

Abstract
Background and Purpose:
DTNBP1 gene variation and lower dysbindin-1 protein are associated with schizophrenia. Previous evidence suggests that downregulated dysbindin-1 expression results in lower expression of copper transporters ATP7A (intracellular copper transporter) and SLC31A1 (CTR1; extracellular copper transporter), which are required for copper transport across the blood brain barrier. However, whether antipsychotic medications used for schizophrenia treatment may modulate these systems is unclear.
Experimental Approach:
The current study measured behavioral indices of neurological function in dysbindin-1 functional knockout (KO) mice and their wild-type (WT) littermates with or without quetiapine treatment. We assessed serum and brain copper levels, ATP7A and CTR1 mRNA, and copper transporter-expressing cellular population transcripts: TTR (transthyretin; choroid plexus epithelial cells), MBP (myelin basic protein; oligodendrocytes), and GJA1 (gap-junction protein alpha-1; astrocytes) in cortex and hippocampus.
Key Results:
Regardless of genotype, quetiapine significantly reduced TTR, MBP, CTR1 mRNA, and serum copper levels. Neurological function of untreated KO mice was abnormal, and ledge instability was rescued with quetiapine. KO mice were hyperactive after 10 minutes in the open-field assay, which was not affected by treatment.
Conclusions and Implications:
Dysbindin-1 KO results in hyperactivity, lower serum copper, and neurological impairment, the last of which is selectively rescued with quetiapine. Antipsychotic treatment modulates specific cellular populations, affecting myelin, the choroid plexus, and copper transport across the blood brain barrier. Together these results indicate the widespread impact of antipsychotic treatment, and that alteration of dysbindin-1 may be sufficient, but not necessary, for specific schizophrenia pathology.
Keywords: dysbindin, copper, schizophrenia, quetiapine, Seroquel, ATP7A, CTR1
INTRODUCTION
Schizophrenia (SZ) is a complex disorder with several risk factors such as genetic vulnerability (DeLisi et al., 2002) and developmental issues (Fatemi and Folsom, 2009). The dystrobrevin binding protein 1 (DTNBP1) gene encodes the dysbindin-1 protein family (dysbindin-1)(Talbot, 2009; Talbot et al., 2009), and is a key part of the BLOC-1 complex involved in SNARE trafficking, neurite outgrowth and development, and lysosomal homeostasis (Ghiani et al., 2010). Furthermore, dysbindin-1 is considered to be a susceptibility gene for SZ (Allen et al., 2008; Bray et al., 2005; Marshall et al., 2017; Straub et al., 2002; Voisey et al., 2010), despite conflicting data (Schizophrenia Working Group of the Psychiatric Genomics, 2014). SZ subjects with DTNBP1 mutations have less cortical, midbrain, and hippocampal dysbindin-1 protein, though findings have varied (Abdolmaleky et al., 2015; Schoonover et al., 2018; Talbot et al., 2009; Weickert et al., 2008; Weickert et al., 2004). However, the implications of lower dysbindin-1 as it relates to SZ pathology remain poorly understood, inspiring the creation of several animal-based models of dysbindin-1 impairment. A spontaneous mutation in DBA/2J mice causing a loss of dysbindin-1 protein expression has been frequently used as such a model (Swank et al.). However, dysbindin-1 knockout (KO) mice with a DBA/2J background often exhibit abnormalities not representative of SZ; therefore, the model was transferred to a C57Bl/6 background to create a more representative model of SZ (Talbot, 2009). For the purpose of this study, only previous studies assessing dysbindin KO of a C57Bl/6 background were considered.
Previous literature has shown that the dysbindin-1 functional KO mice exhibit a 30–50% decrease in the copper transporters ATP7A and SLC31A1 (which produces the protein CTR1) (Gokhale et al., 2015). ATP7A and CTR1 are together responsible for facilitating copper transport across the blood brain barrier (Eisses and Kaplan, 2005; Scheiber et al., 2010; Yamaguchi et al., 1996). Copper is required for many functions including monoamine metabolism, mitochondrial activity, and myelination (Gokhale et al., 2015; Xu et al., 2009). Manipulations which reduce copper in vivo cause demyelination, which is prevented with antipsychotic quetiapine treatment (Gokhale et al., 2015; Gregg et al., 2009; Herring and Konradi, 2011; Xu et al., 2010; Zhang et al., 2008). SZ subjects exhibit lower mitochondrial activity and myelination (Kubicki et al., 2005; Roberts, 2017), and as we previously reported, lower levels of cellular copper in the substantia nigra (Schoonover et al., 2018). Furthermore, we suggested that the deficiency of copper in SZ was a consequence of lower ATP7A and CTR1 protein levels (Schoonover et al., 2018). Therefore, we sought to study the relationship between SZ susceptibility factor dysbindin-1 and copper metabolism. We hypothesized that altered dysbindin-1 expression negatively affects copper transport across the blood brain barrier and speculated that quetiapine treatment would ameliorate this effect.
Therefore, in the current study we quantitatively measured behavioral indices of neurological function and activity in KO mice and their wild-type (WT) littermates with or without quetiapine treatment to determine possible mechanisms that rescue copper deficiency-induced white matter deficits. To determine if KO mice exhibit deficits in copper transport, we measured copper levels in peripheral blood and brain. In cortex and hippocampus, brain areas shown to express less dysbindin-1 protein in SZ subjects (Abdolmaleky et al., 2015; Schoonover et al., 2018; Weickert et al., 2008; Weickert et al., 2004), we measured the transcript levels of the copper transporters ATP7A (ATP7A) and CTR1 (SLC31A1). Furthermore, transcripts of myelin basic protein (MBP), transthyretin (TTR) and gap junction protein alpha 1 (GJA1) were assayed to provide information on the structural integrity/cellular viability of the cell populations that transcribe them, MBP is a marker of myelin integrity, which is disrupted in a copper deficient state (Herring and Konradi, 2011). GJA1 is a marker of astrocytes, which express dysbindin-1 in their end feet surrounding capillaries forming the blood brain barrier (Iijima et al., 2009). Thyroid binding hormone TTR is expressed by the epithelial cells of the choroid plexus, a cellular population that also expresses at least one isoform (1A) of dysbindin-1 (Benson et al., 2001; Brouillette and Quirion, 2008; Spector et al., 2015; Talbot et al., 2009) and is also the key cellular population of the cerebrospinal fluid-brain barrier. These analyses aim to provide expansion of prior studies examining dysbindin-1 KO mice, and elucidate further potential mechanisms of downstream dysbindin-1 alterations in SZ.
METHODS:
Mice:
The mice used in the current study were a gift from Dr. Faundez of Emory University and are detailed in (Larimore et al., 2014). All mice were bred in-house from the Faundez colony founders following protocols approved by the Institutional Animal Care and Use Committee at the University of Alabama at Birmingham and in accordance with the National Institutes of Health guide for the care and use of Laboratory animals. KO mice were on a C57Bl/6J background. Mice were maintained on a 12/12 regular light dark cycle with food and water ad libitum. Female mice (regardless of treatment) were group housed. Due to high incidence of fighting amongst males, males were singly housed. No impact of sex on behavior was observed (data not shown). Breeding pairs were heterozygous males and females. Wildtype (WT) and KO mice were generated at the predicted Mendelian ratios. Mouse genotyping was performed on tailsnips by PCR of genomic DNA. The genotyping protocol was initially optimized by the University of Alabama Transgenic and Genetically Engineered Model Systems Core Facility). The following primers were used: 5’TCCTTGCTTCGTTCTCTGCT3’ (KO or WT forward), 5’CTTGCCAGCCTTCGTATTGT3’ (KO or WT reverse), 5’TGAGCCATTAGGAGATAAGAGCA3’ (KO or WT forward), and 5’AGCTCCACCTGCTGAACATT3’ (KO or WT reverse); this reaction yielded a 580bp and 274bp PCR products for wildtype and mutant, respectively (Figure 1A). q-PCR primers for the exon 2–3 border were used to confirm that the unaffected portion of the DTNBP1 gene was still intact in this model (Figure 1B.1). Knockout of functional dysbindin-1 via loss of the exon 6–7 border in KO mice was verified via q-RT-PCR (Figure 1B.2). The primer/probe sets used for dysbindin-1 assay were: Mm00458744 (exons 6–7) and Mm01328533 (exons 2–3).
Figure 1.
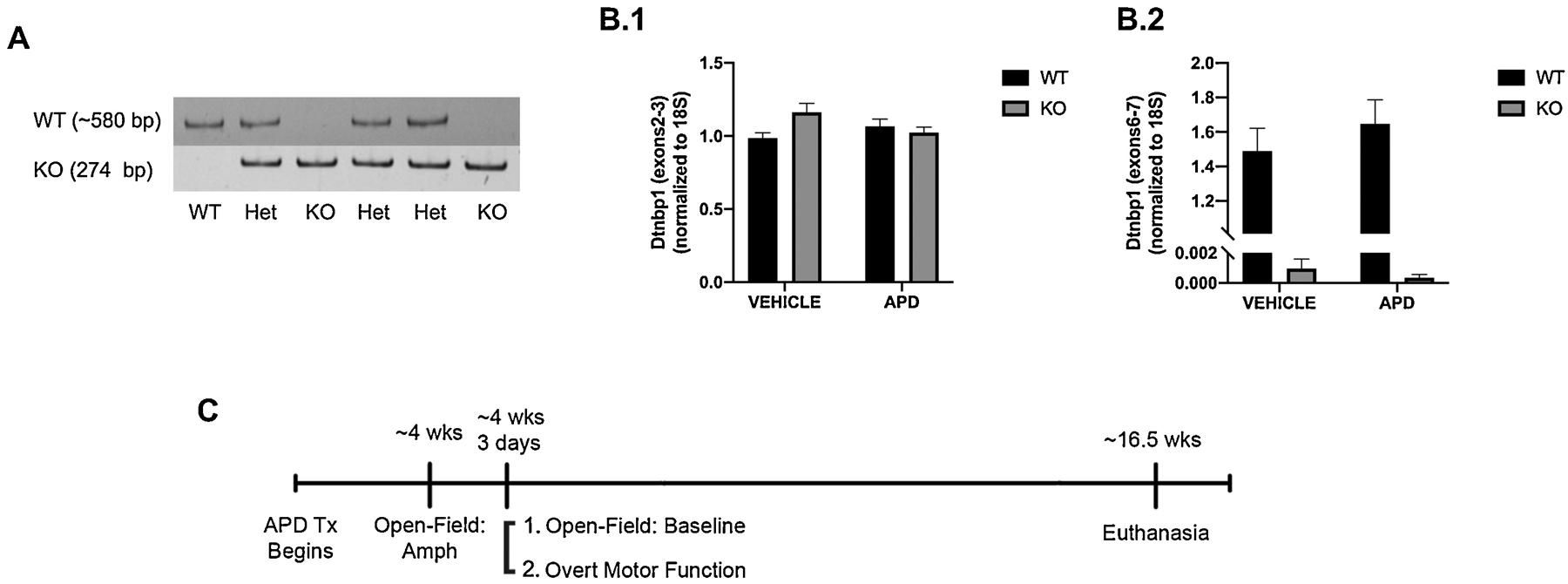
A) Representative genotyping gels. To be identified as a KO mouse, a band was required at the 274bp, but not the 580bp marker. Heterozygous (Het) mice were identified via bands in their respective well at both bp markers. WT mice were identified via a band at 580bp only. All bands were displayed at their expected bp marker. B) PCR validation of the dysbindin-1 KO mouse model. DTNBP1 exons 6–7 are deleted in KO mice, creating a functional knockout of dysbindin-1 protein. B.1) No difference of exons 2–3 levels were observed between KO and WT mice, regardless of treatment. B.2) Levels of exons 6–7 were undetectable in KO mice, regardless of treatment. Normal mRNA levels were present in WT controls. C) Chronological timeline of treatment and behavioral assays for mice. Abbreviations: wks, weeks; APD tx, antipsychotic drug treatment; Amph, amphetamine treatment; overt motor measure of neurodegeneration.
Antipsychotic treatment:
This study utilized the following groups: KO+quetiapine (n=11; Male: n=7; Female: n=4); KO+placebo (n=12; Male: n=7; Female: n=5); WT+quetiapine (n=16; Male: n=8; Female: n=8); WT+placebo (n=12; Male: n=6; Female: n=6). Group composition did not differ based on sex, as revealed by a Chi-squared test (χ2) (p>0.05). Quetiapine was chosen as the drug of treatment due to a previous study that observed quetiapine-induced amelioration of cognitive deficits in a mouse model of copper deficiency (Xuan et al., 2015). KO and WT littermates were 2–6 months of age at the beginning of the experiment. Once behavioral groups of 12 were formed, mice were placed on either antipsychotic treatment of quetiapine at a dosage of 10mg/kg/day or placebo in drinking water for four weeks before behavioral testing (Guan et al., 2000; He et al., 2009). See Figure 1C for timeline of quetiapine treatment and assays. Quetiapine was mixed thoroughly into drinking water; the same type of water bottle was used for all mice regardless of treatment. Mice in the placebo group received plain drinking water. Preliminary assessments showed no significant difference in quantity of water consumed between treated and non-treated animals (data not shown). Water volumes were appropriately adjusted for number of animals housed in a cage. Water was changed weekly.
Behavioral Assays:
We assessed ambulatory activity (with and without amphetamine) using the open-field assay. Four neurological tests were given: ledge test, hindlimb clasping, gait assessment, and kyphosis (Guyenet et al., 2010). Prior to behavioral tasks, mice were handled weekly and given fruit loops by the same lab member who would eventually behaviorally assess the mice. Fruit loops were given to facilitate habituation to handling. No fruit loops were given on the day of or during behavioral assessment. Due to the visual phenotype of KO mice (sandy grey fur), it was not possible for the experimenter to be blind to genotype; however, the experimenter was blinded to drug treatment group. All behavioral equipment was first cleaned with chlorhexidine followed by 70% ethanol prior to testing; during testing and between trials, 70% ethanol was used.
Ambulatory Activity:
Open-field square boxes measured 27.9cm × 27.9cm × 27.9cm with plexiglass sides consisting of 48 infrared beams and tracking software (Med Associates). After four weeks of treatment, all mice were injected intraperitoneally with 5mg/kg of amphetamine and placed individually in an open-field arena for 60 mins. Approximately three days later, mice were again placed in the open-field boxes and run for 30 mins without amphetamine injection for baseline locomotor activity. All open-field tasks, baseline and amphetamine, took place during the day-light cycle between 9am and 5pm under fluorescent lights (~140 lux) with the presence of white noise. Behavior was recorded automatically and analyzed using Activity Monitor Analysis software.
On the same date and after open-field baseline behavioral assay (i.e., on the day-light cycle between 9am and 5pm), the neurological function of mice were assessed using a simple composite phenotype scoring system as published by Jove (Guyenet et al., 2010), consisting of a ledge test, hindlimb clasping, gait assessment, and kyphosis. Each measure is scored on a scale of 0–3, with 0 representing an absence of a relevant phenotype and 3 representing the most severe phenotype. Each test is performed in triplicate to ensure reproducibility and then averaged for a composite score.
The ledge test is a direct measure of coordination. Each mouse is lifted by the base of its tail from the cage, placed on the ledge of the cage, and allowed to walk along the ledge and attempt to descend back into the cage. A score of 0 is given if the mouse will walk along the ledge without losing its balance and lowers itself gracefully back into its cage using its paws. A score of 1 is earned if the mouse loses its footing while walking along the ledge, but otherwise appears coordinated. A score of 2 is given if the mouse does not effectively use its hind legs or lands on its head rather than its paws when descending into the cage. If the mouse falls off the ledge, or nearly so, while walking or attempting to lower itself, it receives a score of 3.
The hindlimb clasping test, an index of neurological function, consists of grasping the base of the tail of the mouse and lifting it clear of all surrounding objects. The hindlimb position is observed for 10 seconds. If the hindlimbs are consistently splayed outward, away from the abdomen, it is given a score of 0. A score of 1 is given if one hindlimb is retracted toward the abdomen for more than 50% of the time suspended. A score of 2 is given if both hindlimbs are partially retracted toward the abdomen for more than 50% of the time suspended. Finally, a score of 3 is given if its hindlimbs are entirely retracted and touching the abdomen for more than 50% of the time suspended.
Gait and kyphosis were assessed as measures of coordination and dorsal curvature of the spine, respectively. To assess gait, each mouse was observed from behind as it walked. If the mouse moved normally, with its body weight supported on all limbs, with its abdomen not touching the ground, and with both hindlimbs participating evenly, it is given a score of 0. All mice walked normally; no mice were given a score other than 0. To assess kyphosis, the mouse is placed on a flat surface and observed while it walks. The mouse is given a score of 0 if the mouse is easily able to straighten its spine as it walks. All mice were given a score of 0 for kyphosis; all mice were able to straighten their spine while walking.
Euthanasia:
After all behavioral tasks were complete, mice were anesthetized using 3% isoflurane. Blood was collected from the heart prior to perfusion with 0.1M Phosphate buffer (pH 7.4). After perfusion was complete, the brain was removed and hemisected. Alternating between hemispheres, one hemisphere was further dissected to obtain prefrontal cortex and hippocampus separately for quantitative RT-PCR; the non-dissected hemisphere was designated for ICP-MS analysis of copper levels. All tissue was snap frozen using dry ice and then transferred to a freezer (−80°C) for storage until use.
Quantitative Real Time-PCR
Quantitative real-time PCR was conducted as described previously (Lucas et al., 2012). Tissue was homogenized in Trizol using an OmniBead Ruptor (Omni International; Kennesaw, GA). RNA was then isolated using the chloroform-isopropanol method. RNA concentration and purity were determined using the NanoDrop One (Thermo Fisher Scientific; Waltham, MA).
Equivalent amounts of RNA (1ug) were then treated with DNase I (Promega; Madison, WI) at 37°C for 30 min and DNase Stop (Promega) solution at 65°C for 15 min and then reverse-transcribed at 37°C for 2 hours using the High-Capacity cDNA Archive Kit (Applied Biosystems; Foster City, CA). Transcripts were measured using mouse-specific primers from Applied Biosystems and JumpStart Taq Readymix (Sigma-Aldrich; Saint Louis, MO) using a protocol with an initial ramp (2min, 50°C; 10 min, 95°C) and 40 subsequent cycles (15 s, 95°C; 1 min, 60°C). Using the calibrator method, relative concentration of transcript was calculated compared to a standard curve generated from pooled cDNA samples (1, 1:5, 1:10, 1:20, 1:40). Values for each gene were normalized to 18S (Hs99999901_s1). The following primer/probe sets for transcripts of interest are as follows: ATP7A (Mm00437663_m1); Slc31a1 (Mm00558247_m1); MBP (Mm01266402_m1); GJA1 (Mm01179639_s1); TTR (Mm00443267_m1).
Assessing Copper via Inductively-Coupled Plasma Mass Spectrometry:
Inductively-coupled plasma mass spectrometry (ICP-MS) was used to determine the concentration of copper in blood and tissue samples. Analysis of all samples was completed on an Agilent ICP-MS 7700 (Santa Clara, CA). All chemicals used in the ICP-MS analysis were trace metal grade. The blood samples were prepared in a basic solution matrix (BSM), a mixture of 4% (w/v) 1-butonal, 0.1% (w/v) ethylenediaminetetraacetic acid (EDTA), 0.1% (w/v) triton X-100, 2% (w/v) ammonium hydroxide and water, to prevent the solidifying of proteins. Three 500 μL aliquots of each blood sample were diluted to 2.5 mL with BSM. Tissue sample masses were obtained prior to digestion, then digested in concentrated nitric acid over a five-day period. Three 50 μL aliquots of each sample were diluted to 2.5 mL of Milli-Q 18 MΩ water to provide a 2% nitric acid matrix. Data analysis was completed using Agilent Masshunter software.
Statistical Analyses:
Data were summarized using means and standard errors (SE). Within each drug-genotype group, any observations that were at least 1.5-times the interquartile range (IQR) less the first quartile or greater than the third quartile were considered to be outliers and were excluded from the analysis. For each outcome, we fit linear models with effects for genotype, drug, and an interaction between drug and genotype. As these models assume equal variances, we evaluated this assumption by calculating the ratio of the largest group variance to the smallest group variance, as recommended by Box (1953); if this value was ≥ 3, random effects for each group were included in the model in order to allow for heterogeneous variances.
If the overall F-test for the model was statistically significant, we then investigated which measures showed a significant interaction between drug and genotype. For measures with a significant interaction, pairwise two-sample t-tests were used to determine which genotype-drug combinations significantly differed. P-values for these post-hoc comparisons were adjusted using the Tukey-Kramer method (Kramer 1956). For all other tests, p-value < 0.05 was considered statistically significant. SAS version 9.4 (SAS Institute Inc., Cary, NC) was used to conduct all statistical analyses.
Availability of Data
The data that support the findings of this study are available from the corresponding author upon request.
RESULTS
mRNA
Hippocampus:
In the hippocampus, no significant interactions between genotype and quetiapine treatment were observed for TTR, MBP, or SLC31A1 (CTR1) (Figure 2 A–C). However, significant main effects of quetiapine treatment were observed for TTR (p=0.008), MBP (p=0.048), and SLC31A1 (CTR1; p=0.008) (Figure 2D): quetiapine treatment decreased the mRNA of the three genes when data were collapsed across genotype. No significant findings were observed for hippocampal ATP7A or GJA1 (supplemental Figure S1 A–B).
Figure 2.
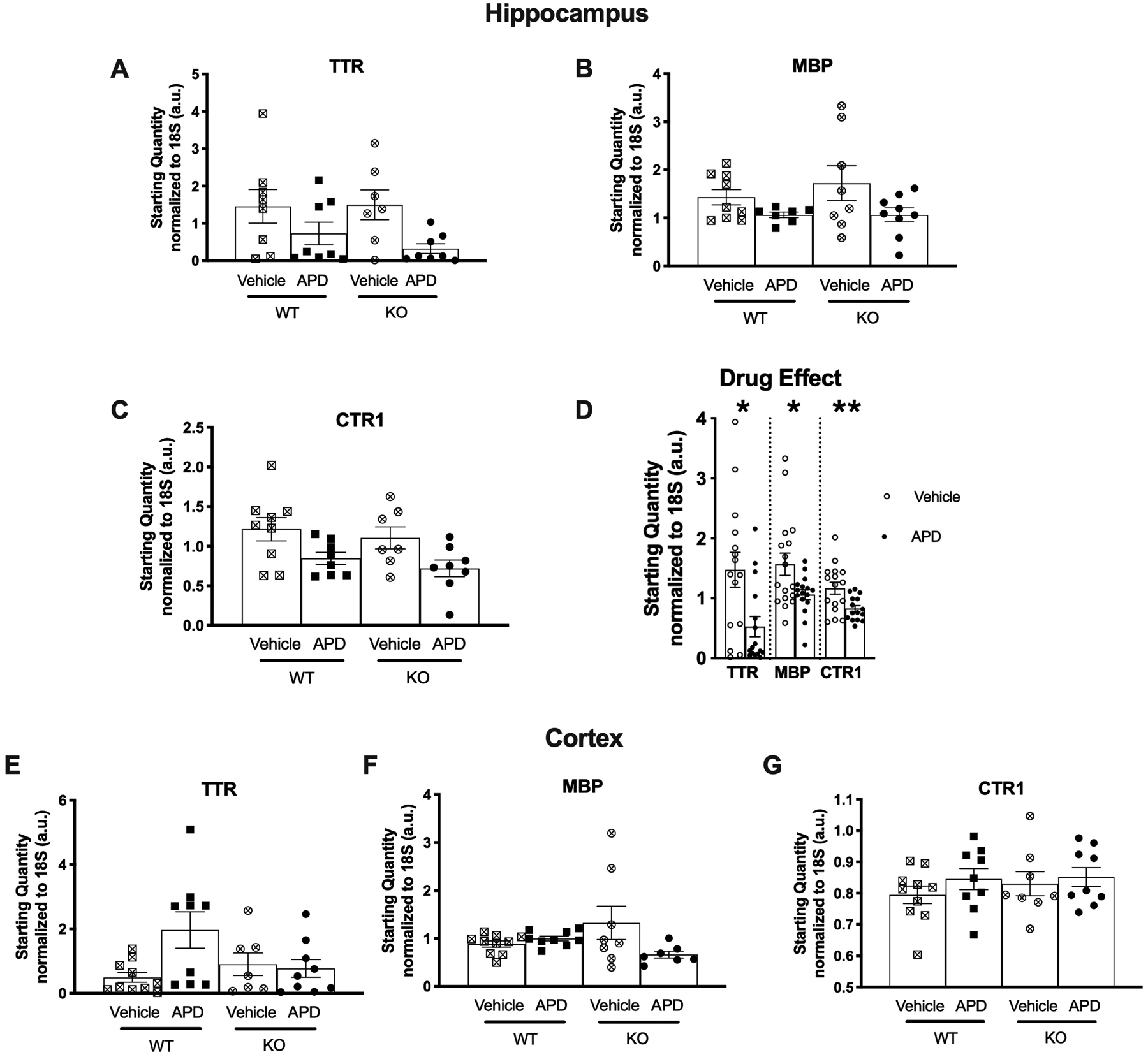
Comparison of hippocampal and cortical TTR, MBP, and SLC31A1 (CTR1) mRNA expression between genotypes and effect of antipsychotic treatment. A-C) mRNA levels of hippocampal TTR, MBP and CTR1 were not significantly different between groups. D) Effect of quetiapine treatment in mice collapsed across genotype for hippocampal TTR, MBP, and CTR1. APD treatment significantly reduced mRNA expression of all three genes. No drug effect was observed for the cortical data (data not shown). E-G) mRNA levels of cortical TTR, MBP and CTR1 were not significantly different between groups. Data are shown as mean and SEM. Significant findings from posthoc analysis: *p<0.05, **p<0.02. Abbreviations: WT, wild type mice; KO, dysbindin-1 knockout mice; APD, antipsychotic drug-treated mice.
Cortex:
No significant interactions or main effects were observed for cortical TTR, MBP or SLC31A1 (CTR1) (Figure 2E–G). Similarly, no significant effects were noted for cortical ATP7A or GJA1 (supplemental Figure S1 C–D). No effect of quetiapine treatment was observed in cortex (data not shown).
Copper Levels:
Analysis of copper content within exsanguinated brain yielded no significant differences between groups (Figure 3A). However, analysis of blood levels of copper revealed a significant between group effect (ANOVA p=0.032; Figure 3B); however, posthoc analysis did not reveal any further significance. Treatment with quetiapine reduced blood copper levels by approximately 50% regardless of genotype (p=0.021; Figure 3C). No correlational relationship between brain and blood copper was observed regardless of genotype or treatment (data not shown).
Figure 3.
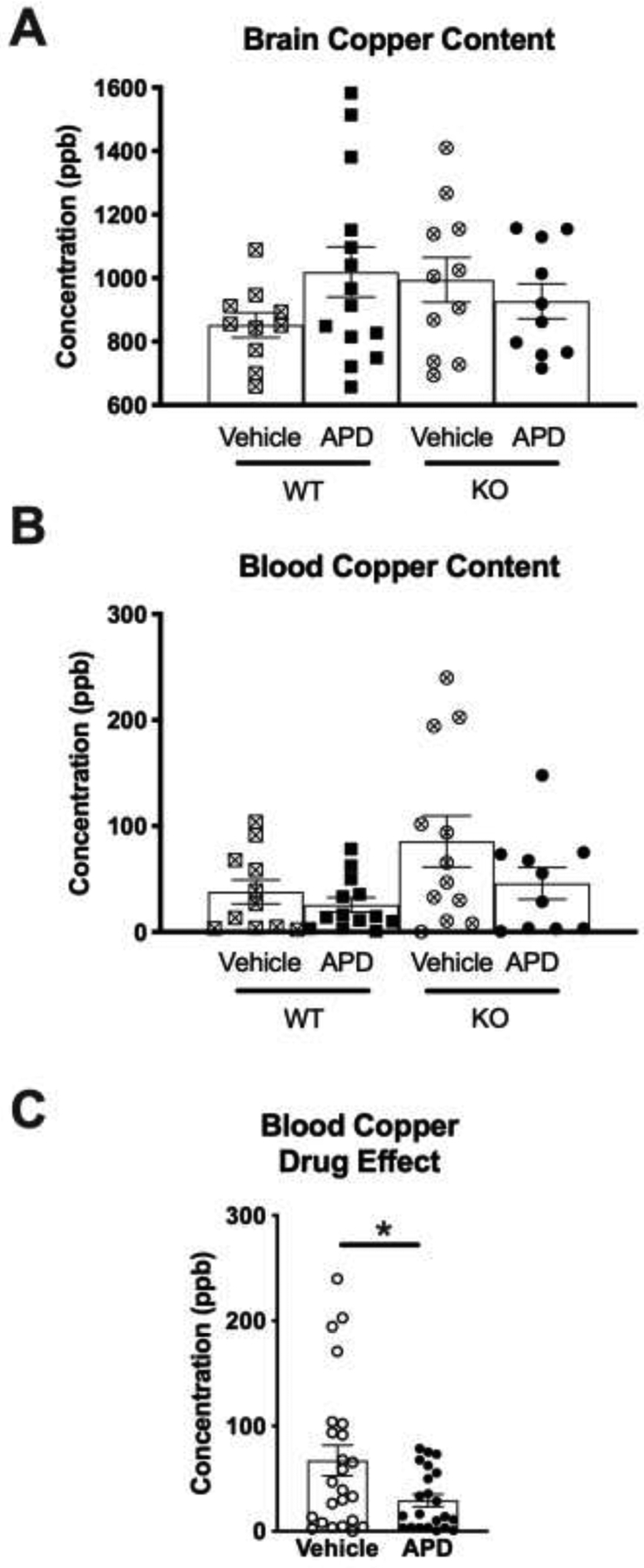
Copper levels as determined by inductively-coupled plasma mass spectrometry. A) Copper levels from exsanguinated brain were not different between groups. B) Copper levels from peripheral blood showed significance between groups (ANOVA, p<0.032); however, posthoc t-tests did not reveal significance. C) Data collapsed across genotype for drug effect showed significantly reduced blood copper levels (p<0.021). Levels expressed as concentration/parts per billion (ppb). Data are shown as mean and SEM. Abbreviations: WT, wild type mice; KO, dysbindin-1 knockout mice; APD, antipsychotic drug-treated mice.
Behavior
Neurological Function:
There were significant between-group differences for ledge instability (ANOVA p=0.023), and subsequent posthoc analysis revealed an increase of ledge instability in untreated KO mice in comparison to WT mice, both untreated (p=0.025) and treated (p=0.032) (Figure 4A). Similarly, a significant ANOVA F test was found for severity of hindlimb clasping (p<0.0001). Post hoc analyses revealed significant increases in severity of hindlimb clasping in untreated KO mice in comparison to untreated WT mice (p=0.0002) and treated WT mice (p=0.011) (Figure 4B).
Figure 4.
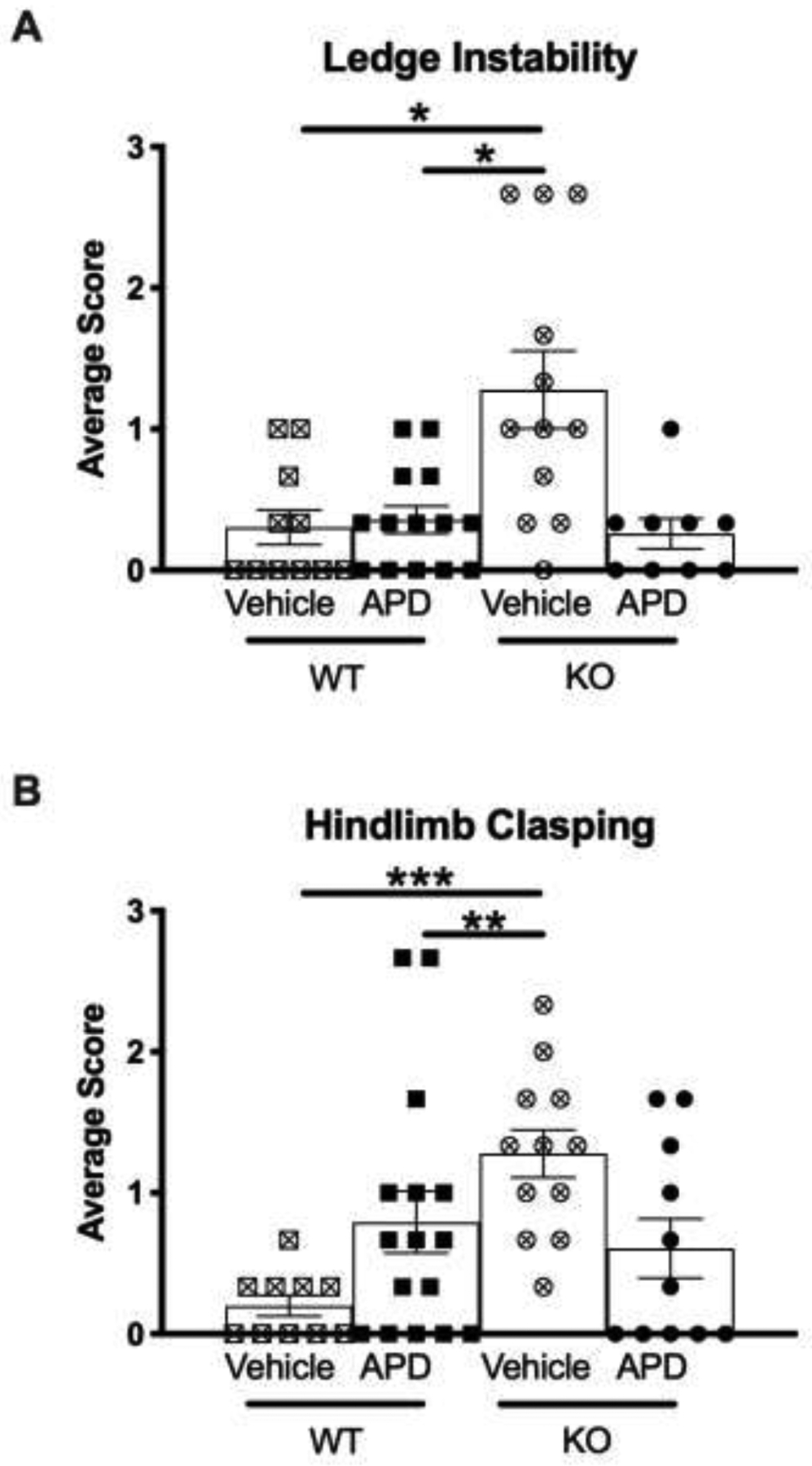
Neurological function assay. Average score is the composite of three trials, where the higher score indicates worse pathology. A) Posthoc analysis revealed that untreated KO mice were significantly more unstable on a ledge than untreated and treated WT mice. B) Untreated KO mice exhibited exacerbated severity of hindlimb clasping when compared to untreated and treated WT mice. Data are shown as mean and SEM. Significant findings from posthoc analysis: *p<0.05, ** p<0.02, *** p<0.001. Abbreviations: WT, wild-type mice; KO, dysbindin-1 knockout mice; APD, antipsychotic drug-treated mice.
Ambulatory Activity:
No interaction of quetiapine treatment with genotype was observed in the total locomotor activity during the baseline open-field task (data not shown). However, untreated dysbindin-1 KO mice were hyperactive in comparison to untreated WT mice at the 10m mark during the baseline assay (p=0.009) (Figure 5A). No differences of locomotor activity were observed among groups after amphetamine challenge (Figure 5B).
Figure 5.
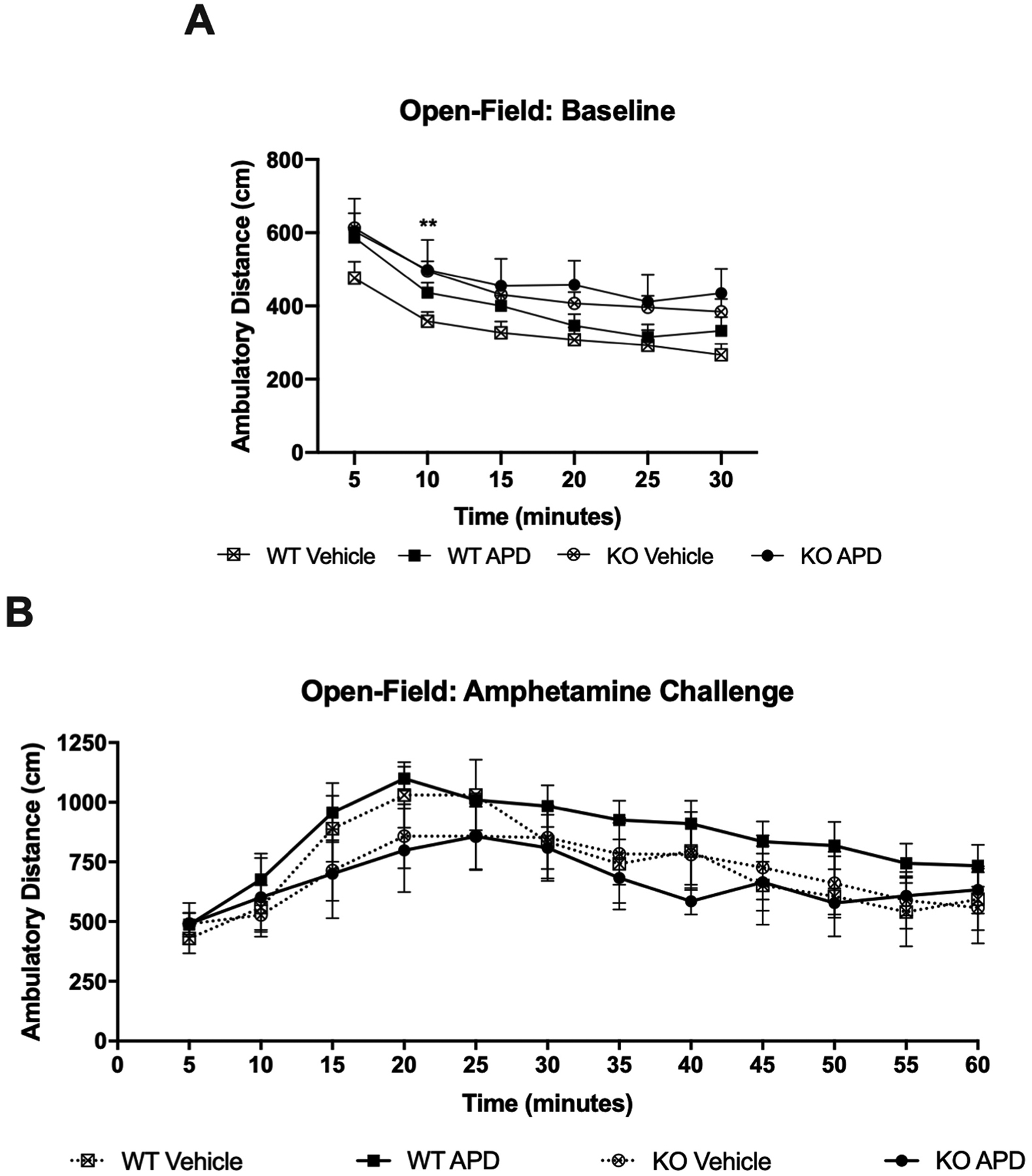
Ambulatory distance from open-field assays are shown in 5 minute bins for A) baseline and B) following intraperitoneal amphetamine injection. Untreated KO mice were significantly more hyperactive than untreated WT mice at the 10 minute mark following start of the assay, indicating a failure to habituate as well as WT mice. No differences in ambulatory distance following amphetamine challenge. Data (mean and SEM) are shown as ambulatory distance in cm for 30 minutes at baseline and 60 minutes for amphetamine challenge. Abbreviations: WT, wild-type mice; KO, dysbindin-1 knockout mice; APD, antipsychotic drug-treated mice.
DISCUSSION
Here we studied several behavioral indices of neurological function, peripheral blood and brain copper content, as well as the genetic expression of several structural integrity/cellular viability markers of specific cellular populations in areas known or suspected to express less dysbindin-1 in SZ. Based on the literature, we anticipated significant decreases in transcript levels of the copper transporters ATP7A, SLC31A1 (CTR1), and MBP in dysbindin-1 KO mice that would be ameliorated with quetiapine. We expected KO mice to be hyperactive in the open-field assay, yet respond equally to WT mice with amphetamine challenge. Furthermore, we sought to determine potential mechanisms of quetiapine rescue of copper deficits (Xu et al., 2010; Zhang et al., 2008).
mRNA
ATP7A and SCL31A1 (CTR1):
Previous literature reported a 30–50% decrease of SCL31A1 (CTR1) and ATP7A mRNA in dysbindin-1 mutant (KO) mice, and suggested that copper dysregulation was a downstream effect of dysbindin abnormalities (Gokhale et al., 2015). However, our study failed to replicate these differences. Upon further investigation, the reasons these differences may exist are twofold and methodological. Our verified (Lucas et al., 2012) data normalization protocol differs from that of Gokhale et al. (2015) in that the present study normalized each measurement to that of an unaltered housekeeping gene. Secondly, the current study and the work of Gokhale et al. (2015) used primers that target different parts of the genetic sequence. The Applied Biosystems primer used in the current study was closer to the 5’ portion of the gene (between exon 6–7) and should detect all the variants of the transcript while the primer used by Gokhale et al. (2015) has the possibility of detecting only some versions of the transcript due to a mismatch at the end of the sequence.
TTR:
Although we did not observe alterations of TTR dependent on brain region or genotype, we did observe a downregulation of its expression with antipsychotic treatment, but only in the hippocampus. Transthyretin, a thyroid hormone binding partner, is endogenously synthesized by the epithelial cells of the choroid plexus as shown by its expression of both TTR protein and mRNA (Herbert et al., 1986). Additionally, TTR is a negative acute phase protein (Doherty et al., 1998), which is downregulated during times of cellular inflammation and stress. It has been repeatedly suggested that SZ is a disease of inflammation (Muller, 2018), which is consistent with the serum and cerebrospinal fluid TTR deficit exhibited by SZ patients (Huang et al., 2006; Wan et al., 2006; Yang et al., 2006). In serum, TTR levels are elevated from baseline in SZ patients after antipsychotic treatment (Wan et al., 2006), which supports studies that have observed an anti-inflammatory effect of antipsychotics (Al-Amin et al., 2013; Kato et al., 2011). Although TTR levels in CSF have yet to be assessed before and after antipsychotic treatment in the same cohort of SZ patients, significantly lower levels of TTR in CSF were observed in antipsychotic treated SZ patients when compared to matched controls (Wan et al., 2006), which is consistent with our findings in hippocampus. Therefore, it is difficult to speculate how antipsychotics affect TTR expression in SZ, and whether such effects are differential between serum and cerebrospinal fluid. However, based on our findings, it may be possible that antipsychotic treatment (and its possible anti-inflammatory effects) affect the TTR production of epithelial cells in the choroid plexus in a manner different than that of the peripheral production of TTR in the liver, and therefore future studies are need to examine such functions. Furthermore, given the discrepancy of our findings in a dysbindin-1 knockout mouse model and the findings in schizophrenia subjects, perhaps dysbindin-1 knockout alone is not sufficient to mimic the cellular inflammation observed in schizophrenia, and therefore we observed no genotype-specific differences.
MBP:
We observed no significant alterations of MBP mRNA in either genotype. Previous literature has shown that decreases in copper function produce deficits in white matter integrity (Herring and Konradi, 2011). Furthermore, abnormalities in white matter diffusion, integrity, oligodendrocytes, and related white matter genes within whole brain, prefrontal cortex, and hippocampus via live imaging, genetic assay, and postmortem analysis have been repeatedly observed in SZ (Chambers and Perrone-Bizzozero, 2004; Hakak et al., 2001; Hof et al., 2002; Lyu et al., 2015; Schoonover et al., 2019), as have decreases in dysbindin (Abdolmaleky et al., 2015; Talbot, 2009; Talbot et al., 2009; Weickert et al., 2008; Weickert et al., 2004). Therefore, our finding of unaltered MBP was unexpected. However, unaltered MBP mRNA does not necessarily indicate unaltered MBP protein. Therefore, an assay correlating MBP mRNA levels with its cognate protein expression in KO mice would ameliorate this conundrum.
Impact of Quetiapine Treatment
Hippocampal TTR, MBP, and SLC31A1 (CTR1) mRNA were decreased by quetiapine regardless of genotype as shown by significant main effects of drug treatment in hippocampus. This brain region-specific drug effect is consistent with the very low density of D2 receptors in the prefrontal cortex (Hall et al., 1994), in comparison to the well characterized contribution of these receptors to hippocampus-based cognitive functions (for review please see (Lisman et al., 2011; Shohamy and Adcock, 2010)). Therefore, it is likely that the brain-region specific antipsychotic effect observed here is due to the binding profile of quetiapine and the inherent receptor densities of each brain region.
Our findings of lower MBP mRNA following antipsychotic drug treatment is not surprising. SZ patients exhibit white matter abnormalities that are consistent across medication status (Alvarado-Alanis et al., 2015; Asami et al., 2014; Holleran et al., 2014; Lee et al., 2013), indicating that antipsychotic treatment does not sufficiently improve white matter integrity in SZ.
Similar to the current results, one study observed lower serum copper levels in SZ after antipsychotic treatment (Chen et al., 2018). Given the suspected anti-inflammatory properties of antipsychotics (Al-Amin et al., 2013; Kato et al., 2011), and the potential inhibitory impact of stressors on protein synthesis and translation factors (Gameiro and Struhl, 2018), one could speculate that a complex battle of disease and drug effect is at play. CTR1 expression is downregulated in the substantia nigra in SZ (Schoonover et al., 2018), perhaps due to the stress sensitivity of SLC31A1 translation. However, once a patient is administered antipsychotics, the cellular stress is lessened via the anti-inflammatory properties of antipsychotics and the rate of protein translation is increased, resulting in a compensatory downregulation of transcription. Such a scenario would result in lower SLC31A1 mRNA, while simultaneously upregulating CTR1 protein to remedy its deficit, which in turn would increase the amount of copper transported across the blood brain barrier. However, such an idea has not been yet been studied, and therefore such events remain speculation.
GJA1 is a marker of astrocytic gap junctions, which express dysbindin-1 in their end feet surrounding capillaries forming the blood brain barrier (BBB) (Iijima et al., 2009). There is an increasing amount of evidence suggesting blood brain barrier alterations in psychosis and in particular, schizophrenia. Studies have implicated a “leaky” BBB in different ways: 1) by altered CSF/serum albumin ratio in patients (Axelsson et al., 1982; Bauer and Kornhuber, 1987; Kirch et al., 1985; Severance et al., 2015); 2) alterations of genes involved in ion transport, cell adhesion, and proliferation in microvascular endothelial cells (Harris et al., 2008); and 3) alterations of the extra-capillary components of the neurovascular unit (Najjar and Pearlman, 2015, Vostrikov et al., 2008, Webster et al., 2001, Webster et al., 2005). Therefore, given the negative impact on white matter stemming from loss of dysbindin-1 (Nickl-Jockschat et al., 2012), and the ultrastructural abnormalities in myelin that develop in GJA1-knockout animals (Magnotti et al., 2011), we suspected that GJA1 and dysbindin-1 may potentially interact either directly or indirectly. However, our findings here instead indicate that there is more than one way to dysfunction; dysbindin-1 knockout does not alter GJA1 expression, indicating different upstream modulators that result in similar pathologies.
Copper
As previously conducted by Gokhale et al. (2015), we assessed brain copper content and as an extension of their work, and also assessed peripheral blood content. Similarly, we observed no significant alterations of brain copper content. Although copper homeostasis appears to be altered as shown by slightly elevated peripheral blood copper in KO mice, brain copper levels could be maintained through an adaptive response or metabolic set point. However, neurons of KO mice exhibit an impaired response to excess extracellular copper (Gokhale et al., 2015), so perhaps an unknown modulator of copper transport exists that would result in these findings. While only a trend, we did observe an elevation of blood copper content in placebo KO mice, and an interesting downregulating effect of quetiapine treatment regardless of genotype. Taken together with previous literature, these results suggest that dysbindin-1 is sometimes but not always associated with changes in copper homeostasis, and that induction of alternate pathways by quetiapine treatment may be involved. Given that this is the first study of peripheral copper in KO mice, these results require replication.
Behavior
We anticipated alterations in measures of hyperactivity, and for quetiapine to rescue the symptomology. Indeed, we observed time-dependent hyperactivity of KO mice at baseline, but no exaggeration of the phenotype with amphetamine treatment, which is consistent with previous literature (Cox et al., 2009; Papaleo et al., 2012). However, we observed no significantly positive influence of quetiapine. This could be attributed to the binding profile of quetiapine, as it has affinity for serotonergic, histaminergic, and dopaminergic receptors, and rapidly dissociates from D2 receptors (Schatzberg and Nemeroff, 2009). Given its quick action at the dopaminergic D2 receptor, its primary affinity for other transmitter systems, and that quetiapine occupies only 30% of D2 receptors at its therapeutic dose (Schatzberg and Nemeroff, 2009), its lack of effect on a primarily dopaminergic-stemming behavior is not surprising. Although quetiapine is not as frequently prescribed as other antipsychotics and not the most affinitive for D2 receptors, it was chosen for its ameliorative effect in a copper chelation model (Xu et al., 2010; Zhang et al., 2008) in hopes to reveal the underlying the mechanisms of its rescue. Specifically, we hypothesized that the mechanism through which quetiapine rescued behavior would be through a positive effect on the structure/viability of specific cellular populations comprising and/or supporting the blood brain barrier.
However, ledge instability and hindlimb clasping, measures of neurological function, were unaffected by quetiapine treatment, but were exacerbated in untreated KO versus untreated WT mice indicating that dysbindin-1 KO impairs neurological function. Specifically, ledge stability is a neurological measure of coordination. Difficulty with balance has been associated with reduced dendritic complexity and abnormal patterns of synapses of the GABAergic Purkinje cells of the cerebellum (Wang et al., 2015), which in turn can impact eye-blink conditioning, procedural learning, and cognitive performance (Andreasen and Pierson, 2008). Indeed, the various regions of the cerebellum have been implicated in several high-level processes, including emotion, memory encoding and retrieval, attention, and social cognition (Andreasen, 1997; Andreasen et al., 1996; Andreasen et al., 1997; Crespo-Facorro et al., 2001a; Crespo-Facorro et al., 2001b; Paradiso et al., 2003), reportedly due to its connections with several regions of the cerebral cortex via a cortico-cerebellar-thalamic-cortical circuit (Andreasen and Pierson, 2008). In point of fact, SZ subjects exhibit reduced dendritic complexity, abnormal synapse patterns, and lower size and linear density of cerebellar Purkinje cells and overall cerebellar volume (Ichimiya et al., 2001; Katsetos et al., 1997; Mavroudis et al., 2017; Reyes and Gordon, 1981), the consequence of which results in an excitatory/inhibitory imbalance and altered cerebellar input and output that modulates behavior and perception (Andreasen and Pierson, 2008). Indeed, the described cerebellar abnormalities can result in abnormalities of coordination, gait, and visual motor disturbances, and the experience of auditory hallucinations, misrepresentation of environmental significance and paranoid delusions, all of which are observed in SZ patients (Andreasen and Pierson, 2008; Gupta et al., 1995; Kinney et al., 1999).
Limb clasping is associated with early developmental stages and is often considered a primitive reflex (Schott and Rossor, 2003). In the weeks immediately following birth, young mouse pups will exhibit limb flexion when picked up by the tail; in contrast, adults will extend all four paws (known as limb extension) in anticipation of contact with the ground. However, the occurrence of primitive reflexes in adults (murine or human) is considered to be a subtle indicator of abnormal neurological function, a consequence of “released” inhibition stemming from prefrontal cortex hypofunction and cerebellar abnormality (Futagi and Suzuki, 2010; Paulson and Gottlieb, 1968; Picard et al., 2008; Schott and Rossor, 2003). Indeed, SZ subjects exhibit elevated frontal release signs (also known as primitive reflexes), as indexed by hand grasp reflex scores, which were positively correlated with number of perseverative errors on the Wisconsin Card Sort task and negatively correlated with IQ (Hyde et al., 2007). Therefore, our findings of prominent hindlimb clasping, as well as ledge instability in dysbindin-1 KO mice indicate neurological abnormalities of the prefrontal cortex and cerebellum that have been associated with overall neurological function, cognition, and several abnormalities observed in SZ.
Here we studied several indices of neurological function, peripheral blood and brain copper content, and the genetic expression of several relevant cellular markers. These findings highlight the sensitivity of behavioral and genetic assays to genotype, and antipsychotic drug interactions, as well as brain regional specificity. Furthermore, they indicate a complexity of downstream alterations of dysbindin-1 deficiency that require further elucidation. Additionally, while looking at individual markers is always of importance, this study highlights that conducting broader circuitry-aimed analyses can yield fascinating data. These data offer significant implications for the influence of brain region, copper homeostasis, and antipsychotic drug treatment in the context of a dysbindin-1 KO model of SZ.
Supplementary Material
Highlights.
Quetiapine lowers myelin, transthyretin, and copper transporter gene expression
Dysbindin-1 knockout and quetiapine treatment alter serum copper levels
Loss of dysbindin-1 expression results in hyperactive mice
Dysbindin-1 loss impairs neurological function; quetiapine selectively rescues it
Funding & Acknowledgments
The current study was supported by NINDS F99NS105208 to Dr. Kirsten E Schoonover, and NIMH R21MH108867 and NIMH R21117434 to Dr. Rosalinda C Roberts. This work was also supported in part by NIH R56MH111459 to Dr. Victor Faundez. The Engineered Model Systems Core Facility was supported by awards NIH P30 CA13148, P30 AR048311, P30 DK074038, P30 DK05336, and P60 DK079626. Behavioral studies were completed in part thanks to the Evelyn F. McKnight Brain Institute Behavioral Assessment Core at the University of Alabama at Birmingham. Also special thanks to Ivis Chapel for conducting a portion of the brain and blood copper assessment.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors have no relevant conflicts of interest to disclose.
REFERENCES
- Abdolmaleky HM, Pajouhanfar S, Faghankhani M, Joghataei MT, Mostafavi A & Thiagalingam S 2015. Antipsychotic Drugs Attenuate Aberrant DNA Methylation Of DTNBP1 (Dysbindin) Promoter In Saliva And Post-Mortem Brain Of Patients With Schizophrenia And Psychotic Bipolar Disorder. Am J Med Genet B Neuropsychiatr Genet, 168, 687–96. [DOI] [PubMed] [Google Scholar]
- Al-Amin MM, Nasir Uddin MM & Mahmud Reza H 2013. Effects Of Antipsychotics On The Inflammatory Response System Of Patients With Schizophrenia In Peripheral Blood Mononuclear Cell Cultures. Clin Psychopharmacol Neurosci, 11, 144–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen NC, Bagade S, Mcqueen MB, Ioannidis JP, Kavvoura FK, Khoury MJ, Tanzi RE & Bertram L 2008. Systematic Meta-Analyses And Field Synopsis Of Genetic Association Studies In Schizophrenia: The Szgene Database. Nat Genet, 40, 827–34. [DOI] [PubMed] [Google Scholar]
- Alvarado-Alanis P, Leon-Ortiz P, Reyes-Madrigal F, Favila R, Rodriguez-Mayoral O, Nicolini H, Azcarraga M, Graff-Guerrero A, Rowland LM & De La Fuente-Sandoval C 2015. Abnormal White Matter Integrity In Antipsychotic-Naive First-Episode Psychosis Patients Assessed By A DTI Principal Component Analysis. Schizophr Res, 162, 14–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasen NC 1997. Linking Mind And Brain In The Study Of Mental Illnesses: A Project For A Scientific Psychopathology. Science, 275, 1586–93. [DOI] [PubMed] [Google Scholar]
- Andreasen NC, O’leary DS, Cizadlo T, Arndt S, Rezai K, Ponto LL, Watkins GL & Hichwa RD 1996. Schizophrenia And Cognitive Dysmetria: A Positron-Emission Tomography Study Of Dysfunctional Prefrontal-Thalamic-Cerebellar Circuitry. Proc Natl Acad Sci U S A, 93, 9985–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasen NC, O’leary DS, Flaum M, Nopoulos P, Watkins GL, Boles Ponto LL & Hichwa RD 1997. Hypofrontality In Schizophrenia: Distributed Dysfunctional Circuits In Neuroleptic-Naïve Patients. Lancet, 349, 1730–1734. [DOI] [PubMed] [Google Scholar]
- Andreasen NC & Pierson R 2008. The Role Of The Cerebellum In Schizophrenia. Biol Psychiatry, 64, 81–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asami T, Hyuk Lee S, Bouix S, Rathi Y, Whitford TJ, Niznikiewicz M, Nestor P, Mccarley RW, Shenton ME & Kubicki M 2014. Cerebral White Matter Abnormalities And Their Associations With Negative But Not Positive Symptoms Of Schizophrenia. Psychiatry Res, 222, 52–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelsson R, Martensson E & Alling C 1982. Impairment Of The Blood-Brain Barrier As An Aetiological Factor In Paranoid Psychosis. Br J Psychiatry, 141, 273–81. [DOI] [PubMed] [Google Scholar]
- Bauer K & Kornhuber J 1987. Blood-Cerebrospinal Fluid Barrier In Schizophrenic Patients. Eur Arch Psychiatry Neurol Sci, 236, 257–9. [DOI] [PubMed] [Google Scholar]
- Benson MA, Newey SE, Martin-Rendon E, Hawkes R & Blake DJ 2001. Dysbindin, A Novel Coiled-Coil-Containing Protein That Interacts With The Dystrobrevins In Muscle And Brain. J Biol Chem, 276, 24232–41. [DOI] [PubMed] [Google Scholar]
- Bray NJ, Preece A, Williams NM, Moskvina V, Buckland PR, Owen MJ & O’donovan MC 2005. Haplotypes At The Dystrobrevin Binding Protein 1 (DTNBP1) Gene Locus Mediate Risk For Schizophrenia Through Reduced DTNBP1 Expression. Hum Mol Genet, 14, 1947–54. [DOI] [PubMed] [Google Scholar]
- Brouillette J & Quirion R 2008. Transthyretin: A Key Gene Involved In The Maintenance Of Memory Capacities During Aging. Neurobiol Aging, 29, 1721–32. [DOI] [PubMed] [Google Scholar]
- Chambers JS & Perrone-Bizzozero NI 2004. Altered Myelination Of The Hippocampal Formation In Subjects With Schizophrenia And Bipolar Disorder. Neurochem Res, 29, 2293–302. [DOI] [PubMed] [Google Scholar]
- Chen X, Li Y, Zhang T, Yao Y, Shen C & Xue Y 2018. Association Of Serum Trace Elements With Schizophrenia And Effects Of Antipsychotic Treatment. Biol Trace Elem Res, 181, 22–30. [DOI] [PubMed] [Google Scholar]
- Cox MM, Tucker AM, Tang J, Talbot K, Richer DC, Yeh L & Arnold SE 2009. Neurobehavioral Abnormalities In The Dysbindin-1 Mutant, Sandy, On A C57BL/6J Genetic Background. Genes Brain Behav, 8, 390–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo-Facorro B, Paradiso S, Andreasen NC, O’leary DS, Watkins GL, Ponto LL & Hichwa RD 2001a. Neural Mechanisms Of Anhedonia In Schizophrenia: A PET Study Of Response To Unpleasant And Pleasant Odors. Jama, 286, 427–35. [DOI] [PubMed] [Google Scholar]
- Crespo-Facorro B, Wiser AK, Andreasen NC, O’leary DS, Watkins GL, Boles Ponto LL & Hichwa RD 2001b. Neural Basis Of Novel And Well-Learned Recognition Memory In Schizophrenia: A Positron Emission Tomography Study. Hum Brain Mapp, 12, 219–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delisi LE, Shaw SH, Crow TJ, Shields G, Smith AB, Larach VW, Wellman N, Loftus J, Nanthakumar B, Razi K, Stewart J, Comazzi M, Vita A, Heffner T & Sherrington R 2002. A Genome-Wide Scan For Linkage To Chromosomal Regions In 382 Sibling Pairs With Schizophrenia Or Schizoaffective Disorder. Am J Psychiatry, 159, 803–12. [DOI] [PubMed] [Google Scholar]
- Doherty NS, Littman BH, Reilly K, Swindell AC, Buss JM & Anderson NL 1998. Analysis Of Changes In Acute-Phase Plasma Proteins In An Acute Inflammatory Response And In Rheumatoid Arthritis Using Two-Dimensional Gel Electrophoresis. Electrophoresis, 19, 355–63. [DOI] [PubMed] [Google Scholar]
- Eisses JF & Kaplan JH 2005. The Mechanism Of Copper Uptake Mediated By Human CTR1: A Mutational Analysis. J Biol Chem, 280, 37159–68. [DOI] [PubMed] [Google Scholar]
- Fatemi SH & Folsom TD 2009. The Neurodevelopmental Hypothesis Of Schizophrenia, Revisited. Schizophr Bull, 35, 528–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futagi Y & Suzuki Y 2010. Neural Mechanism And Clinical Significance Of The Plantar Grasp Reflex In Infants. Pediatr Neurol, 43, 81–6. [DOI] [PubMed] [Google Scholar]
- Gameiro PA & Struhl K 2018. Nutrient Deprivation Elicits A Transcriptional And Translational Inflammatory Response Coupled To Decreased Protein Synthesis. Cell Rep, 24, 1415–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiani CA, Starcevic M, Rodriguez-Fernandez IA, Nazarian R, Cheli VT, Chan LN, Malvar JS, De Vellis J, Sabatti C & Dell’angelica EC 2010. The Dysbindin-Containing Complex (BLOC-1) In Brain: Developmental Regulation, Interaction With SNARE Proteins And Role In Neurite Outgrowth. Mol Psychiatry, 15, 115, 204–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokhale A, Vrailas-Mortimer A, Larimore J, Comstra HS, Zlatic SA, Werner E, Manvich DF, Iuvone PM, Weinshenker D & Faundez V 2015. Neuronal Copper Homeostasis Susceptibility By Genetic Defects In Dysbindin, A Schizophrenia Susceptibility Factor. Hum Mol Genet, 24, 5512–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg JR, Herring NR, Naydenov AV, Hanlin RP & Konradi C 2009. Downregulation Of Oligodendrocyte Transcripts Is Associated With Impaired Prefrontal Cortex Function In Rats. Schizophr Res, 113, 277–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan HJ, Dai J & Zhu XZ 2000. Atypical Antipsychotic Effects Of Quetiapine Fumarate In Animal Models. Acta Pharmacol Sin, 21, 205–10. [PubMed] [Google Scholar]
- Gupta SK, Kunka RL, Metz A, Lloyd T, Rudolph G & Perel JM 1995. Effect Of Alosetron (A New 5-HT 3 Receptor Antagonist) On The Pharmacokinetics Of Haloperidol In Schizophrenic Patients. J.Clin.Pharmacol, 35, 202–207. [DOI] [PubMed] [Google Scholar]
- Guyenet SJ, Furrer SA, Damian VM, Baughan TD, La Spada AR & Garden GA 2010. A Simple Composite Phenotype Scoring System For Evaluating Mouse Models Of Cerebellar Ataxia. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakak Y, Walker JR, Li C, Wong WH, Davis KL, Buxbaum JD, Haroutunian V & Fienberg AA 2001. Genome-Wide Expression Analysis Reveals Dysregulation Of Myelination-Related Genes In Chronic Schizophrenia. Proc Natl Acad Sci U S A, 98, 4746–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall H, Sedvall G, Magnusson O, Kopp J, Halldin C & Farde L 1994. Distribution Of D 1 - And D 2 -Dopamine Receptors, And Dopamine And Its Metabolites In The Human Brain. Neuropsychopharmacology, 11, 245–256. [DOI] [PubMed] [Google Scholar]
- He J, Luo H, Yan B, Yu Y, Wang H, Wei Z, Zhang Y, Xu H, Tempier A, Li X & Li XM 2009. Beneficial Effects Of Quetiapine In A Transgenic Mouse Model Of Alzheimer’s Disease. Neurobiol Aging, 30, 1205–16. [DOI] [PubMed] [Google Scholar]
- Herbert J, Wilcox JN, Pham KT, Fremeau RT Jr., Zeviani M, Dwork A, Soprano DR, Makover A, Goodman DS, Zimmerman EA & et al. 1986. Transthyretin: A Choroid Plexus-Specific Transport Protein In Human Brain. The 1986 S. Weir Mitchell Award. Neurology, 36, 900–11. [DOI] [PubMed] [Google Scholar]
- Herring NR & Konradi C 2011. Myelin, Copper, And The Cuprizone Model Of Schizophrenia. Front Biosci (Schol Ed), 3, 23–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hof PR, Haroutunian V, Copland C, Davis KL & Buxbaum JD 2002. Molecular And Cellular Evidence For An Oligodendrocyte Abnormality In Schizophrenia. Neurochem Res, 27, 1193–200. [DOI] [PubMed] [Google Scholar]
- Holleran L, Ahmed M, Anderson-Schmidt H, Mcfarland J, Emsell L, Leemans A, Scanlon C, Dockery P, Mccarthy P, Barker GJ, Mcdonald C & Cannon DM 2014. Altered Interhemispheric And Temporal Lobe White Matter Microstructural Organization In Severe Chronic Schizophrenia. Neuropsychopharmacology, 39, 944–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JT, Leweke FM, Oxley D, Wang L, Harris N, Koethe D, Gerth CW, Nolden BM, Gross S, Schreiber D, Reed B & Bahn S 2006. Disease Biomarkers In Cerebrospinal Fluid Of Patients With First-Onset Psychosis. Plos Med, 3, E428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyde TM, Goldberg TE, Egan MF, Lener MC & Weinberger DR 2007. Frontal Release Signs And Cognition In People With Schizophrenia, Their Siblings And Healthy Controls. Br J Psychiatry, 191, 120–5. [DOI] [PubMed] [Google Scholar]
- Ichimiya T, Okubo Y, Suhara T & Sudo Y 2001. Reduced Volume Of Cerebellar Vermis In Neuroleptic-Naive Schizophrenia. Biological Psychiatry, 49, 20–27. [DOI] [PubMed] [Google Scholar]
- Iijima S, Masaki H, Wakayama Y, Inoue M, Jimi T, Hara H, Unaki A, Oniki H, Nakano K, Hirayama Y & Kishimoto K 2009. Immunohistochemical Detection Of Dysbindin At The Astroglial Endfeet Around The Capillaries Of Mouse Brain. J Mol Histol, 40, 117–21. [DOI] [PubMed] [Google Scholar]
- Kato TA, Monji A, Mizoguchi Y, Hashioka S, Horikawa H, Seki Y, Kasai M, Utsumi H & Kanba S 2011. Anti-Inflammatory Properties Of Antipsychotics Via Microglia Modulations: Are Antipsychotics A ‘Fire Extinguisher’ In The Brain Of Schizophrenia? Mini Rev Med Chem, 11, 565–74. [DOI] [PubMed] [Google Scholar]
- Katsetos CD, Hyde TM & Herman MM 1997. Neuropathology Of The Cerebellum In Schizophrenia--An Update: 1996 And Future Directions. Biol Psychiatry, 42, 213–24. [DOI] [PubMed] [Google Scholar]
- Kinney DK, Yurgelun-Todd DA & Woods BT 1999. Neurologic Signs Of Cerebellar And Cortical Sensory Dysfunction In Schizophrenics And Their Relatives. Schizophr Res, 35, 99–104. [DOI] [PubMed] [Google Scholar]
- Kirch DG, Kaufmann CA, Papadopoulos NM, Martin B & Weinberger DR 1985. Abnormal Cerebrospinal Fluid Protein Indices In Schizophrenia. Biol Psychiatry, 20, 1039–1046. [DOI] [PubMed] [Google Scholar]
- Kubicki M, Mccarley RW & Shenton ME 2005. Evidence For White Matter Abnormalities In Schizophrenia. Curr Opin Psychiatry, 18, 121–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larimore J, Zlatic SA, Gokhale A, Tornieri K, Singleton KS, Mullin AP, Tang J, Talbot K & Faundez V 2014. Mutations In The BLOC-1 Subunits Dysbindin And Muted Generate Divergent And Dosage-Dependent Phenotypes. J Biol Chem, 289, 14291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Kubicki M, Asami T, Seidman LJ, Goldstein JM, Mesholam-Gately RI, Mccarley RW & Shenton ME 2013. Extensive White Matter Abnormalities In Patients With First-Episode Schizophrenia: A Diffusion Tensor Iimaging (DTI) Study. Schizophr Res, 143, 231–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Grace AA & Duzel E 2011. A Neohebbian Framework For Episodic Memory; Role Of Dopamine-Dependent Late LTP. Trends Neurosci, 34, 536–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas EK, Dougherty SE, Mcmeekin LJ, Trinh AT, Reid CS & Cowell RM 2012. Developmental Alterations In Motor Coordination And Medium Spiny Neuron Markers In Mice Lacking Pgc-1alpha. Plos One, 7, E42878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu H, Hu M, Eyler LT, Jin H, Wang J, Ou J, Guo X, He Z, Liu F, Zhao J & Guo W 2015. Regional White Matter Abnormalities In Drug-Naive, First-Episode Schizophrenia Patients And Their Healthy Unaffected Siblings. Aust N Z J Psychiatry, 49, 246–54. [DOI] [PubMed] [Google Scholar]
- Magnotti LM, Goodenough DA & Paul DL 2011. Deletion Of Oligodendrocyte Cx32 And Astrocyte Cx43 Causes White Matter Vacuolation, Astrocyte Loss And Early Mortality. Glia, 59, 1064–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall CR, Howrigan DP, Merico D, Thiruvahindrapuram B, Wu W, Greer DS, Antaki D, Shetty A, Holmans PA, Pinto D, Gujral M, Brandler WM, Malhotra D, Wang Z, Fajarado KVF, Maile MS, Ripke S, Agartz I, Albus M, Alexander M, Amin F, Atkins J, Bacanu SA, Belliveau RA Jr., Bergen SE, Bertalan M, Bevilacqua E, Bigdeli TB, Black DW, Bruggeman R, Buccola NG, Buckner RL, Bulik-Sullivan B, Byerley W, Cahn W, Cai G, Cairns MJ, Campion D, Cantor RM, Carr VJ, Carrera N, Catts SV, Chambert KD, Cheng W, Cloninger CR, Cohen D, Cormican P, Craddock N, Crespo-Facorro B, Crowley JJ, Curtis D, Davidson M, Davis KL, Degenhardt F, Del Favero J, Delisi LE, Dikeos D, Dinan T, Djurovic S, Donohoe G, Drapeau E, Duan J, Dudbridge F, Eichhammer P, Eriksson J, Escott-Price V, Essioux L, Fanous AH, Farh KH, Farrell MS, Frank J, Franke L, Freedman R, Freimer NB, Friedman JI, Forstner AJ, Fromer M, Genovese G, Georgieva L, Gershon ES, Giegling I, Giusti-Rodriguez P, Godard S, Goldstein JI, Gratten J, De Haan L, Hamshere ML, Hansen M, Hansen T, Haroutunian V, Hartmann AM, Henskens FA, Herms S, Hirschhorn JN, Hoffmann P, Hofman A, Huang H, Ikeda M, Joa I, Kahler AK, et al. 2017. Contribution Of Copy Number Variants To Schizophrenia From A Genome-Wide Study Of 41,321 Subjects. Nat Genet, 49, 27–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavroudis IA, Petrides F, Manani M, Chatzinikolaou F, Ciobica AS, Padurariu M, Kazis D, Njau SN, Costa VG & Baloyannis SJ 2017. Purkinje Cells Pathology In Schizophrenia. A Morphometric Approach. Rom J Morphol Embryol, 58, 419–424. [PubMed] [Google Scholar]
- Muller N 2018. Inflammation In Schizophrenia: Pathogenetic Aspects And Therapeutic Considerations. Schizophr Bull, 44, 973–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickl-Jockschat T, Stöcker T, Markov V, Krug A, Huang R, Schneider F, Habel U, Zerres K, Nöthen MM, Treutlein J, Rietschel M, Shah NJ & Kircher T 2012. The Impact Of A Dysbindin Schizophrenia Susceptibility Variant On Fiber Tract Integrity In Healthy Individuals: A TBSS-Based Diffusion Tensor Imaging Study. Neuroimage, 60, 847–53. [DOI] [PubMed] [Google Scholar]
- Papaleo F, Yang F, Garcia S, Chen J, Lu B, Crawley JN & Weinberger DR 2012. Dysbindin-1 Modulates Prefrontal Cortical Activity And Schizophrenia-Like Behaviors Via Dopamine/D2 Pathways. Mol Psychiatry, 17, 85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradiso S, Andreasen NC, Crespo-Facorro B, O’leary DS, Watkins GL, Boles Ponto LL & Hichwa RD 2003. Emotions In Unmedicated Patients With Schizophrenia During Evaluation With Positron Emission Tomography. Am J Psychiatry, 160, 1775–83. [DOI] [PubMed] [Google Scholar]
- Paulson G & Gottlieb G 1968. Developmental Reflexes: The Reappearance Of Foetal And Neonatal Reflexes In Aged Patients. Brain, 91, 37–52.5643282 [Google Scholar]
- Picard H, Amado I, Mouchet-Mages S, Olie JP & Krebs MO 2008. The Role Of The Cerebellum In Schizophrenia: An Update Of Clinical, Cognitive, And Functional Evidences. Schizophr.Bull, 34, 155–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes MG & Gordon A 1981. Cerebellar Vermis In Schizophrenia. Lancet, 2, 700–1. [DOI] [PubMed] [Google Scholar]
- Roberts RC 2017. Postmortem Studies On Mitochondria In Schizophrenia. Schizophrenia Research, 187, 17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatzberg A & Nemeroff C 2009. The American Psychiatric Publishing Textbook Of Psychopharmacology, Arlington, American Psychiatric Publishing. [Google Scholar]
- Scheiber IF, Mercer JF & Dringen R 2010. Copper Accumulation By Cultured Astrocytes. Neurochem Int, 56, 451–60. [DOI] [PubMed] [Google Scholar]
- Schizophrenia Working Group Of The Psychiatric Genomics, C. 2014. Biological Insights From 108 Schizophrenia-Associated Genetic Loci. Nature, 511, 421–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoonover KE, Farmer CB, Cash AE & Roberts RC 2019. Pathology Of White Matter Integrity In Three Major White Matter Fasciculi: A Post-Mortem Study Of Schizophrenia And Treatment Status. Br J Pharmacol, 176, 1143–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoonover KE, Queern SL, Lapi SE & Roberts RC 2018. Impaired Copper Transport In Schizophrenia Results In A Copper-Deficient Brain State: A New Side To The Dysbindin Story. World J Biol Psychiatry, 1–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schott JM & Rossor MN 2003. The Grasp And Other Primitive Reflexes. J Neurol Neurosurg Psychiatry, 74, 558–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severance EG, Gressitt KL, Alaedini A, Rohleder C, Enning F, Bumb JM, Müller JK, Schwarz E, Yolken RH & Leweke FM 2015. Igg Dynamics Of Dietary Antigens Point To Cerebrospinal Fluid Barrier Or Flow Dysfunction In First-Episode Schizophrenia. Brain Behav Immun, 44, 148–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shohamy D & Adcock RA 2010. Dopamine And Adaptive Memory. Trends Cogn Sci, 14, 464–72. [DOI] [PubMed] [Google Scholar]
- Spector R, Keep RF, Robert Snodgrass S, Smith QR & Johanson CE 2015. A Balanced View Of Choroid Plexus Structure And Function: Focus On Adult Humans. Exp Neurol, 267, 78–86. [DOI] [PubMed] [Google Scholar]
- Straub RE, Jiang Y, Maclean CJ, Ma Y, Webb BT, Myakishev MV, Harris-Kerr C, Wormley B, Sadek H, Kadambi B, Cesare AJ, Gibberman A, Wang X, O’neill FA, Walsh D & Kendler KS 2002. Genetic Variation In The 6p22.3 Gene DTNBP1, The Human Ortholog Of The Mouse Dysbindin Gene, Is Associated With Schizophrenia. Am.J.Hum.Genet, 71, 337–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swank RT, Sweet HO, Davisson MT, Reddington M & Novak EK 1991. Sandy: A New Mouse Model For Platelet Storage Pool Deficiency. Genet Res, 58, 51–62. [DOI] [PubMed] [Google Scholar]
- Talbot K 2009. The Sandy (Sdy) Mouse: A Dysbindin-1 Mutant Relevant To Schizophrenia Research Progress In Brain Research. Elsevier. [DOI] [PubMed] [Google Scholar]
- Talbot K, Ong WY, Blake DJ, Tang J, Louneva N, Carlson GC & Arnold SE 2009. Dysbindin-1 And Its Protein Family Handbook Of Neurochemistry And Molecular Neurobiology. Springer US. [Google Scholar]
- Voisey J, Swagell CD, Hughes IP, Lawford BR, Young RM & Morris CP 2010. Analysis Of Hapmap Tag-Snps In Dysbindin (DTNBP1) Reveals Evidence Of Consistent Association With Schizophrenia. Eur Psychiatry, 25, 314–9. [DOI] [PubMed] [Google Scholar]
- Wan C, Yang Y, Li H, La Y, Zhu H, Jiang L, Chen Y, Feng G & He L 2006. Dysregulation Of Retinoid Transporters Expression In Body Fluids Of Schizophrenia Patients. J Proteome Res, 5, 3213–6. [DOI] [PubMed] [Google Scholar]
- Wang JY, Yu IS, Huang CC, Chen CY, Wang WP, Lin SW, Jeang KT & Chi YH 2015. Sun1 Deficiency Leads To Cerebellar Ataxia In Mice. Dis Model Mech, 8, 957–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weickert CS, Rothmond DA, Hyde TM, Kleinman JE & Straub RE 2008. Reduced DTNBP1 (Dysbindin-1) Mrna In The Hippocampal Formation Of Schizophrenia Patients. Schizophr Res, 98, 105–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weickert CS, Straub RE, Mcclintock BW, Matsumoto M, Hashimoto R, Hyde TM, Herman MM, Weinberger DR & Kleinman JE 2004. Human Dysbindin (DTNBP1) Gene Expression In Normal Brain And In Schizophrenic Prefrontal Cortex And Midbrain. Arch Gen.Psychiatry, 61, 544–555. [DOI] [PubMed] [Google Scholar]
- Xu H, Yang HJ, Mcconomy B, Browning R & Li XM 2010. Behavioral And Neurobiological Changes In C57BL/6 Mouse Exposed To Cuprizone: Effects Of Antipsychotics. Front Behav Neurosci, 4, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Yang HJ, Zhang Y, Clough R, Browning R & Li XM 2009. Behavioral And Neurobiological Changes In C57BL/6 Mice Exposed To Cuprizone. Behav Neurosci, 123, 418–29. [DOI] [PubMed] [Google Scholar]
- Xuan Y, Yan G, Wu R, Huang Q, Li X & Xu H 2015. The Cuprizone-Induced Changes In (1)H-MRS Metabolites And Oxidative Parameters In C57BL/6 Mouse Brain: Effects Of Quetiapine. Neurochem Int, 90, 185–92. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Heiny ME, Suzuki M & Gitlin JD 1996. Biochemical Characterization And Intracellular Localization Of The Menkes Disease Protein. Proc Natl Acad Sci U S A, 93, 14030–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Wan C, Li H, Zhu H, La Y, Xi Z, Chen Y, Jiang L, Feng G & He L 2006. Altered Levels Of Acute Phase Proteins In The Plasma Of Patients With Schizophrenia. Anal Chem, 78, 3571–6. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xu H, Jiang W, Xiao L, Yan B, He J, Wang Y, Bi X, Li X, Kong J & Li XM 2008. Quetiapine Alleviates The Cuprizone-Induced White Matter Pathology In The Brain Of C57BL/6 Mouse. Schizophr Res, 106, 182–91. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request.