

Abstract
Prion diseases are invariably fatal neurodegenerative disorders that have gained much publicity due to their transmissible nature. Sporadic Creutzfeldt-Jakob disease (sCJD) is the most common human prion disorder, with an incidence of 1 in a million. Inherited prion disorders are relatively rare, and associated with mutations in the prion protein gene. More than 50 different point mutations, deletions, and insertions have been identified so far. Most are autosomal dominant and fully penetrant. Prion disorders also occur in animals, and are of major concern because of the potential for spreading to humans. The principal pathogenic event underlying all prion disorders is a change in the conformation of prion protein (PrPC) from a mainly α-helical to a β-sheet rich isoform, PrP-scrapie (PrPSc). Accumulation of PrPSc in the brain parenchyma is the major cause of neuronal degeneration. The mechanism by which PrPSc is transmitted, propagates, and causes neurodegenerative changes has been investigated over the years, and several clues have emerged. Efforts are also ongoing for identifying specific and sensitive diagnostic tests for sCJD and animal prion disorders, but success has been limited. The eye is suitable for these evaluations because it shares several anatomical and physiological features with the brain, and can be observed in vivo during disease progression. The retina, considered an extension of the central nervous system, is involved extensively in prion disorders. Accordingly, Optical Coherence Tomography and electroretinogram have shown some promise as pre-mortem diagnostic tests for human and animal prion disorders. However, a complete understanding of the physiology of PrPC and pathobiology of PrPSc in the eye is essential for developing specific and sensitive tests. Below, we summarize recent progress in ocular physiology and pathology in prion disorders, and the eye as an anatomically accessible site to diagnose, monitor disease progression, and test therapeutic options.
Keywords: Prion protein, glaucoma, extracellular matrix, hepcidin, iron, TGFβ2, oxidative stress
1. Introduction
Prions or PrPSc is a conformational isoform of a normal glycosylphosphatidyl inositol (GPI) linked protein expressed on the plasma membrane of all cells, but most abundant on neuronal cells (Prusiner, 1998). The triggers that initiate its conversion from PrPC to the PrPSc form, the mechanism by which it spreads, and the underlying cause of neuronal death have been the focus of several studies, and significant progress has been made on all fronts (Scheckel and Aguzzi, 2018; Sigurdson et al., 2019; Singh et al., 2014). For example, it is known that point mutations in the PrPC gene increase its susceptibility for conversion to the PrPSc form (Singh et al., 2010). In infectious disorders, exogenous PrPSc initiates the conversion of PrPC to PrPSc, which then spreads to distant sites (Aguzzi and Glatzel, 2006; Prusiner, 1998). Factors that contribute to the susceptibility of a particular PrPC form for conversion to PrPSc are not fully understood, nor is the mechanism by which PrPSc causes neuronal death. These, and other questions need to be answered because of the spread of prion disease in free ranging deer and elk as Chronic Wasting Disease (CWD), and the danger of potential spread to humans (Hannaoui et al., 2017). Although prion disorders have been known in sheep and goats for several years, the spread to farm and wild animals is relatively new. The emergence of Bovine Spongiform Encephalopathy (BSE) in cattle and its onward spread to humans was a rude reminder that protection offered by the species barrier is only partial, and new strains are likely to emerge (Prusiner, 1997). The eye provides an opportunity to address these questions because of structural similarities to the brain, and the ease of access during disease development.
Natural pathways of transmission of PrPSc include the oral route from the gastro-intestinal tract to the brain, peripheral blood, placenta, mucous membranes, and the cornea (Weissmann et al., 2002). Accidental transmission of BSE to humans occurred by eating contaminated beef, and is under control since regulations on the processing of animal feed were put into place. The cornea has been of some concern because of transmission through corneal transplants from subjects exposed to BSE (Maddox et al., 2008). Currently, CWD is spreading among the deer and elk population from contaminated water reservoirs and common grazing grounds (Saunders et al., 2012), and could spread horizontally and to other species from corneal abrasions since corneal epithelium expresses PrPC, and infectious PrPSc has been detected at that site (Ashok et al., 2018).
In addition to expression of PrPC (Büeler et al., 1992), a certain degree of sequence homology is essential for transmission (Hagiwara et al., 2013), and is the basis of species barrier. Other prominent factors that contribute to disease pathogenesis include inflammation and brain iron dyshomeostasis (Carroll et al., 2015; Singh et al., 2009b; Singh et al., 2010). Whether these play a primary role, or amplify the pathology by other secondary mechanisms has been difficult to evaluate. It is likely that several factors contribute to the final outcome, making it difficult to discern which comes first. One of the hypotheses is the loss of physiological function of PrPC combined with gain of toxic’ function by PrPSc in prion disease pathogenesis.
Several functions have been ascribed to PrPC. In the eye, overexpression of PrPC protects the photoreceptors from damaging light (Frigg et al., 2006; Williams et al., 2011), and plays a role in iron transport across retinal pigment epithelial cells that form the outer blood-retinal barrier, and non-pigmented epithelial cells of the ciliary epithelium that form the blood-aqueous barrier. PrPC is known to function as a ferrireductase partner for the Zirt, Irt-like (ZIP) family of iron transporters on the plasma membrane and the endosome membrane (Ehsani et al., 2012; Singh et al., 2015). It is therefore logical to assume that loss of function of PrPC due to conversion to PrPSc will induce a certain degree of iron deficiency, an essential metal necessary for vital enzymatic reactions (Gasperini et al., 2016; Singh et al., 2014). Compensatory upregulation of iron uptake proteins creates iron dyshomeostasis and a potentially toxic microenvironment. PrPC is known to stabilize the extracellular matrix, and loss of this function interferes with neuritogenesis (Alleaume-Butaux et al., 2013; Kleene et al., 2007; Ramljak et al., 2015) in the brain, and induces primary open angle glaucoma in the eye (Ashok 2019). These are a few examples where the eye has helped to confirm physiological functions of PrPC in the brain, and uncover pathological implications of dysfunction in the eye.
An important area of investigation concerns the role of inflammation in PrPSc-mediated neurotoxicity (Carroll et al., 2015). Several studies have demonstrated upregulation of various cytokines during prion disease progression. Most studies are in mouse models, and use different strains of scrapie to demonstrate the universality of the phenomenon (Carroll and Chesebro, 2019; Carroll et al., 2015). Although some cytokines are beneficial, these are known to upregulate hepcidin, a peptide hormone secreted mainly by the liver and known to regulate systemic iron by downregulating ferroportin (Fpn), an iron export protein (Drakesmith et al., 2015). In that respect, identification of extra-hepatic, local expression of hepcidin in the brain and the eye adds a new dimension to iron management (Ashok 2020), organs separated from systemic circulation by the blood-brain and blood-retinal barriers respectively.
Progress on these fronts has been slow, partly because of the structural and functional complexity of the brain. The eye provides a suitable alternative because disease-specific changes can be visualized in vivo during disease progression, and the mechanism of toxicity by PrPSc can be parsed out in a way that is experimentally not possible in the brain. Thus, using the eye as a ‘window’ to the brain, several new observations have emerged. Some reinforce previous findings from human and animal brains, while others provide new perspective on the functional role of PrPC and consequences of dysfunction. Some of these observations are discussed below.
2. The eye as a ‘window’ to the brain
Thus far, the eye has been studied mainly as a site where PrPSc accumulates and causes disease (Greenlee et al., 2016). It is only recently that the physiological function of proteins implicated in diseases of the brain such as PrPC, the main cause of sCJD, amyloid precursor protein (APP) and tau, involved in Alzheimer’s disease (AD), and α-synuclein, the cause of synucleinopathies have been evaluated in the eye. The retina provides a convenient site to examine loss of function and gain of toxic function by these proteins in vivo during disease progression, and has been used successfully for the early diagnosis of some of these disorders. For example, Optical Coherence Tomography (OCT) is used for the early diagnosis of prion disorders, AD, PD, amyotrophic lateral sclerosis, and multiple sclerosis, to mention a few, and the sensitivity and specificity of the test is improving consistently (Adhikary et al., 2010; Doustar et al., 2017; Torrent et al., 2010). Likewise, retinal function measured by electroretinogram (ERG) is emerging as a sensitive test for retinal function in the early stages of these disorders (Ngoo et al., 2019). The retina is more conducive to analysis than the brain, and has yielded important information about the physiological function of PrPC, APP, and a-synuclein. Like the brain, the retina is separated from systemic circulation by the outer and inner blood retinal barriers comprised of retinal pigment epithelial (RPE) cells and capillary endothelial cells. RPE cells form a monolayer between choroidal sinuses and the outer retina, and expresses proteins and receptors in a polarized manner to allow unidirectional transport of select substances. Likewise, capillary endothelial cells form a tight monolayer between the peripheral circulation and the inner retina, and allow selective transport of essential substances to the inner neuroretina. The RPE cell layer is followed by photoreceptors, which rely on these cells for the daily turnover of outer segments by autophagy. This layer is followed by the outer and inner plexiform and nuclear layers, which are traversed by Muller glia. The ganglion cells form the innermost layer of the retina, which form the retinal nerve fiber layer (RNFL) that carries signals to the brain through the optic nerve. PrPC is expressed in all layers of the retina, including the outer and inner blood-retinal barriers, and blood aqueous barrier formed by non-pigmented ciliary epithelial cells. It is also expressed widely in the anterior segment, including the ciliary body, lens epithelium, corneal endothelium and epithelium, and the trabecular meshwork (Ashok et al., 2018; Asthana et al., 2017; Frederikse et al., 2000). One might argue that PrPC is of neuroectodermal origin, and it is not surprising to expect widespread expression in ocular tissues. However, systematic studies on different regions of the eye have revealed important physiological functions of PrPC, and deleterious consequences of loss of this function.
The eye is involved early in prion diseases (Chesebro et al., 2005; Striebel et al., 2019). Several studies have examined the spread of prion disease from the brain to the neuroretina, and total destruction of the latter by end stage disease. Approximately 40% of patients with sCJD develop visual symptoms, providing a window of opportunity for early diagnosis and intervention. PrPC is expressed, and is converted to the PrPSc form in the cornea, sclera, extraocular muscle, choroid, optic nerve, and lens (Figure 1) (Kercher et al., 2007; Orru et al., 2018). Soluble PrPC is present in the aqueous humor and vitreous humor (Ashok 2019). Iatrogenic spread of prions through corneal grafts has been reported, though it is relatively rare (Armitage et al., 2009; Head et al., 2003). In transgenic mice expressing CWD prions, PrPSc was detected in the cornea in addition to all layers of the retina (Asthana et al., 2017). The presence of PrPSc in dust particles, grass blades, and water reservoirs have prompted studies on peripheral routes of entry from the conjunctiva with positive results (Gough et al., 2015). Thus, the eye is a source of PrPSc transmission, all be it at a much slower state (Atkins et al., 2018; Gnanajothy et al., 2013). This is an important question since horizontal spread of animal prions through corneal abrasions is a distinct possibility (Asthana et al., 2017).
Figure 1. Expression of PrPC in the eye.
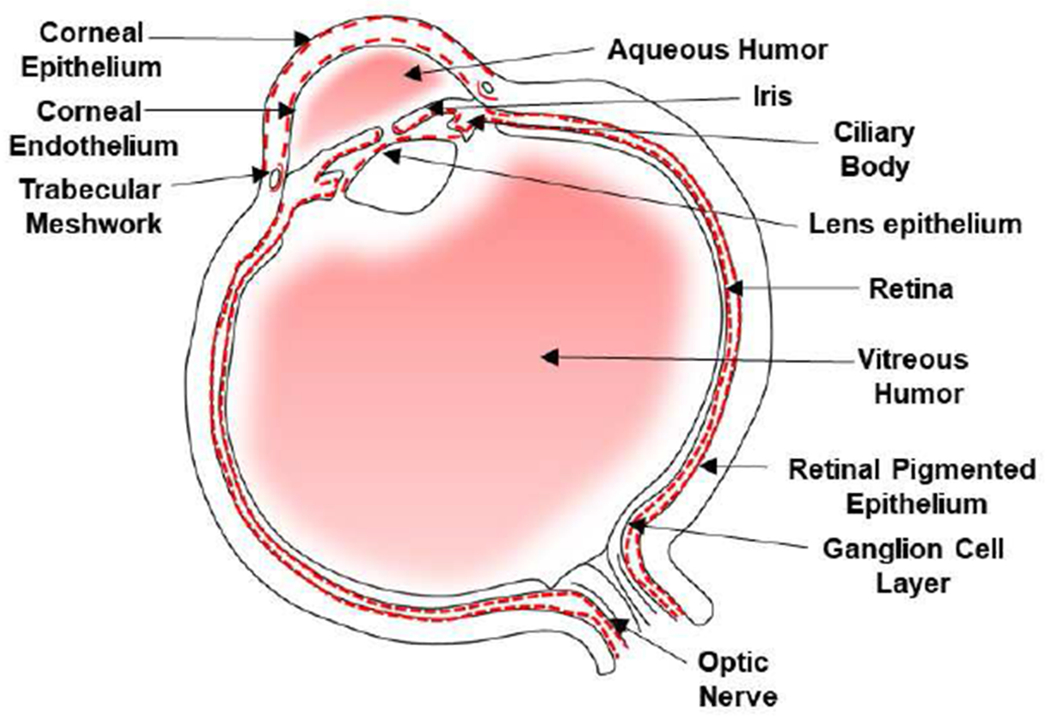
Immunoreaction of human eye shows a positive reaction for PrPC in the cornea epithelium and endothelium, TM, CB, iris, lens epithelium, retinal ganglion cell layer, and RPE cells (Ashok et al., 2018; Asthana et al., 2017). PrPC is expressed on the basolateral surface of NPE and RPE cells. Soluble PrPC is present in the aqueous humor and vitreous humor. Red dotted line represents a positive reaction for PrPC.
The eye offers several other advantages. For example, progression of the disease can be visualized in vivo in mouse models infected with prions, and the specific tissue analyzed after harvesting the eye. This allows a spatiotemporal evaluation of disease progression, and correlation between loss of function and gain of toxic function with disease progression. By generating tissue-specific PrPnull mice, it is possible to evaluate the progression of disease, and the role of PrPC at specific sites. Though several functions of PrPC have been described, implications of their dysfunction varies based on the tissue or organ in question. Some of these are exemplified by the eye because of its unique function in transmitting light signals. We provide some examples below.
3. The loss of physiological function and gain of toxic function hypothesis.
Of the many functions that PrPC participates in, we will focus on those where the eye has helped to resolve outstanding questions or reinforce known facts. Below are some examples.
Iron transport
Transport of iron to the eye has been the subject of several studies because of its vital role in the eye, and the inherent toxicity due to its redox-active nature (García-Castiñeiras, 2010; Song and Dunaief, 2013). The eye is particularly susceptible to iron induced toxicity because of constant exposure to light, which reduces iron from its relatively stable ferric form to the redoxactive ferrous form (Song et al., 2014). Transport of iron to the eye is therefore regulated by the outer and inner blood-retinal barriers, and the blood-aqueous barrier by specific proteins. In addition, relatively high concentrations of citrate, glutathione peroxidase, superoxide dismutase, and other reducing agents and enzymes protect the eye from iron-mediated toxicity (Goralska et al., 2009). It is believed that iron enters the eye from the inner blood retinal barrier, and leaves from the outer blood retinal barrier.
PrPC is a ferrireductase, and facilitates the transport of transferrin (Tf)-bound and non-Tf bound iron across biological membranes. Both forms of iron are in the oxidized, ferric (Fe3+) form, and need reduction to the ferrous (Fe2+) form for transport through metal transporters that span the membrane. Tf-bound iron taken up by cells via the Tf/transferrin receptor (TfR) pathway is released from Tf at the low pH of endosomes, and reduced to the ferrous form by membrane bound reductases such as PrPC or Steap3 before transport to the cytosol across divalent metal transporter 1 (DMT1) (Asthana et al., 2017; Ohgami et al., 2006; Singh et al., 2013). Likewise, ferric iron reduced at the plasma membrane by PrPC or Dcytb is transported through the ZIP family of divalent metal transporters to the cytosol (Ashok and Singh, 2018; Lane et al., 2015; Leung et al., 2008) (Figure 2). Here, it is utilized for metabolic purposes, and excess is transported out through the combined action of Fpn and a ferroxidase such as ceruloplasmin or haphestin. The redundancy in ferrireductase proteins emphasizes the significance of this function to cell viability. PrPC is expressed on capillary endothelial cells that form the inner blood-retinal barrier, RPE cells that form the outer blood-retinal barrier, and non-pigmented epithelial cells that form the blood-aqueous barrier. Absence of PrPC in PrPnull mice creates a phenotype of mild iron deficiency, indicating a role in iron transport (Singh et al., 2009a; Singh et al., 2009b).
Figure 2. PrPC-mediated iron homeostasis.
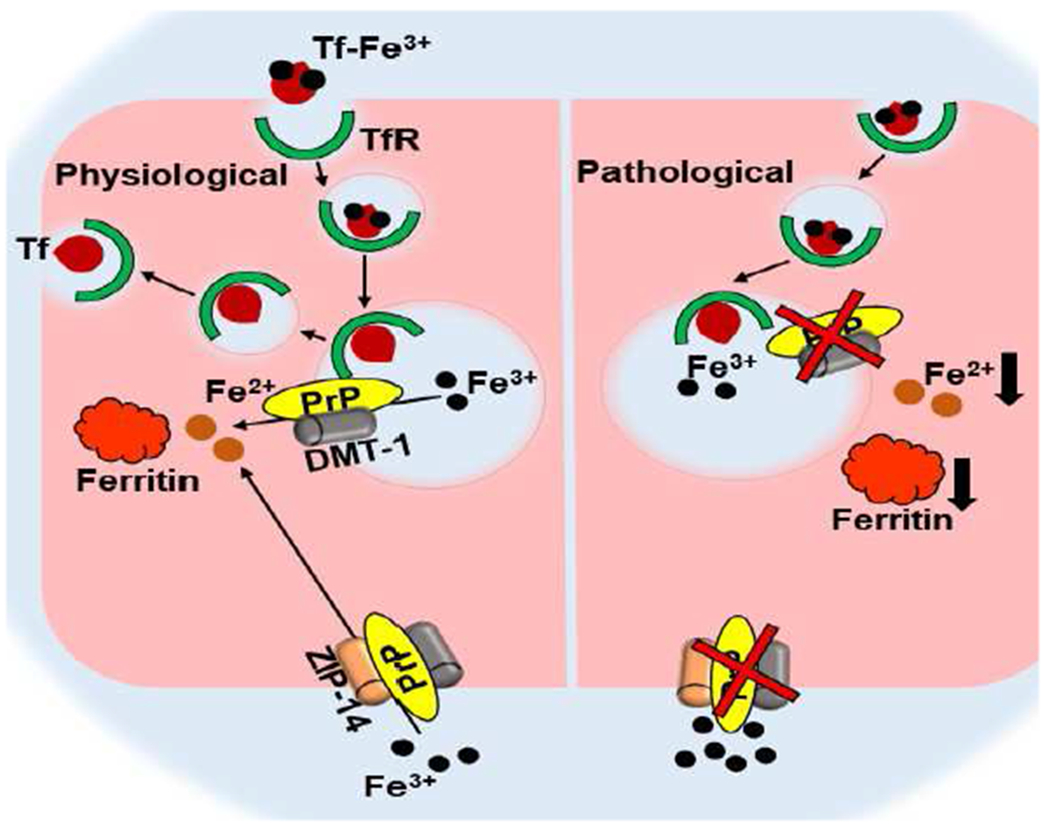
PrPC facilitates uptake of Tf-Fe3+ and non-Tf-bound iron (Fe3+) by its ferrireductase activity. Tf iron released in the endosome is reduced to Fe2+ for transport through DMT1. Non-Tf bound iron is reduced at the plasma membrane for transport through ZIP14 and other divalent metal transporters. Loss of PrPC function reduces iron uptake, and creates iron deficiency Singh 2014).
In RPE cells, PrPC is expressed on the basolateral membrane in vivo and in vitro. In an RPE cell line, polarization increases intracellular ferritin, indicating uptake of iron. Concomitant cleavage of PrPC that releases the N-terminal ferrireductase domain comprising of the octapeptide repeat region suggests regulated uptake of iron (Asthana et al., 2017). It is likely that PrPC plays a dual role in iron uptake or export depending on the iron status of the retina. It is interesting that almost all PrPC in RPE cells is cleaved at the β-site, an event that correlates with polarization and accumulation of ferritin (Ashok et al., 2018; Asthana et al., 2017). It is likely that accumulation of ferritin serves as a signal, and shedding of the ferrireductase domain blocks further uptake of iron. Silencing of PrPC in RPE cells lowers, while overexpression upregulates intracellular ferritin, supporting this claim (Asthana et al., 2017). The reason for β-cleavage of PrPC at amino acid ~90 instead of the physiological cleavage at amino acids 111/112 or the α-site is unclear at present (McDonald et al., 2014; Watt et al., 2005). Although both cleavage events would remove the N-terminal ferrireductase domain, β-cleavage, an event associated with oxidative stress, is evident in most ocular tissues (Haigh et al., 2015; Watt et al., 2005). The significance of this observations is unclear at present.
The presence of PrPC on the basolateral membrane of non-pigmented epithelial (NPE) cells suggests transport of iron from this site as well (Asthana et al., 2017). Local expression of hepcidin, Fpn, and ceruloplasmin on NPE cells support this assumption (Ashok et al., 2020b; Ashok et al., 2018). Moreover, expression of local hepcidin in the ciliary epithelium, corneal endothelium, lens epithelium, and trabecular meshwork further supports local regulation of iron at these sites (Ashok et al., 2020b). This contrasts with the belief that the anterior segment receives iron from the retina by diffusion, and supports the presence of an independent source of iron for the anterior segment (García-Castiñeiras, 2010). It is notable that PrPC is mainly full-length in the corneal endothelium, and β-cleaved in RPE, NPE, and trabecular meshwork (TM) cells (Ashok et al., 2018). Further investigations are necessary to evaluate whether this has functional implications, or is a cell specific phenomenon.
How does loss of function of PrPC and gain of toxic function impact the functioning of the eye? Although iron deficiency or excess alters ocular function in defined ways, the effect of PrPC on iron homeostasis has been difficult to discern. Loss of PrPC in PrPnull mice has minimal effect on functioning of the eye, though a mild phenotype of iron deficiency is noted (Asthana et al., 2017). It is likely that other iron uptake proteins compensate for the loss of PrPC, minimizing the effect of loss of function of PrPC. However, PrPSc results in sequestration of iron in ferritin, which co-aggregates with PrPSc (Singh et al., 2009a). This could result from different scenarios, which are not mutually exclusive. 1) PrPSc is transported to lysosomes for turnover, where it accumulates because it is resistant to lysosomal hydrolases. Ferritin is also transported to lysosomes for releasing stored iron for utilization by the cell. It is likely that the hydrophobic nature of ferritin and PrPSc results in co-aggregation of the two, resulting in PrPSc-ferritin aggregates. Such aggregates have been observed in neuroblastoma cells infected with PrPSc. The ferritin is rich in iron and shows a Perls’ positive reaction, leaving no doubt that iron is indeed increased but biologically unavailable. This creates a phenotype of relative iron deficiency despite excess brain iron, and upregulation of iron uptake proteins Tf and TfR (Singh et al., 2009a). 2) Neuro-inflammation accompanies prion infection, and the cytokines released are toxic of their own accord. In addition, IL-6, IL-1β, and the TGF family of cytokines are transcriptional triggers of hepcidin. Downregulation of Fpn by hepcidin is likely to increase intracellular iron and upregulate ferritin, resulting in the accumulation of iron rich ferritin. This is likely to create a feed-forward loop by further activating microglia, and the release of cytokines. In scrape infected retina, activation of Müller glia occurs prior to accumulation of ferritin, supporting this concept (Asthana et al., 2017; Carroll et al., 2015). Further studies are necessary to understand this phenomenon fully, and determine its role in PrPSc-induced toxicity.
One of the cytokines that upregulate hepcidin is transforming growth factor β2 (TGFβ2). This is of special interest in the TM because of the association of TGFβ2 with primary open angle glaucoma (POAG) (Hill et al., 2018). The cause of POAG is not completely understood, but TGFβ2-induced upregulation of fibrillogenic proteins increases the resistance of aqueous outflow from the TM, elevating intraocular pressure (IOP). A persistent increase in IOP induces ganglion cell death, resulting in glaucoma (Montecchi-Palmer et al., 2017). There are two main pathways by which TGFβ2 brings about this change; the canonical or Smad dependent pathway, and the non-canonical or Smad-independent pathway (Hata and Chen, 2016; Prendes et al., 2013). The Smad-dependent pathway functions through pospho-Smad2/3, which along with co-Smad 4, is translocated to the nucleus and activates various fibrillogenic proteins. These change the characteristics of the extracellular matrix and increase the IOP. One of the genes upregulated by phospho-Smad4 is hepcidin. Since the TM expresses hepcidin locally (Ashok et al., 2020b; Ashok et al., 2019), this raises the intriguing possibility of upregulation of hepcidin by TGFβ2, which downregulates Fpn and causes iron accumulation and upregulation of ferritin. The oxidative stress induced by sequestration of iron in ferritin upregulates TGFβ2, creating a positive feed-forward loop. Heparin, a hepcidin antagonist, and N-acetyl carnosine, an antioxidant, are able to disrupt this loop, bringing iron-mediated oxidative stress to the fore-front of glaucomatous change (Poli et al., 2017) (Figure 3).
Fig 3. TGFβ2-Hepcidin feed-forward loop.
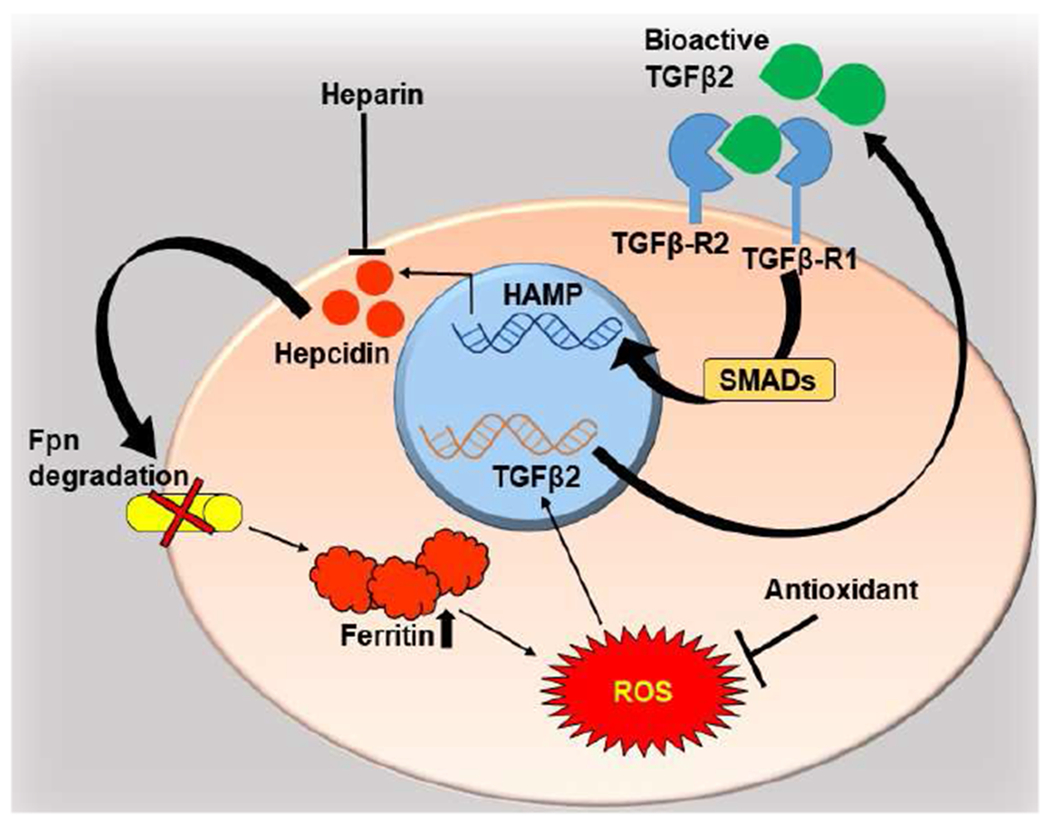
TGFβ2 upregulates transcription of hepcidin by the canonical, Smad-dependent pathway. Autocrine and paracrine activity of hepcidin downregulates Fpn, resulting in intracellular accumulation of iron, compensatory upregulation of ferritin, and increase in ROS, which upregulates TGFβ2. Heparin, a hepcidin antagonist, and antioxidants inhibit upregulation of TGFβ2 by decreasing ROS, disrupting the cycle (Ashok et al., 2020a).
PrPC and extracellular matrix
PrPC plays a prominent role in endothelial to mesenchymal transition (Endo-MT), a change associated with POAG. In contrast to upregulation of hepcidin that occurs through the canonical pathway, Endo-MT is mediated by the non-canonical pathway, and is associated with neurotoxicity by interfering with neuronal polarity and neuritogenesis (Ghodrati et al., 2018; Hajj et al., 2007; Hartmann et al., 2013). PrPC interacts with the extracellular matrix through β1 integrin, and downregulation of PrPC or dysfunction due to change in conformation to the PrPSc form induces increased synthesis of fibrillogenic proteins including fibrinogen, α-smooth muscle actin, collagen 1A, vimentin, and laminin, indicating transition to a mesenchyme-like phenotype (Ashok et al., 2019; Marbiah et al., 2014; Mehrabian et al., 2014).This changes cell-cell interaction to cell-substrate interactions, interfering with neuronal polarity and neuritogenesis in the neurons, and pliability of the extracellular matrix surrounding the TM. This change interferes with fine-tuning of the Ras homolog gene family member A (RhoA)-associated coiled-coil containing kinase (ROCK) signaling pathway, resulting in over-activation of ROCK. This induces a change in the pliability of the TM such that it does not react to stretch, resulting in elevation of the IOP (Arantes et al., 2009; Loubet et al., 2012; Zhong et al., 2018) (Figure 4). Inhibitors of the RhoA-Rock pathway are being suggested as a therapeutic option for both prion disorders and POAG, supporting a key role of PrPC in regulating cytoskeletal homeostasis (Alleaume-Butaux et al., 2013; Alleaume-Butaux et al., 2015; Kim et al., 2017).
Fig 4. PrPC stabilizes the ECM.
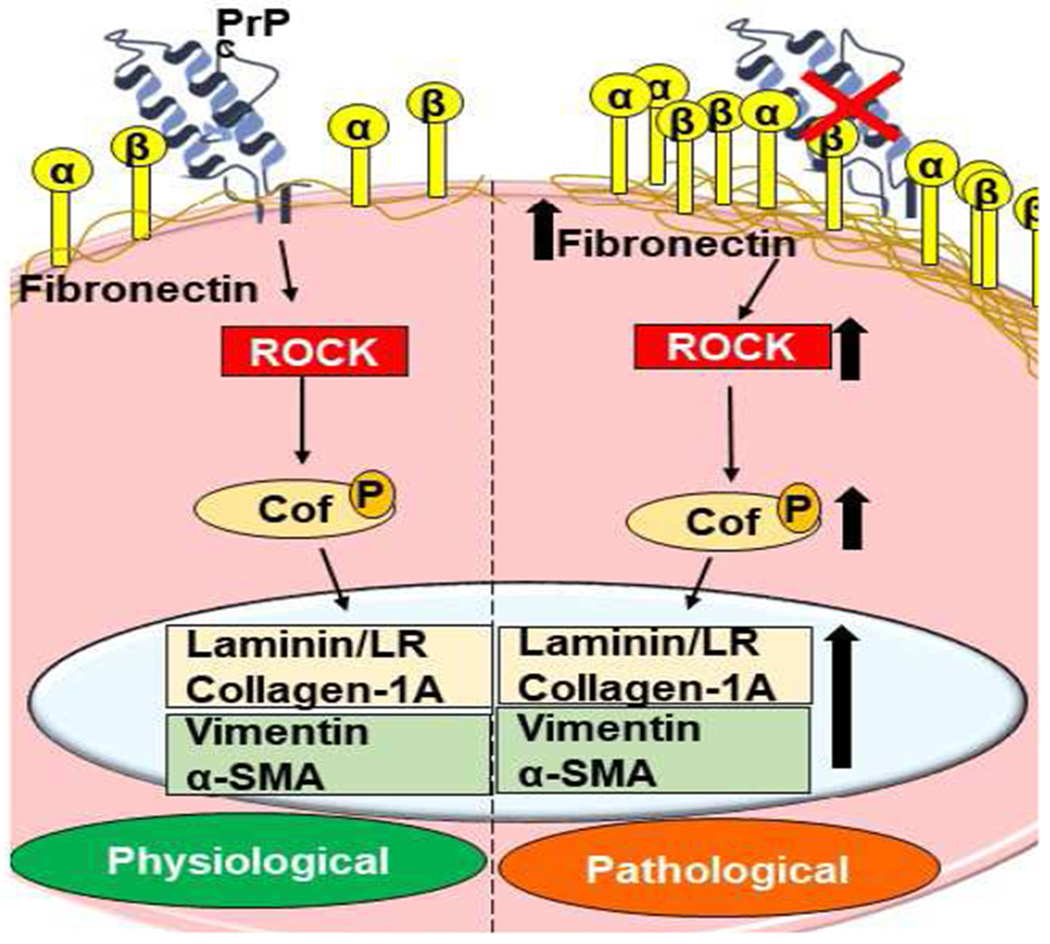
PrPC maintains cell-ECM interactions through β1-integrin and other proteins. Downregulation of PrPC causes aggregation of β1-integrin, activating the RhoA-ROCK pathway, and upregulation of fibronectin, collagen 1A, α-SMA, vimentin, and laminin that deposit in the ECM. It also modulates LIM kinases, which play a major role in maintaining cytoskeletal dynamics via phosphorylation of the cofilin family of proteins (Alleaume-Butaux et al., 2015; Ashok et al., 2019; Kim et al., 2020).
Conclusions
The eye offers several advantages over the brain for evaluating and understanding the pathobiology of prion disorders. The physiological functions of PrPC are reproducible in the eye, and so is their corruption by PrPSc. Iron transport across the outer and inner blood-retinal and blood aqueous barriers is easier to evaluate in the eye relative to the brain because of its less complicated structure, and individual proteins participating in this process can be identified with ease. The anterior segment provides a unique opportunity to study the exchange of iron through monolayers of cells, and determine the directionality of transport. Expression of iron transport proteins and hepcidin, and polarized expression of Fpn, and ceruloplasmin on monolayers is identifiable only in the eye. Upregulation of hepcidin by TGFβ2, and reciprocal upregulation of TGFβ2 by hepcidin in the TM has been possible in the eye, and is likely to have broader implications in the brain. The role of PrPC as an extracellular matrix stabilizing protein and its corruption by PrPSc has been described in the brain, but is easier to understand in the eye where it causes glaucomatous alterations by similar mechanisms. PrPC is likely to perform other functions in the eye, including the cornea, lens epithelium, iris, and other structures. A detailed analysis of downregulation of PrPC and change in conformation to PrPSc is likely to reveal other important functions of PrPC, and the mechanism by which their corruption causes disease. Additional research is necessary to understand how PrPSc causes disease-specific changes in the retina and perhaps other regions of the eye, which have remained unexplored due to the rapid course of this disease. A clear understanding of these processes will help in the development of a specific and sensitive diagnostic test for prion disorders, and therapeutic options in the future.
Highlights.
Prion protein (PrPC) is expressed in the anterior & posterior segment of the eye.
Absence of PrPC promotes endothelial to mesenchymal transition in TM cells.
PrPC helps to maintain iron homeostasis in the anterior segment.
Anterior segment maintains iron homeostasis independent of the retina.
TGFβ2 and hepcidin form a positive feed-forward loop fueled by iron-catalyzed ROS.
Acknowledgements
This work was supported by grants from the National Institutes of health, National Institute of Neurological Disorders and Stroke, R01 NS 092145 to NS.
Glossary
- PrPC
Prion protein
- Fpn
Ferroportin
- sCJD
Sporadic Creutzfeldt-Jakob disease
- PrPSc
Prion protein scrapie
- OCT
Optical Coherence Tomography
- ERG
Electroretinogram
- CWD
Chronic Wasting Disease
- BSE
Bovine Spongiform Encephalopathy
- ZIP
Zirt, Irt-like protein
- APP
Amyloid precursor protein
- RPE
Retinal pigment epithelium
- RNFL
Retinal nerve fiber layer
- NPE
Non-pigmented epithelium
- TM
Trabecular meshwork
- TfR
Transferrin receptor
- DMT1
Divalent metal transporter 1
- TGFβ2
Transforming growth factor beta 2
- POAG
Primary open angle glaucoma
- IOP
Intraocular pressure
- Endo-MT
Endothelial to mesenchymal transition
- RhoA
Ras homolog gene family member A
- ROCK
Rho associated coiled-coil containing kinase
- ECM
Extracellular matrix
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
The authors declare no competing interest.
References
- Adhikary R, Mukherjee P, Krishnamoorthy G, Kunkle RA, Casey TA, Rasmussen MA, Petrich JW, 2010. Fluorescence spectroscopy of the retina for diagnosis of transmissible spongiform encephalopathies. Analytical chemistry 82, 4097–4101. [DOI] [PubMed] [Google Scholar]
- Aguzzi A, Glatzel M, 2006. Prion infections, blood and transfusions. Nat Clin Pract Neurol 2, 321–329. [DOI] [PubMed] [Google Scholar]
- Alleaume-Butaux A, Dakowski C, Pietri M, Mouillet-Richard S, Launay J-M, Kellermann O, Schneider B, 2013. Cellular prion protein is required for neuritogenesis: fine-tuning of multiple singaling pathways involved in focal adhesions and actin cytoskeleton dynamics. Cell Health and Cytoskeleton 5, 1–12. [Google Scholar]
- Alleaume-Butaux A, Nicot S, Pietri M, Baudry A, Dakowski C, Tixador P, Ardila-Osorio H, Haeberle AM, Bailly Y, Peyrin JM, Launay JM, Kellermann O, Schneider B, 2015. Double-Edge Sword of Sustained ROCK Activation in Prion Diseases through Neuritogenesis Defects and Prion Accumulation. PLoS Pathog 11, e1005073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arantes C, Nomizo R, Lopes MH, Hajj GN, Lima FR, Martins VR, 2009. Prion protein and its ligand stress inducible protein 1 regulate astrocyte development. Glia 57, 1439–1449. [DOI] [PubMed] [Google Scholar]
- Armitage WJ, Tullo AB, Ironside JW, 2009. Risk of Creutzfeldt-Jakob disease transmission by ocular surgery and tissue transplantation. Eye (Lond) 23, 1926–1930. [DOI] [PubMed] [Google Scholar]
- Ashok A, Chaudhary S, Kritikos AE, Kang MH, McDonald D, Rhee DJ, Singh N, 2020a. TGFβ2-Hepcidin Feed-Forward Loop in the Trabecular Meshwork Implicates Iron in Glaucomatous Pathology. Investigative Ophthalmology & Visual Science 61,24–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashok A, Chaudhary S, McDonald D, Kritikos A, Bhargava D, Singh N, 2020b. Local synthesis of hepcidin in the anterior segment of the eye: A novel observation with physiological and pathological implications. Experimental Eye Research 190, 107890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashok A, Kang MH, Wise AS, Pattabiraman P, Johnson WM, Lonigro M, Ravikumar R, Rhee DJ, Singh N, 2019. Prion protein modulates endothelial to mesenchyme-like transition in trabecular meshwork cells: Implications for primary open angle glaucoma. Scientific reports 9, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashok A, Karmakar S, Chandel R, Ravikumar R, Dalal S, Kong Q, Singh N, 2018. Prion protein modulates iron transport in the anterior segment: Implications for ocular iron homeostasis and prion transmission. Exp Eye Res 175, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashok A, Singh N, 2018. Prion protein modulates glucose homeostasis by altering intracellular iron. Sci Rep 8, 6556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asthana A, Baksi S, Ashok A, Karmakar S, Mammadova N, Kokemuller R, Greenlee MH, Kong Q, Singh N, 2017. Prion protein facilitates retinal iron uptake and is cleaved at the β-site: Implications for retinal iron homeostasis in prion disorders. Scientific reports 7, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins N, Hodge W, Li B, 2018. A Systematic Review Regarding Tonometry and the Transmission of Infectious Diseases. Journal of clinical medicine research 10, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büeler H, Fischer M, Lang Y, Bluethmann H, Lipp H-P, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C, 1992. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 356, 577–582. [DOI] [PubMed] [Google Scholar]
- Carroll JA, Chesebro B, 2019. Neuroinflammation, Microglia, and Cell-Association during Prion Disease. Viruses 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll JA, Striebel JF, Race B, Phillips K, Chesebro B, 2015. Prion infection of mouse brain reveals multiple new upregulated genes involved in neuroinflammation or signal transduction. J Virol 89, 2388–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesebro B, Race R, Kercher L, 2005. Scrapie pathogenesis in brain and retina: effects of prion protein expression in neurons and astrocytes. Journal of neurovirology 11,476–480. [DOI] [PubMed] [Google Scholar]
- Doustar J, Torbati T, Black KL, Koronyo Y, Koronyo-Hamaoui M, 2017. Optical Coherence Tomography in Alzheimer’s Disease and Other Neurodegenerative Diseases. Front Neurol 8, 701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drakesmith H, Nemeth E, Ganz T, 2015. Ironing out Ferroportin. Cell Metab 22, 777–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehsani S, Mehrabian M, Pocanschi CL, Schmitt-Ulms G, 2012. The ZIP-prion connection. Prion 6, 317–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederikse PH, Zigler JS, Farnsworth PN, Carper DA, 2000. Prion protein expression in mammalian lenses. Current eye research 20, 137–143. [PubMed] [Google Scholar]
- Frigg R, Wenzel A, Samardzija M, Oesch B, Wariwoda H, Navarini AA, Seeliger MW, Tanimoto N, Reme C, Grimm C, 2006. The prion protein is neuroprotective against retinal degeneration in vivo. Exp Eye Res 83, 1350–1358. [DOI] [PubMed] [Google Scholar]
- García-Castiñeiras S, 2010. Iron, the retina and the lens: a focused review. Experimental eye research 90, 664–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasperini L, Meneghetti E, Legname G, Benetti F, 2016. In Absence of the Cellular Prion Protein, Alterations in Copper Metabolism and Copper-Dependent Oxidase Activity Affect Iron Distribution. Front Neurosci 10, 437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghodrati F, Mehrabian M, Williams D, Halgas O, Bourkas MEC, Watts JC, Pai EF, Schmitt-Ulms G, 2018. The prion protein is embedded in a molecular environment that modulates transforming growth factor beta and integrin signaling. Sci Rep 8, 8654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnanajothy R, UMashankeR D, Vega M, Wu BJ, 2013. A case of Creutzfeldt-Jakob disease following cataract surgery: sporadic versus iatrogenic cause. Connecticut medicine 77, 335–337. [PubMed] [Google Scholar]
- Goralska M, Ferrell J, Harned J, Lall M, Nagar S, Fleisher LN, McGahan MC, 2009. Iron metabolism in the eye: a review. Exp Eye Res 88, 204–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough KC, Baker CA, Simmons HA, Hawkins SA, Maddison BC, 2015. Circulation of prions within dust on a scrapie affected farm. Vet Res 46, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenlee MHW, Lind M, Kokemuller R, Mammadova N, Kondru N, Manne S, Smith J, Kanthasamy A, Greenlee J, 2016. Temporal resolution of misfolded prion protein transport, accumulation, glial activation, and neuronal death in the retinas of mice inoculated with scrapie. The American journal of pathology 186, 2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagiwara K, Hara H, Hanada K, 2013. Species-barrier phenomenon in prion transmissibility from a viewpoint of protein science. J Biochem 153, 139–145. [DOI] [PubMed] [Google Scholar]
- Haigh C, McGlade A, Collins S, 2015. MEK1 transduces the prion protein N2 fragment antioxidant effects. Cellular and molecular life sciences 72, 1613–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajj GN, Lopes MH, Mercadante AF, Veiga SS, da Silveira RB, Santos TG, Ribeiro KC, Juliano MA, Jacchieri SG, Zanata SM, Martins VR, 2007. Cellular prion protein interaction with vitronectin supports axonal growth and is compensated by integrins. J Cell Sci 120, 1915–1926. [DOI] [PubMed] [Google Scholar]
- Hannaoui S, Schatzl HM, Gilch S, 2017. Chronic wasting disease: Emerging prions and their potential risk. PLoS Pathog 13, e1006619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann CA, Martins VR, Lima FR, 2013. High levels of cellular prion protein improve astrocyte development. FEBS Lett 587, 238–244. [DOI] [PubMed] [Google Scholar]
- Hata A, Chen Y-G, 2016. TGF-β signaling from receptors to Smads. Cold Spring Harbor perspectives in biology 8, a022061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head MW, Northcott V, Rennison K, Ritchie D, McCardle L, Bunn TJ, McLennan NF, Ironside JW, Tullo AB, Bonshek RE, 2003. Prion protein accumulation in eyes of patients with sporadic and variant Creutzfeldt-Jakob disease. Invest Ophthalmol Vis Sci 44, 342–346. [DOI] [PubMed] [Google Scholar]
- Hill LJ, Mead B, Thomas CN, Foale S, Feinstein E, Berry M, Blanch RJ, Ahmed Z, Logan A, 2018. TGF-β-induced IOP elevations are mediated by RhoA in the early but not the late fibrotic phase of open angle glaucoma. Molecular vision 24, 712. [PMC free article] [PubMed] [Google Scholar]
- Kercher L, Favara C, Striebel JF, LaCasse R, Chesebro B, 2007. Prion protein expression differences in microglia and astroglia influence scrapie-induced neurodegeneration in the retina and brain of transgenic mice. Journal of virology 81, 10340–10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H-J, Kim M-J, Mostafa MN, Park J-H, Choi H-S, Kim Y-S, Choi E-K, 2020. RhoA/ROCK Regulates Prion Pathogenesis by Controlling Connexin 43 Activity. International Journal of Molecular Sciences 21, 1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Choi HS, Park JH, Kim MJ, Lee HG, Petersen RB, Kim YS, Park JB, Choi EK, 2017. Regulation of RhoA activity by the cellular prion protein. Cell Death Dis 8, e2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleene R, Loers G, Langer J, Frobert Y, Buck F, Schachner M, 2007. Prion protein regulates glutamate-dependent lactate transport of astrocytes. J Neurosci 27, 12331–12340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane DJ, Bae D-H, Merlot AM, Sahni S, Richardson DR, 2015. Duodenal cytochrome b (DCYTB) in iron metabolism: an update on function and regulation. Nutrients 7, 2274–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung KW, Liu M, Xu X, Seiler MJ, Barnstable CJ, Tombran-Tink J, 2008. Expression of ZnT and ZIP zinc transporters in the human RPE and their regulation by neurotrophic factors. Investigative ophthalmology & visual science 49, 1221–1231. [DOI] [PubMed] [Google Scholar]
- Loubet D, Dakowski C, Pietri M, Pradines E, Bernard S, Callebert J, Ardila-Osorio H, Mouillet-Richard S, Launay JM, Kellermann O, Schneider B, 2012. Neuritogenesis: the prion protein controls beta1 integrin signaling activity. FASEB J 26, 678–690. [DOI] [PubMed] [Google Scholar]
- Maddox RA, Belay ED, Curns AT, Zou W-Q, Nowicki S, Lembach RG, Geschwind MD, Haman A, Shinozaki N, Nakamura Y, 2008. Creutzfeldt-Jakob disease in recipients of corneal transplants. Cornea 27, 851–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marbiah MM, Harvey A, West BT, Louzolo A, Banerjee P, Alden J, Grigoriadis A, Hummerich H, Kan HM, Cai Y, 2014. Identification of a gene regulatory network associated with prion replication. The EMBO journal 33, 1527–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald AJ, Dibble JP, Evans EG, Millhauser GL, 2014. A new paradigm for enzymatic control of alpha-cleavage and beta-cleavage of the prion protein. J Biol Chem 289, 803–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrabian M, Ehsani S, Schmitt-Ulms G, 2014. An emerging role of the cellular prion protein as a modulator of a morphogenetic program underlying epithelial-to-mesenchymal transition. Front Cell Dev Biol 2, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montecchi-Palmer M, Bermudez JY, Webber HC, Patel GC, Clark AF, Mao W, 2017. TGFβ2 induces the formation of cross-linked actin networks (CLANs) in human trabecular meshwork cells through the Smad and non-Smad dependent pathways. Investigative ophthalmology & visual science 58, 1288–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngoo QZ, Wan Hitam WH, Ab Razak A, 2019. Evaluation of Retinal Nerve Fiber Layer Thickness, Electroretinogram and Visual Evoked Potential in Patients with Alzheimer’s Disease. J Ophthalmol 2019, 6248185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohgami RS, Campagna DR, McDonald A, Fleming MD, 2006. The Steap proteins are metalloreductases. Blood 108, 1388–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orru CD, Soldau K, Cordano C, Llibre-Guerra J, Green AJ, Sanchez H, Groveman BR, Edland SD, Safar JG, Lin JH, Caughey B, Geschwind MD, Sigurdson CJ, 2018. Prion Seeds Distribute throughout the Eyes of Sporadic Creutzfeldt-Jakob Disease Patients. mBio 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli M, Asperti M, Ruzzenenti P, Naggi A, Arosio P, 2017. Non-anticoagulant heparins are hepcidin antagonists for the treatment of anemia. Molecules 22, 598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prendes MA, Harris A, Wirostko BM, Gerber AL, Siesky B, 2013. The role of transforming growth factor β in glaucoma and the therapeutic implications. British Journal of Ophthalmology 97, 680–686. [DOI] [PubMed] [Google Scholar]
- Prusiner SB, 1997. Prion diseases and the BSE crisis. Science 278, 245–251. [DOI] [PubMed] [Google Scholar]
- Prusiner SB, 1998. Prions. Proceedings of the National Academy of Sciences 95, 13363–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramljak S, Schmitz M, Zafar S, Wrede A, Schenkel S, Asif AR, Carimalo J, Doeppner TR, Schulz-Schaeffer WJ, Weise J, Zerr I, 2015. Cellular prion protein directly interacts with and enhances lactate dehydrogenase expression under hypoxic conditions. Exp Neurol 271, 155–167. [DOI] [PubMed] [Google Scholar]
- Saunders SE, Bartelt-Hunt SL, Bartz JC, 2012. Occurrence, transmission, and zoonotic potential of chronic wasting disease. Emerging infectious diseases 18, 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheckel C, Aguzzi A, 2018. Prions, prionoids and protein misfolding disorders. Nature Reviews Genetics 19, 405–418. [DOI] [PubMed] [Google Scholar]
- Sigurdson CJ, Bartz JC, Glatzel M, 2019. Cellular and Molecular Mechanisms of Prion Disease. Annu Rev Pathol 14, 497–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Haldar S, Horback K, Tom C, Zhou L, Meyerson H, Singh N, 2013. Prion protein regulates iron transport by functioning as a ferrireductase. J Alzheimers Dis 35, 541–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Isaac AO, Luo X, Mohan ML, Cohen ML, Chen F, Kong Q, Bartz J, Singh N, 2009a. Abnormal brain iron homeostasis in human and animal prion disorders. PLoS pathogens 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Kong Q, Luo X, Petersen RB, Meyerson H, Singh N, 2009b. Prion protein (PrP) knockout mice show altered iron metabolism: a functional role for PrP in iron uptake and transport. PLoS One 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N, 2014. The role of iron in prion disease and other neurodegenerative diseases. PLoS pathogens 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N, Asthana A, Baksi S, Desai V, Haldar S, Hari S, Tripathi AK, 2015. The prion-ZIP connection: From cousins to partners in iron uptake. Prion 9, 420–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N, Haldar S, Tripathi AK, Horback K, Wong J, Sharma D, Beserra A, Suda S, Anbalagan C, Dev S, Mukhopadhyay CK, Singh A, 2014. Brain iron homeostasis: from molecular mechanisms to clinical significance and therapeutic opportunities. Antioxid Redox Signal 20, 1324–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N, Singh A, Das D, Mohan ML, 2010. Redox control of prion and disease pathogenesis. Antioxidants & redox signaling 12, 1271–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song D, Dunaief JL, 2013. Retinal iron homeostasis in health and disease. Frontiers in aging neuroscience 5, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song D, Zhao L, Li Y, Hadziahmetovic M, Song Y, Connelly J, Spino M, Dunaief JL, 2014. The oral iron chelator deferiprone protects against systemic iron overload-induced retinal degeneration in hepcidin knockout mice. Investigative ophthalmology & visual science 55, 4525–4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striebel JF, Race B, Williams K, Carroll JA, Klingeborn M, Chesebro B, 2019. Microglia are not required for prion-induced retinal photoreceptor degeneration. Acta neuropathologica communications 7, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrent J, Soukkarieh C, Lenaers G, Arndt C, Forster V, Picaud S, Mestre-Francés N, Verdier J-M, 2010. Microcebus murinus retina: a new model to assess prion-related neurotoxicity in primates. Neurobiology of disease 39, 211–220. [DOI] [PubMed] [Google Scholar]
- Wang CY, Babitt JL, 2016. Hepcidin regulation in the anemia of inflammation. Curr Opin Hematol 23, 189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt NT, Taylor DR, Gillott A, Thomas DA, Perera WS, Hooper NM, 2005. Reactive oxygen species-mediated beta-cleavage of the prion protein in the cellular response to oxidative stress. J Biol Chem 280, 35914–35921. [DOI] [PubMed] [Google Scholar]
- Weissmann C, Enari M, Klöhn P-C, Rossi D, Flechsig E, 2002. Transmission of prions. The Journal of infectious diseases 186, S157–S165. [DOI] [PubMed] [Google Scholar]
- Williams SK, Fairless R, Weise J, Kalinke U, Schulz-Schaeffer W, Diem R, 2011. Neuroprotective effects of the cellular prion protein in autoimmune optic neuritis. Am J Pathol 178, 2823–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, Grasso L, Sibilla C, Stevens TJ, Barry N, Bertolotti A, 2018. Prion-like protein aggregates exploit the RHO GTPase to cofilin-1 signaling pathway to enter cells. EMBO J 37. [DOI] [PMC free article] [PubMed] [Google Scholar]