

Abstract
Rationale:
Hereditary hemorrhagic telangiectasia (HHT) is a genetic disease caused by mutations in ENG, ALK1, or SMAD4. Since proteins from all three HHT genes are components of signal transduction of TGF-β family members, it has been hypothesized that HHT is a disease caused by defects in the ENG-ALK1-SMAD4 linear signaling. However, in vivo evidence supporting this hypothesis is scarce.
Objective:
We tested this hypothesis and investigated the therapeutic effects and potential risks of induced-ALK1 or -ENG overexpression for HHT.
Methods and Results:
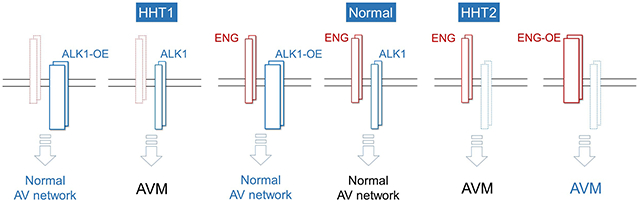
We generated a novel mouse allele (ROSA26Alk1) in which HA-tagged ALK1 and bicistronic eGFP expression are induced by Cre activity. We examined whether ALK1-overexpression (OE) using the ROSA26Alk1 allele could suppress the development of AVMs in wounded adult skin and developing retinas of Alk1- and Eng-inducible knockout (iKO) mice. We also used a similar approach to investigate whether ENG-OE could rescue AVMs. Biochemical and immunofluorescence analyses confirmed the Cre-dependent overexpression of the ALK1-HA transgene. We could not detect any pathological signs in ALK1-OE mice up to 3 months after induction. ALK1-OE prevented the development of retinal AVMs and wound-induced skin AVMs in Eng-iKO as well as Alk1-iKO mice. ALK1-OE normalized expression of SMAD and NOTCH target genes in ENG-deficient endothelial cells (ECs) and restored the effect of BMP9 on suppression of phosphor-AKT levels in these ECs. On the other hand, ENG-OE could not inhibit the AVM development in Alk1-iKO models.
Conclusions:
These data support the notion that ENG and ALK1 form a linear signaling pathway for the formation of a proper arteriovenous network during angiogenesis. We suggest that ALK1 overexpression or activation can be an effective therapeutic strategy for HHT. Further research is required to study whether this therapy could be translated into treatment for humans.
Keywords: Activin receptor-like kinase 1, animal model cardiovascular disease, arteriovenous malformation, endoglin, hereditary hemorrhagic telangiectasia, signal transduction, endothelial cell, Angiogenesis, Animal Models of Human Disease, Growth Factors/Cytokines, Hemodynamics, Vascular Biology
Graphical Abstract
INTRODUCTION
The development of highly organized vascular networks consisting of arteries, capillaries, and veins is crucial for the functions of the circulatory system. A failure of this process results in an arteriovenous malformation (AVM), an abnormal shunt between arteries and veins without intervening capillaries.1 Vessels associated with AVMs are overly dilated, tortuous, and prone to rupture, causing hemorrhagic insults.2, 3 The mechanisms underlying development, progression, and maintenance of AVMs are not clearly understood. Hereditary hemorrhagic telangiectasia (HHT) is a genetic vascular disorder, occurring about 1 in 5,000 people worldwide.2, 4 The clinical features include recurrent epistaxis, telangiectases in the skin and gastrointestinal tract, and large AVMs in multiple organs including the brain, lungs, and liver.4 Brain AVM is a risk factor for hemorrhagic stroke, whereas pulmonary AVMs can cause an ischemic stroke or brain abscess. Liver and visceral AVMs result in both organ damage and high output cardiac failure. Recurrent epistaxis and chronic bleeding from telangiectases of the gastrointestinal tract cause anemia. Management and surgical options are available for these clinical symptoms, but curative therapies are currently unavailable. Genetic studies have shown that heterozygous mutations in ENG, ALK1 (ACVRL1), or SMAD4 genes cause HHT.5–8 HHT is classified into three types depending on the mutant genes: HHT1 (ENG), HHT2 (ALK1) and juvenile polyposis-HHT (SMAD4).2 HHT1 and HHT2 account for over 90% of HHT cases.2
Interestingly, all three HHT gene products are major components of a signaling pathway of the transforming growth factor-beta (TGF-β) and bone morphogenetic protein (BMP) families of growth factors. Recent studies have indicated that BMP9 and BMP10 are likely the physiological ligands for the ENG-ALK1 signaling.9, 10 Both ENG and ALK1 form a high-affinity binding to BMP9 at different binding sites.11, 12 The binding region of BMP9 to ENG overlaps with that to a type II receptor, such as ACVR2B or BMPR2, suggesting that ENG and a type II receptor compete for binding to BMP9.11, 13 These structural studies indicate that ENG in the ternary complex can be replaced with a type II receptor for activating the type I receptor ALK1 for signaling to downstream targets such as SMADs, and support a postulation that deficiency of a linear BMP9/10-ENG-ALK1-SMAD4 pathway underlies the AVM development.
Animal models for HHT are well established. Although global knockout for Eng or Alk1 results in embryonic lethality in the midgestational period, with cardiovascular defects including shunts between the dorsal aortae and cardinal veins,14, 15 heterozygous deletion of Eng or Alk1 causes the development of HHT-like vascular lesions with incomplete penetrance in aged mice.16–18 Because of the limitations of studying the disease mechanism with homozygous and heterozygous KO mice, the most widely used HHT mouse models are homozygous for the deletion of Eng or Alk1. These mice may have a global or cell-type-specific conditional knockout in which they exhibit AVM formations in a reproducible manner. It has been shown that endothelial cells (ECs) are the primary cell type in which ENG, ALK1, and SMAD4 function for proper development of an arteriovenous network.19–22 Common HHT phenotypes in HHT1 and HHT2 models include AVMs in the brain and developing retinas in neonates, and wound-induced skin AVMs, and VEGF-induced brain AVMs in adults.19, 21, 23–27 However, there are differences in HHT-associated phenotypes between HHT1 and HHT2 models. For instance, tamoxifen-induced EC-specific Alk1 deletion in adult mice leads to anemia and hemorrhages in the gastrointestinal (GI) tract, whereas the same mode of Eng deletion does not cause GI bleeding or anemia.19 In zebrafish models of HHT, alk1 deletion causes AVM development in cranial vessels and death around 48 hours postfertilization,28. In contrast, eng mutant fishes survive to adult stages with AVM development in regenerating fins.29 Genotype-phenotype correlation studies in HHT have shown that the brain and pulmonary AVMs are more prevalent in HHT1, and liver and GI AVMs, in HHT2.30–32 These phenotype differences may be consistent with the linear signaling pathway of ENG and ALK1, and explained by differences in the availability of ligands, ALK1 or type II receptors in local tissue milieu. Alternatively, these differences may indicate parallel independent roles for ENG and ALK1, which then share a common set of downstream genes that play important roles in the formation of the arteriovenous network.
We used a mouse strain with conditional ALK1- or ENG-overexpression (OE) to investigate whether ALK1-OE can compensate for the loss of ENG and whether ENG-OE can compensate for the loss of ALK1. The data presented here support the postulation of the ENG-ALK1 linear pathway for the development of a proper arteriovenous network. They also indicate that stimulation of ALK1 expression could be an effective and safe therapeutic strategy for HHT1 in these mice.
METHODS
All supporting data are available within the article and its online data supplement. For details on the experimental procedures, see the materials and methods section in the online data supplement.
RESULTS
Generation of mice with Alk1 overexpression allele.
We have generated a novel mouse allele (ROSA26Alk1; R26Alk1) in which mouse ALK1 expression can be induced by Cre activity (Figure 1 A and B; Online Figure I) to investigate the effect of cell-type-specific, time-dependent induction of ALK1 overexpression in normal and pathological vasculature. HA–epitope-tagged mouse ALK1 (ALK1-HA) expression is designed to be controlled by CMV/chick β-actin promoter/enhancer when the Cre recombinase excises the upstream polyadenylation sequences (transcription stop sequences) flanked by the loxP sequences (Figure 1 A and B). Bicistronic insertion of green fluorescence protein (GFP) would allow us to monitor the Cre activity and to identify the cells where ALK1-HA is expressed.
Figure 1. Endothelial-specific induction of exogenous ALK1 in L1Cre; ALK1-OE mice.
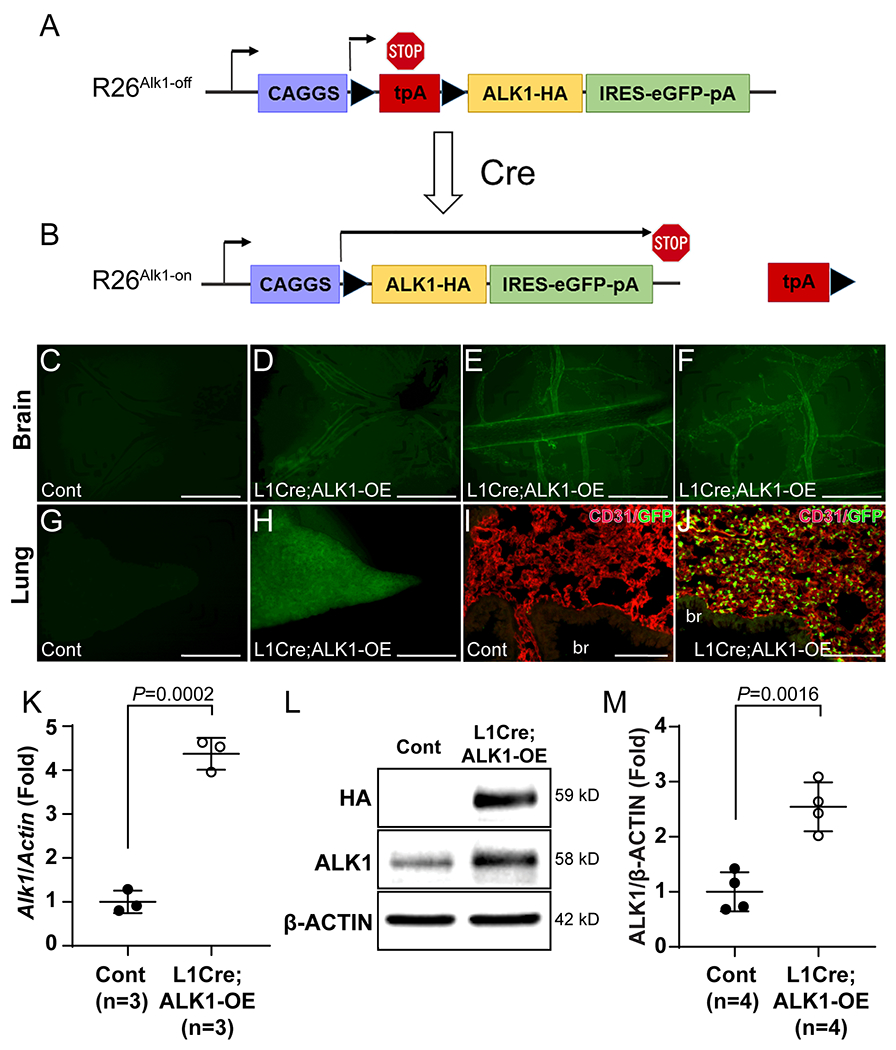
A and B, Schematic diagram of the R26Alk1-off (A), and R26Alk1-on (B) alleles. Mouse ALK1 carrying an HA tag at the C-terminus is bicistronically transcribed with eGFP by the CMV/chick β-actin (CAGGS) promoter from the R26Alk1-on allele due to the excision of transcription stop sequences in the presence of Cre recombinase. LoxP sequences are indicated by arrowheads. tpA, transcription stop sequences. C-J, Fluorescence stereomicroscopic images on brains (C-F) and lungs (G and J) of control (L1Cre-negative ALK1-OE, C, G, and I) and L1Cre;ALK1-OE (D-F and H and J) mice. I and J, CD31 (red) immunostaining on control (I) and L1Cre;ALK1-OE (J) lung section. GFP is detected in CD31-positive pulmonary ECs, but not in bronchial epithelial or smooth muscle cells (J). br, bronchus. K. Quantitative RT-PCR on Alk1 transcripts in control and L1Cre;ALK1-OE mouse lungs. β-Actin was used for normalization. n=3 per each group. Unpaired t-test. L. Western blot analyses against HA and ALK1 proteins in control and L1Cre;ALK1-OE lung. M. Quantification of ALK1 levels normalized with β-ACTIN. n=4 per each group. Unpaired t-test. All data are means ± SDs. Scale bars: C-H, 1 mm; I and M, 100 μm.
For testing ALK1 overexpression in mice, R26Alk1 (ALK1-OE) mice were intercrossed with L1Cre mice, in which Cre recombinase is predominantly expressed in endothelial cells by the Alk1 promoter.33 Fluorescence stereomicroscopy of the brain of L1Cre;ALK1-OE mice showed GFP expression in the blood vessels (Figure 1 C–F). The lungs of the bigenic mice were also GFP positive (Figure 1H), and most GFP-expressing cells in the lungs were found to be CD31-positive endothelial cells (Figure 1 I and J; Online Figure II). Real-time reverse transcriptase-polymerase chain reaction (RT-PCR) analysis showed about a 4-fold increase in the Alk1 transcript level in L1Cre;ALK1-OE mouse lungs compared to that in the control lungs (Figure 1K). ALK1-HA protein was detected in ALK1-OE mouse lungs, and the ALK1 protein level was increased about 3-fold in L1Cre;ALK1-OE lungs (Figure 1 L and M). Interestingly, 2-month-old L1Cre;ALK1-OE mice did not exhibit any visible pathological symptoms.
Pan endothelial ALK1-overexpression does not disturb the development of normal arteriovenous networks.
Using an Alk1-lacZ knock-in reporter allele (Alk1lacZ), we have previously shown that low levels of Alk1 expression in the arterial endothelium in the subdermal skin of healthy adult mice are markedly increased following wounding.34 To examine Alk1-lacZ expression during wound healing in adult mice with and without Alk1 gene deletion, we stained the wounded skins of R26+/+;Alk12loxP/lacZ and R26+/CreER;Alk12loxP/lacZ mice with X-gal. As previously shown, AVMs formed in the vessels surrounding the wound in Alk1 deleted mice and were maintained over 9 days post–Alk1-gene deletion and wounding.25 Dilated and tortuous arteries and veins were visible in wounded skin area of mutant mice, indicating the presence of AVMs (Online Figure III D and G). Arteries feeding the wounds were X-gal-positive in the control and mutant mouse skin specimens (Online Figure III H and I). Because NOTCH downstream genes are regulated by ALK1 signaling,21, 35, 36 it has been proposed that ALK1 is important for arterialization and that ALK1-OE itself could cause AVMs by upregulation of NOTCH signaling.37–40 To determine whether ALK1 overexpression causes arterialization, altered arteriovenous network formation, or AVMs, we examined ALK1-OE mice in which ALK1 expression was induced in venous, capillary, and arterial ECs in adult mice using the SclCreER driver,41 or in smooth muscle cells using Myh11CreER.42 As shown in Online Figure IV A–C, GFP expression was induced in vascular but not lymphatic ECs in the wounded skin of SclCreER;AKL1-OE mice 9 days after tamoxifen injections and wounding. GFP expression in SclCreER;ALK1-OE was detected in blood vessels of the brain and lungs and mostly overlapped with ERG-positive arterial and venous endothelial cells in the subdermal vasculature (Online Figure V). SclCreER;AKL1-OE and Myh11CreER;AKL1-OE mice had no apparent signs of illness. Hemoglobin levels were unchanged in these ALK1-OE mice (Online Figure IV D). Latex perfusion via the left ventricle revealed no sign of AVMs in the wounded skin of these EC- or SMC-specific ALK1-OE mice (Online Figure IV F–K and Online Figure VI). The quantification of the vascular area did not show significant differences compared to the controls (Online Figure IV E). We also investigated whether long-term ALK1 overexpression in ECs or SMCs for 5 weeks had any adverse effects (Online Figure VII A). These ALK1-OE mice did not exhibit any signs of sickness. Hemoglobin levels in these mice were normal (Online Figure VII B), and no AVM in visceral organs or wounded skin was detected (Online Figures VII C–H). Furthermore, AVMs were not observed in mice with global ALK1-OE driven by R26CreER (Online Figure VIII). In addition, we also examined various physiological parameters such as blood pressure, oxygen saturation, heart rates (Online Figure IX), complete blood counts (Online Figure X), histology (Online Figure XI), presence of AVMs using latex dye perfusion (Online Figure XII), proliferation and apoptosis (Online Figure XIII) in SclCreER;ALK1-OE and control mice 3 months after five tamoxifen injections. We did not find significant differences between ALK1-OE and age-matched control mice, suggesting that overexpression of ALK1 protein does not cause vascular abnormalities.
To examine the molecular property of ALK1-OE in ECs, we isolated pulmonary ECs from SclCreER;ALK1-OE mice. Hydroxytamoxifen (OH-TM) treatment deleted the stop cassette (Online Figure XIV A) and induced ALK1 expression to about 2.5-fold (Online Figure XIV B and C). OH-TM-treated cells showed significantly higher sensitivity to BMP9 for SMAD1/5/8 phosphorylation (Online Figure XIV D–G). In the steady-state condition with normal media, however, there was no significant difference in SMAD1/5/8 phosphorylation (Online Figure XIV H and I). To examine the transcript levels of BMP and Notch downstream gene in retinal ECs, we compared them by isolating retinal ECs from control and SclCreER;ALK1-OE mice. Consistent with the results of the steady-state condition, there was no difference in the transcript levels of BMP and Notch downstream genes (Online Figure XIV J). In addition, we also examined the difference of pSMAD1,5,8 in pulmonary ECs of control and SclCreER;ALK1-OE mice 3 months after induction. As shown in Online Figure XV, the number of pSMAD1,5,8-positive pulmonary ECs in SclCreER;ALK1-OE was similar to that in the controls.
ALK1-overexpression rescues AVM phenotypes in Eng- as well as Alk1-inducible KOs.
To determine whether ALK1-OE can rescue Alk1-iKO phenotypes, we intercrossed the two lines to generate Alk1-iKO;ALK1-OE mice and investigated the effect of ALK1-OE on HHT phenotypes that we had established for Alk1-iKO mice in previous studies.24, 25, 43 Eight days after tamoxifen administration and wound infliction on the dorsal skin, EC-specific Alk1-iKO mice had lower hemoglobin levels (Figure 2 A), GI hemorrhage (Figure 2 B), and wound-induced skin AVM formation (Figures 2 C and D). These Alk1-iKO phenotypes were fully rescued in Alk1-iKO;ALK1-OE mice: there was no reduction in hemoglobin level (Figure 2 A), no GI bleeding (Figure 2B), and no wound-induced AVMs (Figure 2 C; Online Figure XVI). Quantification of the density of vessels containing the latex dye showed normalization of the vessel density in Alk1-iKO;ALK1-OE mice compared to that of controls (Figure 2 D). This result indicates that transgenic ALK1-HA is functional and can fully compensate for the loss of endogenous ALK1.
Figure 2. ALK1-OE rescues the vascular phenotypes of ENG- as well as ALK1-deficient mice.
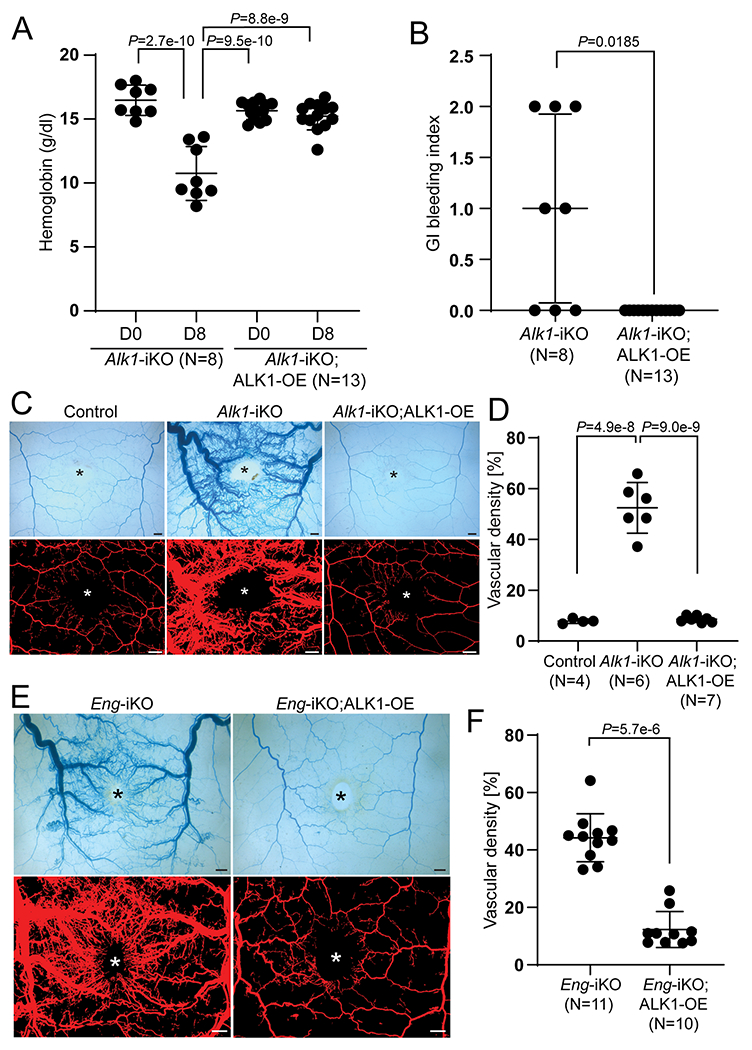
A, Alteration of hemoglobin levels in SclCreER;Alk1-iKO (n=8) and SclCreER;Alk1-iKO;ALK1-OE (n=13) on the first day (D0) and 8 days (D8). Two-way ANOVA with Tukey’s correction. B, Gastrointestinal hemorrhage index of SclCreER;Alk1-iKO (n=8) and SclCreER;Alk1-iKO;ALK1-OE (n=13) mice. The unpaired t-test was performed for statistical analysis. Welch t-test. C, Representative images of latex dye-perfused blood vessels (upper panels) and processed images (lower panels) on the dorsal skin of control (SclCreER-negative Alk1-iKO;ALK1-OE (n=4), SclCreER;Alk1-iKO (n=6), and SclCreER;Alk1-iKO;ALK1-OE (n=7) mice on 8 days after wounding. D, Quantification of the vascular area containing latex with the processed images. One-way ANOVA with Tukey’s correction. E, Latex perfusion images in SclCreER;Eng-iKO (n=11) and SclCreER;Eng-iKO;ALK1-OE (n=10). F, Quantification of vascular density containing latex. Mann-Whitney test. All data are means ± SDs. The wound sites are marked by asterisks. Scale bars: 1 mm.
We then examined whether ALK1-OE could compensate for the loss of ENG. As previously shown19, EC-specific Eng-iKO mice developed wound-induced skin AVMs (Figure 2 E). The skin AVMs were remarkably suppressed in Eng-iKO;ALK1-OE mice (Figures 2 E and F; Online Figure XVII). This result indicates that ALK1-OE is sufficient to overcome AVM development caused by ENG-deficiency.
ALK1-overexpression inhibits AVM development in the retinal vasculature of Eng- as well as Alk1-iKOs.
We further investigated the effects of ALK1-OE in retinal vascular development of control and mutant mice. Unlike arterial-specific expression patterns in subdermal skin (Online Figure XVIII A), Alk1 expression was detected in the veins and capillaries as well as arteries in the central and proximal retina of Alk1-GFP reporter (Alk1+/GFP) mice (Online Figure XVIII B). Consistent with previous reports,21, 23 ALK1 immunostaining also showed panendothelial expression of ALK1 in the developing retinas of control mice (Online Figures XIX A and B). Tamoxifen treatment-induced ALK1-HA expression in venous and capillary ECs as well as arterial ECs of SclCreER;ALK1-OE mice (Online Figure XIX C), and the patterns of ALK1 overexpression overlapped with those of GFP (Online Figures XIX D). Consistent with subdermal vessels, the retinal vasculature in ALK1-OE mice was indistinguishable from that in the control mice (Online Figures XIX E–G), demonstrating that panendothelial ALK1-OE does not disturb retinal vascular development.
About 4-5 AVMs were found in the developing retinas of SclCreER;Alk1-iKOs by postnatal day 5 (Figure 3 B and D), around 2 days after tamoxifen administration on day 3. However, no AVMs were detected in SclCreER;Alk1-iKO;ALK1-OE retinas (Figure 3 C, E, and F). ALK1-OE also suppressed AVM development and increased vascular density in SclCreER;Eng-iKO retinas (Figures 3G–L). Furthermore, delayed radial expansion of the retinal vasculature in Alk1-iKO and Eng-iKO neonates was restored by ALK1-OE (Online Figure XX).
Figure 3. ALK1-OE in retinal ECs suppresses the development of AVMs caused by ALK1- or ENG-deficiency.
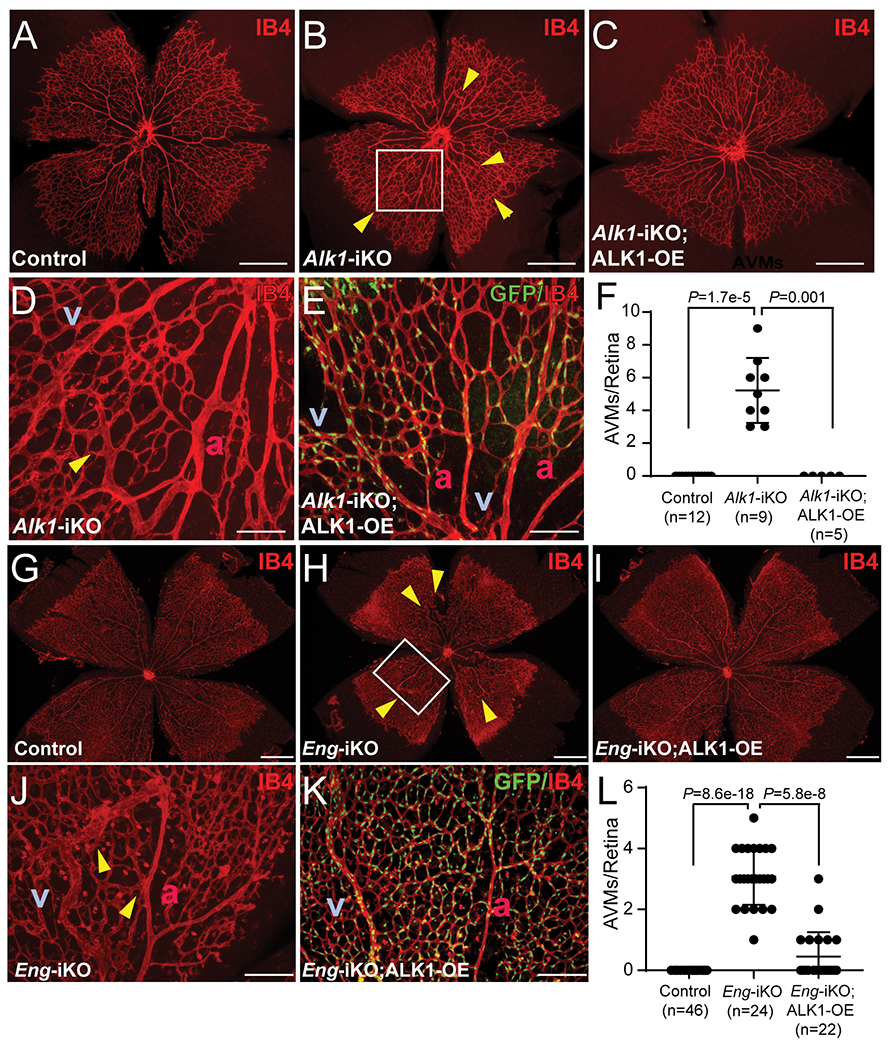
A-E, IB4 staining of postnatal day 5 (PN5) retinas from control (A, n=12), SclCreER;Alk1-iKO (B and D, n=9), and SclCreER;Alk1-iKO;ALK1-OE (C and E, n=5) mice. The boxed region in B is a magnified image showing direct connections between arteries and veins (D). SclCreER-mediated GFP reporter expression in SclCreER;Alk1-iKO;ALK1-OE retinas (E). F, Quantification of AVM numbers in PN5 retinal vasculature. Kruskal–Wallis test with Dunn’s correction. G-K, IB4 staining of PN7 retinas from control (G, n=46), SclCreER;Eng-iKO (H and J, n=24), and SclCreER;Eng-iKO;ALK1-OE (I and K, n=22) mice. The boxed region in H is a magnified image showing arteriovenous shunts (J). Bicistronic GFP reporter expression in SclCreER;Eng-iKO;ALK1-OE retinas (K). L, Quantification of the number of AVMs. Yellow arrowheads indicate AVMs in the developing retinas. Kruskal–Wallis test with Dunn’s correction. All data are means ± SDs. a, artery; v, vein. Scale bars: A-C and G-I, 500 μm; D, E, J, and K, 100 μm.
ENG protein detected in ECs of arteries, veins, and capillaries of postnatal day 7 control retinas (Figure 4 A and D) was undetectable in most of the blood vessels, including AVM lesions of Eng-iKO retinas (Figure 4 B and E). ENG-deficiency was similarly maintained in Eng-iKO; ALK1-OE retinas (Figure 4 C and F), indicating that inhibition of AVMs in Eng-iKO;ALK1-OE mice was not due to incomplete deletion of Eng gene or increased ENG expression from an incompletely deleted Eng allele in response to ALK1-OE. As it was previously shown,22 artery-specific smooth muscle actin staining in control retinas (Figure 4 G) is altered in Eng-iKO retinas, where smooth muscle actin staining was found in arteriovenous shunts and connecting veins as well as the arteries (Figure 4 H). Such aberrant smooth muscle α actin staining in veins, an indicator of the presence of AVMs, is undetectable in Eng-iKO;ALK1-OE mice (Figure 4 I).
Figure 4. ALK1-OE restores aberrant SMC coverage observed in Eng-iKO retinal vasculature.
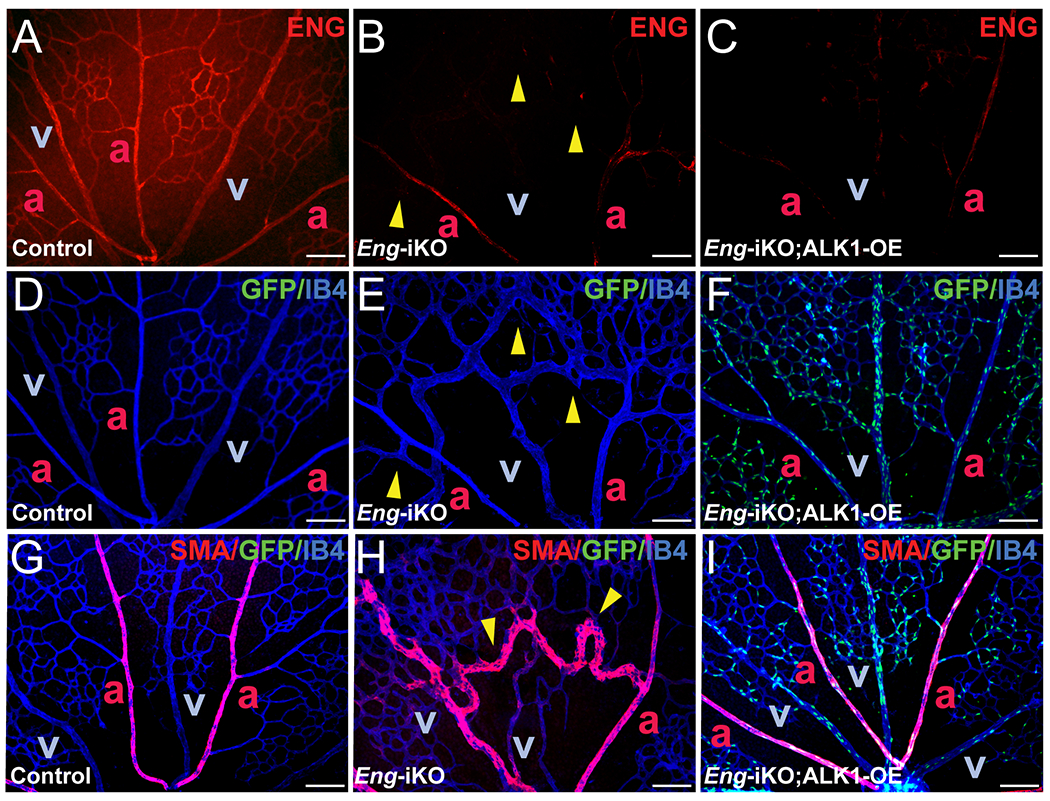
A-F, ENG (red) and IB4 (blue) fluorescence staining with retinas isolated from PN7 control (SclCreER-negative Eng-iKO, A and D), SclCreER;Eng-iKO (B and E), and SclCreER;Eng-iKO;ALK1-OE (C and F) mice. G-I. Double staining of IB4 (blue) and SMA (red) in PN7 retinas of controls (G), SclCreER;Eng-iKO (H), and SclCreER;Eng-iKO;ALK1-OE (I) mice. GFP reporter expression in IB4-positive retinal ECs in SclCreER;Eng-iKO;ALK1-OE (F and I). Yellow arrowheads indicate developed retinal AVMs. a, artery; v, vein. Scale bars: 100 μm.
Disrupted vessel type-specific gene expressions in ENG-deficient ECs are recovered by enhanced ALK1 signaling.
Endomucin (EMCN) is predominantly expressed in the ECs of veins and capillaries but not in arterial branches of the retinas of control mice (Figure 5 A and D). In Eng-iKO mouse retinas, however, EMCN was found in arteries near the junction of arteriovenous shunts (Figure 5 B and E), but this aberrant EMCN expression in arterial branches was not observed in Eng-iKO;ALK1-OE mouse retinas (Figure 5 C and F). Jagged1 (JAG1), mainly expressed in arterial ECs (Figure 5 G and J), was undetectable in Eng-iKO retinas (Figure 5 H and K). ALK1-OE restored JAG1 expression in Eng-depleted arterial ECs (Figure 5 I and L).
Figure 5. Disrupted arteriovenous EC identities in Eng-deficient vasculature are rescued by ALK1-OE.
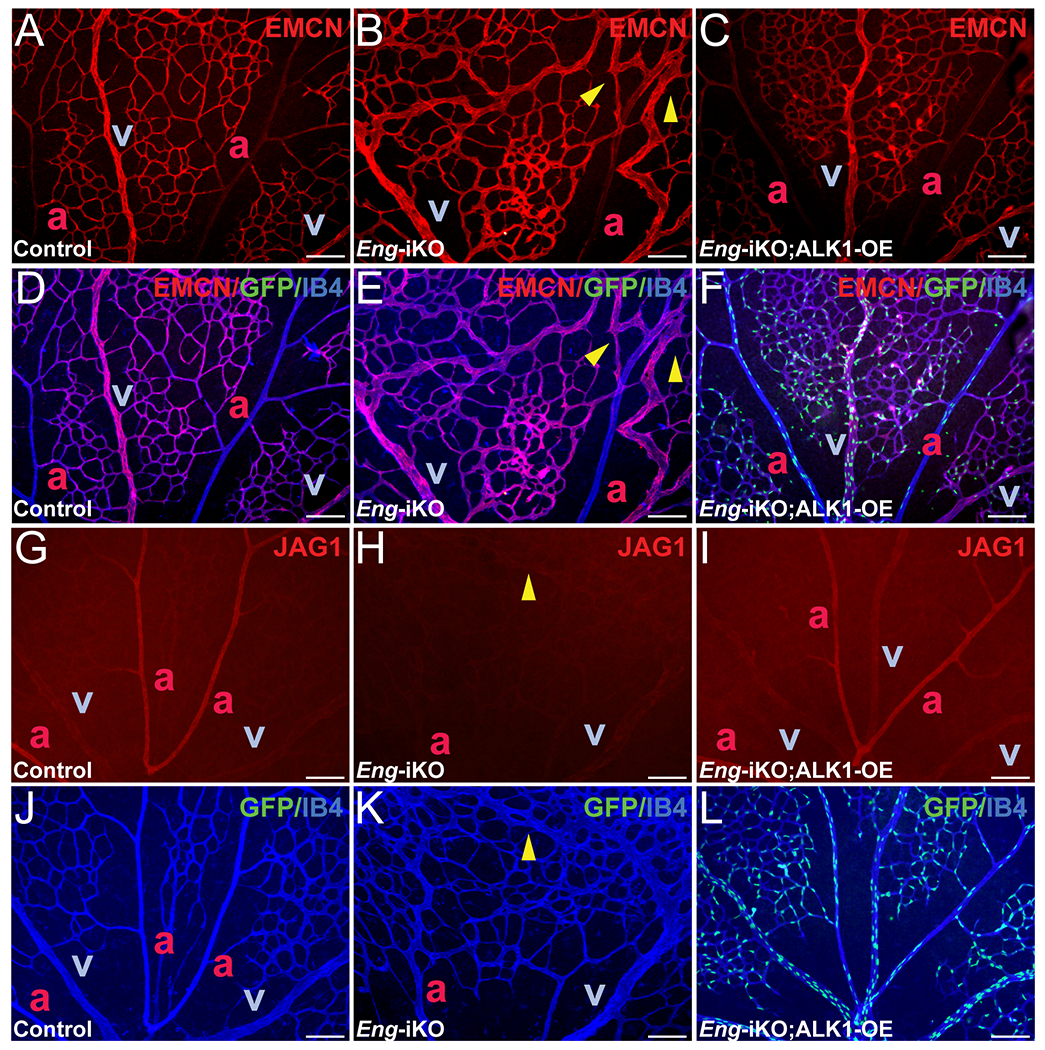
A-F, Immunostaining on EMCN (red) and IB4 (blue) in PN 7 retinas of control (SclCreER-negative Eng-iKO, A and D), SclCreER;Eng-iKO (B and E), and SclCreER;Eng-iKO;ALK1-OE (C and F) mice. Merged images of EMCN, GFP, and IB4 (D-F). G-L, JAG1 (red) expression in PN7 retinas of controls (G and J), SclCreER;Eng-iKO (H and K), and SclCreER;Eng-iKO;ALK1-OE (I and L) mice. ECs are marked with IB4 (blue) staining. GFP signals in IB4-stained retinal ECs (J-L). Yellow arrowheads indicate AVMs in the retinal vasculature. a, artery; v, vein. Scale bars: 100 μm.
To determine the extent to which ALK1-OE overcomes the ENG-deficiency on signaling pathways, we examined transcript levels of genes, including BMP and Notch downstream targets in retinal ECs. RT-PCR analysis demonstrated that about 20% of Eng remained in the retinal ECs of P7 Eng-iKO and Eng-iKO;ALK1-OE pups (Figure 6 A). The Alk1 transcript level was also decreased to about 60% of that of the control in Eng-iKO ECs, and it was increased about 6-fold in Eng-iKO;ALK1-OE retinal ECs compared to that of controls (Figure 6 B). Notch targets (Hey1, Jag1, and Jag2) and BMP downstream gene (Id1) expression were decreased in Eng-iKO retinal ECs but appeared to be normalized in Eng-iKO;ALK1-OE retinal ECs (Figure 6 C–F). These results indicate that elevated ALK1 expression could overcome ENG-deficiency for regulating the expression of some downstream genes.
Figure 6. Altered NOTCH and SMAD downstream genes in Eng-iKO ECs are restored by ALK1-OE.
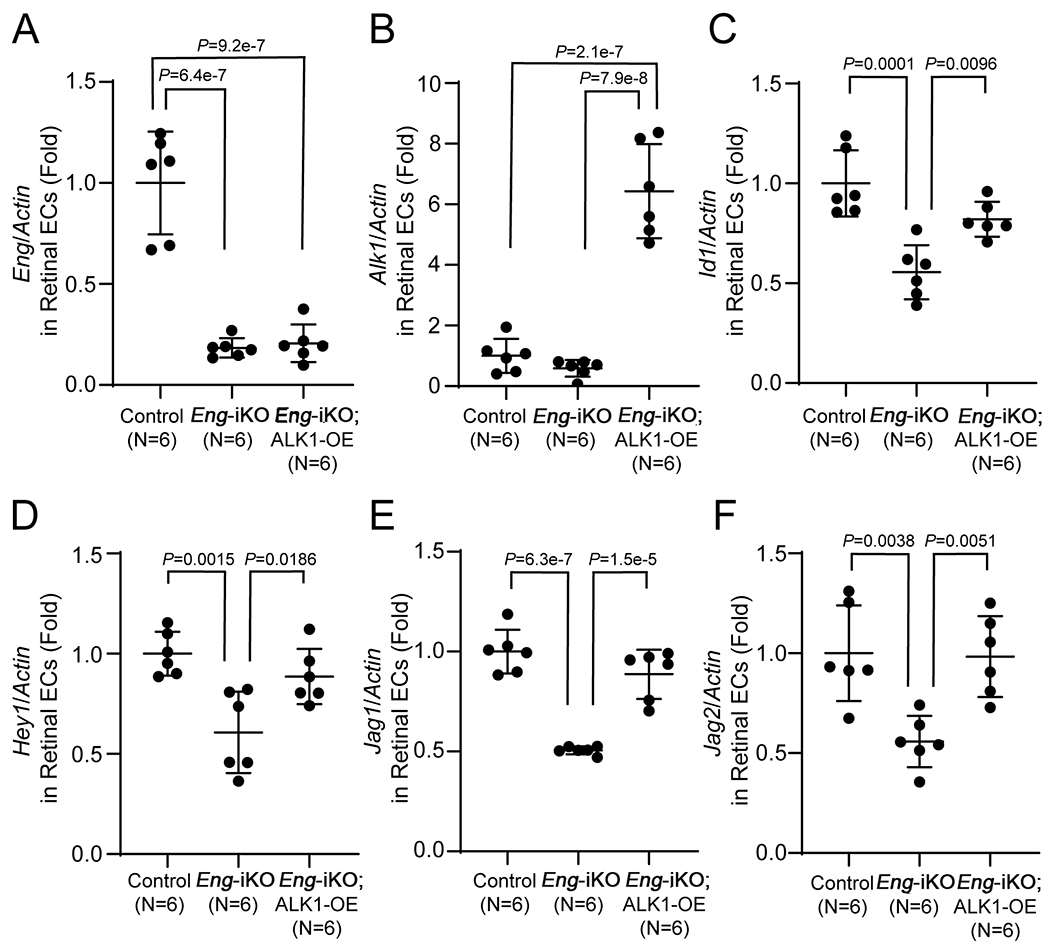
Quantitative RT-PCR analysis of Eng (A) and Alk1 (B), and Id1 (C)and NOTCH downstream genes [Hey1 (D), Jag1 (E), and Jag2 (F)] in retinal ECs purified from control (SclCreER-negative Eng-iKO, n=6), SclCreER;Eng-iKO (n=6), and SclCreER;Eng-iKO;ALK1-OE (n=6) mice. The mRNA levels of the genes are normalized with actin. One-way ANOVA with Tukey’s correction. All data are means ± SDs.
To test the effects of ALK1-OE on SMAD and AKT pathways in ECs, we employed pulmonary ECs isolated from R26CreER/+;Eng-iKO and R26CreER/+;Eng-iKO;ALK1-OE mice. Eng gene deletion and ALK1-OE were induced by 4-hydroxy-tamoxifen treatment. To examine the level of SMAD1/5/8 phosphorylation, we added BMP9 (0.5 ng/mL) to serum-starved pulmonary ECs for 45 min in static or flow conditions. Significantly depressed SMAD1/5/8 phosphorylation in ENG-deficient (OH-TM -treated Eng-iKO) cells was restored in ENG-deficient;ALK1-OE cells to the level similar to ENG-intact cells (OH-TM untreated Eng-iKO) in both static and flow conditions (Figure 7 A–C). It has been shown that BMP9 suppressed AKT phosphorylation induced by VEGF or flow. This BMP9 effect on AKT regulation was blunted in ALK1 or SMAD4-deficient ECs.20, 23, 44 PI3K/AKT inhibitors or Akt1-deletion in ECs could prevent AVM development in Alk1- or Smad4 mutant retinas, indicating that dysregulation of PI3K/AKT plays a pivotal role in AVM development.20, 23 In ENG-intact ECs, the flow increased AKT phosphorylation. ENG-deficiency itself increased phospho-AKT (pAKT) levels in both static and flow conditions. While BMP9 treatment suppressed AKT phosphorylation in ENG-intact pulmonary ECs, such a BMP9 response was blunted in ENG-deficient pulmonary ECs (Figure 7 D, E, and G). In contrast, ALK1-OE reduced AKT phosphorylation in ENG-deficient pulmonary ECs in the static condition to the level similar to that seen with BMP9 treatment (Figure 7 D and F). Interestingly, ALK1-OE restored the BMP9 mediated suppression of AKT phosphorylation in ENG-deficient;ALK1-OE pulmonary ECs under the flow conditions (Figure 7 D and H), which indicates that ALK1-OE can normalize the aberrantly activated endothelial AKT signaling caused by ENG-deficiency.
Figure 7. ALK1–OE leads to the rescue of disrupted SMAD and AKT signaling in Eng-iKO ECs.
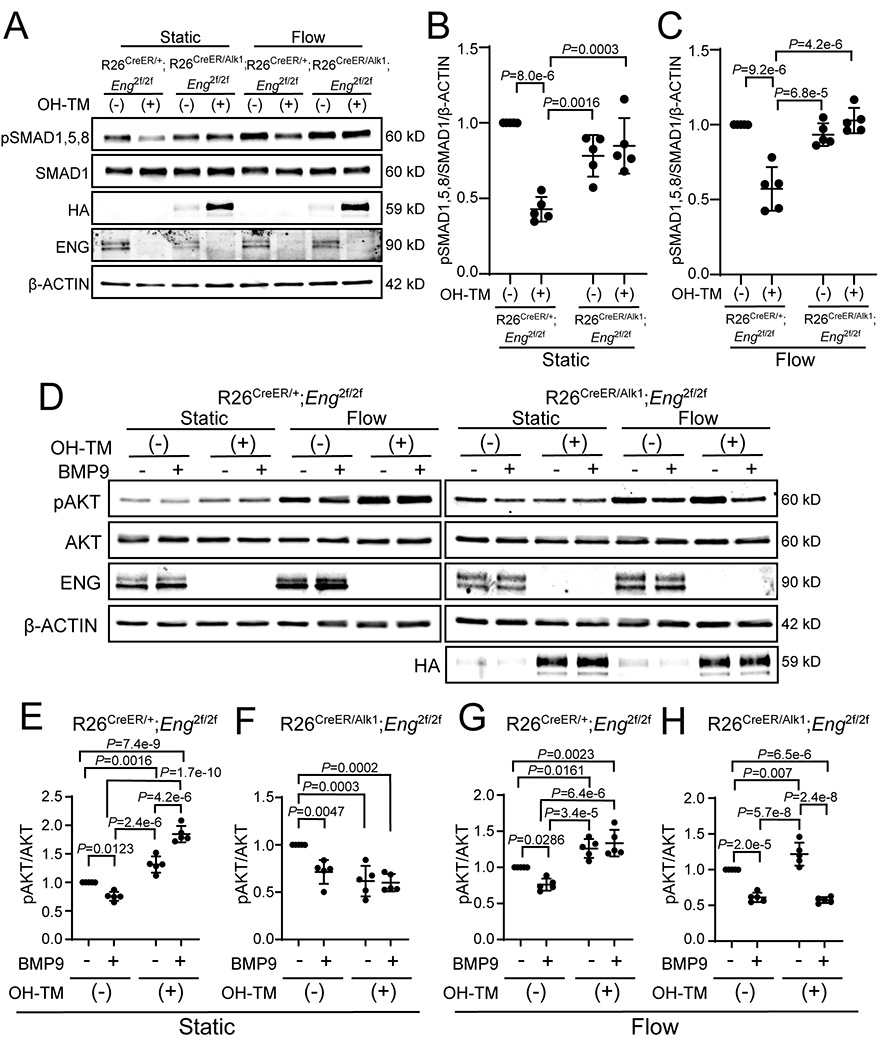
A, Western blot analyses of phosphorylated SMAD1,5,8 (pSMAD1,5,8), ENG, and HA in pulmonary ECs from R26CreER;Eng-iKO or R26CreER;Eng-iKO;ALK1-OE. HA indicates transgenic ALK1 expression. One μM of 4-hydroxy-tamoxifen was treated to delete the Eng gene and to induce HA-ALK1 expression. BMP9 (0.5 ng/mL) was added to serum-starved pulmonary ECs for 45 min in the static or flow condition. B and C, Quantification of SMAD1,5,8 phosphorylation levels in static condition (B) and flow stimulation (C). pSMAD1,5,8 levels were normalized with total SMAD1 and β-ACTIN. n=5 per each group. Two-way ANOVA with Tukey’s correction. D, Western blot analyses of phosphorylated AKT (pAKT) and ENG in pulmonary ECs of R26CreER;Eng-iKO (left panel) or R26CreER;Eng-iKO;ALK1-OE (right panel). HA indicates ALK1 overexpression. One μM of OH-TM was used to delete the Eng gene. BMP9 (10 ng/mL) was treated for 90 min, followed by 30 min exposure to flow. E-H, Quantification of relative phosphor-AKT levels normalized with total AKT in static condition (E and F) and flow stimulation (G and H). n=5 per each group. Two-way ANOVA with Tukey’s correction. All data are means ± SDs.
hENG-OE fails to inhibit wound-induced skin AVMs and retinal AVMs caused by ALK1-deficiency.
As a complementary experiment, we tested whether overexpression of human ENG could rescue the AVM phenotypes caused by ALK1-deficiency. We used a transgenic mouse strain in which hENG expression could be induced by Cre recombinase.45 This hENG overexpression has previously been shown to rescue the retinal AVM phenotype of the Eng-iKO mouse.46 We first tested if hENG-OE could affect Alk1 or Eng transcript levels in EC-specific Alk1-iKO mice. The Alk1 transcript level in R26CreER/+;Alk1-iKO lungs was almost undetectable, and hENG-OE did not change the Alk1 transcript level (Online Figure XXI A). The Eng transcript level in R26CreER/+;Alk1-iKO mouse lungs was reduced to 40% of the control level, and hENG-OE did not affect it (Online Figure XXI B). Human ENG mRNA and protein were detected in R26CreER/+;Alk1-iKO;ENG-OE mouse lungs (Online Figure XXI C and Figure 8). To test the functionality of hENG, we examined the wound-induced skin AVMs in SclCreER;Eng-iKO;hENG-OE mice. Latex dye-perfused vascular images on the wound skin area showed a remarkable reduction of AVMs and latex dye-containing vascular density in SclCreER;Eng-iKO;hENG-OE mice compared to those in SclCreER;Eng-iKO mice (Figures 8 A–D), indicating that hENG can compensate for the loss of mouse ENG. We then tested if hENG can compensate for the loss of ALK1 on R26+/CreER;Alk1-iKO and SclCreER;Alk1-iKO backgrounds. As shown in Figure 8 E–I, hENG-OE could not inhibit the wound-induced AVM development caused by global or EC-specific Alk1 deletions. Consistently, hENG-OE could not inhibit retinal AVM development in Alk1-iKO mice (Figure 8 J–M). Reduced level of pSMAD 1/5/8 in retina vascular ECs of Alk1-iKO was not affected by ENG-OE (Figure 8 N–P).
Figure 8. ENG-OE does not overcome the vascular phenotype of Alk1-iKO mice.
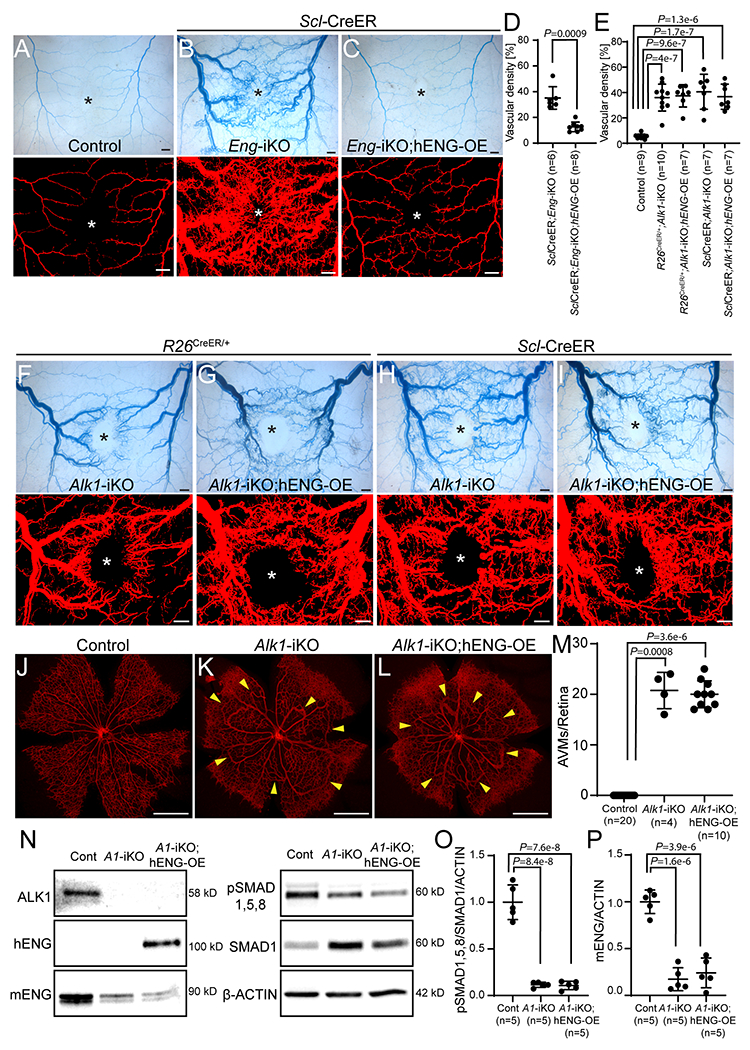
A-C, Latex perfusion images in control (A, n=9), SclCreER;Eng-iKO (B, n=6), and SclCreER;Eng-iKO;hENG-OE (C, n=8) on 8 days after wounding. D and E, Quantification of vascular density containing latex using the processed images. Welch t-test (D). One-way ANOVA with Tukey’s correction (E). F-I, Visualization of wound-induced skin AVMs using latex infusion in R26CreER;Alk1-iKO (F, n=10), R26CreER;Alk1-iKO;hENG-OE (G, n=7), SclCreER;Alk1-iKO (H, n=7) and SclCreER;Alk1-iKO;hENG-OE (I, n=7) mice. J-L, IB4 (red) staining of PN5 retinal vasculature from control (J, n=20), R26CreER;Alk1-iKO (K, n=4), and R26CreER;Alk1-iKO;hENG-OE (L, n=10) mice. Yellow arrowheads indicate AVMs in the retinal vasculature. M, Quantification of the number of AVMs per retina. Kruskal–Wallis test with Dunn’s correction. N, Western blot analyses in the effects of hENG-OE on the SMAD1,5,8 phosphorylation and endogenous mENG level. O and P, Quantification of pSMAD1,5,8 levels normalized to normalized with SMAD1 and β-ACTIN (O) and mouse ENG levels normalized with β-ACTIN (P). N=5 per each group. One-way ANOVA with Tukey’s correction. All data are means ± SDs. Scale bars: A-C and F-I, 1 mm; J-L, 500 μm.
DISCUSSION
In the HHT mouse models, we showed that mucocutaneous AVMs that form in the skin of adult mice requires two factors: deficiency of ALK1 or ENG, and a secondary insult represented by wounding.19, 24 With these results, we may infer two therapeutic axes for inhibiting de novo AVMs. One is to overcome the ALK1 or ENG-deficiency, and the other is to block a molecular pathway derived from the secondary insults, such as angiogenesis, inflammation, or oxidative stress. Bevacizumab47, 48 and pazopanib (both anti-angiogenic),49 thalidomide (anti-inflammatory),50 and N-acetyl cysteine (antioxidant)51 are effective in some cases of anemia and epistaxis in HHT patients. Parallel with the therapeutic approaches targeting the secondary insults, therapies targeting ENG/ALK1 signaling are emerging.
We report several novel findings. First, overexpression of ALK1 globally or in pan endothelial cells at normal physiological conditions did not cause vascular malformations. ALK1 signaling regulates Notch ligands and target genes, but ALK1-OE in normal ECs does not generate any changes in Notch target expression.35 Activation, as well as repression of Notch signaling, have been implicated in AVM development. While impaired Notch signaling caused AVMs52, endothelial-specific activation of Notch4 also induced AVMs in the brain and lungs36–38, 53, albeit by a reversible vasodilation mechanism.54 Deletion of Mgp, a BMP antagonist, caused AVM development in multiple organs partly by upregulating ALK1 expression.40 MGP depletion in ECs increased expression of Notch signaling components by an ALK1-dependent manner.39 Furthermore, reduction of Notch activities by heterozygous deletion of Jag1 or Jag2 suppressed AVM development in Mgp-null mice.39 Collectively, these findings raise the possibility that overexpression of ALK1 itself in the normal vasculature may elicit vascular malformation. They also raise a concern about the potential risks of ALK1 overexpression for HHT therapy. In our systemic yet limited analyses, we demonstrated that ALK1 overexpression in ECs (SclCreER), SMCs (Myh11CreER), or nonspecific cells (R26CreER) did not affect vitality, nor cause obvious vascular pathology. However, the safety of ALK1 overexpression in the long-term (longer than 3 months) and various stress conditions has yet to be determined. In this study, we used the wild-type ALK1, which may require ligands for activation. The results might have been different if we had used a constitutively active form of ALK1.
Second, overexpression of ALK1 could rescue phenotypes caused by ENG- as well as ALK1-deficiency. ALK1-OE could inhibit all phenotypes shown in Alk1-iKO mice, including retinal vascular expansion and AVMs in neonates and GI hemorrhages, anemia, and wound-induced skin AVMs in adult mice. This result not only demonstrates that the ALK1-OE system worked as designed but also that the Alk1-iKO phenotypes are actually due to ALK1-deficiency. ALK1-OE could also rescue the AVM phenotype caused by ENG-deficiency, indicating that ALK1-OE can compensate for the loss of ENG. In other words, while ENG is necessary for signaling in normal physiological conditions, it could be dispensable when ALK1 is overexpressed.
Third, overexpression of ENG could not rescue phenotypes caused by ALK1-deficiency. Since all three identified genes associated with HHT, i.e., ENG, ALK1, and SMAD4, are components of signal transduction of TGF-β/BMP family members, HHT has been considered a disease caused by defects in signaling mediated by ENG-ALK1. Because the cellular functions of ENG are shown to be broader than just mediating TGF-β/BMP signals,55 and ENG and ALK1 interact with multiple ligands, it has not been entirely clear whether HHT is caused by the defect of a linear or parallel signaling pathways of ENG and ALK1. Genotype-phenotype correlation studies have shown that HHT1 has a higher prevalence of AVMs in the brain and lungs while AVMs in HHT2 are found more frequently in the liver and GI tract.30–32 This miscorrelation also raised the possibility of a parallel pathway of ENG and ALK1. However, our data showed that ALK1-OE rescued ENG-deficiency. In contrast, ENG-OE could not rescue ALK1-deficiency, which supports the notion of the linear pathway of ENG and ALK1 where ALK1 is downstream of ENG. Human ENG was used for overexpression. The same system was previously used for rescuing the retinal AVM phenotype in Eng-iKO.46 We have demonstrated that hENG could prevent wound-induced AVM development in Eng-iKO, indicating that hENG can function similarly to mENG. However, it is possible that hENG may not function efficiently enough to overcome ALK1 deficiency as mENG would.
Since the mechanisms of AVM development by ALK1- or ENG-deficiency are still unclear, it is difficult to discern the mechanism by which ALK1-OE rescues the phenotypes caused by ENG-deficiency. One possibility could be that ALK1-OE normalizes altered expression of BMP targets, including NOTCH ligands or targets and angiopoietin 2. An alternative mechanism could be the normalization of elevated PI3K/AKT pathway responses in ALK1-deficient or ENG-deficient ECs. Elevated PI3K/AKT activities are shown to be closely associated with AVM development and progression.20, 23 We showed that ENG-deficiency blunted BMP9-mediated repression of AKT phosphorylation under flow conditions, similar to ALK1-deficiency.20, 23 Normalization of PI3k/AKT response may explain ALK1-OE rescue of the AVM phenotype, but further investigation is required to explore other contributory mechanisms, including abnormal migration behavior of ALK1-deficient cells in response to flow46, 56 and the enlargement of cell size.29 All these abnormal responses in ALK1-deficient ECs in vivo occur in the context of developmental angiogenesis, tissue wounds, or other similar pathology. VEGF plays a significant role in driving vascular growth and remodeling under these circumstances and activates the PI3K/AKT pathway. Therefore, targeting VEGF, by VEGF-neutralizing antibody treatment, could reduce established skin AVMs.25 In addition, treatments with a PI3K inhibitor or an angiopoietin 2 antibody neutralizer reversed retinal AVMs in Alk1-iKO and Smad4-iKO models, respectively.23, 57
The overexpression system in our study has a major limitation. It did not allow for testing of whether or not overexpression of ALK1 could reverse preestablished AVMs. Our results suggest that viral delivery of wild-type ALK1 could hold therapeutic promise for HHT in humans for the prevention of forming de novo AVMs in adults, such as telangiectases in the nasal mucosa, skin, and GI tract. However, considering the cell-autonomous nature of ALK1 function and the resultant requirement to rescue a vast majority, if not all ALK1 or ENG-deficient ECs, gene delivery of ALK1 to all ECs would be impractical. Drugs that can increase the expression of ALK1 or enhance ALK1 signaling could be more feasible approaches. Recently several drugs have shown to be useful for mitigating some HHT symptoms in small scale clinical studies. Among these, some drugs, including raloxifene, bazedoxifene, FK506, atorvastatin, and tranexamic acid, could stimulate the expression of ENG or ALK1 or both.58–63 Although it remains questionable whether or not the transcriptional stimulation of ALK1 expression is the mechanism of action of these drugs, our results support such a postulation. Recently, somatic mutations of the wild-type copy of the causal ENG or ALK1 germline mutation have been identified from telangiectases of HHT patients, suggesting that a loss of heterozygosity is a necessary event for AVM genesis in HHT,64 and is consistent with evidence from preclinical genetic studies.65 If this is a general mechanism of HHT pathogenesis, the therapeutic strategy of transcriptional stimulation of ALK1 expression would be more suited for HHT1 than HHT2.
Our findings establish the importance of the linear ENG/ALK1 signaling pathway in protecting the vasculature against AVM formation during development and tissue repair. Further investigation of the factors involved in regulating the formation and stability of AVMs is critical for the development of therapeutics for AVM regression and protection against life-threatening AVM rupture.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Hereditary hemorrhagic telangiectasia (HHT) is a genetic vascular disease caused by mutations in components of signal transduction of TGF-β family members, namely ENG, ALK1, and SMAD4.
The underlying pathogenesis of HHT is arteriovenous malformations (AVMs), abnormal shunts between arteries and veins without intervening capillaries. However, the detailed molecular mechanism of this disease is not clearly understood.
Management and surgical options are available for clinical symptoms of HHT, but curative therapies are currently unavailable.
What New Information Does This Article Contribute?
ALK1 overexpression in normal mice does not induce vascular malformation.
ALK1 overexpression can prevent AVM formation in ENG-deficient mice, whereas ENG overexpression fails to inhibit AVM formation in ALK1-deficient mice.
ALK1 overexpression or activation could be a potential therapeutic strategy for HHT.
Hereditary hemorrhagic telangiectasia (HHT) is a genetic disease caused by mutations in ENG, ALK1, or SMAD4 that encode components of signal transduction of TGF-β family members. Despite the strong implication of a linear pathway of ENG-ALK1-SMAD4 in HHT pathogenesis, it has been suspected that parallel pathways of ENG and ALK1 might be associated with this malady because of the promiscuity of these receptors and miscorrelations of HHT genotype-phenotype. In addition, there is speculation that ALK1 overexpression itself could cause vascular malformation by upregulating NOTCH signaling genes. We generated a novel transgenic mouse line in which ALK1 expression can be induced. We could not detect any signs of vascular malformation in the ALK1-OE mice up to 3 months after induction of ALK1 overexpression in endothelial cells. We found that ALK1 overexpression could suppress the development of AVMs in wounded adult skin and developing retinas of ENG-deficient mice, whereas ENG overexpression could not inhibit the AVM development in ALK1-deficient mice. These in vivo results support the notion of a linear pathway of ENG and ALK1 in which ALK1 is downstream of ENG in HHT pathogenesis. The study findings show that ALK1 overexpression or activation appears to be safe; thus, it could be developed as a potential therapeutic strategy for HHT.
ACKNOWLEDGMENT
We thank Lynda Orescanin of Neuroscience Publications at Barrow Neurological Institute for editorial assistance.
SOURCES OF FUNDING
This work was supported by the US Department of Defense (PR161205), the US National Institutes of Health (R01 HL128525), the Leducq Foundation (ATTRACT), and Barrow Neurological Foundation to S.P.O; and the Korea Mouse Phenotyping Project (NRF-2014M3A9D5A01073528) of the Ministry of Science and ICT through the National Research Foundation (NRF) to Y.J.L., in part by an NRF grant funded by the Ministry of Science and ICT (NRF-2017R1A2B4003322) to Y.J.L., by the US National Institutes of Health (R01 HL139887) to C.P.H.V., and by the British Heart Foundation (PG/14/86/31177) to H.M.A.
Nonstandard Abbreviations and Acronyms:
- AVM
arteriovenous malformation
- BMP
bone morphogenetic protein
- ECs
endothelial cells
- GFP
green fluorescence protein
- GI
gastrointestinal
- HA
human influenza hemagglutinin
- HHT
hereditary hemorrhagic telangiectasia
- iKO
inducible knockout
- OE
overexpression
- TGF-β
transforming growth factor beta
Footnotes
DISCLOSURES
Nothing to disclose.
REFERENCES
- 1.Marchuk DA, Srinivasan S, Squire TL and Zawistowski JS. Vascular morphogenesis: tales of two syndromes. Human molecular genetics. 2003;12 Spec No 1:R97–112. [DOI] [PubMed] [Google Scholar]
- 2.Govani FS and Shovlin CL. Hereditary haemorrhagic telangiectasia: a clinical and scientific review. Eur J Hum Genet. 2009;17:860–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guttmacher AE, Marchuk DA and White RI, Jr. Hereditary hemorrhagic telangiectasia. N Engl J Med. 1995;333:918–24. [DOI] [PubMed] [Google Scholar]
- 4.Shovlin CL. Hereditary haemorrhagic telangiectasia: pathophysiology, diagnosis and treatment. Blood Rev. 2010;24:203–19. [DOI] [PubMed] [Google Scholar]
- 5.Gallione CJ, Repetto GM, Legius E, Rustgi AK, Schelley SL, Tejpar S, Mitchell G, Drouin E, Westermann CJ and Marchuk DA. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4). Lancet. 2004;363:852–9. [DOI] [PubMed] [Google Scholar]
- 6.Gallione C, Aylsworth AS, Beis J, Berk T, Bernhardt B, Clark RD, Clericuzio C, Danesino C, Drautz J, Fahl J, Fan Z, Faughnan ME, Ganguly A, Garvie J, Henderson K, Kini U, Leedom T, Ludman M, Lux A, Maisenbacher M, Mazzucco S, Olivieri C, Ploos van Amstel JK, Prigoda-Lee N, Pyeritz RE, Reardon W, Vandezande K, Waldman JD, White RI Jr., Williams CA and Marchuk DA. Overlapping spectra of SMAD4 mutations in juvenile polyposis (JP) and JP-HHT syndrome. Am J Med Genet A. 2010;152A:333–9. [DOI] [PubMed] [Google Scholar]
- 7.Johnson DW, Berg JN, Baldwin MA, Gallione CJ, Marondel I, Yoon SJ, Stenzel TT, Speer M, Pericak-Vance MA, Diamond A, Guttmacher AE, Jackson CE, Attisano L, Kucherlapati R, Porteous ME and Marchuk DA. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nature genetics. 1996;13:189–95. [DOI] [PubMed] [Google Scholar]
- 8.McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, Helmbold EA, Markel DS, McKinnon WC, Murrell J and et al. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nature genetics. 1994;8:345–51. [DOI] [PubMed] [Google Scholar]
- 9.Tillet E and Bailly S. Emerging roles of BMP9 and BMP10 in hereditary hemorrhagic telangiectasia. Front Genet. 2014;5:456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laux DW, Young S, Donovan JP, Mansfield CJ, Upton PD and Roman BL. Circulating Bmp10 acts through endothelial Alk1 to mediate flow-dependent arterial quiescence. Development. 2013;140:3403–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saito T, Bokhove M, Croci R, Zamora-Caballero S, Han L, Letarte M, de Sanctis D and Jovine L. Structural Basis of the Human Endoglin-BMP9 Interaction: Insights into BMP Signaling and HHT1. Cell Rep. 2017;19:1917–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lawera A, Tong Z, Thorikay M, Redgrave RE, Cai J, van Dinther M, Morrell NW, Afink GB, Charnock-Jones DS, Arthur HM, Ten Dijke P and Li W. Role of soluble endoglin in BMP9 signaling. Proc Natl Acad Sci U S A. 2019;116:17800–17808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Townson SA, Martinez-Hackert E, Greppi C, Lowden P, Sako D, Liu J, Ucran JA, Liharska K, Underwood KW, Seehra J, Kumar R and Grinberg AV. Specificity and structure of a high affinity activin receptor-like kinase 1 (ALK1) signaling complex. J Biol Chem. 2012;287:27313–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oh SP, Seki T, Goss KA, Imamura T, Yi Y, Donahoe PK, Li L, Miyazono K, ten Dijke P, Kim S and Li E. Activin receptor-like kinase 1 modulates transforming growth factor-beta 1 signaling in the regulation of angiogenesis. Proc Natl Acad Sci U S A. 2000;97:2626–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Urness LD, Sorensen LK and Li DY. Arteriovenous malformations in mice lacking activin receptor-like kinase-1. Nature genetics. 2000;26:328–31. [DOI] [PubMed] [Google Scholar]
- 16.Srinivasan S, Hanes MA, Dickens T, Porteous ME, Oh SP, Hale LP and Marchuk DA. A mouse model for hereditary hemorrhagic telangiectasia (HHT) type 2. Human molecular genetics. 2003;12:473–82. [DOI] [PubMed] [Google Scholar]
- 17.Bourdeau A, Dumont DJ and Letarte M. A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest. 1999;104:1343–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Torsney E, Charlton R, Diamond AG, Burn J, Soames JV and Arthur HM. Mouse model for hereditary hemorrhagic telangiectasia has a generalized vascular abnormality. Circulation. 2003;107:1653–7. [DOI] [PubMed] [Google Scholar]
- 19.Garrido-Martin EM, Nguyen HL, Cunningham TA, Choe SW, Jiang Z, Arthur HM, Lee YJ and Oh SP. Common and distinctive pathogenetic features of arteriovenous malformations in hereditary hemorrhagic telangiectasia 1 and hereditary hemorrhagic telangiectasia 2 animal models--brief report. Arterioscler Thromb Vasc Biol. 2014;34:2232–6. [DOI] [PubMed] [Google Scholar]
- 20.Ola R, Kunzel SH, Zhang F, Genet G, Chakraborty R, Pibouin-Fragner L, Martin K, Sessa W, Dubrac A and Eichmann A. SMAD4 Prevents Flow Induced Arterial-Venous Malformations by Inhibiting Casein Kinase 2. Circulation. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tual-Chalot S, Mahmoud M, Allinson KR, Redgrave RE, Zhai Z, Oh SP, Fruttiger M and Arthur HM. Endothelial depletion of Acvrl1 in mice leads to arteriovenous malformations associated with reduced endoglin expression. PLoS One. 2014;9:e98646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mahmoud M, Allinson KR, Zhai Z, Oakenfull R, Ghandi P, Adams RH, Fruttiger M and Arthur HM. Pathogenesis of arteriovenous malformations in the absence of endoglin. Circ Res. 2010;106:1425–33. [DOI] [PubMed] [Google Scholar]
- 23.Ola R, Dubrac A, Han J, Zhang F, Fang JS, Larrivee B, Lee M, Urarte AA, Kraehling JR, Genet G, Hirschi KK, Sessa WC, Canals FV, Graupera M, Yan M, Young LH, Oh PS and Eichmann A. PI3 kinase inhibition improves vascular malformations in mouse models of hereditary haemorrhagic telangiectasia. Nat Commun. 2016;7:13650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park SO, Wankhede M, Lee YJ, Choi EJ, Fliess N, Choe SW, Oh SH, Walter G, Raizada MK, Sorg BS and Oh SP. Real-time imaging of de novo arteriovenous malformation in a mouse model of hereditary hemorrhagic telangiectasia. J Clin Invest. 2009;119:3487–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han C, Choe SW, Kim YH, Acharya AP, Keselowsky BG, Sorg BS, Lee YJ and Oh SP. VEGF neutralization can prevent and normalize arteriovenous malformations in an animal model for hereditary hemorrhagic telangiectasia 2. Angiogenesis. 2014;17:823–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi EJ, Chen W, Jun K, Arthur HM, Young WL and Su H. Novel brain arteriovenous malformation mouse models for type 1 hereditary hemorrhagic telangiectasia. PLoS One. 2014;9:e88511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walker EJ, Su H, Shen F, Choi EJ, Oh SP, Chen G, Lawton MT, Kim H, Chen Y, Chen W and Young WL. Arteriovenous malformation in the adult mouse brain resembling the human disease. Ann Neurol. 2011;69:954–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Corti P, Young S, Chen CY, Patrick MJ, Rochon ER, Pekkan K and Roman BL. Interaction between alk1 and blood flow in the development of arteriovenous malformations. Development. 2011;138:1573–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sugden WW, Meissner R, Aegerter-Wilmsen T, Tsaryk R, Leonard EV, Bussmann J, Hamm MJ, Herzog W, Jin Y, Jakobsson L, Denz C and Siekmann AF. Endoglin controls blood vessel diameter through endothelial cell shape changes in response to haemodynamic cues. Nat Cell Biol. 2017;19:653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bayrak-Toydemir P, McDonald J, Markewitz B, Lewin S, Miller F, Chou LS, Gedge F, Tang W, Coon H and Mao R. Genotype-phenotype correlation in hereditary hemorrhagic telangiectasia: mutations and manifestations. Am J Med Genet A. 2006;140:463–70. [DOI] [PubMed] [Google Scholar]
- 31.Canzonieri C, Centenara L, Ornati F, Pagella F, Matti E, Alvisi C, Danesino C, Perego M and Olivieri C. Endoscopic evaluation of gastrointestinal tract in patients with hereditary hemorrhagic telangiectasia and correlation with their genotypes. Genet Med. 2014;16:3–10. [DOI] [PubMed] [Google Scholar]
- 32.Karlsson T and Cherif H. Mutations in the ENG, ACVRL1, and SMAD4 genes and clinical manifestations of hereditary haemorrhagic telangiectasia: experience from the Center for Osler’s Disease, Uppsala University Hospital. Ups J Med Sci. 2018;123:153–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hong KH, Lee YJ, Lee E, Park SO, Han C, Beppu H, Li E, Raizada MK, Bloch KD and Oh SP. Genetic ablation of the BMPR2 gene in pulmonary endothelium is sufficient to predispose to pulmonary arterial hypertension. Circulation. 2008;118:722–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seki T, Yun J and Oh SP. Arterial endothelium-specific activin receptor-like kinase 1 expression suggests its role in arterialization and vascular remodeling. Circ Res. 2003;93:682–9. [DOI] [PubMed] [Google Scholar]
- 35.Larrivee B, Prahst C, Gordon E, del Toro R, Mathivet T, Duarte A, Simons M and Eichmann A. ALK1 signaling inhibits angiogenesis by cooperating with the Notch pathway. Dev Cell. 2012;22:489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carlson TR, Yan Y, Wu X, Lam MT, Tang GL, Beverly LJ, Messina LM, Capobianco AJ, Werb Z and Wang R. Endothelial expression of constitutively active Notch4 elicits reversible arteriovenous malformations in adult mice. Proc Natl Acad Sci U S A. 2005;102:9884–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murphy PA, Kim TN, Huang L, Nielsen CM, Lawton MT, Adams RH, Schaffer CB and Wang RA. Constitutively active Notch4 receptor elicits brain arteriovenous malformations through enlargement of capillary-like vessels. Proc Natl Acad Sci U S A. 2014;111:18007–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murphy PA, Lam MT, Wu X, Kim TN, Vartanian SM, Bollen AW, Carlson TR and Wang RA. Endothelial Notch4 signaling induces hallmarks of brain arteriovenous malformations in mice. Proc Natl Acad Sci U S A. 2008;105:10901–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yao Y, Yao J, Radparvar M, Blazquez-Medela AM, Guihard PJ, Jumabay M and Bostrom KI. Reducing Jagged 1 and 2 levels prevents cerebral arteriovenous malformations in matrix Gla protein deficiency. Proc Natl Acad Sci U S A. 2013;110:19071–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yao Y, Jumabay M, Wang A and Bostrom KI. Matrix Gla protein deficiency causes arteriovenous malformations in mice. J Clin Invest. 2011;121:2993–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gothert JR, Gustin SE, van Eekelen JA, Schmidt U, Hall MA, Jane SM, Green AR, Gottgens B, Izon DJ and Begley CG. Genetically tagging endothelial cells in vivo: bone marrow-derived cells do not contribute to tumor endothelium. Blood. 2004;104:1769–77. [DOI] [PubMed] [Google Scholar]
- 42.Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horvath B, Maser-Gluth C, Greiner E, Lemmer B, Schutz G, Gutkind JS and Offermanns S. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med. 2008;14:64–8. [DOI] [PubMed] [Google Scholar]
- 43.Kim YH, Kim MJ, Choe SW, Sprecher D, Lee YJ and S PO. Selective effects of oral antiangiogenic tyrosine kinase inhibitors on an animal model of hereditary hemorrhagic telangiectasia. J Thromb Haemost. 2017;15:1095–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alsina-Sanchis E, Garcia-Ibanez Y, Figueiredo AM, Riera-Domingo C, Figueras A, Matias-Guiu X, Casanovas O, Botella LM, Pujana MA, Riera-Mestre A, Graupera M and Vinals F. ALK1 Loss Results in Vascular Hyperplasia in Mice and Humans Through PI3K Activation. Arterioscler Thromb Vasc Biol. 2018;38:1216–1229. [DOI] [PubMed] [Google Scholar]
- 45.Mancini ML, Verdi JM, Conley BA, Nicola T, Spicer DB, Oxburgh LH and Vary CP. Endoglin is required for myogenic differentiation potential of neural crest stem cells. Dev Biol. 2007;308:520–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jin Y, Muhl L, Burmakin M, Wang Y, Duchez AC, Betsholtz C, Arthur HM and Jakobsson L. Endoglin prevents vascular malformation by regulating flow-induced cell migration and specification through VEGFR2 signalling. Nat Cell Biol. 2017;19:639–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davidson TM, Olitsky SE and Wei JL. Hereditary hemorrhagic telangiectasia/avastin. Laryngoscope. 2010;120:432–5. [DOI] [PubMed] [Google Scholar]
- 48.Guilhem A, Fargeton AE, Simon AC, Duffau P, Harle JR, Lavigne C, Carette MF, Bletry O, Kaminsky P, Leguy V, Lerolle N, Roux D, Lambert M, Chinet T, Bonnet D, Dupuis-Girod S and Riviere S. Intra-venous bevacizumab in hereditary hemorrhagic telangiectasia (HHT): A retrospective study of 46 patients. PLoS One. 2017;12:e0188943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Faughnan ME, Gossage JR, Chakinala MM, Oh SP, Kasthuri R, Hughes CCW, McWilliams JP, Parambil JG, Vozoris N, Donaldson J, Paul G, Berry P and Sprecher DL. Pazopanib may reduce bleeding in hereditary hemorrhagic telangiectasia. Angiogenesis. 2019;22:145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lebrin F, Srun S, Raymond K, Martin S, van den Brink S, Freitas C, Breant C, Mathivet T, Larrivee B, Thomas JL, Arthur HM, Westermann CJ, Disch F, Mager JJ, Snijder RJ, Eichmann A and Mummery CL. Thalidomide stimulates vessel maturation and reduces epistaxis in individuals with hereditary hemorrhagic telangiectasia. Nat Med. 2010;16:420–8. [DOI] [PubMed] [Google Scholar]
- 51.de Gussem EM, Snijder RJ, Disch FJ, Zanen P, Westermann CJ and Mager JJ. The effect of N-acetylcysteine on epistaxis and quality of life in patients with HHT: a pilot study. Rhinology. 2009;47:85–8. [PubMed] [Google Scholar]
- 52.Nielsen CM, Cuervo H, Ding VW, Kong Y, Huang EJ and Wang RA. Deletion of Rbpj from postnatal endothelium leads to abnormal arteriovenous shunting in mice. Development. 2014;141:3782–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Miniati D, Jelin EB, Ng J, Wu J, Carlson TR, Wu X, Looney MR and Wang RA. Constitutively active endothelial Notch4 causes lung arteriovenous shunts in mice. Am J Physiol Lung Cell Mol Physiol. 2010;298:L169–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Murphy PA, Kim TN, Lu G, Bollen AW, Schaffer CB and Wang RA. Notch4 normalization reduces blood vessel size in arteriovenous malformations. Sci Transl Med. 2012;4:117ra8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rossi E, Bernabeu C and Smadja DM. Endoglin as an Adhesion Molecule in Mature and Progenitor Endothelial Cells: A Function Beyond TGF-beta. Front Med (Lausanne). 2019;6:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rochon ER, Menon PG and Roman BL. Alk1 controls arterial endothelial cell migration in lumenized vessels. Development. 2016;143:2593–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Crist AM, Zhou X, Garai J, Lee AR, Thoele J, Ullmer C, Klein C, Zabaleta J and Meadows SM. Angiopoietin-2 Inhibition Rescues Arteriovenous Malformation in a Smad4 Hereditary Hemorrhagic Telangiectasia Mouse Model. Circulation. 2019;139:2049–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Albinana V, Bernabeu-Herrero ME, Zarrabeitia R, Bernabeu C and Botella LM. Estrogen therapy for hereditary haemorrhagic telangiectasia (HHT): Effects of raloxifene, on Endoglin and ALK1 expression in endothelial cells. Thromb Haemost. 2010;103:525–34. [DOI] [PubMed] [Google Scholar]
- 59.Albinana V, Sanz-Rodriguez F, Recio-Poveda L, Bernabeu C and Botella LM. Immunosuppressor FK506 increases endoglin and activin receptor-like kinase 1 expression and modulates transforming growth factor-beta1 signaling in endothelial cells. Mol Pharmacol. 2011;79:833–43. [DOI] [PubMed] [Google Scholar]
- 60.Zarrabeitia R, Ojeda-Fernandez L, Recio L, Bernabeu C, Parra JA, Albinana V and Botella LM. Bazedoxifene, a new orphan drug for the treatment of bleeding in hereditary haemorrhagic telangiectasia. Thromb Haemost. 2016;115:1167–77. [DOI] [PubMed] [Google Scholar]
- 61.Zemankova L, Varejckova M, Dolezalova E, Fikrova P, Jezkova K, Rathouska J, Cerveny L, Botella LM, Bernabeu C, Nemeckova I and Nachtigal P. Atorvastatin-induced endothelial nitric oxide synthase expression in endothelial cells is mediated by endoglin. J Physiol Pharmacol. 2015;66:403–13. [PubMed] [Google Scholar]
- 62.Fernandez LA, Garrido-Martin EM, Sanz-Rodriguez F, Ramirez JR, Morales-Angulo C, Zarrabeitia R, Perez-Molino A, Bernabeu C and Botella LM. Therapeutic action of tranexamic acid in hereditary haemorrhagic telangiectasia (HHT): regulation of ALK-1/endoglin pathway in endothelial cells. Thromb Haemost. 2007;97:254–62. [PubMed] [Google Scholar]
- 63.Ruiz-Llorente L, Gallardo-Vara E, Rossi E, Smadja DM, Botella LM and Bernabeu C. Endoglin and alk1 as therapeutic targets for hereditary hemorrhagic telangiectasia. Expert Opin Ther Targets. 2017;21:933–947. [DOI] [PubMed] [Google Scholar]
- 64.Snellings DA, Gallione CJ, Clark DS, Vozoris NT, Faughnan ME and Marchuk DA. Somatic Mutations in Vascular Malformations of Hereditary Hemorrhagic Telangiectasia Result in Bi-allelic Loss of ENG or ACVRL1. Am J Hum Genet. 2019;105:894–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tual-Chalot S, Oh SP and Arthur HM. Mouse models of hereditary hemorrhagic telangiectasia: recent advances and future challenges. Front Genet. 2015;6:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Park SO, Lee YJ, Seki T, Hong KH, Fliess N, Jiang Z, Park A, Wu X, Kaartinen V, Roman BL and Oh SP. ALK5- and TGFBR2-independent role of ALK1 in the pathogenesis of hereditary hemorrhagic telangiectasia type 2. Blood. 2008;111:633–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nguyen HL, Lee YJ, Shin J, Lee E, Park SO, McCarty JH and Oh SP. TGF-beta signaling in endothelial cells, but not neuroepithelial cells, is essential for cerebral vascular development. Lab Invest. 2011;91:1554–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Badea TC, Wang Y and Nathans J. A noninvasive genetic/pharmacologic strategy for visualizing cell morphology and clonal relationships in the mouse. J Neurosci. 2003;23:2314–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim YH, Choe SW, Chae MY, Hong S and Oh SP. SMAD4 Deficiency Leads to Development of Arteriovenous Malformations in Neonatal and Adult Mice. J Am Heart Assoc. 2018;7:e009514. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.