular docking studies were also applied to help define the structural requirements responsible for the anti-HIV activity of betulinic acid derivatives. In 3D-QSAR studies, both the CoMFA and CoMSIA methods were satisfactory based on the statistical validation results as well as the contour map analysis. Molecular docking was used to explore the binding mode between these derivatives and HIV gp120. The correlation of the results obtained from 3D-QSAR and docking studies can serve as useful guidelines for the further modification of betulinic acid to produce useful anti-HIV agents.67
Two A-seco lupane derivatives (65 and 66) (Figure 9) showed weak inhibitory activity against HIV-1 protease (IC50 15.7 and 25.4 μM, respectively).68 In another report, the synthesis and evaluation of anti-HIV activity of mono- and diamides of 2,3-secolupane acids were reported, and compound 67 (Figure 10) showed anti-HIV activity.69 Lupane triterpene oximes (68 and 69) (Figure 11) were synthesized using oxonitrile recyclization and the ThorpeeZiegler cyclization. Both 68 and 69 effectively suppressed the reproduction of HIV-1 in vitro (EC50 0.06 μM).70 In a concurrent study, lupeol and betulin derivatives were prepared and evaluated for their ability to perturb X4- and R5-tropic HIV-1-envelope (Env)-mediated fusion membrane in a cell-to-cell fusion model. Compounds 70–75 (Table 1 and Figure 12) showed a wide arrange of inhibitory activity.71 Recently, six novel lupane-type C-3 triterpenones with C-28 heterocyclic moieties were synthesized. Compound 76 (Figure 13) (EC50 < 10 nM) showed significant anti-HIV potency in MT2 cells infected with 92HT599 in a 40% serum binding assay.72
FIGURE 9.
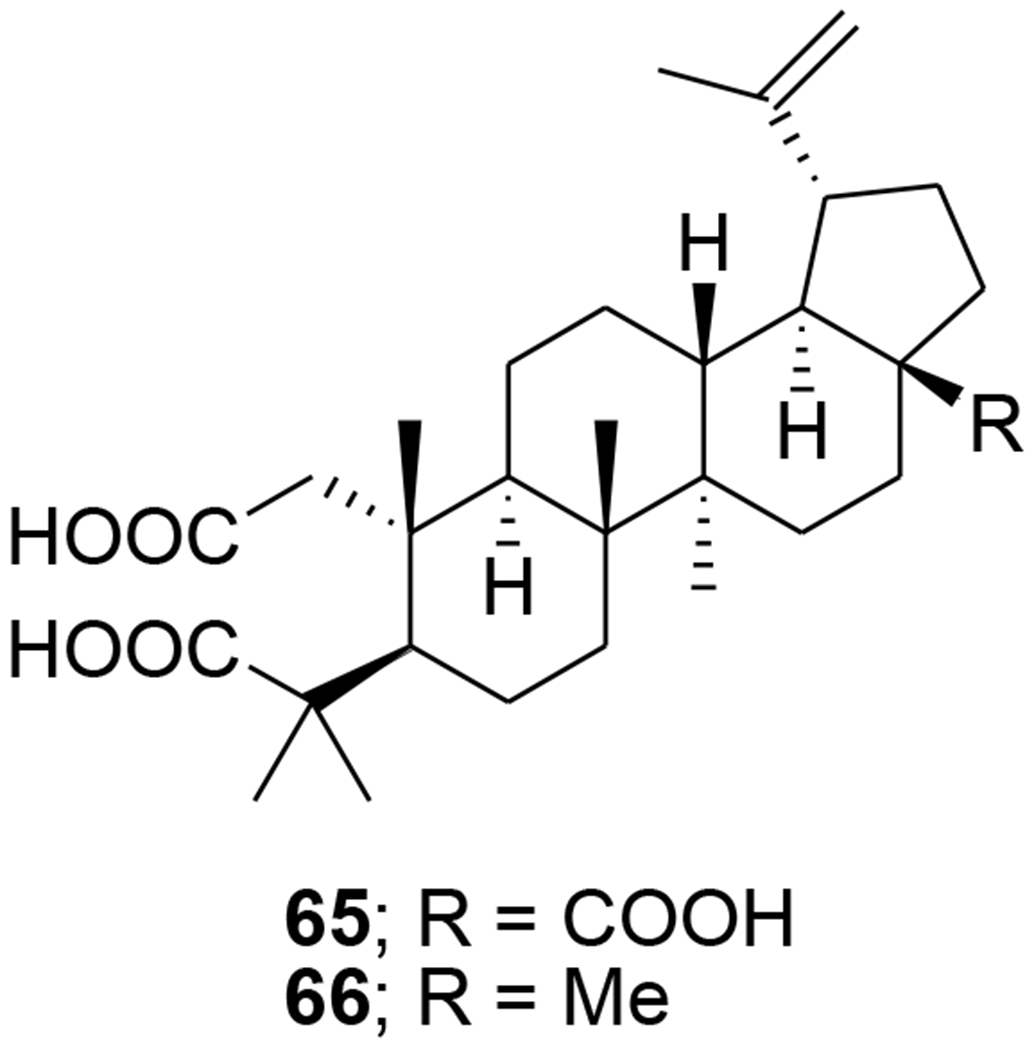
Chemical structures of compounds 65 and 66
FIGURE 10.
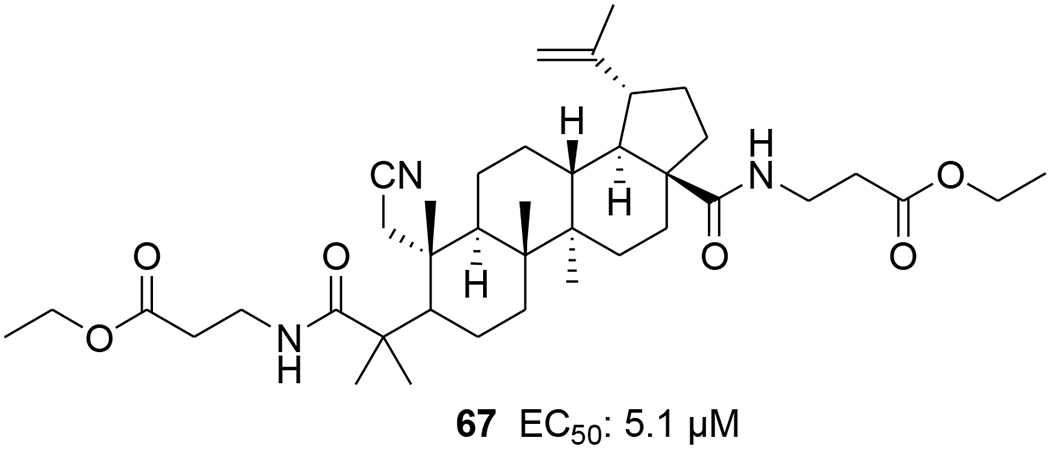
Chemical structure of compound 67
FIGURE 11.
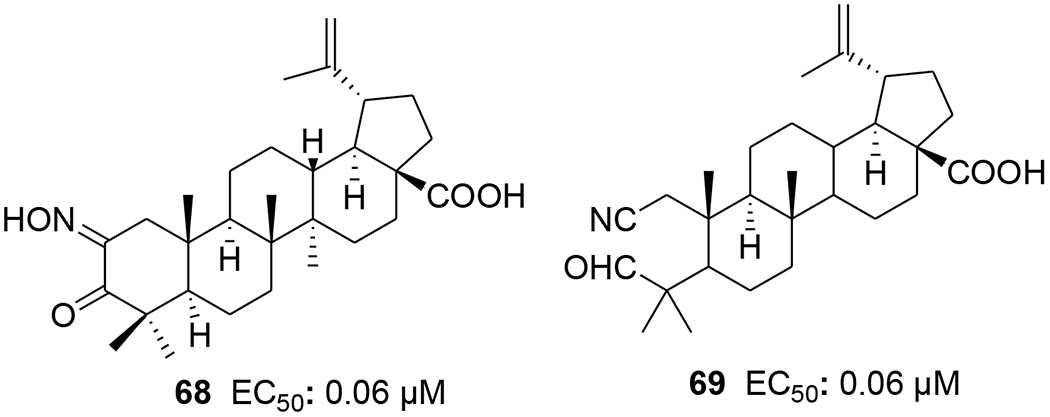
Chemical structures of compounds 68 and 69
FIGURE 12.
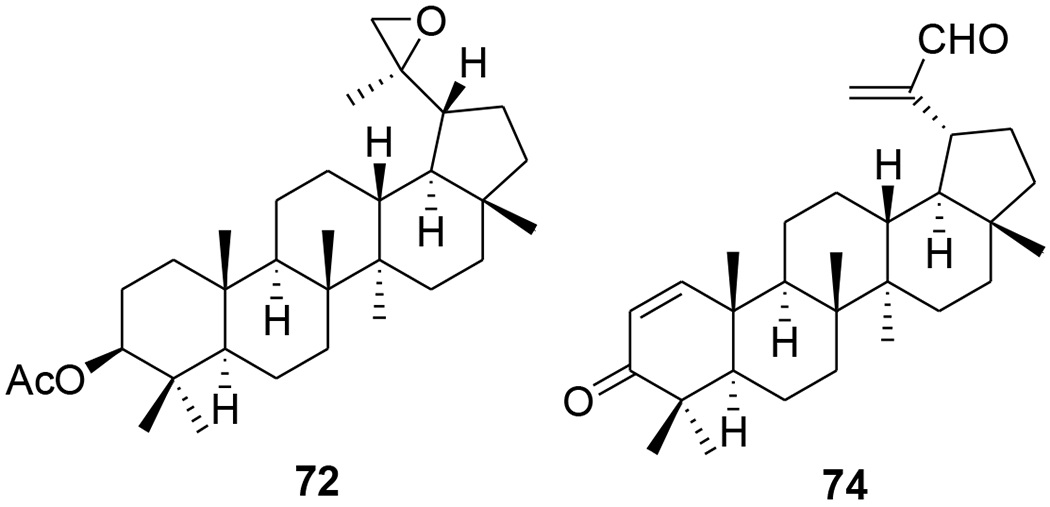
Chemical structures of compounds 72 and 74
FIGURE 13.
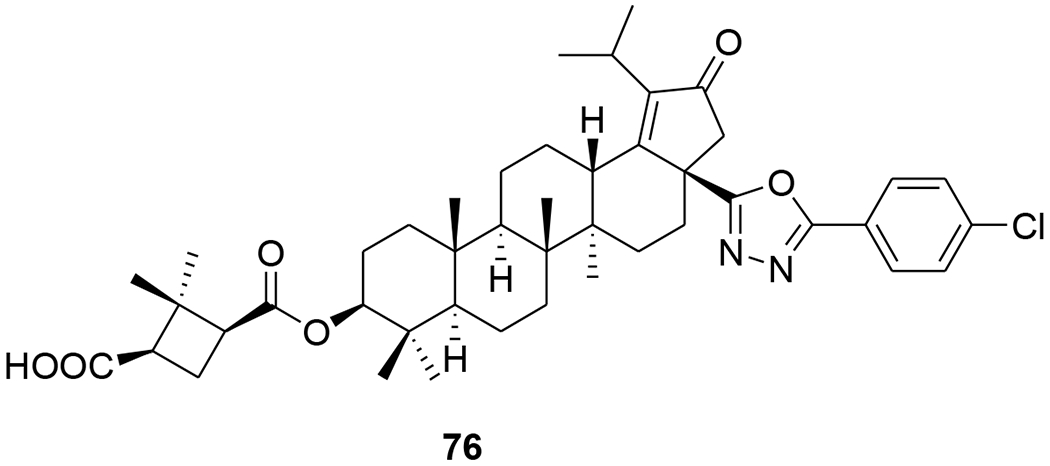
Chemical structure of compound 76
30-Oxo-calenduladiol (77) (Figure 14) is a specific CCR5 antagonist that inhibits CCR5-mediated HIV-1 infection. It binds to the CCR5 receptor without triggering cell signaling or receptor internalization, and inhibits RANTES (regulated on activation normal T cell expressed and secreted)-mediated CCR5 internalization, intracellular calcium mobilization, and cell chemotaxis.73 Six new and 20 known lupane triterpenoids were isolated from the stem of Cassine xylocarpa and root bark of Maytenus cuzcoina. In addition, some derivatives were prepared by chemical modification of the isolates. Sixteen compounds, including 78, from this series displayed inhibitory effects on HIV-1 replication with IC50 values in the micromolar range (Table 1).74 2-Acetoxyalphitolic acid (79) and 3-acetoxyalphitolic acid (80) (Figure 15) were isolated from the leaves and twigs of Garcinia hanburyi. They displayed anti-HIV-1 activities in anti-HIV-1 reverse transcriptase (IC50 values 116.9 and 16.3 μg/mL, respectively) and syncytium assays (EC50 5.6 and 6.0 μg/mL, SI 2.8 and 3.3).75
FIGURE 14.
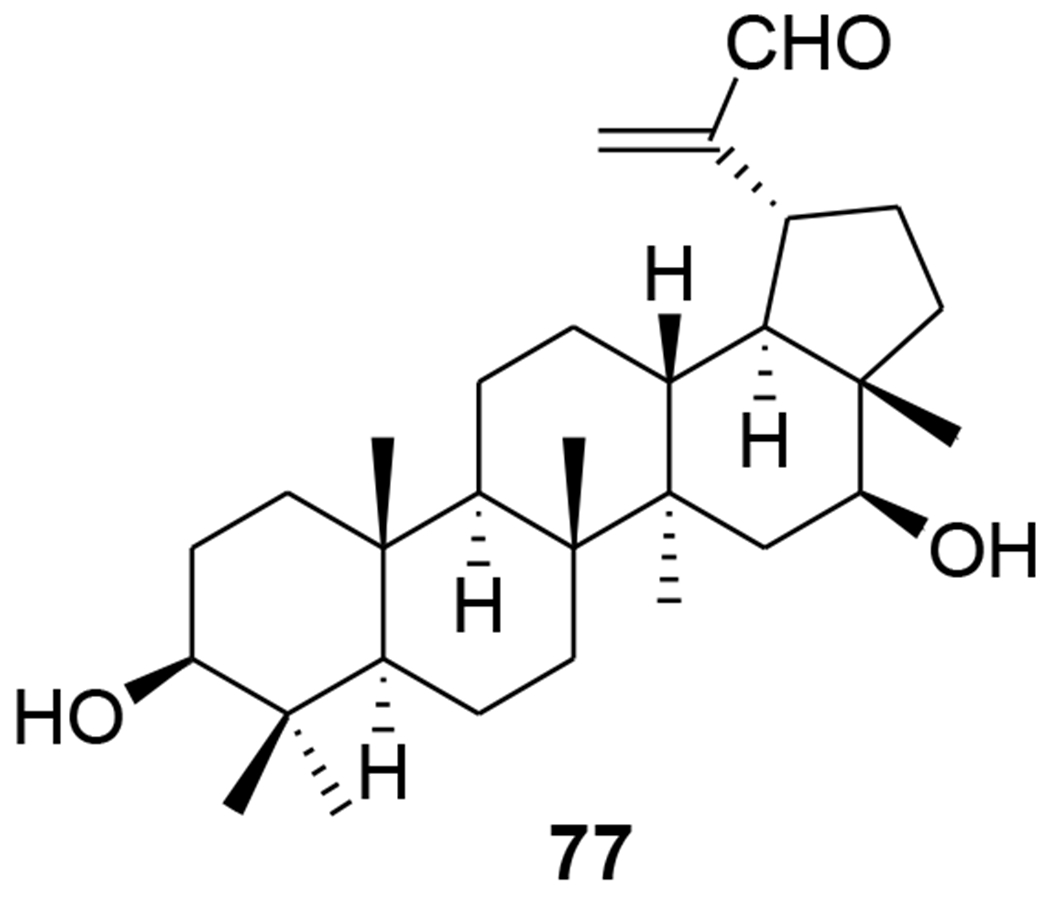
Chemical structure of compound 77
FIGURE 15.
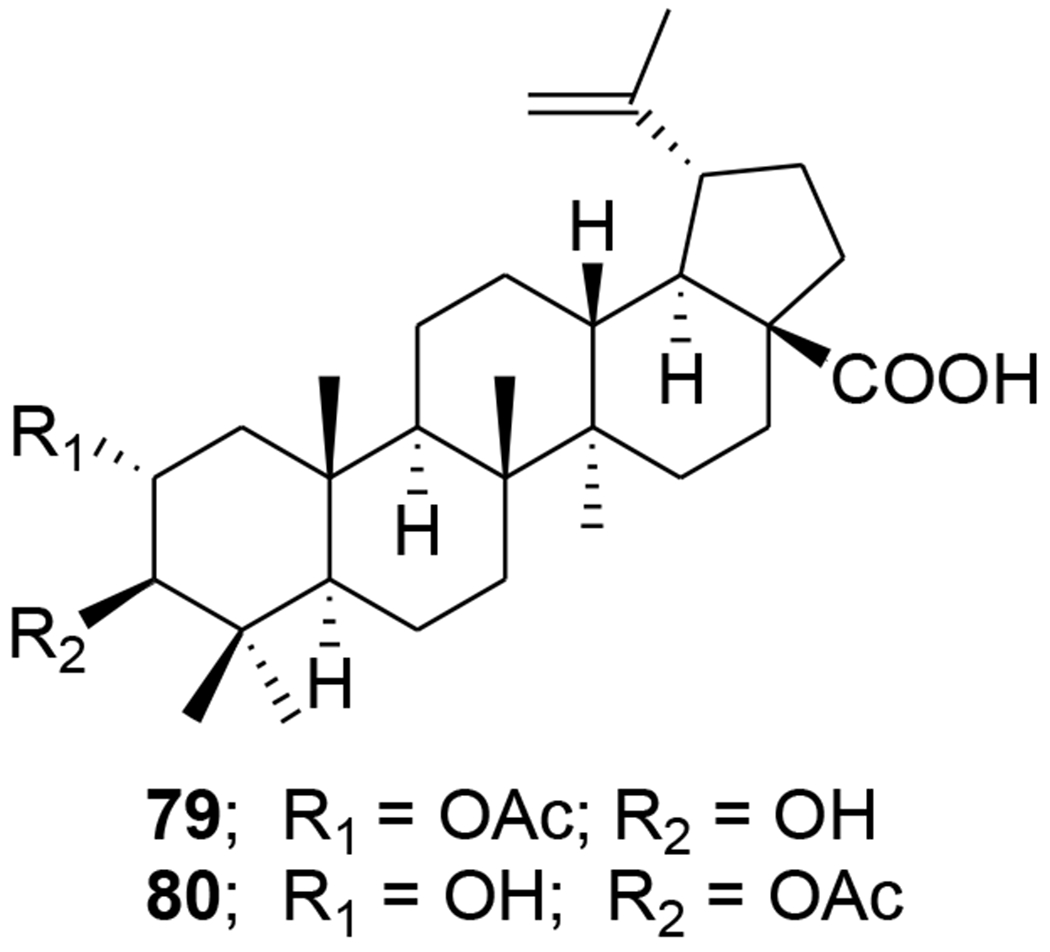
Chemical structures of compounds 79 and 80
2.2 |. The oleanane and ursane groups
Acylated oleanolic (81) and maslinic (86) acid derivatives were synthesized through solution and solid-phase procedures. Some acylated derivatives (82–85 and 87) (Figure 16) proved to be potent inhibitors of the HIV-1-protease with IC50 values between 0.31 and 0.8 μM.76 Oleanene derivatives 89 and 90 were prepared from germanicol (88). Compounds 88–90 (Figure 17) were assessed for their ability to perturb X4- and R5-tropic HIV-1-envelope (Env)-mediated fusion membrane in a cell-to-cell fusion model. However, the olean-18-ene derivatives were inactive.77 Glycyrrhizic acid was conjugated with tert-butyl esters of amino acids or benzyl esters of dipeptides containing two residues of l-amino acids (Met, Phe, Pro, and Ile or dipeptides Gly-Leu and Gly-Phe). Conjugates 91 and 92 (Figure 18) containing dipeptide fragments -Gly-Leu-OH and -Gly-Phe-OH, respectively, expressed anti-HIV-1 activity in cultures of MT-4 cells and were 90–70 times less cytotoxic than AZT. The selectivity indexes of the compounds exceeded those of glycyrrhizic acid by 110 and 34 times, respectively.78 In another report, a series of maslinic acid derivatives containing amino acid or peptide at C-28 were synthesized using solution and solid-phase synthetic procedures. Compounds 93 and 94 (Figure 19) showed anti-HIV-1 activity in MT-2 cells infected with viral clones carrying the luciferase gene as a reporter.79
FIGURE 16.
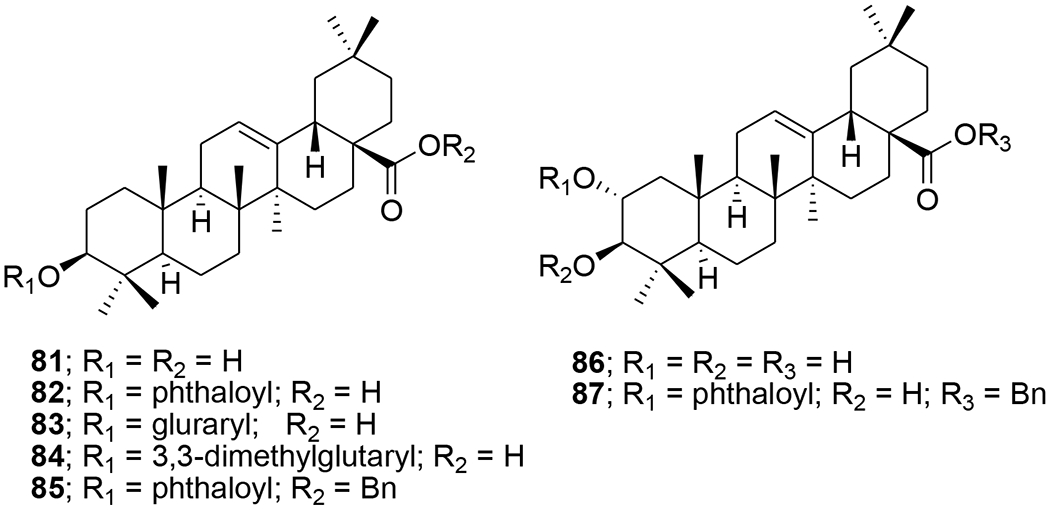
Chemical structures of compounds 81–87
FIGURE 17.
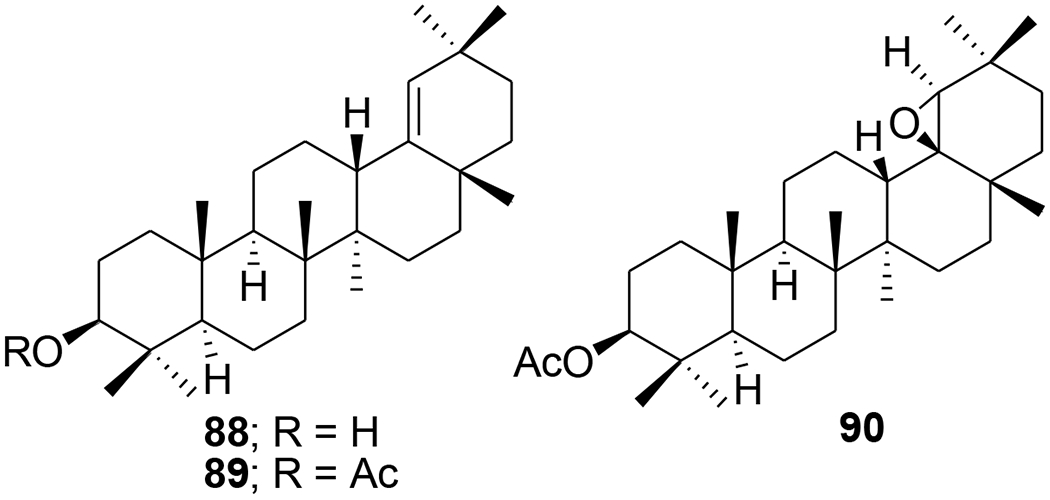
Chemical structures of compounds 88–90
FIGURE 18.
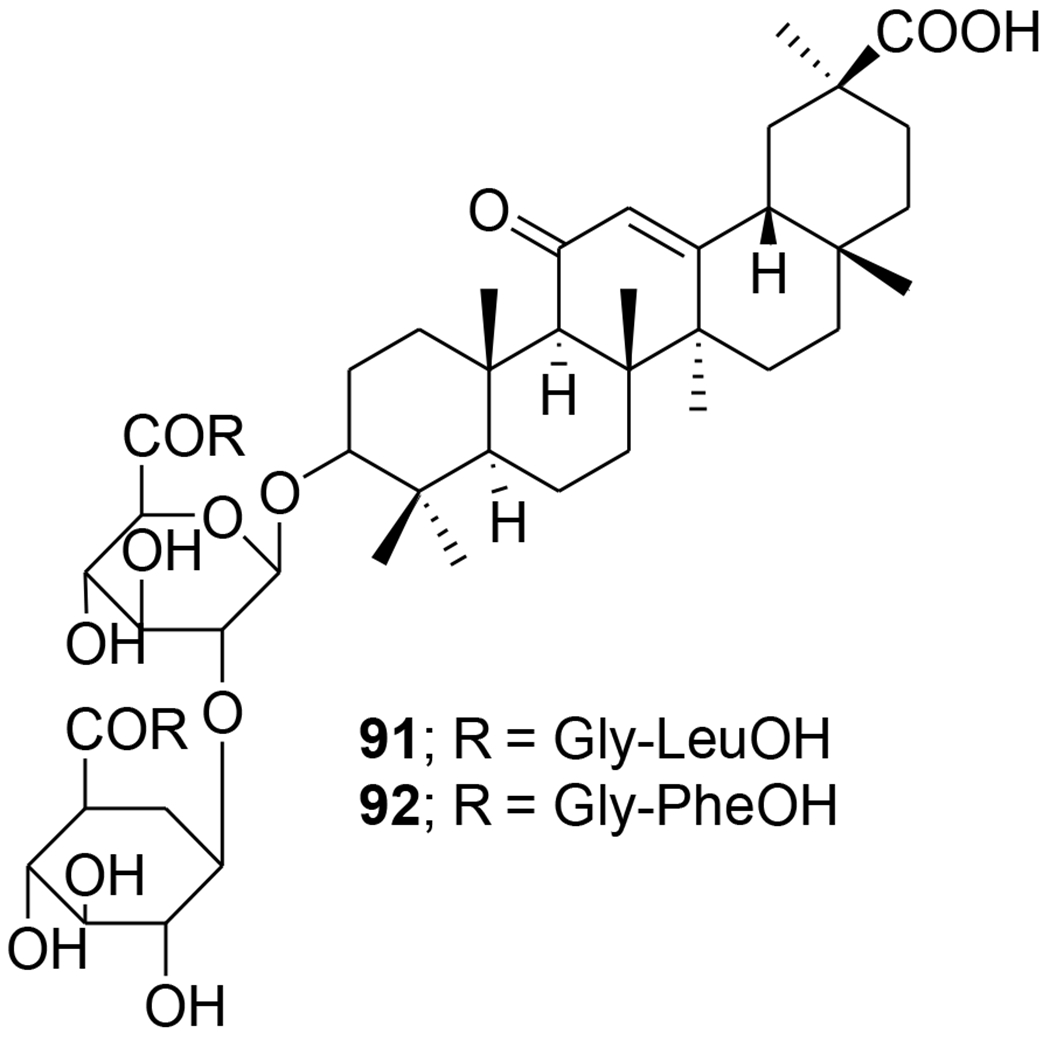
Chemical structures of compounds 91 and 92
FIGURE 19.
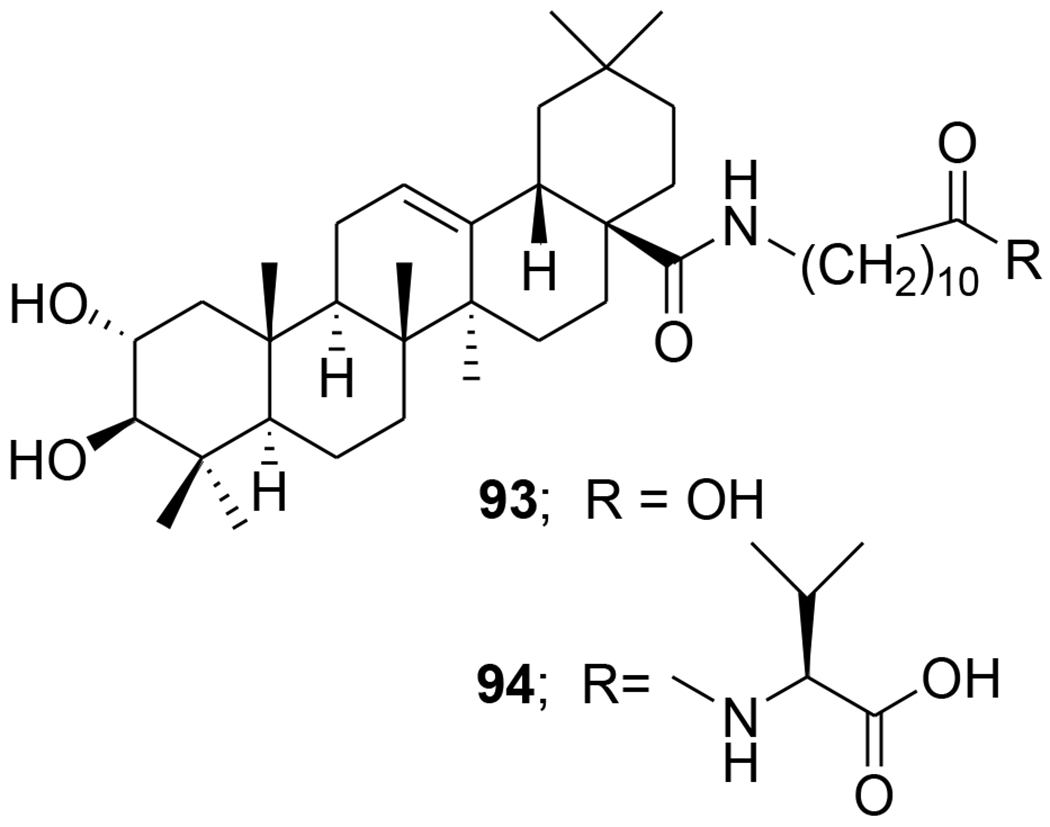
Chemical structures of compounds 93 and 94
Di- and trisubstituted amides of glycyrrhizic acid containing fragments of heterocyclic and aromatic amines (2-aminopyridine, 4-aminopyridine, 5-aminouracil, sulfadimezine, sulfapyridazine, and L-histidine methyl ester) were synthesized using the dicyclohexylcarbodiimide method. Compound 96 exhibited marked anti-HIV activity, efficiently inhibiting the accumulation of virus-specific protein p24 (ID50 = 52.8 μM) and total viral antigens, decreasing RT activity (ID50 = 52.8 μM), and effectively protecting cells from death (102–123%) and from viral infection. Compound 95 was less active than 96 (Figure 20).80 The newly developed method for synthesizing glycyrrhizic acid 3-O-galactosides produced primarily 3-O-α-D- (97) or β-D-galactopyranoside (98) (Figure 21) depending on the reaction conditions. When tested for inhibition of accumulated HIV-1-specific protein p24, compound 97 exhibited an index of selectivity 2.9-fold greater than that of glycyrrhizic acid. Compound 98 was more cytotoxic toward MT-4 cells and exhibited only weak anti-HIV-1 activity.81
FIGURE 20.
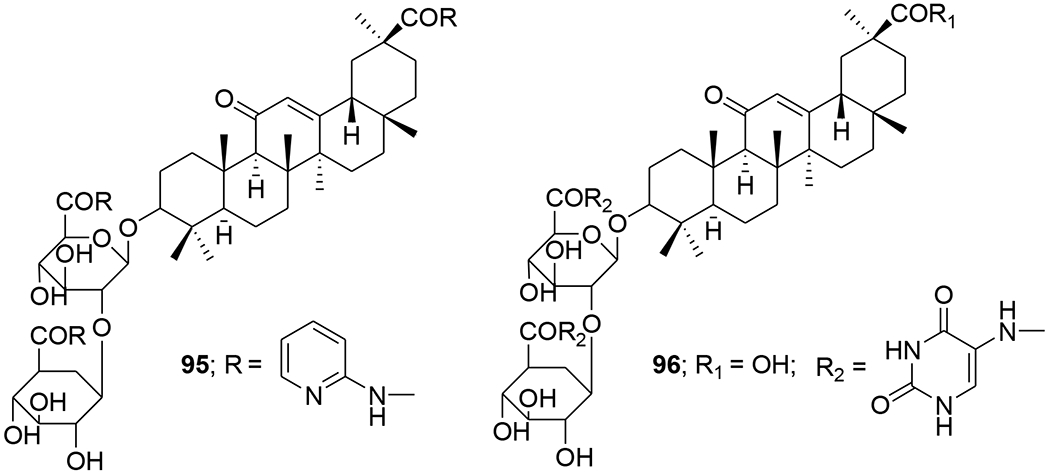
Chemical structures of compounds 95 and 96
FIGURE 21.
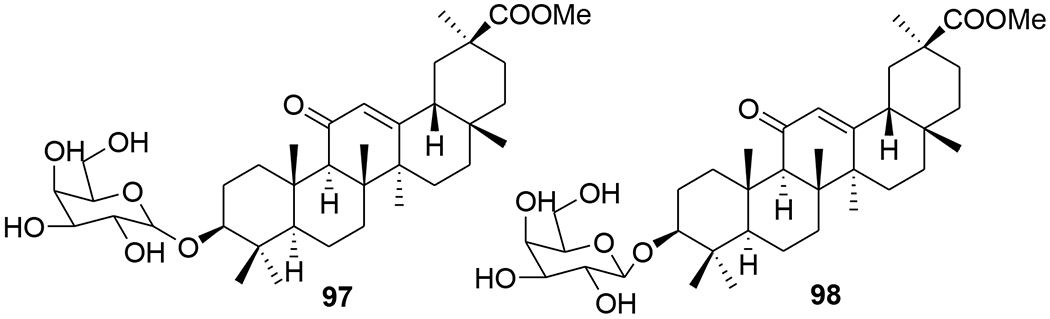
Chemical structures of compounds 97 and 98
A-seco-triterpenoids with a methylketone group were synthesized via a Grignard reaction. 3-Methyl-1-cyano-19β,28-epoxy-2,3-seco-2-nor-18αH-olean-3-one (99) (Figure 22) inhibited in vitro reproduction of HIV-1 (EC50: 7.2 μg/mL).82 Morolic acid derivatives (100–102) (Figure 23) showed anti-HIV-1 activity with EC50 values of 15, 14 and 39 μM, respectively.40 C-3 modified moronic acid analogues 103–111 (Figure 24) diverse substitutions at the 3β position were tested against HIV-1IIIB; however, none of them showed significant anti-HIV activity.41
FIGURE 22.
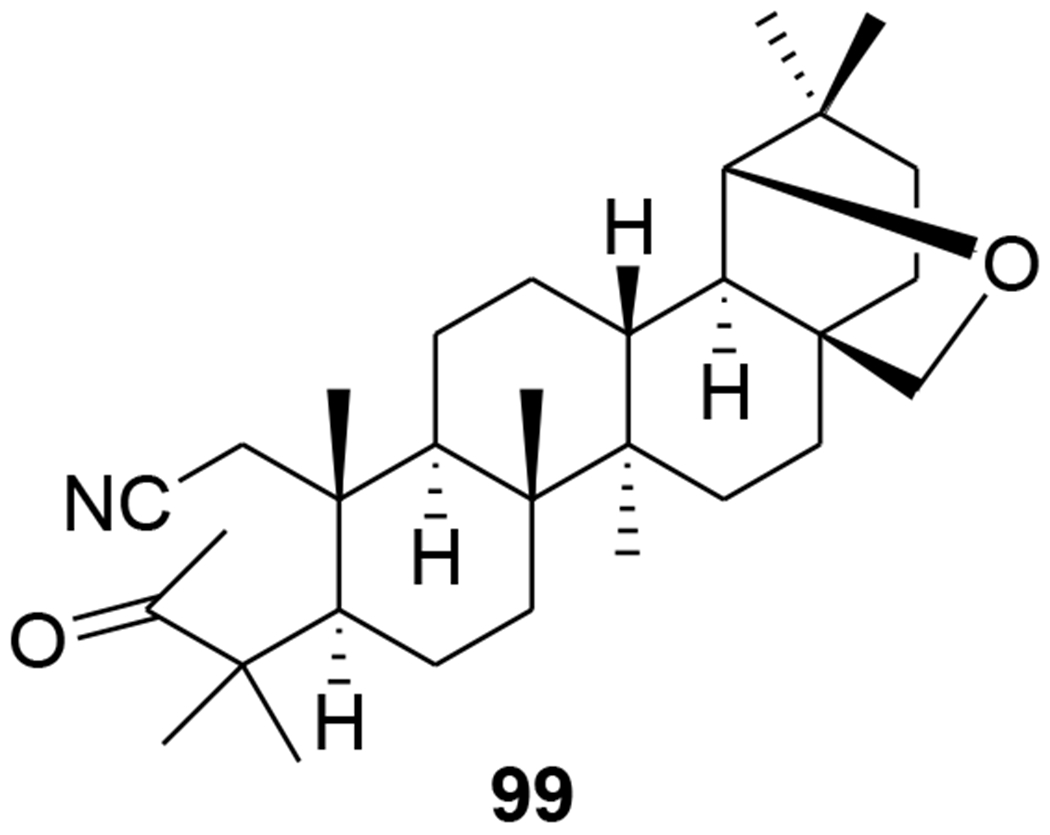
Chemical structure of compound 99
FIGURE 23.
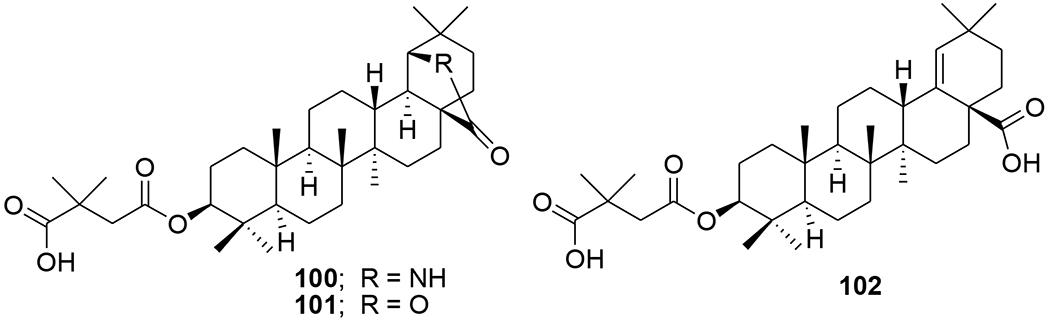
Chemical structures of compounds 100–102
FIGURE 24.
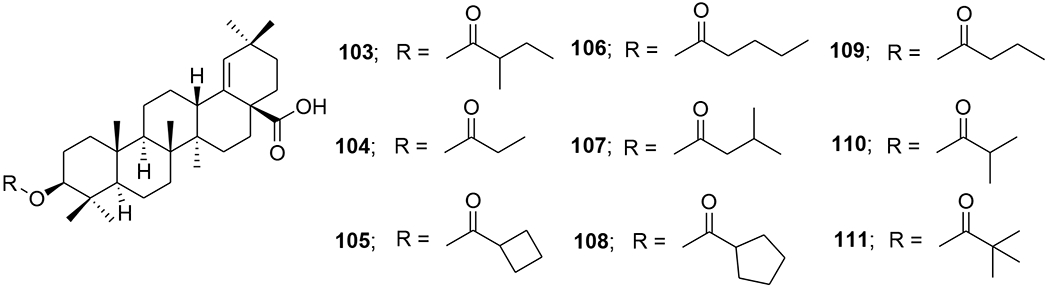
Chemical structures of compounds 103–111
Nine new and three known olean-18-ene triterpenes were isolated from Cassine xylocarpa and Maytenus jelskii. Five compounds (112–116) from this series displayed potent antiviral activity with IC50 values in the micromolar range; 112 and 116 being the most active compounds (Figure 25). Compared with currently licensed antiretroviral drugs, these compounds have a different target; they act as inhibitors of enhancer-dependent transcription.83
FIGURE 25.
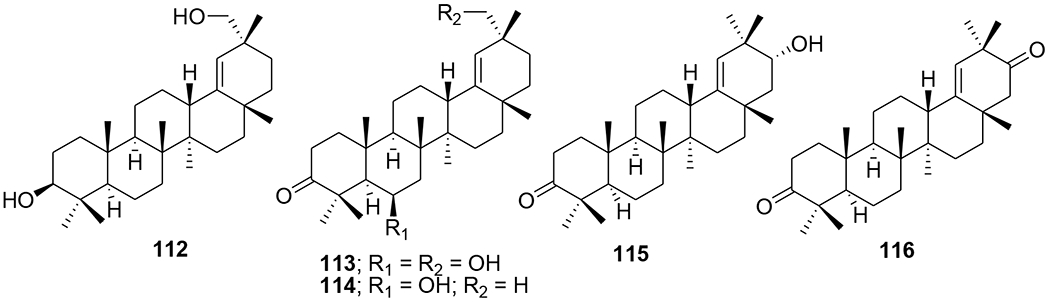
Chemical structures of compounds 112–116
Correspondingly, transactivator of transcription (Tat), an early virus-encoded protein required for the efficient transcription of the HIV genome, could be developed as a target for small molecular therapeutics. Celastrol (117) (Figure 26) isolated from Tripterygium wilfordii exhibited high inhibitory activity against Tat. By covalently modifying the cysteine thiols, celastrol inhibits Tat transactivation function as well as the transcription elongation of the HIV proviral genome through mechanisms other than Tat–TAR (transactivation-responsive region) interaction.84 22β-Acetoxyglycyrrhizin (118) and 3-O-β-D-glucuronopyranosylglycyrrhetinic acid (119) (Figure 27) showed anti-HIV activities with IC50 values of 29.5 and 41.7 μM, respectively.85
FIGURE 26.
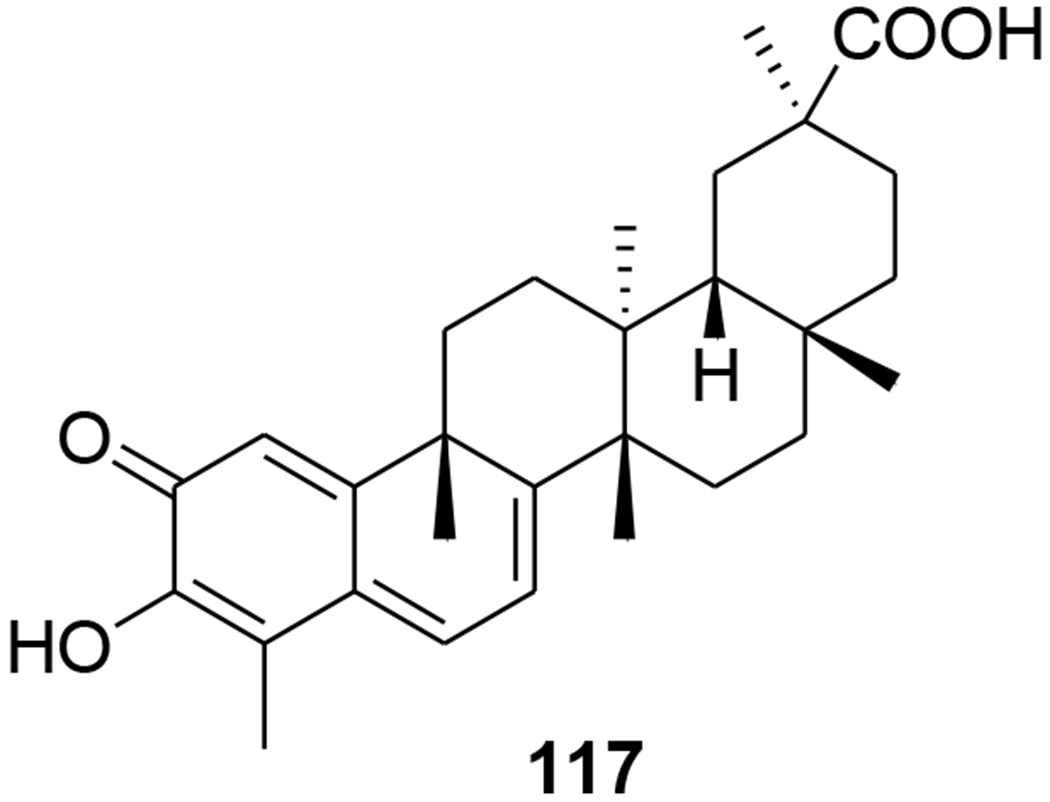
Chemical structure of compound 117
FIGURE 27.
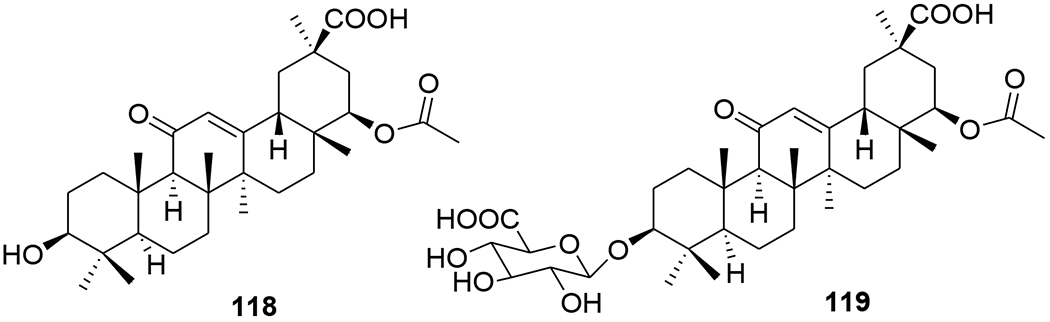
Chemical structures of compounds 118 and 119
Recently, the anti-HIV and other pharmacological activities of ursolic acid were reviewed.86 A new series of HIV-1 protease inhibitors with pentacyclic triterpenoids as P2 ligands and phenylsulfonamide as P2′ ligands were synthesized. These compounds exhibited micromolar inhibitory potency. Among them, compound T1c (120) (Figure 28) exhibited HIV-1 protease inhibition with an IC50 value of 0.12 μM, thus, showing 67 times the inhibitory activity of its raw material ursolic acid (8.0 μM).87 A-seco ursolic acid derivatives (121–124) (Figure 29) showed inhibitory activity against HIV-1 protease (IC50 5.7, 17.6, 3.9 and 88.1 μM, respectively).68
FIGURE 28.
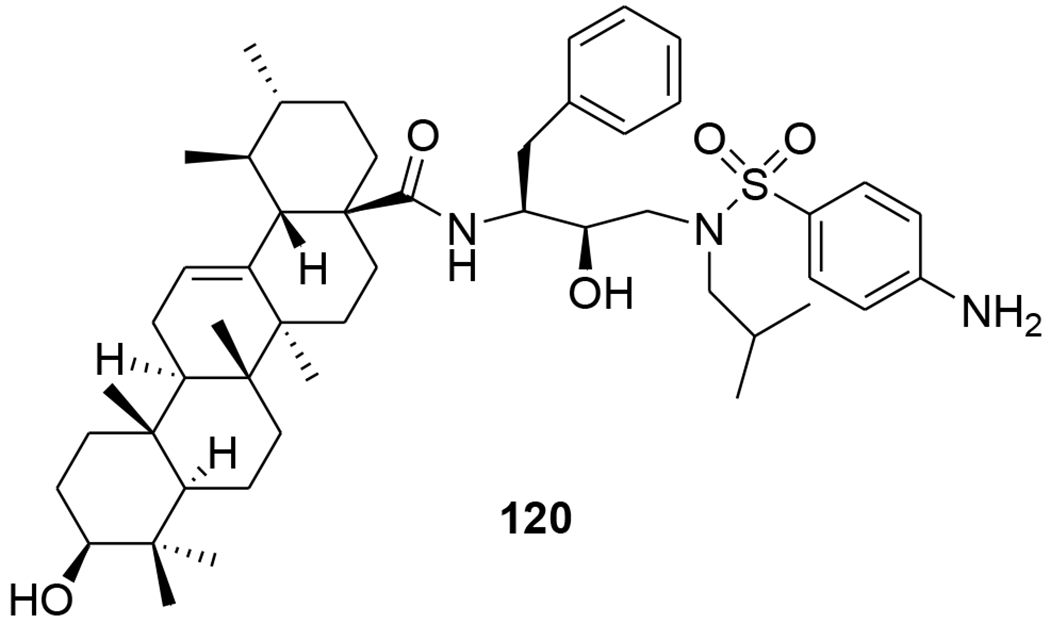
Chemical structure of compound 120
FIGURE 29.
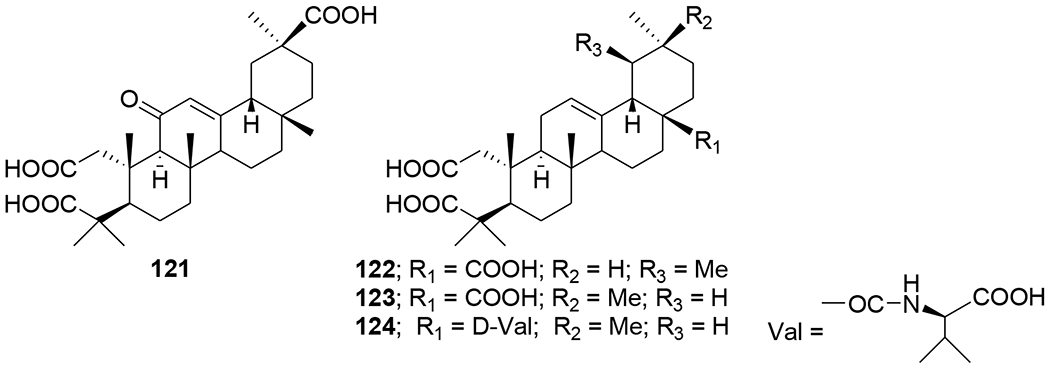
Chemical structures of compounds 121–124
2.3 |. Others
3a-Methoxyserrat-14-en-21β-ol (125) and 3β-methoxyserrat-14-en-21β-ol (126) and their curcumin, kojic acid, quercetin, and baicalein conjugates were evaluated for in vitro anti-HIV-1 RT activity in infected C8166-CCR5 cells (Figure 30). Compound 127, the conjugate of two molecules of 126 and one molecule of kojic acid, exerted significant anti-HIV activity with an EC50 value of 0.12 μg/mL.88 An efficient synthesis of epiceanothic acid (128) (Figure 31) starting from betulin (3) was accomplished in 12-steps with a total yield of 10%. Epiceanothic acid (EC50: 15.6 μM) exhibited moderate HIV-1 inhibition in acutely HIV-1NL4–3 infected MT-4 cells.89
FIGURE 30.
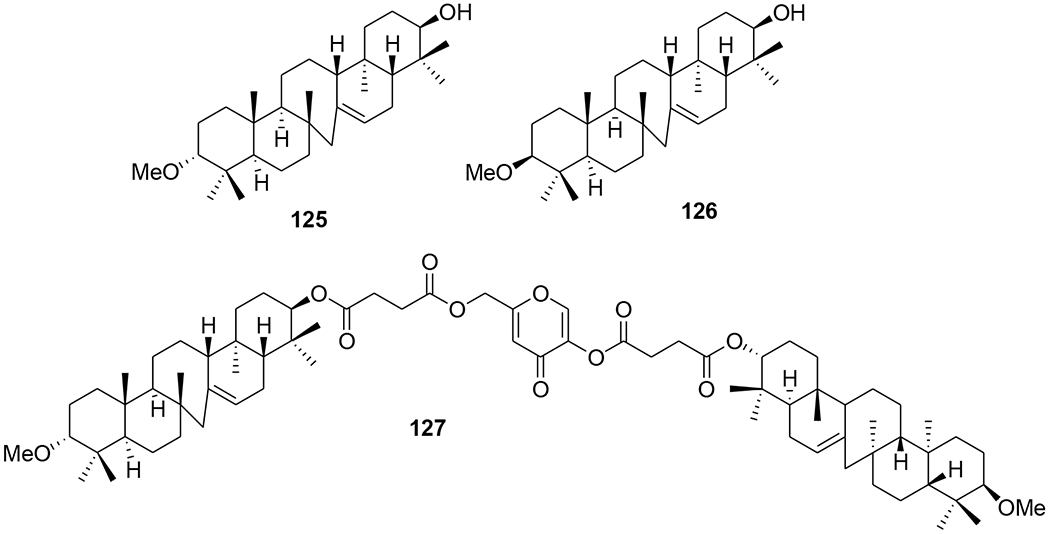
Chemical structures of compounds 125–127
FIGURE 31.
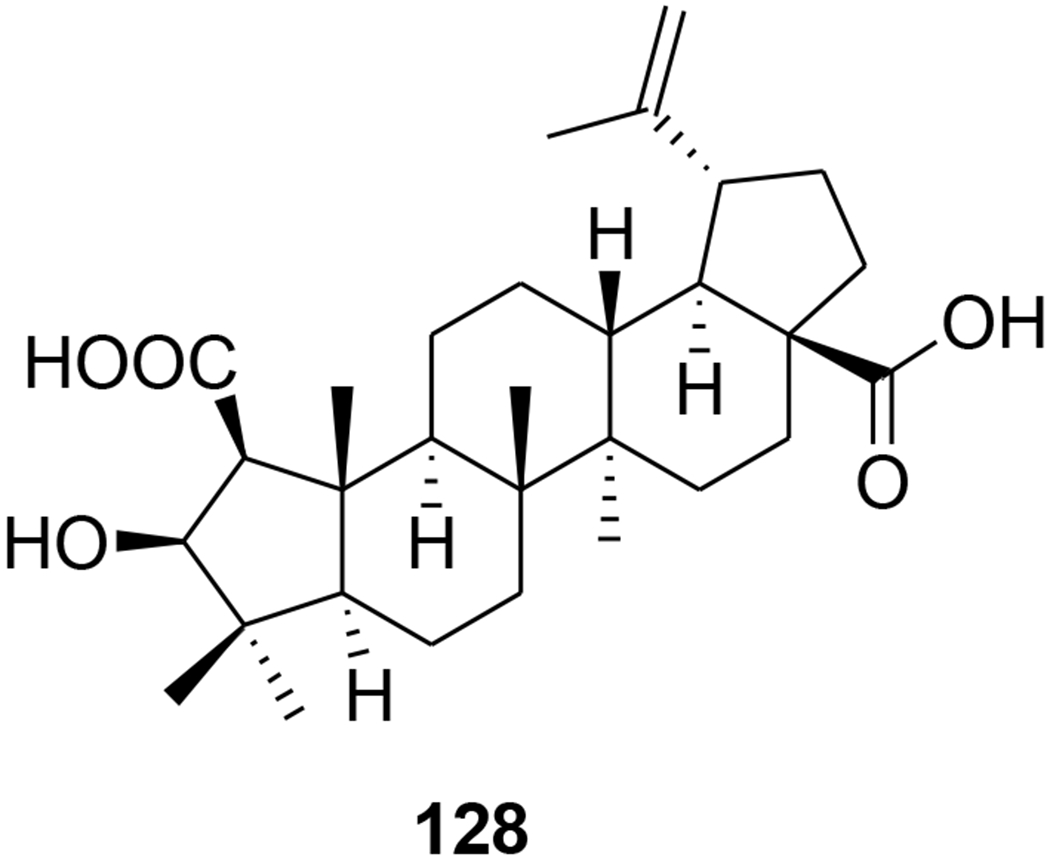
Chemical structure of compound 128
3 |. TETRACYCLIC TRITERPENOID
3.1 |. The lanostane group
Five highly oxygenated lanostane-type triterpenoids, ganoderic acid GS-1 (129), ganoderic acid GS-2 (130), ganoderic acid GS-3 (131), 20(21)-dehydrolucidenic acid N (132) and 20-hydroxylucidenic acid A (133), as well as several known compounds were isolated from the fruiting body of Ganoderma sinense (Figure 32). Compounds 130, 132, and 20-hydroxylucidenic acid N (134) and ganoderiol F (135) inhibited HIV-1 protease with IC50 values of 30, 48, 25, 22 μM, respectively.90 Three new tricyclic rearranged lanostane triterpenoids, kadcotriones A–C (136–138), together with a biogenetically related lanostane-type triterpenoid (139), were isolated from Kadsura coccinea (Figure 33). Compounds 137 and 139 exhibited anti-HIV-1 activities with EC50 values of 30.29 and 54.81 μM, respectively.91 Later, 11 triterpene acids including kadcoccinic acids A–J (140–149) and 150 (Figure 34) were also isolated from the stems of Kadsura coccinea by the same group. Except for 149, these compounds feature a rearranged lanostane skeleton with a 6/6/5/6 tetracyclic ring system. Compounds 140 and 141 are the first triterpenoids with a 2,3-seco-6/6/5/6-fused tetracyclic skeleton. Among them, compounds 143 and 146 demonstrated anti-HIV-1 activity with respective EC50 values of 62.0 and 58.7 μM.92 In a computational study on Ganoderma lucidum triterpenoids, ganoderat acid-B (151) (Figure 35) showed the best affinity to HIV-1 protease (binding energy= −7.49 kcal/mol and Ki= 0.001 mM), better than that of nelfinavir.93 Another lanostane triterpene, garcinuntine (152) (Figure 36), isolated from the roots of Garcinia nuntasaenii Ngerns. & Suddee was inactive.94
FIGURE 32.
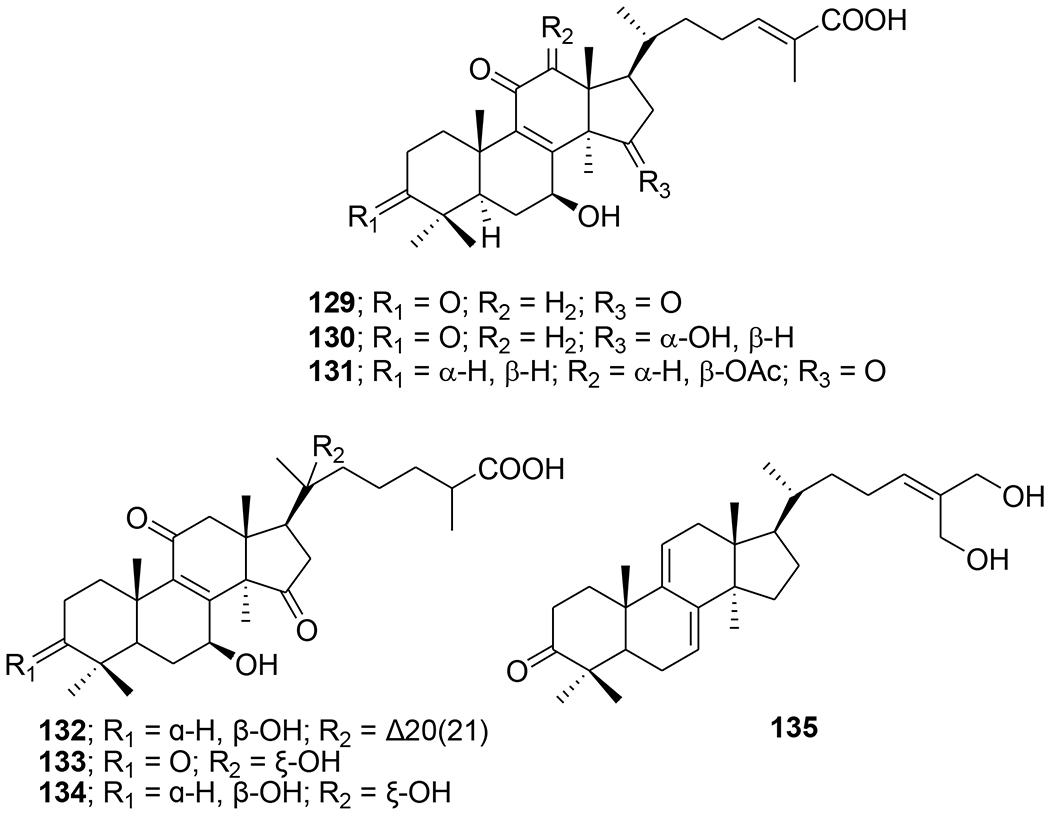
Chemical structures of compounds 129–135
FIGURE 33.
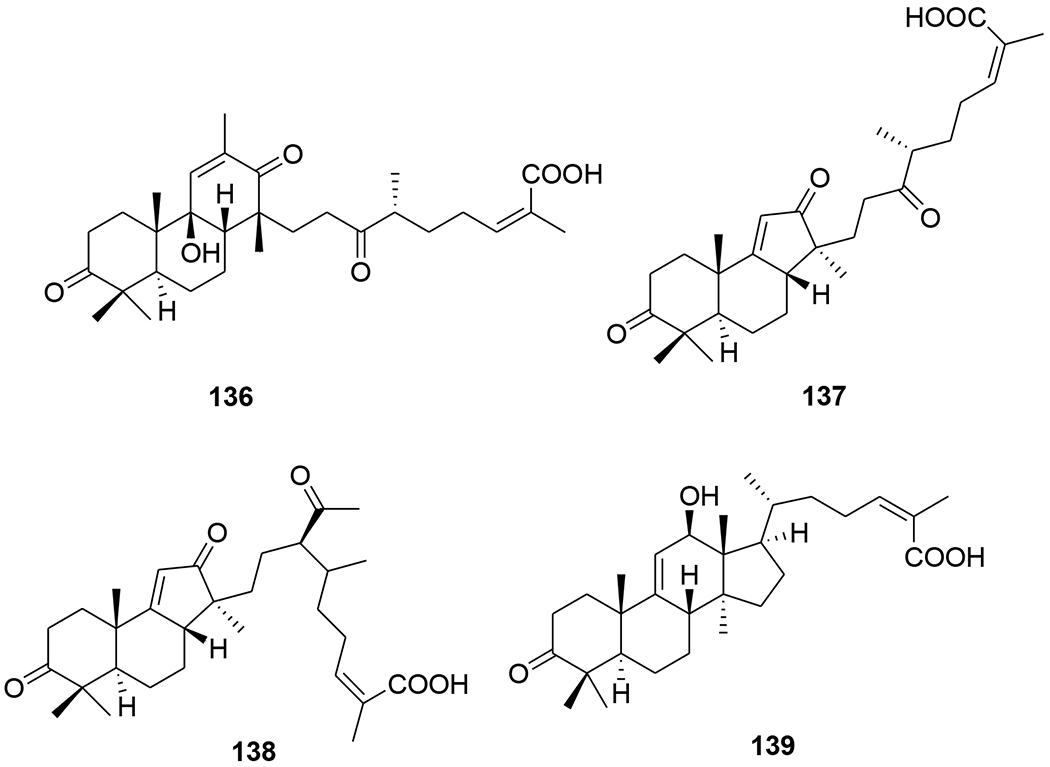
Chemical structures of compounds 136–139
FIGURE 34.
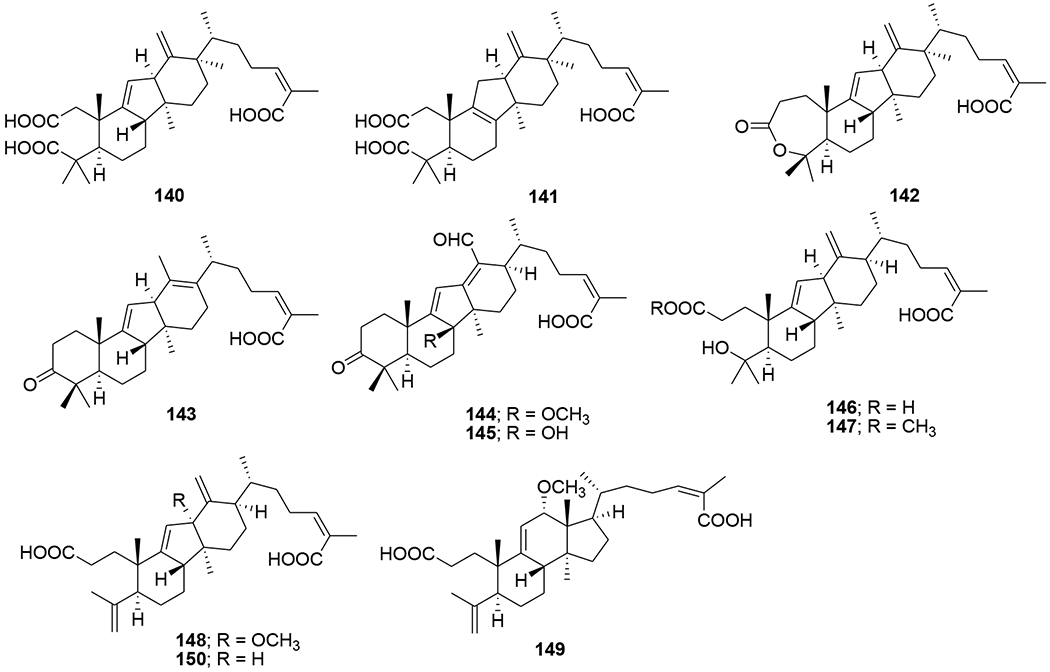
Chemical structures of compounds 140–150
FIGURE 35.
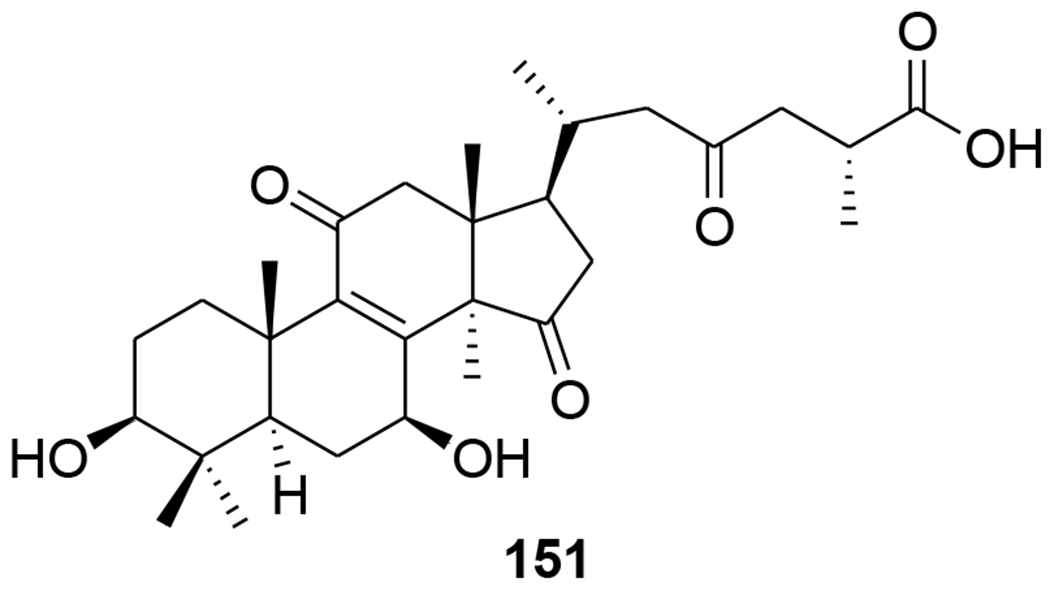
Chemical structure of compound 151
FIGURE 36.
Chemical structure of compound 152
3.2 |. The dammarane group
Several dammarane triterpene derivatives were synthesized and evaluated for HIV-1 protease inhibitory activity. The mono- and di-succinyl derivatives 153–157 (Figure 37) were significant inhibitors of HIV-1 protease with EC50 values of 2.7, 6.5, 3.9, 2.7 and 5.4 μM.95 Three other triterpenoids, (20R)-20,25-epoxy-dammaran-2-en-6a,12β-diol (158), (20R)- 20,25-epoxy-3-methyl-28-nordammaran-2-en-6a,12β-diol (159) and isodehydroprotopanaxatriol (160), isolated from an acidic hydrolysate of Panax ginseng (Figure 38), showed inhibitory activity against HIV-1 protease with EC50 values of 10.5, 10.3, and 12.3 μM, respectively.96 In the screening of a panel of purified compounds isolated from Aglaia sp. (Meliaceae) for inhibition of early steps in the lentiviral replication cycle, the 3,4-secodammarane triterpenoid 161 (Figure 39), exhibited potent inhibition of HIV-1 infection (IC50 = 0.48 μg/mL), while cytotoxic effects and inhibition of cell proliferation were observed only at concentrations exceeding 10.69 μg/mL.97
FIGURE 37.
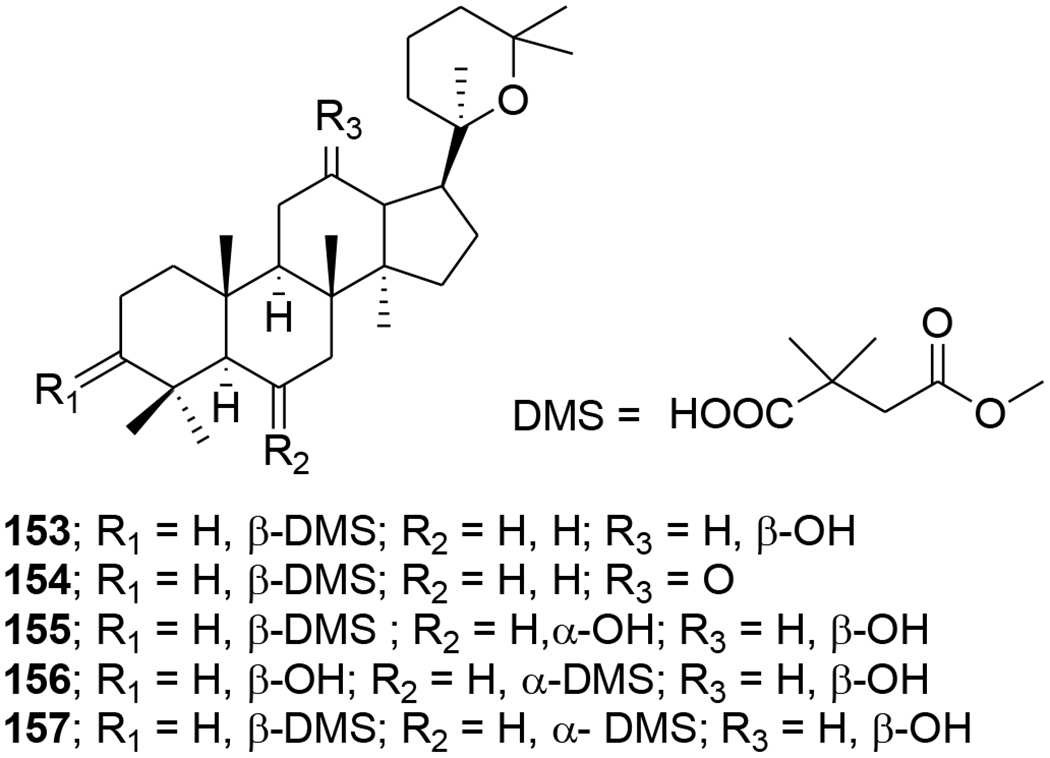
Chemical structures of compounds 153–157
FIGURE 38.
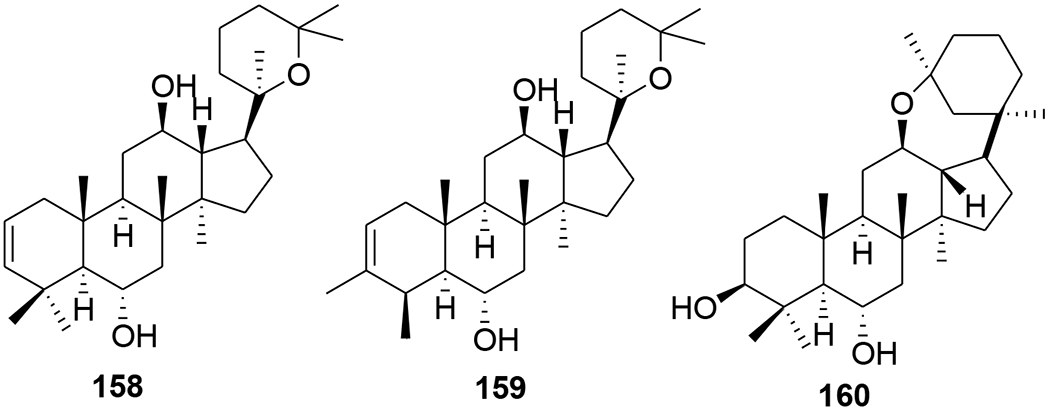
Chemical structures of compounds 158–160
FIGURE 39.
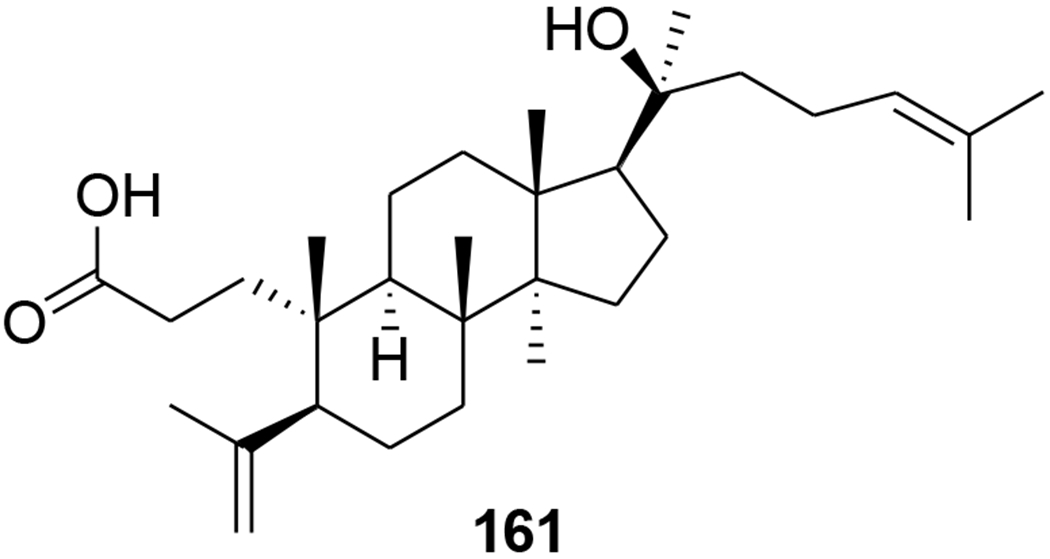
Chemical structure of compound 161
3.3 |. Cycloartane group
Four cycloartane triterpenoids, angustific acid A (162), angustific acid B (163), angustifodilactone A (164) and angustifodilactone B (165) (Figure 40), were isolated from the branches of Kadsura angustifolia. Compound 162, characterized by the presence of a C-16/C-17, C-20/C-21 conjugated diene and a C-1/C-7 ester bridge formed in rings A and B, exemplified a novel structural skeleton for 3,4-secocycloartane triterpenoids. Compound 162 exhibited the most potent anti-HIV activity with an EC50 value of 6.1 μg/mL in infected C8166 cells and a therapeutic index of more than 32.8.98 Two cycloartane triterpenoids, cycloccidentalic acids A and B (166 and 167), and five related saponins, cycloccidentalisides I–V (168–172) (Figure 41), were isolated from Cassia occidentalis. Compounds 167 and 170 showed modest anti-HIV-1 activities with EC50 values of 2.23 μM and 4.36 μM, respectively.99
FIGURE 40.
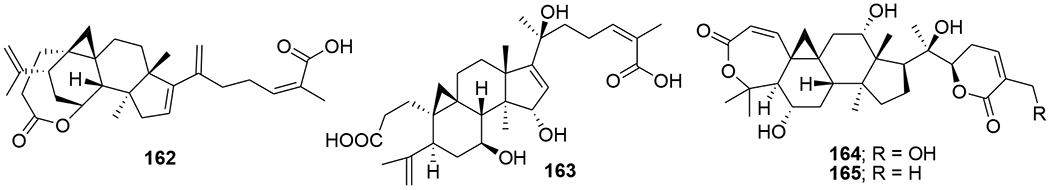
Chemical structures of compounds 162–165
FIGURE 41.
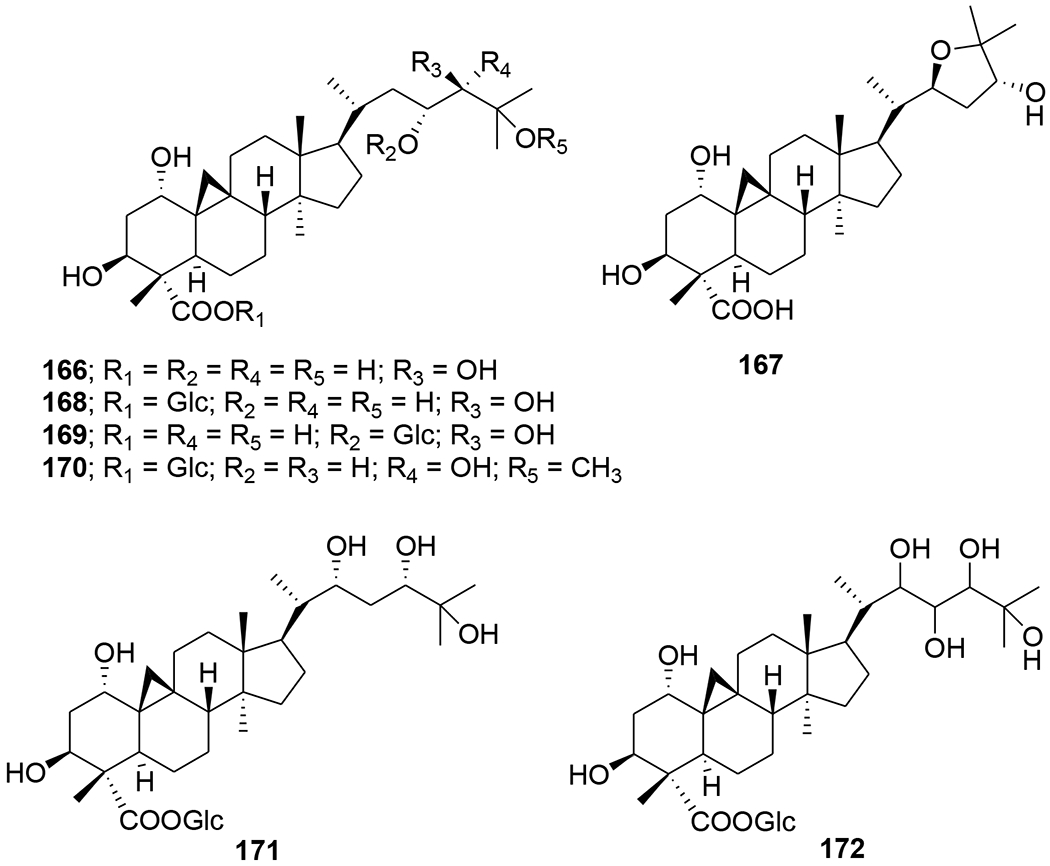
Chemical structures of compounds 166–172
Eight cycloartane triterpenoids carinatins A–H (173–180), secaubryolide (181) and dikamaliartane D (182) (Figure 42) were isolated from the leaves and twigs of Gardenia carinata and evaluated for anti-HIV-1 activities using HIV-1 reverse transcriptase and syncytium inhibition assays using the ΔTat/RevMC99 virus and 1A2 cell line system. Compounds 173, 174, 177–179, and 182 exhibited significant inhibitory activities by significantly reducing the number of syncytium formations in the syncytium inhibition assay. Compound 182 showed the most potent anti-HIV-1 activity (EC50 < 8.3 μM). In the reverse transcriptase assay, only compounds 175 and 182 were active against HIV-1 reverse transcriptase, with IC50 values of 85.7 and 68.7 μM, respectively.100
FIGURE 42.
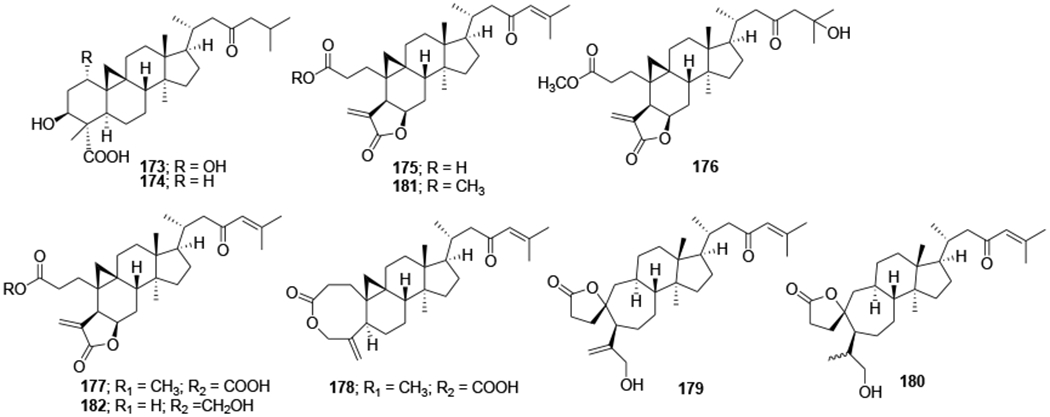
Chemical structures of compounds 173–182
Two metabolites (184 and 185) were obtained by microbial transformation of the triterpene nigranoic acid (183) in a culture of Trichoderma sp. JY-1, a fungus obtained from the branches of Kadsura angustifolia. Compound 184 was characterized with an unusual 17(20),17(E)-ene structure, while compound 185 featured an unprecedented 18(13→17β)-abeo-secocyloartane skeleton. Additionally, compounds 183–185 (Figure 43) showed weak anti-HIV activity with EC50 values of 10.5, 8.8 and 7.6 μg/mL and therapeutic index values of 8.48, 9.12 and 10.1, respectively.101 In another report, the microbiological transformation of the triterpene nigranoic acid (183) to 3,4-secocycloarta-4(28),17(20),24(Z)-triene-7β-hydroxy-16β,26-lactone-3-oic acid (186) and 3,4-secocycloarta-4(28), 17(20)(Z),24(Z)-triene-7β-hydroxy-16β-methoxy-3,26-dioic acid (187) (Figure 44) by the freshwater fungus Dictyosporium heptasporum YMF1.01213 was demonstrated. Compound 186, characterized by the presence of a formed C-16/C-26 ester bridge, exemplifies a novel nine-membered lactone ring structural skeleton for 3,4-secocycloartane triterpenoid. In addition, compounds 186 and 187 exhibited weak in vitro anti-HIV activity with respective EC50 values of 15.3 and 18.1 μg/mL.102 In a recent report, a novel cycloartane triterpenoid alkaloid, kleinhospitine E (188), and six cycloartane triterpenoids (189–194) (Figure 45) were isolated from Kleinhovia hospita. Compound 188 is the first cycloartane alkaloid possessing an unusual γ-lactam with an oxopropylidene side chain. Compounds 189, 190, and 194 were assigned as cycloartane triterpenoids with a 9α,10α-cyclopropyl ring, which is found rarely among naturally occurring compounds, while 192 and 193 were established as isomers of 190 containing a 21,23-diacetal side chain. Compound 194 exhibited anti-HIV activity with an EC50 value of 0.8 μM.103
FIGURE 43.
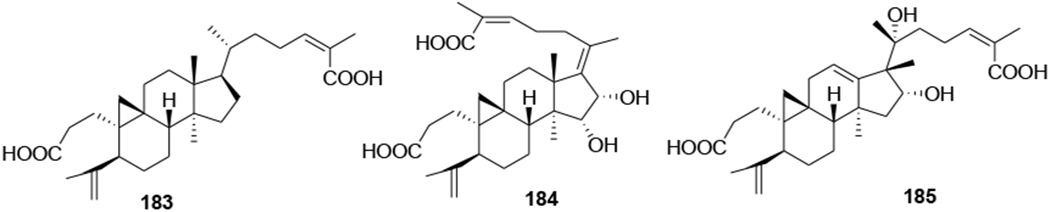
Chemical structures of compounds 183–185
FIGURE 44.
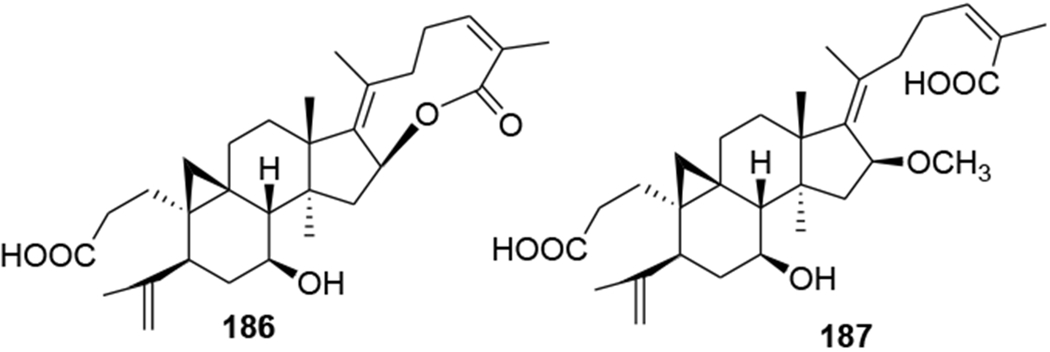
Chemical structures of compounds 186 and 187
FIGURE 45.
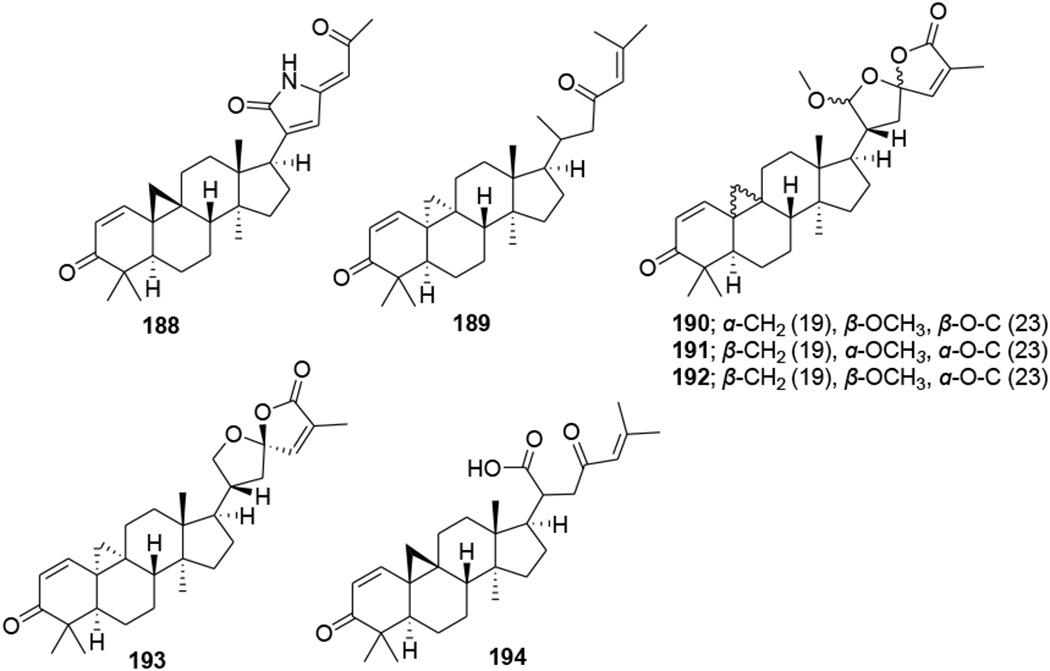
Chemical structures of compounds 188–194
In our Natural Products Research Laboratories, 12 known cycloartane triterpenoids (195–206) (Figure 46) with four different skeletons were isolated from the roots of Souliea vaginata and screened for their anti-HIV activity. Among these compounds, beesioside I (195) showed the highest potency against HIV-1NL4–3 with an EC50 value of 2.32 μM. Further modification at the C-3 position of the aglycone (207) of 195 led to a series of derivatives (208–221) (Figure 47). Among them, compound 214 was the most potent with an EC50 value of 0.025 μM and TI value greater than 800, comparable to those of BVM. Other analogues exhibited strong to weak inhibition of HIV-1 replication in MT-4 cells.104
FIGURE 46.
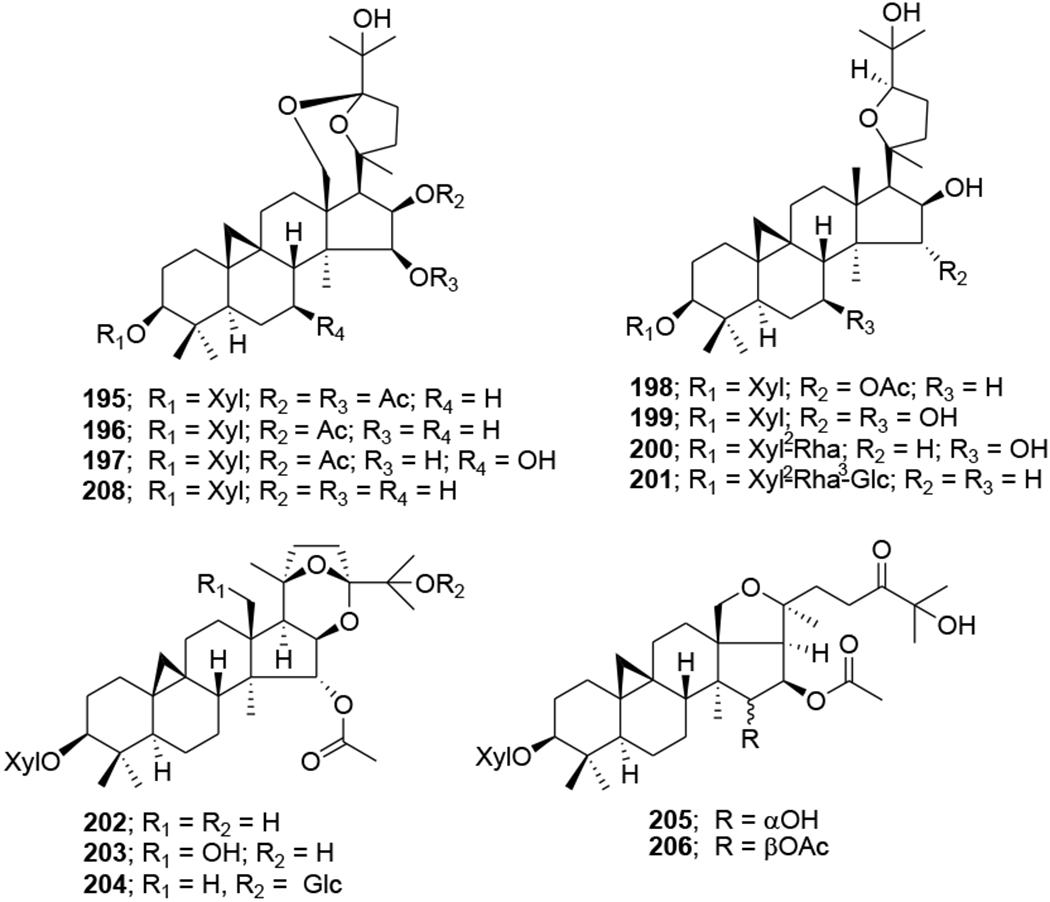
Chemical structures of compounds 195–206 and 208
FIGURE 47.
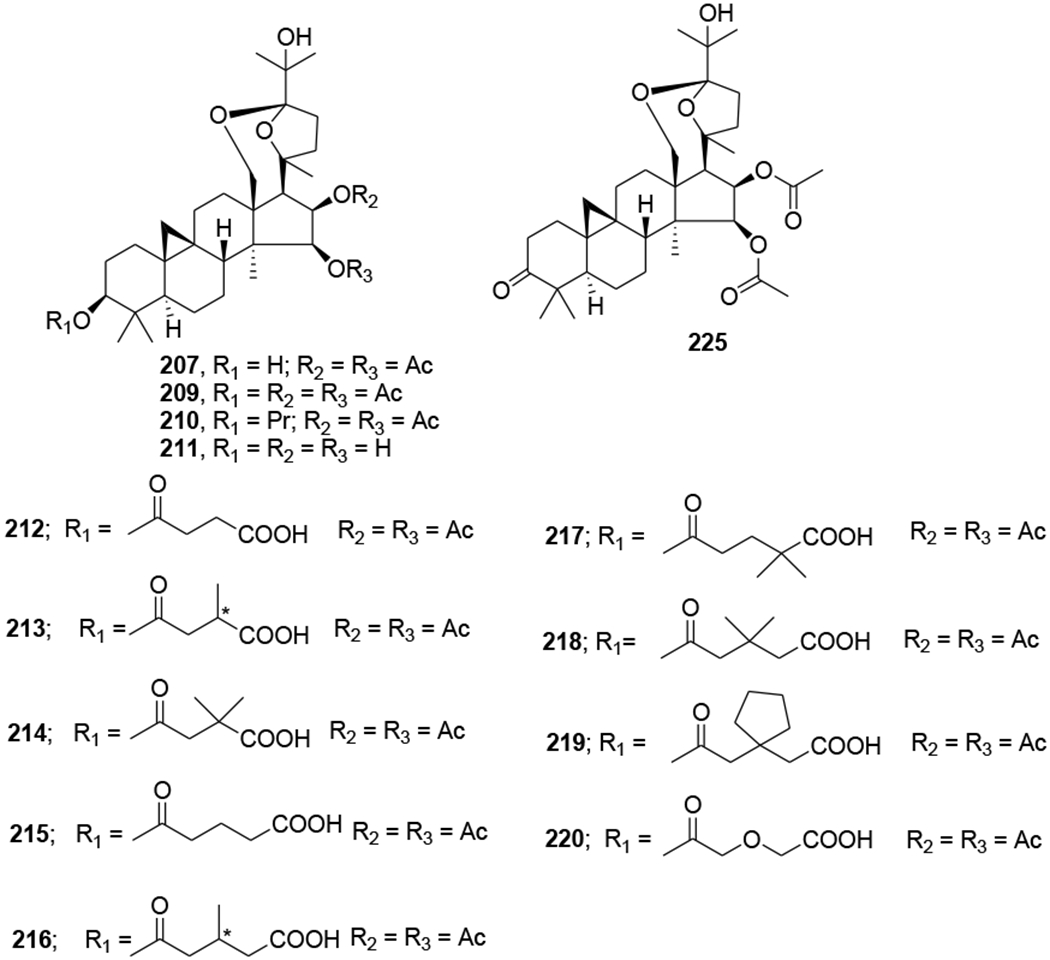
Chemical structures of compounds 207 and 209–221
3.4 |. Limonoid group
Three limonoids, trichiconin A (222) with a unique carbon skeleton featuring a rearranged A,B-ring system and trichiconins B (223) and C (224) (Figure 48) with an unprecedented A,B,D-seco skeleton, were isolated from the twigs of Trichilia connaroides. Compounds 223 and 224 showed modest anti-HIV activity with EC50 values of 5.9 and 3.6 μM, respectively, while 222 was inactive.105 Sixteen limonoids, ciparasins A–P (225–240) (Figure 49), were isolated from Cipadessa cinerascens. Ciparasins E–G (229–231) contain a rare gamma-hydroxylbutenolide moiety at C-17. Ciparasins B (230) and P (240) showed significant anti-HIV activity with EC50 values of 5.5 and 6.1 μM, respectively.106 Five limonoids, sundarbanxylogranins A–E (241–245) (Figure 50), were isolated from the seeds of Xylocarpus granatum. Sundarbanxylogranin A (241) is a rare limonoid containing a bicyclo[5.2.1]dec-3-en-8-one scaffold as the ring A/B-fused core; whereas sundarbanxylogranin B (242) is a typical mexicanolide with an 8a,30a-epoxy ring. Sundarbanxylogranins C–E (243–245) belong to a small group of limonoids containing a C1-O-C29 oxygen bridge; both former compounds have a 29-OMe group but with different orientations. Compound 242 exhibited moderate anti-HIV activity with an IC50 value of 23.14 μM.107
FIGURE 48.
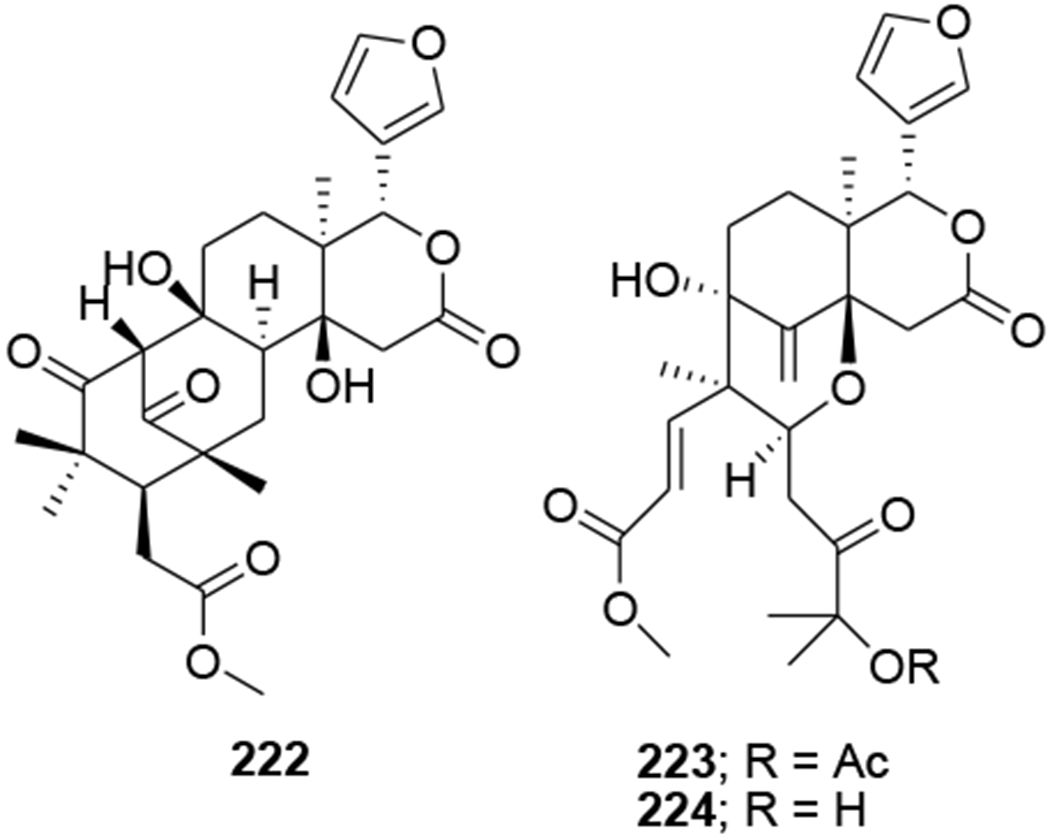
Chemical structures of compounds 222–224
FIGURE 49.
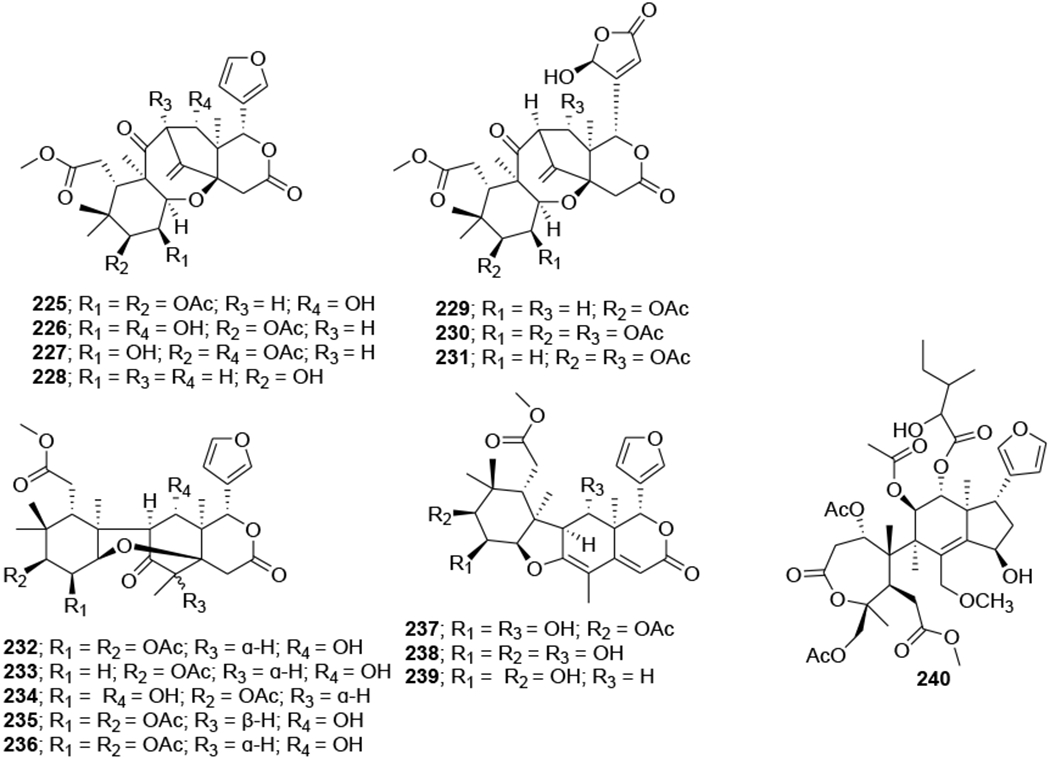
Chemical structures of compounds 225–240
FIGURE 50.
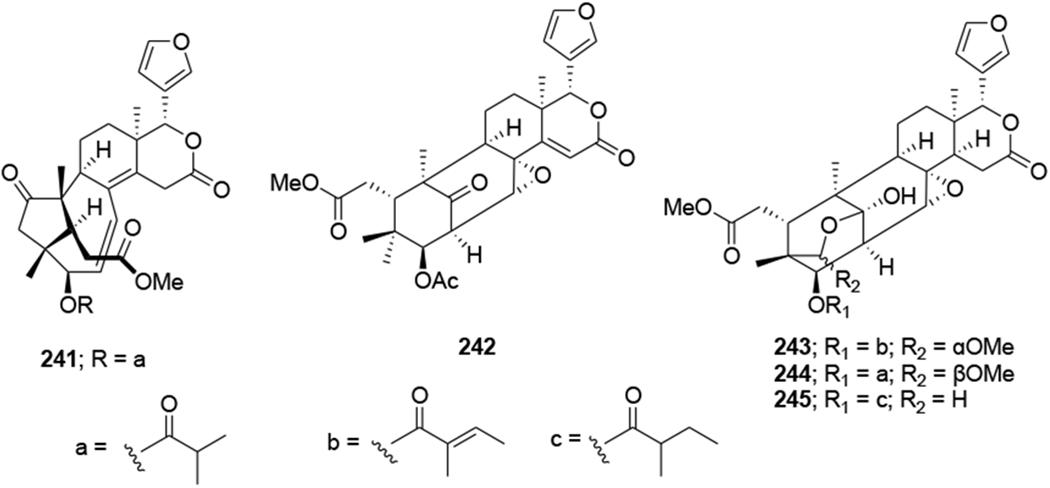
Chemical structures of compounds 241–245
Krishnolides A–D (246–249) (Figure 51), four khayanolide-type limonoids with a 2-carbonyl group, were isolated from the seeds of Xylocarpus moluccensis. Compounds 246–249 are unique khayanolides containing two large ester substituents of five or four carbon atoms at the C-3 and C-30 positions, respectively. Compound 246 with an 8,14-epoxy group exhibited moderate anti-HIV activity with an IC50 value of 17.45 μM.108
FIGURE 51.
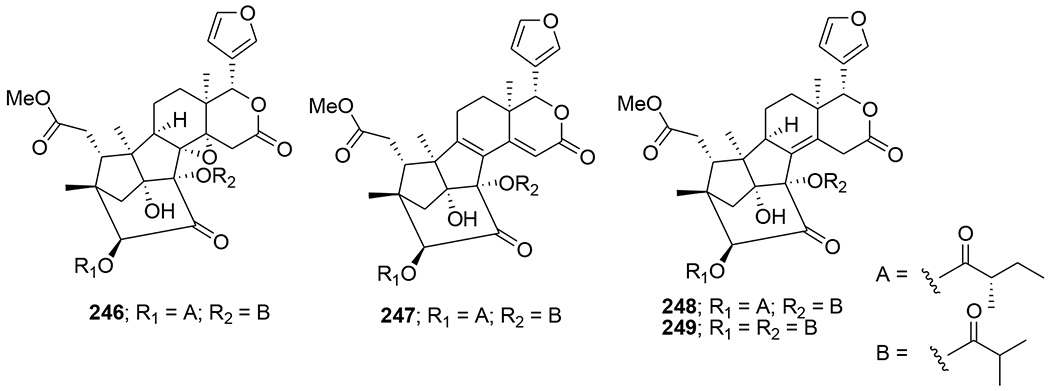
Chemical structures of compounds 246–249
3.5 |. Nortriterpenoids of the Schisandraceae
Three nortriterpenoids, schigrandilactones A–C (250–252) (Figure 52), were isolated from Schisandra grandiflora. Compounds 250 and 251 contain a spirocyclic moiety, and compound 252 has a new oxygenated pattern. Compounds 250–252 displayed EC50 values of 80.2, 20.8, and 5.1 μg/mL in infected C8166 cells.109 A nortriterpenoid, 20-hydroxymicrandilactone D (253) (Figure 53) was isolated from S. lancifolia. Compound 257 showed anti-HIV-1 activities with EC50 value of 99.0 μg/mL.110 Six nortriterpenoids, schirubridilactones A–F (254–259) (Figure 54) were isolated from S. rubriflora. Compounds 254–259 showed anti-HIV-1 activity with EC50 values of 30.1, 15.2, 14.3, 80.8, 66.8, and 50.1 μg/mL and a selectivity index of 5.1, 9.0, 8.7, 2.2, 3.1, and 3.5, respectively.111
FIGURE 52.
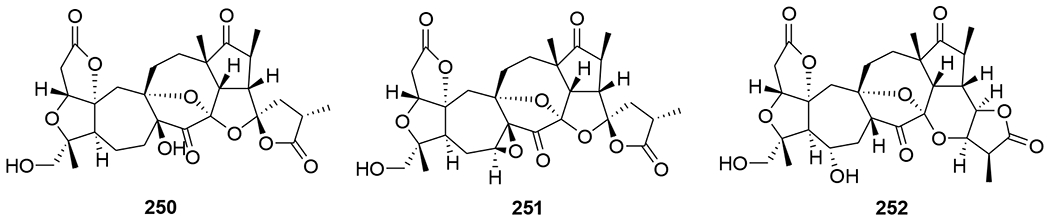
Chemical structures of compounds 250–252
FIGURE 53.
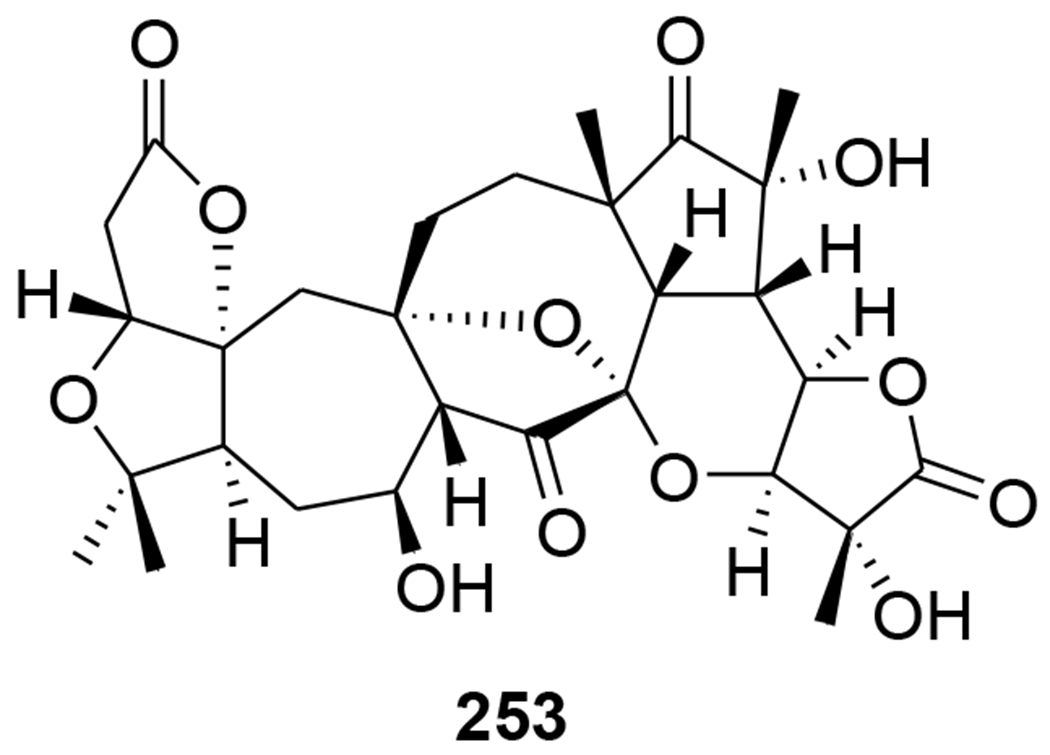
Chemical structure of compound 253
FIGURE 54.
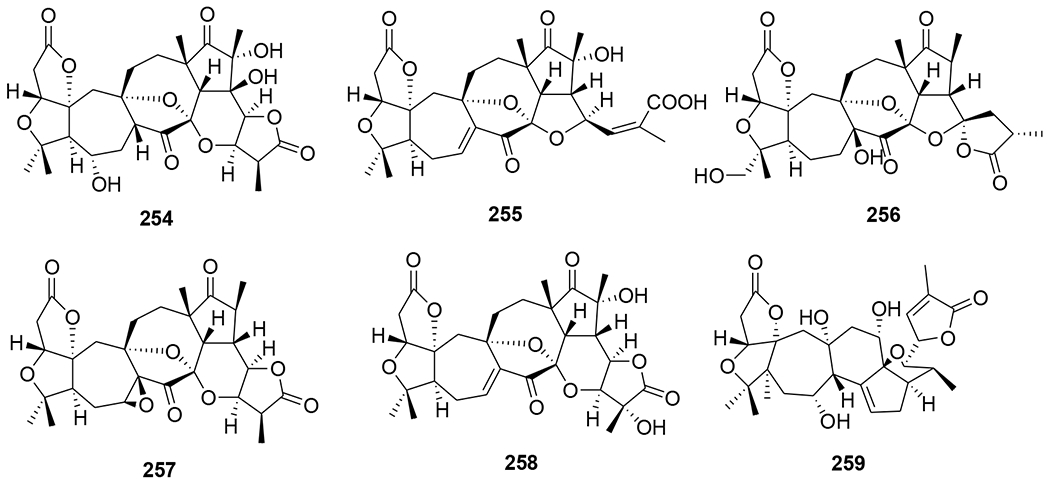
Chemical structures of compounds 254–259
A novel triterpenoid, schinarisanlactone A (260) (Figure 55), with an unprecedented skeleton of a 5/7/7/5/7/5/6/5-fused octacyclic ring system was isolated from Schisandra arisanensis. Compound 260 showed significant inhibition (11.8% survival rate) against the HIV virus at 10 μM.112 Three unique nortriterpenoids, schilancitrilactones A–C (261–263) (Figure 56), were isolated from S. lancifolia. Compound 261 has a 5/5/7/5/5/5-fused hexacyclic ring system with a C29 backbone, while 262 and 263 feature a 5/7/5/5/5-fused pentacyclic ring system with a C27 skeleton. Compound 263 showed anti-HIV-1 activity with an EC50 value of 27.54 μg/mL, while 261 and 262 were not active (EC50 >100 μg/mL).113 Two new highly oxygenated nortriterpenoids, schilancidilactones V and W (264 and 265) (Figure 57), were isolated from S. wilsoniana. They showed moderate anti-HIV-1 activity with EC50 values of 3.05 and 2.87 μg/mL, respectively.114 Schisphendilactones A and B (266 and 267) (Figure 58) isolated from S. sphenanthera exhibited anti-HIV-1 activity with EC50 values of 8.79 and 1.09 μg/mL, respectively.115 Schisarisanlactones A (268) and B (269) (Figure 59), were isolated from S. arisanensis. Compounds 268 and 269 have an unprecedented 5/5/7/5/5-fused pentacyclic ring system. Compound 268 showed significant inhibition (15.5% survival rate), while compound 269 exhibited moderate activity (39.8% survival rate) against the HIV virus at 10 μM.116
FIGURE 55.
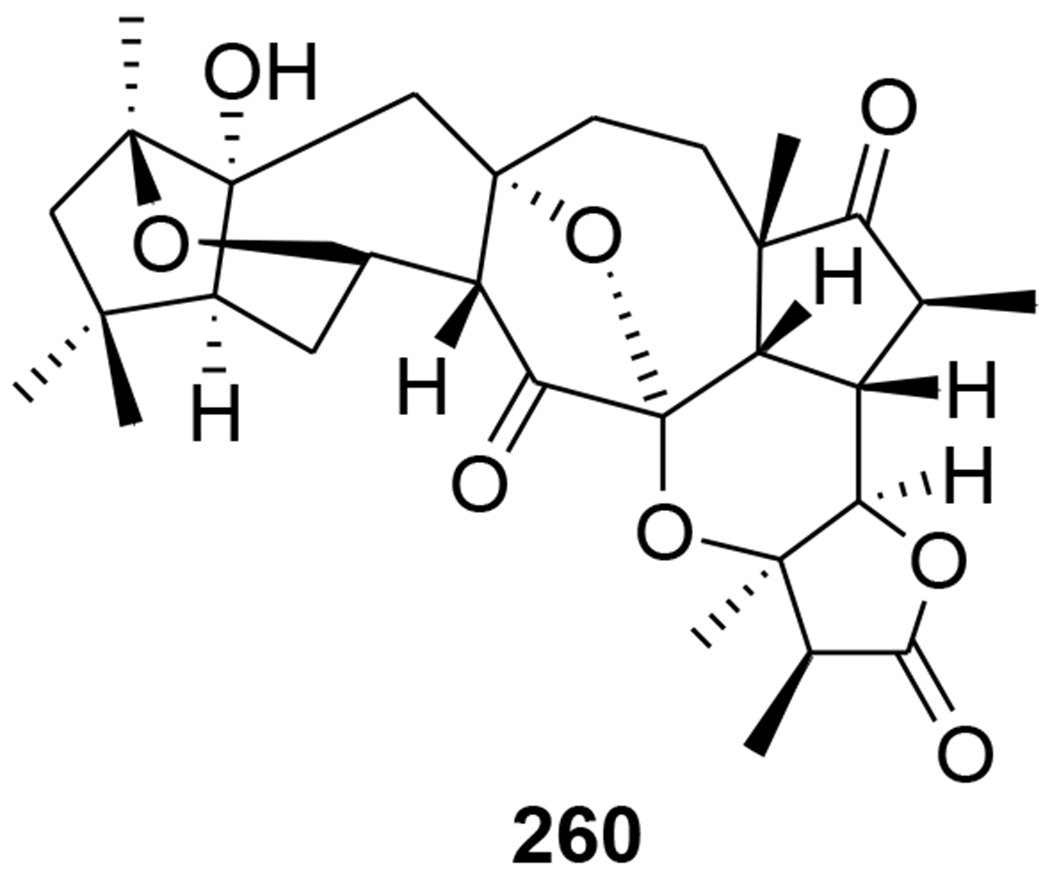
Chemical structure of compound 260
FIGURE 56.
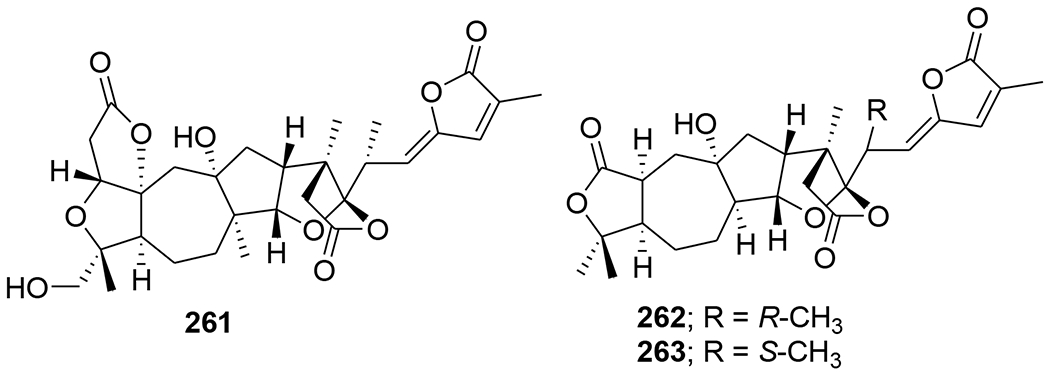
Chemical structures of compounds 261–263
FIGURE 57.
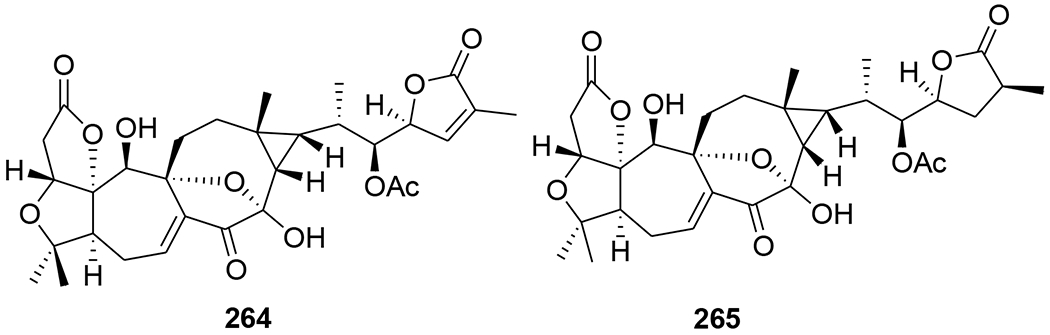
Chemical structures of compounds 264 and 265
FIGURE 58.
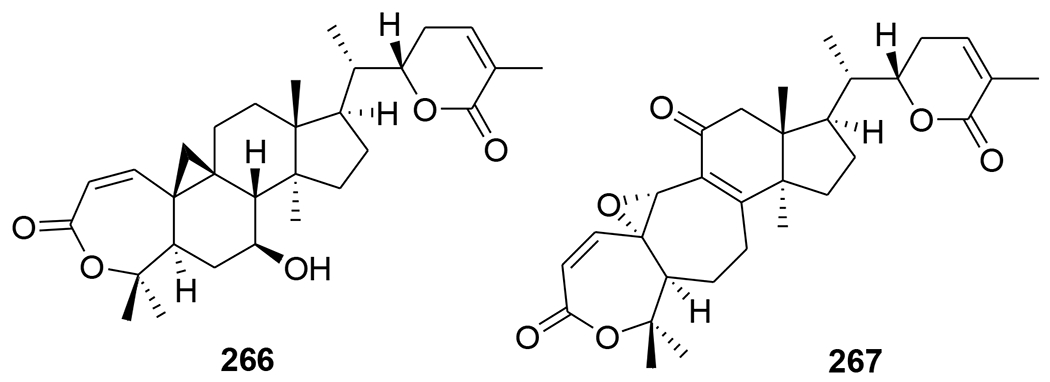
Chemical structures of compounds 266 and 267
FIGURE 59.
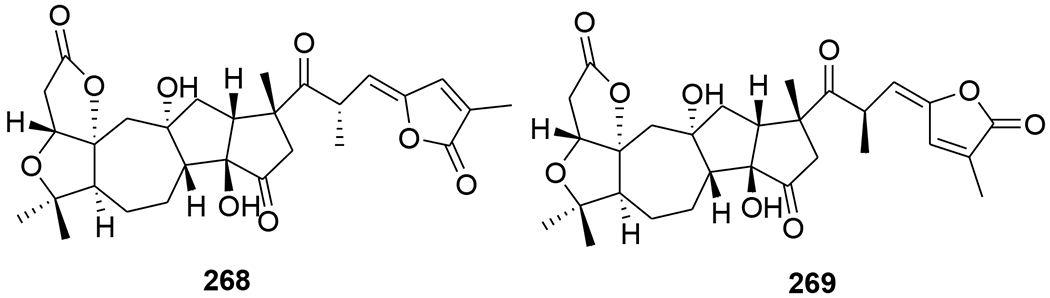
Chemical structures of compounds 268 and 269
3.6 |. Others
Four types of piscidinol A derivatives were synthesized and evaluated for their ability to inhibit HIV-1 protease (PR). Among these tirucallane-type triterpene derivatives, an A-seco derivative (270) moderately inhibited HIV PR (IC50 38.2 μM). However, the 2,2-dimethylsuccinic acid (DMS) acylated tirucallane derivatives (271–273, ranging from 50 to 100 μM) (Figure 60) were more inhibitory against the enzyme.117 Chemical investigation of the vines and leaves of Momordica charantia resulted in the isolation of 14 cucurbitane triterpenoids, kuguacins F–S (274–287) (Figure 61). Compounds 285 and 287 showed anti-HIV-1 activity with EC50 values of 7.2 and 3.7 μg/mL, respectively.118
FIGURE 60.
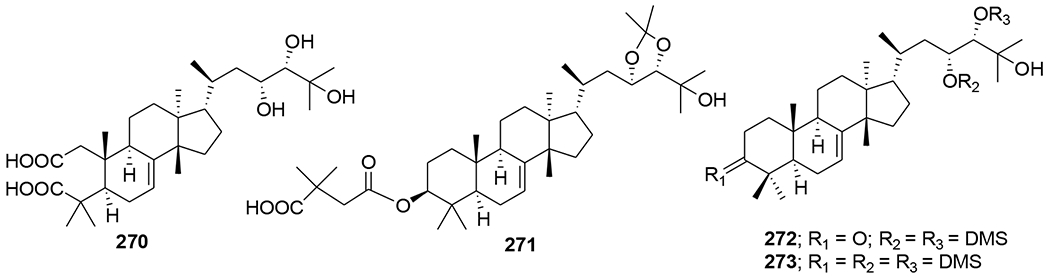
Chemical structures of compounds 270–273
FIGURE 61.
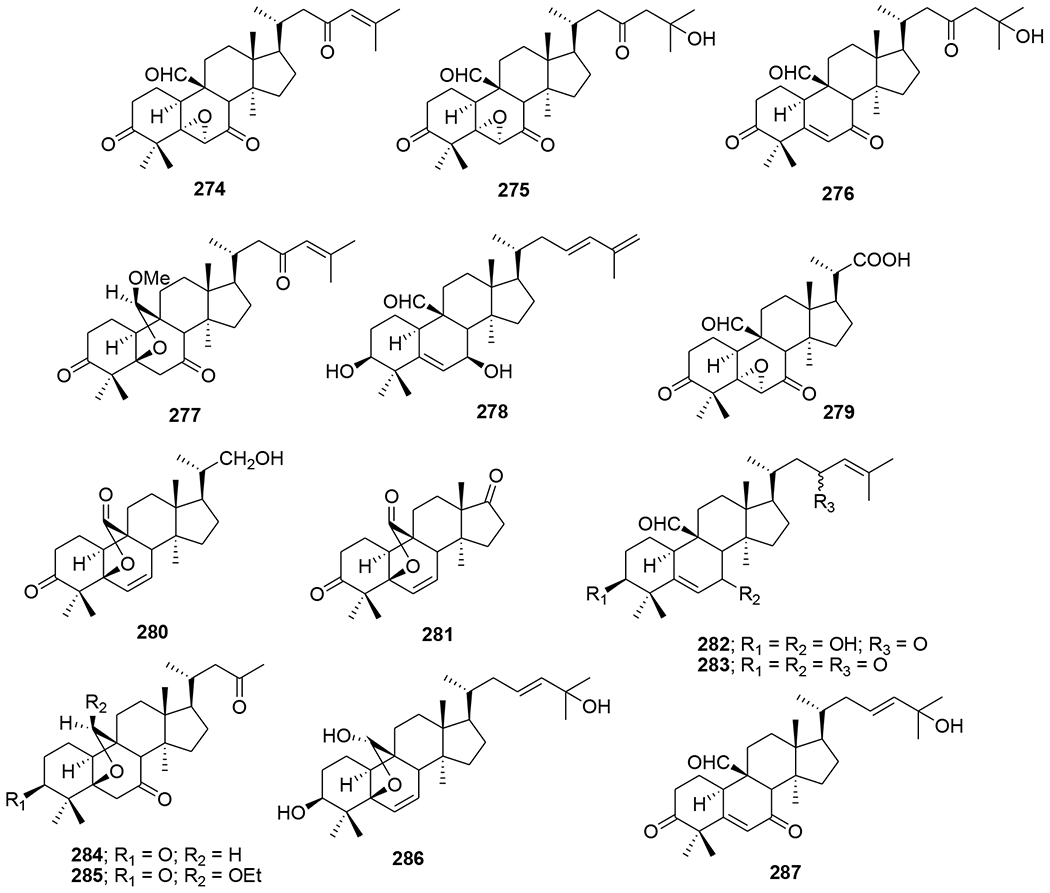
Chemical structures of compounds 274–287
Kadheterilactone A (288) and kadheterilactone B (289) (Figure 62) were isolated from Kadsura heteroclite. Compounds 288 and 289 were inactive against HIV-1 PR and RT.119 A pair of triterpenoid epimers, kadcoccitones A (290) and B (291) (Figure 63), together with a biogenetically related compound, kadcoccitone C (292), were isolated from K. coccinea. These epimers featured an unprecedented carbon skeleton with a 6/6/5/5-fused tetracyclic ring system unit and a C9 side chain. Compounds 290 and 292 showed anti-HIV-1 activity with EC50 values of 47.9 and 32.7 μg/mL, respectively.120
FIGURE 62.
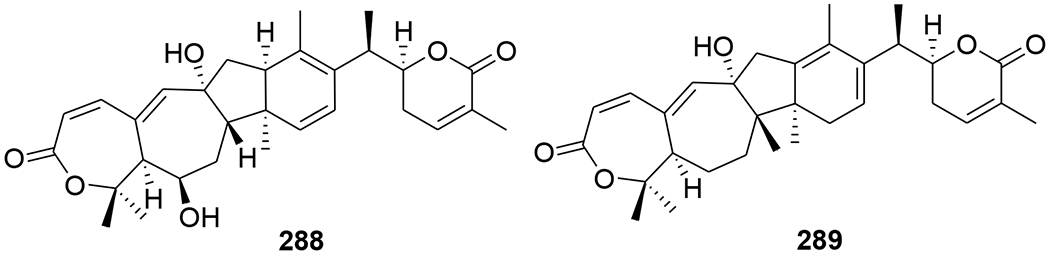
Chemical structures of compounds 288 and 289
FIGURE 63.
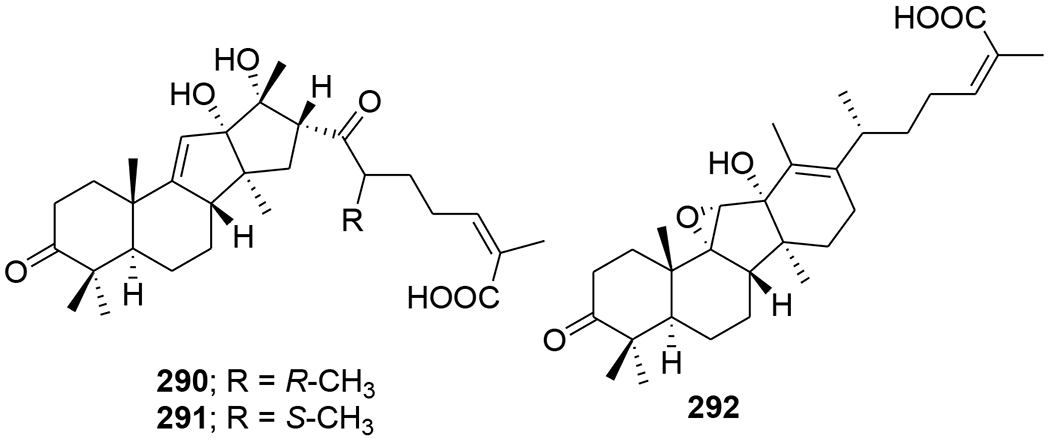
Chemical structures of compounds 290–292
Viral protein R (Vpr) is an accessory protein that plays important roles in the viral pathogenesis of HIV-1. In an anti-Vpr assay, the CHCl3-soluble extract of Picrasma javanica bark exhibited potent anti-Vpr activity. Furthermore, related research on quassinoids previously isolated from the extract demonstrated that compounds 293–307 (Figure 64) exhibit anti-Vpr activity. Among these compounds, javanicin I (307) exhibited the most potent anti-Vpr activity in comparison with that of the positive control damnacanthal. The structure-activity relationship analysis suggested that, in 2,12,14-triene-1,11,16-trione-2,12-dimethoxy-18-norpicrasane quassinoids, a methyl group at C-13 is the critical factor for a potent inhibitory effect in TREx-HeLa-Vpr cells.121
FIGURE 64.
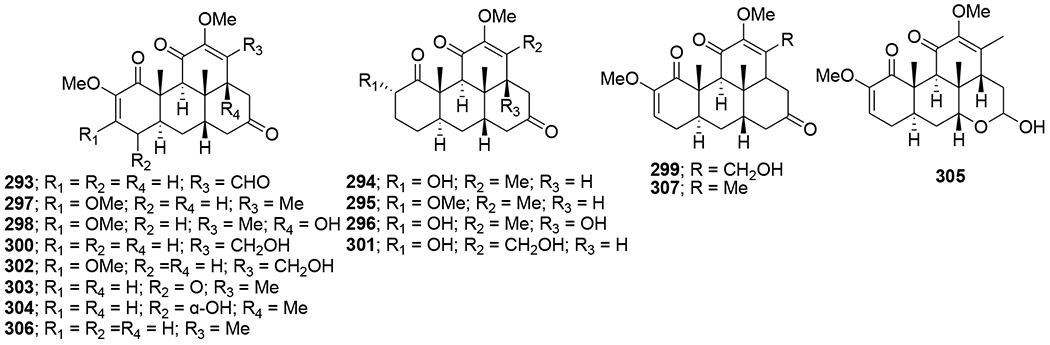
Chemical structures of compounds 293–307
4 |. PERSPECTIVES AND CONCLUSIONS
HIV infection remains a major threat worldwide, especially in developing countries. Although antiretroviral multi-drug treatment can provide durable repression of HIV, these drugs have limited clinical benefit due to the emergence of drug resistance and severe side effects, which have hampered their usefulness to most people suffering from AIDS. Therefore, finding new drugs with novel targets is of clinical significance to treat infected persons and furthermore to ultimately achieve the goal of complete eradication of HIV. Natural products are an important potential source for HIV-AIDS drug discovery. Over the past ten years, extensive research on natural products has revealed that triterpenoids and their derivatives are an important reservoir of anti-HIV drug leads. It appears that pentacyclic triterpenes are the most promising lead compounds for further development of novel antiviral drugs. The rational design of substituents linked to the C-3 and C-28 positions of betulinic acid and analogues is critical in attaining new derivatives with improved anti-HIV activity. Although the development of BVM as the first-in-class HIV-1 maturation inhibitor was halted due to the high baseline drug resistance of HIV-1 variants with polymorphism, further studies identified the second-generation maturation BMS-955176 (64), which showed significantly improved anti-HIV activity toward BVM-resistant variants. Unfortunately, the development of BMS-955176 was also discontinued by the pharmaceutical company GSK for gastrointestinal intolerance and treatment-emergent drug resistance by patients. However, due to the proven biological properties demonstrated by betulinic acid derivatives, many studies involving them are still reported in the literature. These studies conclude that artful modification both at C-3 and C-28 can effectively overcome HIV-1 resistance and improve antiviral activity in the presence of human serum. However, the effect of functionalization at other positions such as C-30 should not be neglected. Besides antiviral activity, solubility and microsomal stability should be considered. The corresponding SAR from the prior studies is a powerful prototype that affords optimal lead compounds to be emphasized for future endeavors. Even though betulinic acid is an interesting scaffold for developing antiviral agents and has attracted more attention, other classes of triterpenoids also offer unprecedented opportunities for the discovery of novel antiviral therapy. These results make it clear that natural triterpenoids and their synthetic derivatives will continue to be promising candidates for the development of novel anti-HIV drugs.
ACKNOWLEDGMENTS
This investigation was supported by NIH grant AI33066 from the NIAID awarded to K.H. Lee, the CAMS Innovation Fund for Medical Sciences (CIFS, No. 2019-I2M-1-005, 2017-I2M-1-013), the Foundation of Hunan Double First-rate Discipline Construction Projects of Bioengineering (No. YYZW2019-36) and the State Scholarship Fund from China Scholarship Council.
Funding information
US NIH National Institute of Allergies and Infectious Diseases Grant/Award Number: AI-033066; CAMS Innovation Fund for Medical Sciences, Grant/Award Numbers: CIFS 2019-I2M-1-005, 2017-I2M-1-013; the Foundation of Hunan Double First-rate Discipline Construction Projects of Bioengineering, Grant/Award Number: YYZW2019-36; the State Scholarship Fund from China Scholarship Council.
Biographies
AUTHORS’ BIOGRAPHIES
Hai-Feng Wu received his B.S. degree (2001) in biochemical engineering from Beijing University of Chemical Engineering, China and his Ph.D. degree (2009) in medicinal chemistry from Northwest Institute of Plateau Biology, Chinese Academy of Sciences, China. His post-doctoral research focused mainly on the isolation of bioactive natural products. He is currently working at the Institute of Medicinal Plant Development, Chinese Academy of Medicinal Sciences & Peking Union Medical College. The research area of Dr. Wu is natural product isolation and synthesis. He has published more than 60 research and review articles in peer-reviewed journals and written several books/book chapters.
Susan L. Morris-Natschke received her B.S. in chemistry from the University of Maryland-College Park in 1975 and her Ph.D. in organic chemistry from the University of North Carolina-Chapel Hill (UNC-CH) in 1982. She is currently Research Professor in the Division of Chemical Biology and Medicinal Chemistry, UNC Eshelman School of Pharmacy, UNC-CH, where she has been on the faculty since 1983. Her interests include scientific writing/editing, as well as the synthesis and structure-activity relationships of bioactive natural products.
Xu-Dong Xu received his PhD degree in medicinal chemistry from Peking Union Medical College in 1999. He undertook post-doctoral research at Chicago State University, Illinois (2001–2003). Since 2003, he has worked at the Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences and Peking Union Medical College. His research interests include the isolation, structural elucidation, and structural modification of bioactive compounds from natural products. He has published about 100 articles.
Mei-Hua Yang received her PhD degree in pharmacognosy from the School of Pharmaceutical Sciences, Peking University. She undertook post-doctoral research with Prof. Zuze Wu at the Beijing Institute of Radiation Medicine (2001–2003). Since 2003, she has worked as a scientist at the Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences, and Peking Union Medical College, where she is currently a professor.
Yung-Yi Cheng received her B.S. in pharmacy, M.S. in Medicinal Chemistry, and Ph.D. in Medicinal chemistry from China Medical University, Taiwan. She had worked as a postdoctoral scholar whose research related to pharmacokinetics in the National Yang-Ming University. She is currently an assistant researcher in China Medical University Hospital and also a visiting scholar at the UNC Eshelman School of Pharmacy. Her research interests include drug metabolism, drug design, and developing liquid chromatography tandem mass spectrometry methods for medicinal pharmacokinetics and food safety. Currently, her research is related to developing drug-like novel antitumor and anti-HIV agents from natural products and their derivatives.
Shi-Shan Yu received his Ph.D. degree in 1993. He has worked as a professor at the Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College. His research interests focus on bioactive substance from toxic natural medicines. He has published more than 60 scientific papers in top peer-reviewed journals.
Kuo-Hsiung Lee received his B.S. in pharmacy from Kaohsiung Medical University, Taiwan (1961), M.S. in pharmaceutical chemistry from Kyoto University, Japan (1965), and Ph.D. in medicinal chemistry from University of Minnesota, Minneapolis (1968). He joined the faculty of UNC Eshelman School of Pharmacy, University of North Carolina-Chapel Hill, in 1970 and is now Kenan Distinguished Professor of Medicinal Chemistry and Director of the Natural Products Research Laboratories. He has published over 940 research articles, has been granted over 121 patents, and has received numerous awards, including the Third Cheung On Tak International Award for Outstanding Achievement in Chinese Medicine from Hong Kong Baptist University in 2016.
Footnotes
CONFLICT OF INTEREST
The authors declare that there are no conflicts of interest.
REFERENCES
- 1.World Health Organization. 2018. www.kff.org/global-health-policy/fact-sheet/the-global-hivaids-epidemic/ [Google Scholar]
- 2.Girum T, Wasie A, Worku A. Trend of HIV/AIDS for the last 26 years and predicting achievement of the 90-90-90 HIV prevention targets by 2020 in Ethiopia: a time series analysis. BMC Infect Dis. 2018;18:320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bhattacharya S, Osman H. Novel targets for anti-retroviral therapy. J Infect. 2009;59:377–386. [DOI] [PubMed] [Google Scholar]
- 4.Marchand C, Maddali K, Metifiot M, Pommier Y. HIV-1 IN inhibitors: 2010 update and perspectives. Curr Top Med Chem. 2009;9:1016–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin LT, Hsu WC, Lin CC. Antiviral natural products and herbal medicines. J Tradit Complement Med. 2014;4:24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Salehi B, Kumar NVA, Şener B, Sharifi-Rad M, Kılıç M, Mahady GB, Vlaisavljevic S, Iriti M, Kobarfard F, Setzer WN, Ayatollahi SA, Ata A, Sharifi-Rad J. Medicinal plants used in the treatment of human immunodeficiency virus. Int J Mol Sci. 2018;19:1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kurapati KRV, Atluri VS, Samikkannu T, Garcia G, Nair MPN. Natural products as anti-HIV agents and role in HIV-associated neurocognitive disorders (HAND): a brief overview. Front Microbiol. 2015;6:1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Habibi P, Daniell H, Soccol CR, Grossi-de-Sa MF. The potential of plant systems to break the HIV-TB link. Plant Biotech J. 2019;17:1868–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee KH. Discovery and development of natural product-derived chemotherapeutic agents based on a medicinal chemistry approach. J Nat Prod. 2010;73:500–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Si LL, Meng K, Tian ZY, Sun JQ, Li HQ, Zhang ZW, Soloveva V, Li HW, Fu G, Xia Q, Xiao SL, Zhang LH, Zhou DM. Triterpenoids manipulate a broad range of virus-host fusion via wrapping the HR2 domain prevalent in viral envelopes. Sci Adv. 2018;4:eaau8408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fujioka T, Kashiwada Y, Kilkuskie RE, Cosentino LM, Ballas LM, Jiang JB, Janzen WP, Chen IS, Lee KH. Anti-AIDS agents, 11. Betulinic acid and platanic acid as anti-HIV principles from Syzigium claviflorum, and the anti-HIV activity of structurally related triterpenoids. J Nat Prod. 1994;57:243–247. [DOI] [PubMed] [Google Scholar]
- 12.Yu D, Morris-Natschke SL, Lee KH. New developments in natural products-based anti-AIDS research. Med Res Rev. 2007;27:108–132. [DOI] [PubMed] [Google Scholar]
- 13.Qian K, Morris-Natschke SL, Lee KH. HIV entry inhibitors and their potential in HIV therapy. Med Res Rev. 2009;29:369–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kashiwada Y, Hashimoto F, Cosentino LM, Chen CH, Garrett PE, Lee KH. Betulinic acid and dihydrobetulinic acid derivatives as potent anti-HIV agents. J Med Chem. 1996;39:1016–1017. [DOI] [PubMed] [Google Scholar]
- 15.Dang Z, Huang L, Chen CH. HIV-1 maturation inhibitors: an update. Drug Future. 2009;34:797–802. [Google Scholar]
- 16.Van Baelen K, Salzwedel K, Rondelez E, Van Eygen V, De Vos S, Verheyen A, Steegen K, Verlinden Y, Allaway GP, Stuyver LJ. Susceptibility of human immunodeficiency virus type 1 to the maturation inhibitor bevirimat is modulated by baseline polymorphisms in Gag spacer peptide 1. Antimicrob Agents Chemother. 2009;53:2185–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu W, Salzwedel K, Wang D, Chakravarty S, Freed EO, Wild CT, Li F. A single polymorphism in HIV-1 subtype C SP1 is sufficient to confer natural resistance to the maturation inhibitor bevirimat. Antimicrob Agents Chemother. 2011;55:3324–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adamson CS, Sakalian M, Salzwedel K, Freed EO. Polymorphisms in Gag spacer peptide 1 confer varying levels of resistance to the HIV-1 maturation inhibitor bevirimat. Retrovirology. 2010;7:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Margot NA, Gibbs CS, Miller MD. Phenotypic susceptibility to bevirimat in isolates from HIV-1-infected patients without prior exposure to bevirimat. Antimicrob Agents Chemother. 2010; 54:2345–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCallister S, Lalezari J, Richmond G, Thompson M, Harrigan R, Martin D, Salzwedel K, Allaway G. HIV-1 Gag polymorphisms determine treatment response to bevirimat (PA-457). Antivir Ther. 2008;13:A10. [Google Scholar]
- 21.Margot NA, Gibbs CS, Miller MD. Phenotypic susceptibility to bevirimat in isolates from HIV-1-infected patients without prior exposure to bevirimat. Antimicrob Agents Chemother. 2010;54:2345–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuo RY, Qian KD, Morris-Natschke SL, Lee KH. Plant-derived triterpenoids and analogues as antitumor and anti-HIV agents. Nat Prod Rep. 2009;26:1321–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Singh IP, Bodiwala HS. Recent advances in anti-HIV natural products. Nat Prod Rep. 2010;27:1781–1800. [DOI] [PubMed] [Google Scholar]
- 24.Cassels BK, Asencio M. Anti-HIV activity of natural triterpenoids and hemisynthetic derivatives 2004–2009. Phytochem Rev. 2011;10:545–564. [Google Scholar]
- 25.Patel RV, Park WS. Journey describing the discoveries of anti-HIV triterpene acid families targeting HIV-entry/fusion, protease functioning and maturation stages. Curr Topics Med Chem. 2014;14:1940–1966. [DOI] [PubMed] [Google Scholar]
- 26.Bednarczyk-Cwynar B, Zaprutko L. Recent advances in synthesis and biological activity of triterpenic acylated oximes. Phytochem Rev. 2015;14:203–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bednarczyk-Cwyna B, Günther A. Advances in chemistry and pharmacology of triterpenoid synthetic dimers. Curr Med Chem. 2017;24:2205–2240. [DOI] [PubMed] [Google Scholar]
- 28.Zhao M, Goedecke T, Gunn J, Duan JA, Che CT. Protostane and fusidane triterpenes: a mini-review. Molecules. 2013;18:4054–4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Verma SP. HIV: A Raft-targeting approach for prevention and therapy using plant-derived compounds (review). Curr Drug Targets. 2009;10:51–59. [DOI] [PubMed] [Google Scholar]
- 30.Hattori M, Ma CM, El Dine RS, Sato N. Survey of anti-HIV and anti-HCV compounds from natural sources. Can Chem Trans. 2013;1:116–140. [Google Scholar]
- 31.Bhatti HN, Khera RA. Biotransformations of diterpenoids and triterpenoids: a review. J Asian Nat Prod Res. 2014;16:70–104. [DOI] [PubMed] [Google Scholar]
- 32.Paduch R, Kandefer-Szerszen M. Antitumor and antiviral activity of pentacyclic triterpenes. Mini-Rev Org Chem. 2014;11:262–268. [Google Scholar]
- 33.Hordyjewska A, Ostapiuk A, Horecka A, Kurzepa J. Betulin and betulinic acid: triterpenoids derivatives with a powerful biological potential. Phytochem Rev. 2019;18:929–951. [Google Scholar]
- 34.Yogeeswari P, Sriram D. Betulinic acid and its derivatives: a review on their biological properties. Curr Med Chem. 2005;12:657–666. [DOI] [PubMed] [Google Scholar]
- 35.Shi W, Tang N, Yan WD. Research and development in betulin and betulinic acid derived triterpenoids. Mini-Rev Org Chem. 2014;11:343–354. [Google Scholar]
- 36.Mayaux JF, Bousseau A, Pauwels R, Huet T, Henin Y, Dereu N, Evers M, Soler F, Poujade C, De Clercq E, Le Pecq JB. Triterpene derivatives that block entry of human immunodeficiency virus type 1 into cells. Proc Natl Acad Sci USA. 1994;91:3564–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang L, Ho P, Lee KH, Chen CH. Synthesis and anti-HIV activity of bi-functional betulinic acid derivatives. Bioorg Med Chem. 2006;14:2279–2289. [DOI] [PubMed] [Google Scholar]
- 38.Sousa JLC, Freire CSR, Silvestre AJD, Silva AMS. Recent developments in the functionalization of betulinic acid and its natural analogues: a route to new bioactive compounds. Molecules. 2019;24:355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bedoya LM, Beltrán M, García-Pérez J, Obregón-Calderón P, Callies O, Jímenez IA, Bazzocchi IL, Alcamí J. Promiscuous, multi-target lupane-type triterpenoids inhibits wild type and drug resistant HIV-1 replication through the interference with several targets. Front Pharmacol. 2018;9:358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dorr CR, Yemets S, Kolomitsyna O, Krasutsky P, Mansky LM. Triterpene derivatives that inhibit human immunodeficiency virus type 1 replication. Bioorg Med Chem Lett. 2011;21:542–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qian KD, Kuo RY, Chen CH, Huang L, Morris-Natschke SL, Lee KH. Anti-AIDS agents 81. Design, synthesis and structure-activity relationship study of betulinic acid and moronic acid derivatives as potent HIV maturation inhibitors. J Med Chem. 2010;53:3133–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang C, Lu L, Na HY, Li XP, Wang Q, Jiang XF, Xu XY, Yu F, Zhang TH, Li JL, Zhang ZQ, Zheng BH, Liang GD, Cai LF, Jiang SB, Liu KL. Conjugation of a nonspecific antiviral sapogenin with a specific HIV fusion inhibitor: a promising strategy for discovering new antiviral therapeutics. J Med Chem. 2014;57:7342–7354. [DOI] [PubMed] [Google Scholar]
- 43.Goff RD, Thorson JS. Enhancing the divergent activities of betulinic acid via neoglycosylation. Org Lett. 2009;11:461–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu Z, Swidorski JJ, Nowicka-Sans B, Terry B, Protack T, Lin ZY, Samanta H, Zhang S, Li ZF, Parker DD, Rahematpura S, Jenkins S, Beno BR, Krystal M, Meanwell NA, Dicker IB, Regueiro-Ren A. C-3 benzoic acid derivatives of C-3 deoxybetulinic acid and deoxybetulin as HIV-1 maturation inhibitors. Bioorg Med Chem. 2016;24:1757–1770. [DOI] [PubMed] [Google Scholar]
- 45.Dang Z, Qian KD, Ho P, Zhu L, Lee KH, Huang L, Chen CH. Synthesis of betulinic acid derivatives as entry inhibitors against HIV-1 and bevirimat-resistant HIV-1 variants. Bioorg Med Chem Lett. 2012;22:5190–5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dang Z, Lai WH, Qian KD, Ho P, Lee KH, Chen CH, Huang L. Betulinic acid derivatives as human immunodeficiency virus type 2 (HIV-2) inhibitors. J Med Chem. 2009;52:7887–7891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao H, Holmes SS, Baker GA, Challa S, Bose HS, Song ZY. Ionic derivatives of betulinic acid as novel HIV-1 protease inhibitors. J Enzyme Inhib Med Chem. 2012;27:715–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qian KD, Yu DL, Chen CH, Huang L, Morris-Natschke SL, Nitz TJ, Salzwedel K, Reddick M, Allaway GP, Lee KH. Anti-AIDS agents 78. Design, synthesis, metabolic stability assessment, and antiviral evaluation of novel betulinic acid derivatives as potent anti-human immunodeficiency virus (HIV) agents. J Med Chem. 2009;52:3248–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Urano E, Ablan SD, Mandt R, Pauly GT, Sigano DM, Schneider JP, Martin DE, Nitz TJ, Wild CT, Freed EO. Alkyl amine bevirimat derivatives are potent and broadly active HIV-1 maturation inhibitors. Antimicrob Agents Chemother. 2016; 60:190–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qian KD, Bori ID, Chen CH, Huang L, Lee KH. Anti-AIDS agents 90. Novel C-28 modified bevirimat analogues as potent HIV maturation inhibitors. J Med Chem. 2012;55:8128–8136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhao Y, Gu Q, Morris-Natschke SL, Chen CH, Lee KH. Incorporation of privileged structures into bevirimat can improve activity against wild-type and bevirimat-resistant HIV-1. J Med Chem. 2016;59:9262–9268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dang Z, Ho P, Zhu L, Qian KD, Lee KH, Huang L, Chen CH. New betulinic acid derivatives for bevirimat-resistant human immunodeficiency virus type-1. J Med Chem. 2013;56:2029–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Coric P, Turcaud S, Souquet F, Briant L, Gay B, Royer J, Chazal N, Bouaziz S. Synthesis and biological evaluation of a new derivative of bevirimat that targets the Gag CA-SP1 cleavage site. European J Med Chem. 2013;62:453–465. [DOI] [PubMed] [Google Scholar]
- 54.Tang J, Jones SA, Jeffrey JL, Miranda SR, Galardi CM, Irlbeck DM, Brown KW, McDanal CB, Johns BA. Discovery of a novel and potent class of anti-HIV-1 maturation inhibitors with improved virology profile against gag polymorphisms. Bioorg Med Chem Lett. 2017;27:2689–2694. [DOI] [PubMed] [Google Scholar]
- 55.Johnson M, Jewell RC, Peppercorn A, Gould E, Xu J, Lou Y, Davies M, Baldwin S, Tenorio AR, Burke M, Jeffrey J, Johns BA. The safety, tolerability, and pharmacokinetic profile of GSK2838232, a novel 2nd generation HIV maturation inhibitor, as assessed in healthy subjects. Pharmacol Res Perspect. 2018;6:e00408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dang ATT, Pham CT, Le TA, Truong HH, Vu HTT, Soldatenkov AT, Nguyen TV. New hybrids between triterpenoid acids and nucleoside HIV-RT inhibitors. Mendeleev Commun. 2015;25:96–98. [Google Scholar]
- 57.Bori ID, Hung HY, Qian KD, Chen CH, Morris-Natschke SL, Lee KH. Anti-AIDS agents 88. Anti-HIV conjugates of betulin and betulinic acid with AZT prepared via click chemistry. Tetrahedron Lett. 2012;53:1987–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Spivak AY, Nedopekina DA, Galimshina ZR, Khalitova RR, Sadretdinova ZR, Gubaidullin RR, Odinokov VN. Click chemistry-assisted synthesis of novel C-2 triazole-linked betulinic acid conjugates with azidothymidine as potential anti-HIV agents. Arkivoc. 2018;part vii:1–19. [Google Scholar]
- 59.Xiong J, Kashiwada Y, Chen CH, Qian KD, Morris-Natschke SL, Lee KH, Takaishi Y. Conjugates of betulin derivatives with AZT as potent anti-HIV agents. Bioorg Med Chem. 2010;18:6451–6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li JZ, Goto M, Yang XM, Morris-Natschke SL, Huang L, Chen CH, Lee KH. Fluorinated betulinic acid derivatives and evaluation of their anti-HIV activity. Bioorg Med Chem Lett. 2016;26:68–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Swidorski JJ, Liu Z, Sit SY, Chen J, Chen Y, Sin N, Venables BL, Parker DD, Nowicka-Sans B, Terry BJ, Protack T, Rahematpura S, Hanumegowda U, Jenkins S, Krystal M, Dicker IB, Meanwell NA, Regueiro-Ren A. Inhibitors of HIV-1 maturation: development of structure–activity relationship for C-28 amides based on C-3 benzoic acid-modified triterpenoids. Bioorg Med Chem Lett. 2016;26:1925–1930. [DOI] [PubMed] [Google Scholar]
- 62.Chen Y, Sit SY, Chen J, Swidorski JJ, Liu Z, Sin N, Venables BL, Parker DD, Nowicka-Sans B, Lin ZY, Li ZF, Terry BJ, Protack T, Rahematpura S, Hanumegowda U, Jenkins S, Krystal M, Dicker ID, Meanwel NA, Regueiro-Ren A. The design, synthesis and structure-activity relationships associated with C28 amine-based betulinic acid derivatives as inhibitors of HIV-1 maturation. Bioorg Med Chem Lett. 2018;28:1550–1557. [DOI] [PubMed] [Google Scholar]
- 63.Regueiro-Ren A, Swidorski JJ, Liu Z, Chen Y, Sin N, Sit SY, Chen J, Venables BL, Zhu JL, Nowicka-Sans B, Protack T, Lin ZY, Terry B, Samanta H, Zhang SR, Li ZF, Easter J, Beno BR, Arora V, Huang XHS, Rahematpura S, Parker DD, Haskell R, Santone KS, Cockett MI, Krystal M, Meanwell NA, Jenkins S, Hanumegowda U, Dicker IB. Design, synthesis, and SAR of C-3 benzoic acid, C-17 triterpenoid derivatives. identification of the HIV-1 maturation inhibitor 4-((1R,3aS,5aR,5bR,7aR,11aS,11bR,13aR,13bR)-3a-((2-(1,1-dioxidothiomorpholino)ethyl)amino)-5a,5b,8,8,11a-pentamethyl-1-(prop-1-en-2-yl)-2,3,3a,4, 5,5a,5b,6,7,7a,8,11,11a,11b,12,13,13a,13boctadecahydro-1H-cyclopenta[a]chrysen-9-yl) benzoic acid (GSK3532795, BMS-955176). J Med Chem. 2018;61:7289–7313. [DOI] [PubMed] [Google Scholar]
- 64.Nowicka-Sans B, Protack T, Lin ZY, Li ZF, Zhang S, Sun YN, Samanta H, Terry B, Liu Z, Chen Y, Sin N, Sit SY, Swidorski JJ, Chen J, Venables BL, Healy M, Meanwell NA, Cockett M, Hanumegowda U, Regueiro-Ren A, Krystal M, Dicker IB. Identification and characterization of BMS-955176, a second-generation HIV-1 maturation inhibitor with improved potency, antiviral spectrum, and gag polymorphic coverage. Antimicrob Agents Chemother. 2016;60:3956–3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ortiz A, Soumeillant M, Savage SA, Strotman NA, Haley M, Benkovics T, Nye J, Xu ZM, Tan YC, Ayers S, Gao Q, Kiau S. Synthesis of HIV-maturation inhibitor BMS-955176 from betulin by an enabling oxidation strategy. J Org Chem. 2017;82:4958–4963. [DOI] [PubMed] [Google Scholar]
- 66.Collins S GSK discontinues development of maturation inhibitor BMS-955176. HIV i-Base website, 29 November 2016. Available at: http://i-base.info/htb/30865. Last accessed on 13 December 2017.
- 67.Lan P, Chen WN, Huang ZJ, Sun PH, Chen WM. Understanding the structure-activity relationship of betulinic acid derivatives as anti-HIV-1 agents by using 3D-QSAR and docking. J Mol Model. 2011;17:1643–1659. [DOI] [PubMed] [Google Scholar]
- 68.Wei Y, Ma CM, Hattori M. Synthesis and evaluation of A-seco type triterpenoids for anti-HIV-1 protease activity. European J Med Chem. 2009;44:4112–4120. [DOI] [PubMed] [Google Scholar]
- 69.Tolmacheva IA, Igosheva EV, Vikharev YB, Grishko VV, Savinova OV, Boreko EI, Eremin VF. Synthesis and biological activity of mono and diamides of 2,3-secotriterpene acids. Russian J Bioorg Chem. 2013;39:186–193. [DOI] [PubMed] [Google Scholar]
- 70.Grishko VV, Galaiko NV, Tolmacheva IA, Kucherov II, Eremin VF, Boreko EI, Savinova OV, Slepukhin PA. Functionalization, cyclization and antiviral activity of A-secotriterpenoids. European J Med Chem. 2014;83:601–608. [DOI] [PubMed] [Google Scholar]
- 71.Gutiérrez-Nicolás F, Gordillo-Román B, Oberti JC, Estévez-Braun A, Ravelo ÁG, Joseph-Nathan P. Synthesis and anti-HIV activity of lupane and olean-18-ene derivatives. Absolute configuration of 19,20-epoxylupanes by VCD. J Nat Prod. 2012;75:669–676. [DOI] [PubMed] [Google Scholar]
- 72.Kargbo RB. Novel triterpenone for treatment of viral diseases-HIV inhibitors. ACS Med Chem Lett. 2018; 9:298–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Barroso-González J, Jaber-Vazdekis NE, García-Expósito L, Machado J, Zárate R, Ravelo ÁG, Estévez-Braun A, Valenzuela-Fernández A. The lupane-type triterpene 30-oxo-calenduladiol is a CCR5 antagonist with anti-HIV-1 and anti-chemotactic activities. J Biol Chem. 2009;284:16609–16620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Callies O, Bedoya LM, Beltrán M, Muñoz A, Calderón PO, Osorio AA, Jiménez IA, Alcamí J, Bazzocchi IL. Isolation, structural modification, and HIV inhibition of pentacyclic lupane-type triterpenoids from Cassine xylocarpa and Maytenus cuzcoina. J Nat Prod. 2015;78:1045–1055. [DOI] [PubMed] [Google Scholar]
- 75.Reutrakul V, Anantachoke N, Pohmakotr M, Jaipetch T, Yoosook C, Kasisit J, Napaswa C, Panthong A, Santisuk T, Prabpai S, Kongsaeree P, Tuchinda P. Anti-HIV-1 and anti-inflammatory lupanes from the leaves, twigs, and resin of Garcinia hanburyi. Planta Med. 2010;76:368–371. [DOI] [PubMed] [Google Scholar]
- 76.Parra A, Martin-Fonseca S, Rivas F, Reyes-Zurita FJ, Medina-O’Donnell M, Martinez A, Garcia-Granados A, Lupiañez JA, Albericio F. Semi-synthesis of acylated triterpenes from olive-oil industry wastes for the development of anticancer and anti-HIV agents. European J Med Chem. 2014;74:278–301. [DOI] [PubMed] [Google Scholar]
- 77.Gutiérrez-Nicolás F, Gordillo-Román B, Oberti JC, Estévez-Braun A, Ravelo ÁG, Joseph-Nathan P. Synthesis and anti-HIV activity of lupane and olean-18-ene derivatives. Absolute configuration of 19,20-epoxylupanes by VCD. J Nat Prod. 2012;75:669–676. [DOI] [PubMed] [Google Scholar]
- 78.Baltina LA Jr, Kondratenko RM, Baltina LA, Baschenko NZ, Pl’yasunova OA. Synthesis and biological activity of new glycyrrhizic acid conjugates with amino acids and dipeptides. Russian J Bioorg Chem. 2009;35:510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Parra A, Rivas F, Lopez PE, Garcia-Granados A, Martinez A, Albericio F, Marquez N, Muñoz E. Solution- and solid-phase synthesis and anti-HIV activity of maslinic acid derivatives containing amino acids and peptides. Bioorg Med Chem. 2009;17:1139–1145. [DOI] [PubMed] [Google Scholar]
- 80.Kondratenko RM, Baltina LA, Baltina LA Jr, Plyasunova OA, Pokrovskii AG, Tolstikov GA, Synthesis of new hetero- and carbocyclic aromatic amides of glycyrrhizic acid as potential anti-HIV agents. Pharm Chem J. 2009;43:383–388. [Google Scholar]
- 81.Baltina LA Jr, Baltina LA, Kondratenko RM, Plyasunova OA, Nepogodiev SA, Field RA. Synthesis and anti-HIV activity of triterpene 3-O-galactopyranosides, analogs of glycyrrhizic acid. Chem Nat Comp. 2010;46:576–582. [Google Scholar]
- 82.Pereslavtseva AV, Tolmacheva IA, Slepukhin PA, Elťsov OS, Kucherov II, Eremin VF, Grishko VV. Synthesis of A-pentacyclic triterpene-alkenentitriles. Chem Nat Comp. 2014;49:1059–1066. [Google Scholar]
- 83.Osorio AA, Muñóz A, Torres-Romero D, Bedoya LM, Perestelo NR, Jiménez IA, Alcamí J, Bazzocchi IL. Olean-18-ene triterpenoids from Celastraceae species inhibit HIV replication targeting NF-kB and Sp1 dependent transcription. European J Med Chem. 2012;52:295–303. [DOI] [PubMed] [Google Scholar]
- 84.Narayan V, Ravindra KC, Chiaro C, Cary D, Aggarwal BB, Henderson AJ, Prabhu KS. Celastrol inhibits Tat-mediated human immunodeficiency virus (HIV) transcription and replication. J Mol Biol. 2011;410:972–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Song W, Si LL, Ji S, Wang H, Fang XM, Yu LY, Li RY, Liang LN, Zhou DM, Ye M. Uralsaponins M–Y, antiviral triterpenoid saponins from the roots of Glycyrrhiza uralensis. J Nat Prod. 2014;77:1632–1643. [DOI] [PubMed] [Google Scholar]
- 86.Woźniak Ł, Skapska S, Marszałek K. Ursolic acid—a pentacyclic triterpenoid with a wide spectrum of pharmacological activities. Molecules. 2015;20:20614–20641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhu M, Du XN, Li YG, Zhang GN, Wang JX, Wang YC. Design, synthesis and biological evaluation of novel HIV-1 protease inhibitors with pentacyclic triterpenoids as P2-ligands. Bioorg Med Chem Lett. 2019;29:357–361. [DOI] [PubMed] [Google Scholar]
- 88.Tanaka R, Tsujii H, Yamada T, Kajimoto T, Amano F, Hasegawa J, Hamashima Y, Node M, Katoh K, Takebe Y. Novel 3α-methoxyserrat-14-en-21β-ol (PJ-1) and 3β-methoxyserrat-14-en-21β-ol (PJ-2)-curcumin, kojic acid, quercetin, and baicalein conjugates as HIV agents. Bioorg Med Chem. 2009;17:5238–5246. [DOI] [PubMed] [Google Scholar]
- 89.Zhang P, Xu L, Qian KD, Liu J, Zhang LY, Lee KH, Sun HB. Efficient synthesis and biological evaluation of epiceanothic acid and related compounds. Bioorg Med Chem Lett. 2011;21:338–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sato N, Zhang Q, Ma CM, Hattori M. Anti-human immunodeficiency virus-1 protease activity of new lanostane-type triterpenoids from Ganoderma sinense. Chem Pharm Bull. 2009;57:1076–1080. [DOI] [PubMed] [Google Scholar]
- 91.Liang CQ, Shi YM, Li XY, Luo RH, Li Y, Zheng YT, Zhang HB, Xiao WL, Sun HD. Kadcotriones A–C: tricyclic triterpenoids from Kadsura coccinea. J Nat Prod. 2013;76:2350–2354. [DOI] [PubMed] [Google Scholar]
- 92.Liang CQ, Shi YM, Wang WG, Hu ZX, Li Y, Zheng YT, Li XN, Du X, Pu JX, Xiao WL, Zhang HB, Sun HD. Kadcoccinic acids A–J, triterpene acids from Kadsura coccinea. J Nat Prod. 2015;78:2067–2073. [DOI] [PubMed] [Google Scholar]
- 93.Kang D, Mutakin M, Levita J. Computational study of triterpenoids of Ganoderma lucidum with aspartic protease enzymes for discovering HIV-1 and plasmepsin inhibitors. International J Chem. 2015;7:62–68. [Google Scholar]
- 94.Chaturonrutsamee S, Kuhakarn C, Surawatanawong P, Prabpai S, Kongsaeree P, Jaipetch T, Piyachaturawat P, Jariyawat S, Akkarawongsapat R, Suksen K, Limthongkul J, Napaswad C, Nuntasaen N, Reutrakul V. Polycyclic polyprenylated acylphloroglucinols and biphenyl derivatives from the roots of Garcinia nuntasaenii Ngerns. & Suddee. Phytochemistry. 2018;146:63–74. [DOI] [PubMed] [Google Scholar]
- 95.Wei Y, Ma CM, Hattori M. Synthesis of dammarane-type triterpene derivatives and their ability to inhibit HIV and HCV proteases. Bioorg Med Chem. 2009;17:3003–3010. [DOI] [PubMed] [Google Scholar]
- 96.Wei Y, Ma CM, Hattori M. Anti-HIV protease triterpenoids from the acid hydrolysate of Panax ginseng. Phytochem Lett. 2009;2:63–66. [Google Scholar]
- 97.Esimone CO, Eck G, Nworu CS, Hoffmann D, Überla K, Proksch P. Dammarenolic acid, a secodammarane triterpenoid from Aglaia sp. shows potent anti-retroviral activity in vitro. Phytomed. 2010;17:540–547. [DOI] [PubMed] [Google Scholar]
- 98.Sun R, Song HC, Wang CR, Shen KZ, Xu YB, Gao YX, Chen YG, Dong JY. Compounds from Kadsura angustifolia with anti-HIV activity. Bioorg Med Chem Lett. 2011;21:961–965. [DOI] [PubMed] [Google Scholar]
- 99.Li SF, Di YT, Luo RH, Zheng YT, Wang YH, Fang X, Zhang Y, Li L, He HP, Li SL, Hao XJ. Cycloartane triterpenoids from Cassia occidentalis. Planta Med. 2012;78:821–827. [DOI] [PubMed] [Google Scholar]
- 100.Kongkum N, Tuchinda P, Pohmakotr M, Reutrakul V, Piyachaturawat P, Jariyawat S, Suksen K, Akkarawongsapat R, Kasisit J, Napaswad C. Cytotoxic, antitopoisomerase IIα, and anti-HIV-1 activities of triterpenoids isolated from leaves and twigs of Gardenia carinata. J Nat Prod. 2013;76:530–537. [DOI] [PubMed] [Google Scholar]
- 101.Yang YH, Sun R, Song HC, Xu YB, Yang P, Yang DY, Shen ZK, Wang AR, Chen YG, Dong JY. Microbial transformation of the triterpene nigranoic acid in Trichoderma sp. Phytochem Lett. 2012;5:123–127. [Google Scholar]
- 102.Sun R, Song HC, Yang YH, Yang P, Yang DY, Shen KZ, Xu YB, Gao YX, Chen YG, Dong JY. Microbiological transformation of the triterpene nigranoic acid by the freshwater fungus Dictyosporium heptasporum. J Asian Nat Prod Res. 2013;15:433–440. [DOI] [PubMed] [Google Scholar]
- 103.Rahim A, Saito YH, Miyake K, Goto M, Chen CH, Alam G, Morris-Natschke S, Lee KH, Nakagawa-Goto K. Kleinhospitine E and cycloartane triterpenoids from Kleinhovia hospital. J Nat Prod. 2018;81:1619–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wu HF, Ma GX, Yang QW, Zhu YD, Huang L, Tian Y, Yang XM, Zhang MH, Chen CH, Morris-Natschke SL, Yang MH, Xu XD, Lee KH. Discovery and synthesis of novel beesioside I derivatives with potent anti-HIV activity. European J Med Chem. 2019;166:159–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Liu CP, Xu JB, Han YS, Wainberg MA, Yue JM. Trichiconins A-C, limonoids with new carbon skeletons from Trichilia connaroides. Org Lett. 2014;16:5478–5481. [DOI] [PubMed] [Google Scholar]
- 106.Yu JH, Wang GC, Han YS, Wu Y, Wainberg MA, Yue JM. Limonoids with anti-HIV activity from Cipadessa cinerascens. J Nat Prod. 2015;78:1243–1252. [DOI] [PubMed] [Google Scholar]
- 107.Dai YG, Wu J, Padmakumar KP, Shen L. Sundarbanxylogranins A-E, five new limonoids from the Sundarban mangrove, Xylocarpus granatum. Fitoterapia. 2017;122:85–89. [DOI] [PubMed] [Google Scholar]
- 108.Zhang Q, Satyanandamurty T, Shen L, Wu J. Krishnolides A-D: new 2-ketokhayanolides from the Krishna mangrove, Xylocarpus moluccensis. Mar Drugs. 2017;15:333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Xiao WL, Gong YQ, Wang RR, Weng ZY, Luo X, Li XN, Yang GY, He F, Pu JX, Yang LM, Zheng YT, Lu Y, Sun HD. Bioactive nortriterpenoids from Schisandra grandiflora. J Nat Prod. 2009;72:1678–1681. [DOI] [PubMed] [Google Scholar]
- 110.Xiao WL, Yang LM, Zhang HB, Xue YB, Yang GY, Pu JX, Wang RR, Zheng YT, Sun HD. Chemical constituents from the leaves and stems of Schisandra lancifolia. Chem Pharm Bull. 2010;58:852–855. [DOI] [PubMed] [Google Scholar]
- 111.Xiao WL, Yang SY, Yang LM, Yang GY, Wang RR, Zhang HB, Zhao W, Pu JX, Lu Y, Zheng YT, Sun HD. Chemical constituents from the leaves and stems of Schisandra rubriflora. J Nat Prod. 2010;73:221–225. [DOI] [PubMed] [Google Scholar]
- 112.Lin YC, Lo IW, Chen SY, Lin PH, Chien CT, Chang SY, Shen YC. Schinarisanlactone A, a new bisnortriterpenoid from Schisandra arisanensis. Org Lett. 2011;13:446–449. [DOI] [PubMed] [Google Scholar]
- 113.Luo X, Shi YM, Luo RH, Luo SH, Li XN, Wang RR, Li SH, Zheng YT, Du X, Xiao WL, Pu JX, Sun HD. Schilancitrilactones A-C: three unique nortriterpenoids from Schisandra lancifolia. Org Lett. 2012;14:1286–1289. [DOI] [PubMed] [Google Scholar]
- 114.Gao XM, Li YQ, Shu LD, Shen YQ, Yang LY, Yang LM, Zheng YT, Sun HD, Xiao WL, Hu QF. New triterpenoids from the fruits of Schisandra wilsoniana and their biological activities. Bull Korean Chem Soc. 2013;34:827–830. [Google Scholar]
- 115.Liang CQ, Luo RH, Yan JM, Li Y, Li XN, Shi YM, Shang SZ, Gao ZH, Yang LM, Zheng YT, Xiao WL, Zhang HB, Sun HD. Structure and bioactivity of triterpenoids from the stems of Schisandra sphenanthera. Arch Pharm Res. 2014;37:168–174. [DOI] [PubMed] [Google Scholar]
- 116.Lo IW, Lin YC, Liao TC, Chen SY, Lin PH, Chien CT, Chang SY, Shen YC. Schisarisanlactones A and B: a new class of nortriterpenoids from the fruits of Schisandra arisanensis. Food Chem. 2013;136:1095–1099. [DOI] [PubMed] [Google Scholar]
- 117.Wei Y, Ma CM, Jiang TB, Du J, Zhou X, Liu GQ, Hattori M. Synthesis of piscidinol A derivatives and their ability to inhibit HIV-1 protease. J Asian Nat Prod Res. 2015;17:1079–1090. [DOI] [PubMed] [Google Scholar]
- 118.Chen JC, Liu WQ, Lu L, Qiu MH, Zheng YT, Yang LM, Zhang XM, Zhou L, Li ZR. Kuguacins F-S, cucurbitane triterpenoids from Momordica charantia. Phytochem. 2009;70:133–140. [DOI] [PubMed] [Google Scholar]
- 119.Xu LJ, Peng ZG, Chen HS, Wang J, Xiao PG. Bioactive triterpenoids from Kadsura heteroclite. Chem Biodivers. 2010;7:2289–2295. [DOI] [PubMed] [Google Scholar]
- 120.Liang CQ, Shi YM, Luo RH, Li XY, Gao ZH, Li XN, Yang LM, Shang SZ, Li Y, Zheng YT, Zhang HB, Xiao WL, Sun HD. Kadcoccitones A and B, two new 6/6/5/5-fused tetracyclic triterpenoids from Kadsura coccinea. Org Lett. 2012;14:6362–6365. [DOI] [PubMed] [Google Scholar]
- 121.Win NN, Ito T, Win YY, Ngwe H, Kodama T, Abe I, Morita H. Quassinoids: viral protein R inhibitors from Picrasma javanica bark collected in Myanmar for HIV infection. Bioorg. Med. Chem. Lett. 2016;26:4620–4624. [DOI] [PubMed] [Google Scholar]