

Abstract
Iron is a key element for normal cellular function and plays a role in many cellular processes including mitochondrial respiration. The role of iron deficiency (ID) in heart failure (HF) has been a subject of debate amid increasing advocacy for intravenous (IV) supplementation. Both the definition and the approach to treatment of ID in HF have been adapted from the experience in patients with chronic kidney disease (CKD). In this review, we highlight the differences in regulatory mechanisms as well as pathophysiology of ID in CKD and HF population both at the systemic and cellular levels. We will review the major clinical trials in HF patients that have shown symptomatic benefit from IV iron supplementation but without effect on clinical outcomes. Intravenous iron loading bypasses the mechanisms that tightly regulate iron uptake and can potentially cause myocardial and endothelial damage by releasing reactive oxygen species. By contrast, newer oral iron preparations do not have similar toxicity concerns and might have a role in heart failure.
Keywords: Iron deficiency, Intravenous iron, Heart failure, Chronic kidney disease
1. Iron homeostasis & regulation
Iron is an essential micronutrient in humans. It is abundantly present in the environment, albeit in the oxidized ferric form (Fe3+) that has limited bioavailability. Despite dietary variation, the homeostatic mechanisms, finessed by evolution maintain a narrow range of iron level in our cells to limit toxicity and provide a steady supply. Two-thirds of the iron in the body is found in hemoglobin, lending oxygen carrying capacity to RBCs, and another 25% is stored in hepatocytes or macrophages, bound to proteins like ferritin and hemosiderin. Iron is also found in myoglobin, intracellular enzymes and the mitochondria of human cells. A very small fraction of the total body iron is found extracellularly (0.1%) and this fraction mostly remains bound to transferrin (Tf). Regulation of iron occurs at the sites of iron absorption (intestine) and iron recycling (RBCs, macrophages).1 There is no known mechanism to regulate iron excretion from the body and the only pathways to remove excess iron are either bleeding or phlebotomy.
Human stores of iron range from 3 to 4 grams. On average, the daily human diet contains ~10–20 mg of iron.2 Of this, about 1–2 mg is absorbed via the intestinal epithelial cells of the duodenum and proximal jejunum. A similar amount is lost every day through obligatory losses such as shedding of the lining epithelial cells. Daily demand of about 20–25 mg of iron is mostly met via recycling of older RBCs by macrophages. When the demand for iron is increased, such as during GI bleeding, iron absorption can increase by up to thirty percent.
1.1. Iron absorption and transport
Dietary iron exists in two forms: heme iron (from animal sources) and the predominant form, non-heme iron (from plant-based sources). Non-heme dietary iron exists mostly in the ferric state, which is non-bioavailable and should be reduced for absorption. Reduction of the dietary iron is mediated by the brush-border enzyme ferrireductase or other reducing agents in our diet, such as ascorbic acid or certain amino acids (cysteine, histidine). The acidic environment of the stomach is important for the action of dietary reducing agents and hence, acid-blockers such as PPIs can inhibit iron absorption. The reduced form of iron (Fe2+) enters duodenal enterocytes through a divalent metal transporter 1 (DMT 1), located on the apical membrane. Subsequently, the export of iron out of enterocytes occurs via a transmembrane protein called ferroportin-1 (Fpn1), which is located on the basal membrane of enterocytes. Of note, Fpn1 is the only known iron export protein in human cells.
The main regulator of iron absorption is a liver-derived hormone called hepcidin, which is transcribed from a gene called hepcidin antimicrobial peptide (HAMP). HAMP transcription increases in states of serum iron excess, resulting in high hepcidin levels. Hepcidin binds to and induces degradation of Fpn1, thereby indirectly reducing absorption of iron in enterocytes and similarly reducing export of iron from iron-laden macrophages. This limits the availability of iron to erythroid precursors and protects from harmful effects of iron excess in circulation (Fig. 1).
Figure 1. Iron absorption and regulation.
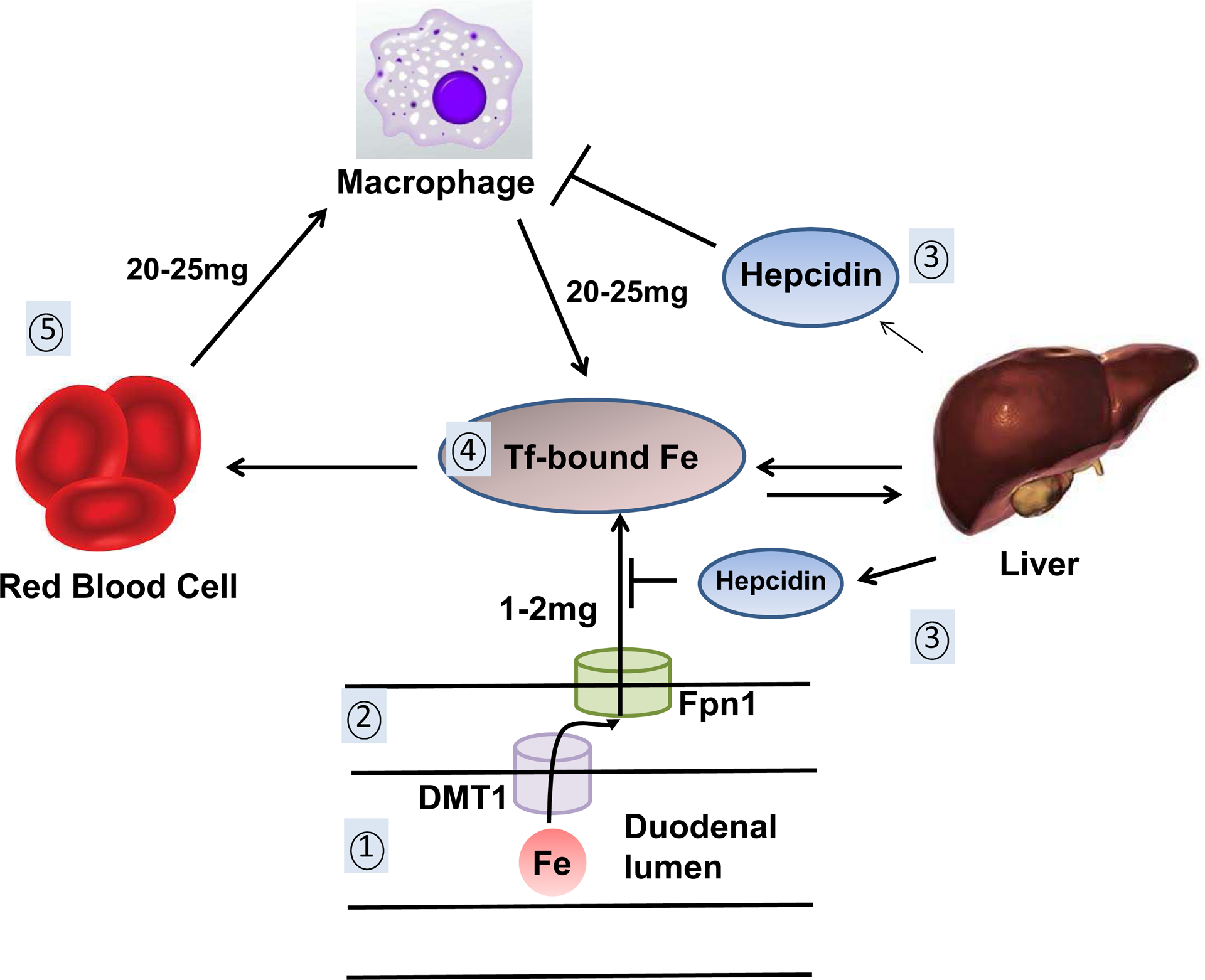
1. Iron (Fe) is absorbed from the duodenal lumen and transported via the apically located divalent-metal transporter 1 (DMT-1) into the enterocytes. 2. Ferroportin 1 (Fpn1), located on the basal membrane of the enterocytes, transports iron out of the enterocytes. 3. Hepcidin, released by the liver, induces the degradation of Fpn1 if iron levels are in excess, thereby limiting iron transport out of enterocytes and macrophages. 4. In circulation, iron is bound to transferrin and is taken up by RBCs, liver and other tissues. 5. Breakdown of senescent RBCs releases about 20–25mg of iron daily that is rapidly taken up by scavenging macrophages and made available to the circulating iron pool.
Circulating iron is largely bound to the transport protein Tf, which delivers iron to tissues in a redox-inert state. Transferrin-receptor 1 (TfR1) facilitates the uptake of transferrin-bound iron (TBI) into the cell. The bound complex of holotransferrin (i.e. iron-bound Tf) and TfR1 is internalized via DMT-1 lined clathrin-coated pits. Upon endocytosis, a proton pump within the endosome turns the pH acidic, which leads to iron release from the holotransferrin-TfR1 complex. Iron is then transported out of the endosome into the cytoplasm via the DMT-1 transporter. Finally, the apotransferrin-TfR1 complex is returned to the cell surface and dissociates at a neutral pH outside of the cell, releasing apotransferrin (i.e. iron-devoid Tf) into circulation for further iron loading.3
A soluble form of the transferrin receptor (sTfR) is detectable in the serum. Since most of the TfR1 expression occurs in erythropoietic cells, an increase in levels of sTfR correlates with increased marrow activity. Additionally, expression of TfR1 increases in states of reduced iron supply, allowing more uptake of iron by cells. Therefore, serum sTfR level increases in states of increased erythropoiesis and iron deficiency.4, 5
1.2. Role of hepcidin in health and disease
Hepcidin levels decline rapidly in iron deficiency (ID) and increase even after a single dose of oral iron, suggesting exquisite sensitivity to serum iron levels.6 Iron is part of the immune defense mechanism for several species. After infection, the host’s immune system switches to a hypoferremic state through an increase in hepcidin level,1 resulting in increased scavenging of iron and higher iron storage in macrophages. This low iron state can limit the growth of foreign microorganisms and protect against infection. In chronic inflammatory states, increased hepcidin level induces a lockdown on iron release and effectively traps iron in macrophages, hepatocytes and enterocytes, resulting in an “inflammatory blockade” of iron or a state of “iron restriction”. When this is associated with anemia, the condition is referred to as anemia of chronic disease (AOCD).
Hepcidin has a molecular weight of 2.7kDa and is renally cleared. In chronic kidney disease (CKD), the serum level of hepcidin increases, both due to reduced glomerular filtration rate (GFR) that decreases hepcidin clearance,7 and due to chronic inflammatory state of CKD. Clearance by hemodialysis is mild-to-intermediate, depending on the use of low-flux or high-flux dialyzers respectively, but dialysis-dependent (DD) CKD patients frequently have bloodstream infections or inflammation from dialysis membrane exposure and continue to have elevated level of hepcidin production.8 Therefore, iron restriction due to elevated levels of hepcidin is seen in both DD and non-DD CKD population. In contrast, studies have shown that systemic level of hepcidin is diminished in HF population9, highlighting the distinct pathophysiologic mechanism of ID in HF and CKD patients (Fig. 2).
Figure 2. Hepcidin levels in CKD and HF.
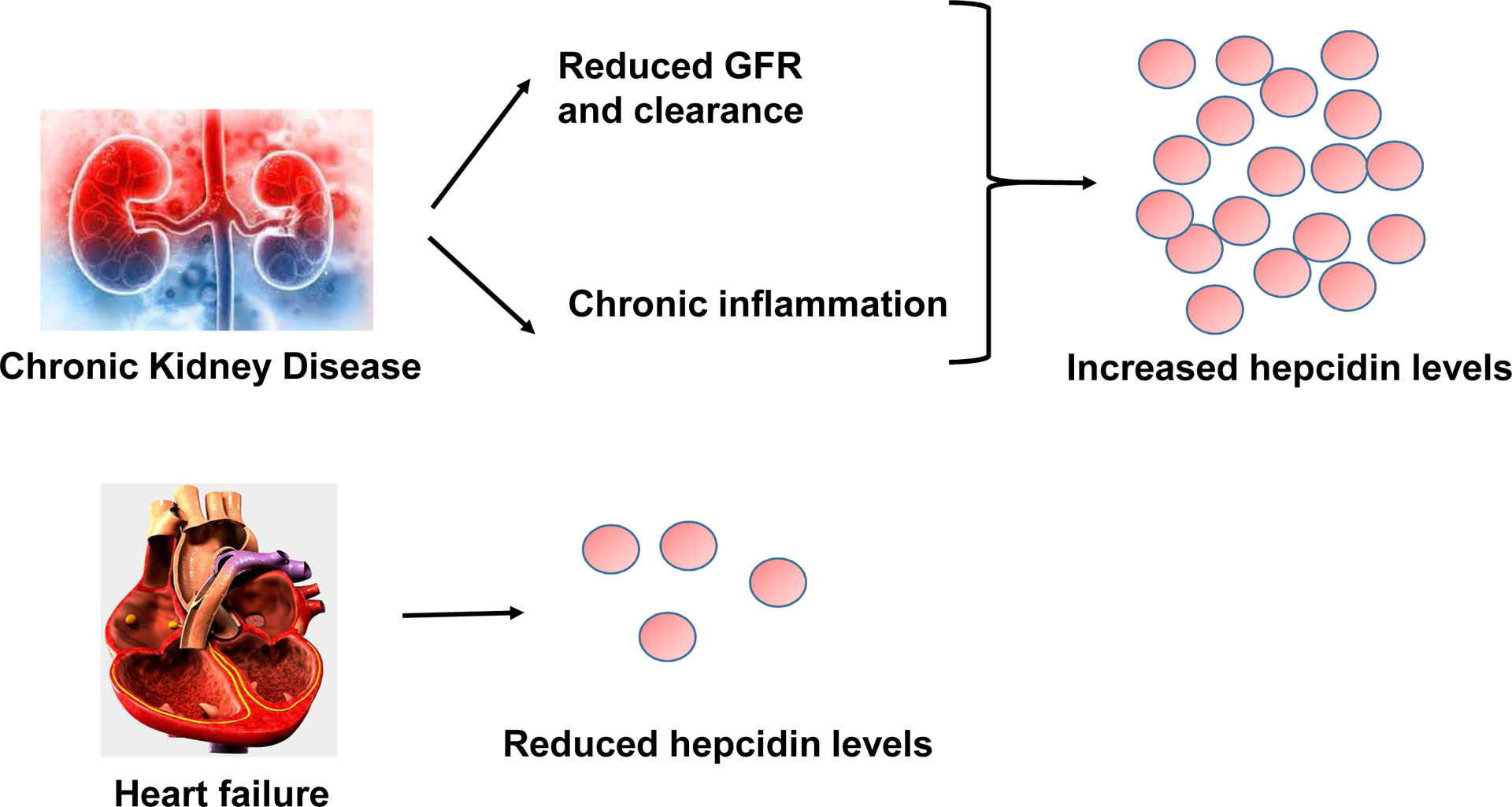
In CKD, the serum level of hepcidin is higher due to reduced renal clearance and chronic inflammation. In contrast, systemic level of hepcidin is lower in HF.
1.3. Iron homeostasis in the heart
Cardiac myocytes require adequate supply of iron for proper function of their iron-containing mitochondrial enzymes in producing energy for contractility.10 At the same time, they are uniquely vulnerable to iron overload conditions because there are several routes for iron entry into the cardiomyocytes (some of which are not regulated by homeostatic mechanisms), but only one pathway for iron export.
Tf-based entry of iron is the principal pathway of iron influx into cardiomyocytes. Tf-knockout mice develop severe cardiomyopathy and die within weeks with severe metabolic derangements and inability to release iron from the mitochondria.11 Other pathways for the entry of non-transferrin bound iron (NTBI) into the cardiomyocytes include DMT-1 transporters,12 calcium channels13 and zinc transporters.14 Among these pathways, only Tf and DMT-1 pathways are regulated by cellular homeostatic mechanisms.
In cardiomyocytes, iron is mainly stored in the form of ferritin, which protects against oxidative stress that can result from free forms of iron. Iron is also stored in heme, Fe/S clusters and mitochondrial ferritin or can remain as labile iron pool (LIP). The only known pathway for iron efflux from cardiomyocyte is via Fpn1.15 Hepcidin is also expressed in the heart and exerts local autocrine effects.16 As opposed to systemic hepcidin response, cardiac hepcidin rises in the setting of ID, a protective response to cellular iron deficiency. Additionally, cardiomyocyte-specific HAMP knockout mice develop severe cardiac dysfunction and cardiomyocyte ID without systemic ID.16
At the cellular level, iron homeostasis is maintained by iron regulatory proteins (IRP-1 & −2). These proteins sense iron levels in the cell and help regulate function of proteins involved in iron metabolism. In states of ID, IRPs stabilize the mRNA of TfR1, which increases iron import into cells. At the same time, IRPs binding to the mRNAs of ferritin and Fpn1 inhibit their translation, thus making iron more available inside the cell and decreasing iron export.
Another key regulatory pathway that causes iron conservation is the mammalian target of rapamycin (mTOR) pathway. mTOR is a protein kinase that becomes activated during conditions of metabolic stress and inactivated during starvation. Our group has shown that ID activates a tandem zinc finger protein called tristetraprolin (TTP) through mTOR. TTP binds to AU-rich regions in the 3’-UTR of mRNA molecules, resulting in their degradation.17 Among the mRNAs TTP targets, our group has shown that TTP binds to the 3’-UTR of TfR1, leading to its degradation and simultaneously fine-tunes iron import into cells during severe ID. This will ensure that iron is available for other vital organs in the setting of severe ID.17 Additionally, TTP targets mRNAs of certain proteins in the mitochondrial electron transport chain to ensure that reduced Fe/S cluster availability does not cause oxidative stress and cell death.18
2. Iron deficiency in chronic kidney disease and heart failure
2.1. Absolute & functional ID
ID could result from depletion of iron stores (absolute iron deficiency) or from insufficient iron uptake into erythroid precursors despite adequate iron stores (functional iron deficiency). This condition could exist in isolation or in concomitance with anemia. The gold standard for diagnosis of ID is bone marrow biopsy and iron staining19,20 however, this invasive test is seldom justified for routine clinical use. Therefore, diagnosis of ID in clinical practice commonly relies on surrogate serum markers such as ferritin, serum iron, transferrin saturation (Tsat) and iron-binding capacity.
In patients with CKD, a normocytic anemia is present due to several factors including chronic inflammation and decreased erythropoietin (EPO) production by the failing kidneys which decreases erythropoietic stimulation.21 As stated above, CKD patients also suffer from ID primarily due to inflammation22 and increased hepcidin.
With the goal of addressing anemia in CKD and stressing its impact on mortality, the KDIGO (Kidney Disease: Improving Global Outcomes) guideline recommends measurement of ferritin and Tsat as markers for assessing ID in the CKD population.23 Since uremia and inflammation raise ferritin level in CKD, the cutoff of ferritin to diagnose ID is higher in CKD patients compared to those with preserved renal function. Absolute ID in CKD patients has been defined as serum ferritin values <100ng/ml in non-DD CKD patients and <200ng/ml in DD-CKD patients with Tsat levels <20%. Functional ID is defined as Tsat levels <20% with normal/high ferritin values, which can frequently result from use of erythropoiesis-stimulating agents (ESAs). ESAs induce increased erythropoiesis, but often, iron availability to the erythroid precursors is not able to match the demand. Furthermore, in CKD patients, uremia leads to chronic inflammation and raises the level of hepcidin. Hepcidin, in turn, induces an inflammatory blockade on the iron export from enterocytes and reticuloendothelial cells and limits iron availability to erythroid lines.6–8
In HF population, potential mechanisms for absolute ID include dietary deficiency, reduced GI absorption due to gut edema or PPI use, and blood loss due to antiplatelets or anticoagulant use.24 The serologic definitions of absolute and functional ID in HF have been adapted from CKD population. However, the two disease processes are drastically distinct. For example, in contrast to CKD patients, hepcidin level is depressed in HF.9, 25
Several studies have suggested that iron homeostasis in cardiomyocytes is abnormal in HF. Melenovsky et al. demonstrated a reduction in myocardial iron content and abnormal mitochondrial function in the explanted hearts of advanced HF patients.26 This could be a result of a dysregulated cardiomyocyte iron homeostasis rather than true systemic ID. On the other hand, our group has shown that heme levels and mitochondrial iron levels are increased in advanced HF.27 We have also shown an increase in mitochondrial iron after ischemia/reperfusion injury in mice and similarly in ischemic heart disease in human hearts.28 The increased iron levels could have detrimental effects on the cardiomyocyte via reactive oxygen species (ROS) generation and oxidative stress injury. Further work is needed to better delineate whether changes in myocardial iron content in HF are adaptations to injury, and hence protective, or causative of injury and therefore, harmful.
2.2. Novel biomarkers to detect ID
Clinical methods to detect myocardial ID are quite limited. Serum ferritin and Tsat do not correlate with myocardial iron stores. In one study, Leszek et al. showed that among several markers, sTfR had the best correlation with depletion of myocardial iron.29 In a study of 165 acute HF patients, Jankowska et al. showed that a combination of high sTfR (reflecting unmet iron needs) and low serum hepcidin (reflecting depleted iron stores) predicted worse outcomes compared to other serum markers. In this study, based on the sTfR-hepcidin definition, only 37% of patients were ID as opposed to 65% based on the serum transferrin-ferritin definition. Furthermore, only 31% of the study population met both definitions of ID, suggesting that prevalence of ID in HF might be overestimated if this condition is defined based on serologic criteria that were used in kidney disease.30
Despite the enthusiasm about use of sTfR and hepcidin in diagnosis of ID, they are yet to be embraced commercially in clinical practice. Additionally, in CKD patients, the utility of sTfR is limited, as the test does not differentiate between increased erythropoietic activity, which can be seen with ESAs use, and systemic ID.
3. Iron supplementation in HF
In past decade, the notion of ID in HF has led to several studies about iron supplementation in HF, both in the oral and IV form. In the following sections, we highlight the key studies and their significance. Additionally, we contrast it with IV Iron supplementation trials in CKD, and highlight the risks of IV versus oral supplementation and the role of the latter in HF.
3.1. IV iron in HF
To date, there have been 3 major randomized controlled trials (RCTs) that have evaluated the role of IV iron supplementation in HF. There are additional RCTs assessing the role of IV Iron in HF that are currently in progress. All three trials involved patients with HF with reduced ejection fraction (HFrEF) using ID criteria of serum ferritin <100ng/ml, or serum ferritin 100–299ng/ml with Tsat <20%.
FAIR-HF was a double-blinded, placebo-controlled, multi-center design trial that enrolled 459 patients with ambulatory HFrEF, of NYHA Class II or III, with LVEF 40% or less (for Class II) or 45% or less (for Class III).31 All participants met criteria for ID as listed above, with hemoglobin ranging from 9.5–13.5g/dl. They were randomized in a 2:1 fashion to receive 200mg of weekly IV ferric carboxymaltose (FCM) or placebo (saline). Dosing was continued in a weekly manner until repletion was achieved (correction phase) and then monthly (maintenance phase). Primary endpoints were self-reported Patient Global Assessment (PGA) and NYHA Class at week 24. Fifty percent of the intervention group compared to 28% of the placebo group reported improvement in the PGA while the NYHA Class for the intervention group improved by 47% compared with 30% for the placebo arm. Secondary outcomes were the distance walked in 6 minutes (6MWT) and the health-related quality of life, both of which showed significant improvement. The treatment effects were similar for those with and without anemia. No mortality benefit or reduction in hospitalization was noted in the FCM arm and adverse events were similar. Of interest, subjects showed an improvement in the primary outcomes as early as 4 weeks although needing a median of 6 weeks for correction of parameters. The rapidness of subjective improvement and mechanism of such change is unclear. Furthermore, study subjects were predominantly white (99%), had ischemic cardiomyopathy (80%) with NYHA class III (82%), which might not be the demographic for all HF patients. While this trial helps focus on patient-centered outcomes, the follow up period was fairly short (at 24weeks) and long-term benefits of this therapy, including effects on LV remodeling, hospitalization and survival are not clear.
CONFIRM-HF was a multi-center, double-blinded, placebo-controlled RCT.32 It enrolled 304 ambulatory HF patients with ID and NYHA class II/III, LVEF ≤45% and elevated natriuretic peptides. Participants were randomized to FCM or placebo in a 1:1 fashion, stratified by Hb <12g/dl or >12g/dl. In the therapy phase, participants received a weight-based total of 500–2000mg of FCM at baseline and week 6. In the maintenance phase, 500mg of FCM was dosed at weeks 12, 24 and 36 if ID was still present. The primary endpoint was a change in the 6MWT distance from baseline to week 24. Secondary outcomes included NYHA class, PGA, fatigue score and health-related quality-of-life surveys at weeks 6, 12, 24, 36 and 52. At week 24, the 6MWT distance dropped in the placebo group and rose in the FCM group, rendering a significant change. Significant improvement was also noted in NYHA class, PGA, fatigue score and quality of life survey score. The demographics of the CONFIRM-HF trial are similar to FAIR-HF: predominantly white (99%) with ischemic cardiomyopathy (83%). In subgroup analyses, it was noted that the magnitude of treatment effects interacted significantly with impaired renal function. Notably, up to a third of the FCM group had anemia and a third had a GFR <60ml/min. Thus, the correction of anemia rather than iron replacement itself might explain the effect on the primary outcome, which should be considered when interpreting the results.
Lastly, the EFFECT-HF was an open-label, multi-center RCT that enrolled 174 stable, ambulatory HF patients (EF ≤45%) with mild-to-moderate symptoms (NYHA Class II-III) and ID using the aforementioned parameters, and randomized them in a 1:1 fashion to FCM vs standard of care (SoC, which could include oral iron) to assess change in exercise capacity.33 Participants received FCM in a 500–2000mg dose (weight & Hb based) at baseline and week 6, followed by another dose of 500mg FCM at week 12 if ID was still present. Primary outcome was the change in peak VO2 at week 24 from baseline. Secondary outcomes included self-reported NYHA class and PGA score. At week 24, peak VO2 in the intervention arm remained unchanged despite a 1g/dl increase in Hb. By comparison, the peak VO2 in the SoC arm dropped by 1.19 ± 0.38 mL/kg/min. Improvement in the NYHA class and PGA were also reported. There are a few points of note here that merit mention. In general, the controlled but open label design of this trial might subject it to bias, especially in self-reporting of outcomes like NYHA class and PGA score. Despite an increase in Hb by 1g/dl at week 24, it is surprising that the peak VO2 did not change in the FCM group since it is a result of change in Hb according to the Fick principle. There were 4 deaths in the SoC group and none in the FCM group. The authors assigned a value of zero to peak VO2 for these patients who died before week 24, which explains the drop in peak VO2 at 24 weeks noted in the SoC group. The authors note that without the imputation of these deaths, the peak VO2 change was not significant between the two groups (p=0.23).
It is noteworthy that all 3 RCTs mentioned above (FAIR-HF, CONFIRM-HF, EFFECT-HF) were sponsored by Vifor Pharma, the maker of FCM (Ferinject/ Injectafer). Additionally, the company was involved in the trial design, implementation and oversight in FAIR-HF and CONFIRM-HF trials, the two trials with the greater impact amongst the three.
4. Iron supplementation in renal failure
As previously mentioned, DD-CKD patients are at increased risk of ID from several factors. Monthly oral uptake of iron in dialysis patients can be as little as 50 mg due to limited absorption from elevated hepcidin levels. Concern for excessive use of ESAs and attendant cardiovascular risks thereof have prompted a call for more conservative dosing of ESAs and use of IV iron to mitigate the increased use. Moreover, ID in CKD might be exacerbated by use of ESAs without adequate iron repletion.34
The 2012 KDIGO guideline on management of anemia in CKD recommends considering a trial of IV or oral iron where an increase in Hb is desired, if Tsat is less than 30% and a serum ferritin < 500 ng/ml is present (Level 2C).23 For CKD without dialysis, the route of iron repletion is deferred to the provider based on severity of anemia while the parenteral route is recommended for DD-CKD patients. In the dialysis population an improved Hb response with IV rather than oral iron has been demonstrated. Additionally, IV iron therapy seems to improve responsiveness to ESAs as well as reduce ESA dosing by up to a third.35
A few points related to the iron repletion guidelines merit consideration. Targets and parameters for iron repletion in chronic kidney disease have been long unclear and have traditionally been based on limited data. The serum ferritin target (<500 ng/ml) has been challenged by trials like the DRIVE trial, which showed a reduction in ESA dosing with IV iron treatment for serum ferritin values of >500 ng/ml, suggesting that in some CKD patients, this cutoff might be arbitrary.36 In CKD patients, improving Hb and Tsat in a short period of time and reducing the cost of ESA use make IV iron an attractive option. However, data on long-term outcomes of repetitive, high-dose IV iron therapy are often conflicting and concerns for oxidative stress, increased atherosclerosis, putative risk of infections and organ toxicities still abound.37 While trials like the REVOKE trial showed concerns for increased incident risk for bacterial infections with IV iron use compared to oral iron, others like FIND-CKD did not show any difference.38, 39 Furthermore, newer agents as discussed below and better-absorbed oral iron formulations, such as ferric citrate (FC), have been introduced, which might yet alter the narrative.
The PIVOTAL trial was a multi-center, prospective, open-label RCT amongst 2141 hemodialysis patients in the UK that assigned patients to high-dose IV iron administered proactively or low-dose IV iron administered reactively to show non-inferiority of the high-dose regimen.40 Included patients had a Tsat ≤ 30% and ferritin <400ng/ml. Advanced HF patients (NYHA IV) were amongst those excluded. In the proactive group, IV iron was delivered monthly and held for ferritin of >700ng/ml or Tsat >40%. The reactive group received IV iron monthly to keep ferritin >200ng/ml and Tsat >20%. The primary endpoint was time to all-cause death or a composite of non-fatal cardiovascular events (MI, stroke and hospitalization for heart failure) over a median follow up period of 2 years. The study concluded the non-inferiority of high-dose IV iron delivered proactively without increased risk of death or major cardiovascular events and seemed to validate some of the findings of the DRIVE trial. Use of more IV iron was associated with lower ESA dosing.
These findings differ from the heart failure trials in the high amounts of iron losses that are endured in the dialysis population. Even the reactive group in PIVOTAL needed a mean of 1.8g of iron in the first year and then an average 165–200mg per month after. Additionally, advanced heart failure patients (NYHA Class IV) were excluded from the study and these findings cannot be generalized to the HF population.
5. Oral iron versus intravenous iron
Oral iron therapies deliver iron in the absorbable ferrous form to the gut but are limited by gastrointestinal symptoms. Response to treatment can be detected as early as a week although full replenishment of stores can take several months. There have been two randomized-controlled trials assessing the role of oral iron in HF patients.
The IRON-HF trial was a randomized, double-blinded, placebo-controlled trial with 23 anemic HF patients, LVEF <40%, NYHA II-IV with ID (Tsat <20% and ferritin <500 ng/ml) that allocated patients to 3 arms: oral iron, oral placebo + IV iron or oral and IV placebo. It would be the first trial to allow a head-to-head comparison of IV and oral iron.41 Unfortunately, the trial was terminated early due to lack of funding. Preliminary results from 23 patients showed improvement in peak VO2 in the IV iron group and increases in serum ferritin and Tsat were seen in both iron groups.
The IRONOUT-HF trial was a double-blinded, placebo-controlled trial comparing oral iron with placebo in 225 HF patients with EF ≤40% and ID defined as serum ferritin 15–100ng/ml or ferritin 100–299ng/ml with a Tsat <20%.42 The treatment arm received oral iron polysaccharide over 16 weeks. The primary endpoint of change in peak VO2 from baseline to 16 weeks was not significantly different between the two groups. The authors concluded that use of oral iron therapy could not be recommended for HF patients with reduced ejection fraction. However, it is unclear if this duration of therapy is enough to bring about meaningful change given that oral iron takes longer to replenish stores. Additionally, although oral iron did not improve peak VO2 at 16 weeks, this effect has not been demonstrated by IV iron in HF patients other than those with anemia (FERRIC-HF). No head-to-head comparison of IV vs oral iron was made in this study. Median values for ferritin and Tsat in this study were also high enough to question if the patients were truly iron deficient. Interestingly, the patients with the lowest quartiles of hepcidin values seemed to respond better to oral iron therapy in terms of improvement in Tsat and ferritin values. Since hepcidin closely regulates iron uptake in the GI tract and lowers itself in ID, this finding might suggest a marker more truly reflective of ID in HF patients and of response to oral iron therapy.
A retrospective cohort study by Niehaus et al. (2015) suggested that oral iron therapy might work for HF patients. This study evaluated 105 HF patients with ID (defined as serum ferritin <100ng/ml or as ferritin 100–300ng/ml with Tsat <20%) with LVEF <45% and the change in iron biomarkers with 164 days of oral iron therapy.43 Favorable and statistically significant increases in serum iron, ferritin, Tsat and Hb were noted, although more studies are needed to assess an effect, if any, on clinical status.
6. Newer oral iron agents
There are currently several forms of parenteral iron agents that are commercially available (Table 1). On the other hand, traditional oral iron agents are beset with problems of poor absorption and significant GI side effects. Thus, their role in chronic inflammatory conditions like CKD has been limited where significant iron losses are not met by the low bioavailability of traditional oral iron preparations.44 The introduction of novel oral iron agents is an exciting change. These agents may have an increased role in iron repletion with their particular properties that enhance absorption and improve tolerance.
Table 1.
Parenteral Iron Preparations
| Parenteral iron formulation | Trade name(s) - manufacturer |
|---|---|
| Low- molecular weight iron dextran | INFeD - Allergan |
| Ferric gluconate | Ferrlecit - Sanofi-Aventis Inc. |
| Iron sucrose | Venofer - Luitpold Pharmaceuticals Inc |
| Ferumoxytol (polyglucose sorbitol carboxymethyl ether) | Feraheme - AMAG Pharmaceuticals |
| Iron isomaltoside | Monofer - Pharmacosmos A/S |
| Ferric carboxymaltose (FCM) | Ferinject - Vifor |
| PharmaInjectafer - Luitpold Pharmaceuticals Inc | |
| Iron polymaltose | Ferrosig- Sigma Company Ltd |
Ferric citrate (FC) is an oral ferric-iron based agent that has some unique properties. It remains soluble over a wide pH range and forms coordination complexes between the ferric and citrate ions that inhibit precipitation, as well as oligomeric or mononuclear complexes at acidic and alkaline pH respectively. This property allows FC to be a phosphate binder in the stomach (low pH, insoluble oligomeric complex) and be a source for ferric absorption in the duodenum (high pH, soluble mononuclear complex).
FC was first explored as a phosphate binder for DD-CKD patients and later found to improve iron indices in this population. Use of FC was then extended to the non-DD CKD patients with IDA where it was noted to replete iron and improve anemia.45 A phase 3 RCT with non-DD CKD patients with an inadequate response or intolerance to prior oral iron showed higher likelihood of achieving >1g/dl increase in Hb from baseline with FC and led to its FDA-approval in Nov 2017. More encouragingly, FC use in the dialysis patients has been associated with lower ESA dependence, with up to 25% lower ESA dose in some trials.46 Also, over a 52-week treatment period, IV iron use dropped in this open-label trial from 57mg in weeks 0–12 to 3.6mg in weeks 28–52. FC has also been shown to reduce fibroblast-growth factor (FGF)-23 levels in non-dialysis CKD population.45 Elevated FGF-23 levels are associated with cardiovascular mortality.47 Unlike with FCM, the risk of hypophosphatemia due to decrease in FGF-23 is low with FC. Long-term effects of FC on time to death or dialysis are currently being studied.
Ferric maltol is a novel oral iron preparation approved for the treatment of IDA in adults. It has both hydrophilic and lipophilic properties from bonds between ferric and maltose groups, allowing high bioavailability compared with standard ferrous iron preparations. Sucrosomial iron is another novel iron preparation that consists of ferric pyrophosphate protected within a phospholipid bilayer, called a sucrosome. The sucrosome allows for increased intestinal absorption, improved Hb and iron status without increase in hepcidin values. An open-label randomized trial of oral sucrosomial iron versus IV ferrous gluconate concluded that short-term oral sucrosomial iron was as effective as IV ferrous gluconate at correcting anemia in non-DD CKD patients and was tolerated well.48
Lastly, emerging evidence suggests that the conventional dosing schedule of oral iron three times daily might be linked with rapid increase in hepcidin levels and limited absorption of the subsequent dose.49 Data from a randomized clinical trial suggests that dosing in single doses and on alternate days might be the preferred modality.50
7. Ongoing clinical trials with IV iron in HF
Several trials are being conducted on the use of IV iron supplementation in HF patients. The FAIR-HF2 trial is a randomized, placebo-controlled trial of FCM or placebo in HF patients with a combined primary endpoint of recurrent hospitalization for HF and cardiovascular death. The IRONMAN is an open-label RCT to assess whether IV iron in high-risk HF patients (NYHA II-IV) with ID (Tsat <20% and/or ferritin <100ng/ml) would lower cardiovascular mortality or hospitalization for worsening HF (primary outcome). It is expected to complete in 2021. HEART-FID is a randomized, double-blind, placebo-controlled trial to assess the effect of IV FCM compared to placebo in HF patients with ID, on 12-month mortality or hospitalization for worsening HF as well as the 6-month change in the 6MWT. It has an expected completion date of 2022. Finally, AFFIRM-HF is a randomized, double-blind, placebo-controlled trial to analyze the effect of IV FCM on hospitalization and mortality in HF patients with ID, admitted for acute HF, with an expected completion date of 2020.
8. Side effects of IV iron
Early IV preparations of iron differed in their pharmacokinetics and caused large pools of labile iron and increased oxidative stress. Side effects of traditional parenteral iron preparations ranged from mild symptoms like nausea, vomiting to more worrisome like hypotension and even anaphylactic shock. With the advent of newer preparations that have a more stable carbohydrate shell, the risk of infusion reactions is much lower. The different IV iron preparations have been tabulated below. Of those listed, FCM has been used in HF trials. In the dialysis population, IV iron use in the early 1990s was limited to iron dextran preparation and has gradually changed to ferric gluconate or iron sucrose.
Aside from infusion reactions, IV iron preparations have been associated with rapid increase in NTBI. Via the Fenton reaction, non-bound iron can interact with hydrogen peroxide and form hydroxyl radicals, which can cause oxidative injury to tissues. Iron sucrose preparations have been shown to cause oxidative stress to in-vitro endothelial cells.51 Similarly, iron dextran causes oxidative stress to cardiovascular tissues of rats with kidney failure.52 In a study of iron sucrose administration to healthy volunteers, attenuated flow-mediated dilatation of the brachial artery was noted, suggesting transient endothelial dysfunction.53 Additionally, in dialysis patients, an increase in lipid peroxidation has been noted after IV iron infusion.54
Several case studies and trials have reported hypophosphatemia in conjunction with the use of IV iron55. This has been proposed to be due to increased levels of FGF-23, a peptide with phosphaturic actions and released from bone, which can cause osteomalacia and rickets.56 FGF-23 levels are elevated in CKD patients and normal in those with preserved renal function. Hence, impaired kidney function is protective against the phosphate-wasting effects of IV iron.
9. Conclusions
Some patients with HFrEF may experience symptomatic benefits from IV iron, albeit to varying objective degrees and with no demonstrated effects on clinical outcomes. It remains to be understood how this occurs and for which subset of HF patients in particular. The physiologic processes resulting in ID in CKD are different from HF, hence the same criteria for defining ID in HF as in CKD might not be appropriate and could overestimate the burden of ID in HF. With the advent of newer, better-absorbed oral iron preparations, there may be a safer way to manage ID in HF patients to avoid the oxidative stress and potential side effects of IV iron loading. Moreover, IV iron might have detrimental effects in cardiovascular patients, especially in those with coronary artery disease where it might result in endothelial dysfunction. We believe that as more longterm outcomes-based research becomes available, future recommendations for the use of IV iron in HF patients might alter, reflecting better markers for identifying true iron-deficiency, and suitability of novel oral iron preparations in certain situations. For the clinician, this would ideally help to better identify the subsets of HF patients that may putatively benefit from IV iron use.
Funding sources
HA is supported by the NIH grants NHLBI HL127646, HL140973, and HL138982
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ganz T, Nemeth E. Iron homeostasis in host defence and inflammation. Nat Rev Immunol. 2015; 15: 500–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abbaspour N, Hurrell R, Kelishadi R. Review on iron and its importance for human health. J Res Med Sci. 2014; 19: 164–174. [PMC free article] [PubMed] [Google Scholar]
- 3.Andrews NC. Iron homeostasis: insights from genetics and animal models. Nat Rev Genet. 2000; 1: 208–217. [DOI] [PubMed] [Google Scholar]
- 4.Punnonen K, Irjala K, Rajamäki A. Iron-deficiency anemia is associated with high concentrations of transferrin receptor in serum. Clin Chem. 1994; 40: 774–776. [PubMed] [Google Scholar]
- 5.Huebers HA, Beguin Y, Pootrakul P, Einspahr D, Finch CA. Intact transferrin receptors in human plasma and their relation to erythropoiesis. Blood. 1990; 75: 102–107. [PubMed] [Google Scholar]
- 6.Ganz T, Olbina G, Girelli D, Nemeth E, Westerman M. Immunoassay for human serum hepcidin. Blood. 2008; 112: 4292–4297. [DOI] [PubMed] [Google Scholar]
- 7.Zaritsky J, Young B, Wang HJ et al. Hepcidin--a potential novel biomarker for iron status in chronic kidney disease. Clin J Am Soc Nephrol. 2009; 4: 1051–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weiss G, Theurl I, Eder S et al. Serum hepcidin concentration in chronic haemodialysis patients: associations and effects of dialysis, iron and erythropoietin therapy. Eur J Clin Invest. 2009; 39: 883–890. [DOI] [PubMed] [Google Scholar]
- 9.Jankowska EA, Kasztura M, Sokolski M et al. Iron deficiency defined as depleted iron stores accompanied by unmet cellular iron requirements identifies patients at the highest risk of death after an episode of acute heart failure. European heart journal. 2014; 35: 2468–2476. [DOI] [PubMed] [Google Scholar]
- 10.Tanne Z, Coleman R, Nahir M, Shomrat D, Finberg JP, Youdim MB. Ultrastructural and cytochemical changes in the heart of iron-deficient rats. Biochem Pharmacol. 1994; 47: 1759–1766. [DOI] [PubMed] [Google Scholar]
- 11.Xu W, Barrientos T, Mao L, Rockman HA, Sauve AA, Andrews NC. Lethal Cardiomyopathy in Mice Lacking Transferrin Receptor in the Heart. Cell Rep. 2015; 13: 533–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ke Y, Chen YY, Chang YZ et al. Post-transcriptional expression of DMT1 in the heart of rat. J Cell Physiol. 2003; 196: 124–130. [DOI] [PubMed] [Google Scholar]
- 13.Tsushima RG, Wickenden AD, Bouchard RA, Oudit GY, Liu PP, Backx PH. Modulation of iron uptake in heart by L-type Ca2+ channel modifiers: possible implications in iron overload. Circ Res. 1999; 84: 1302–1309. [DOI] [PubMed] [Google Scholar]
- 14.Taylor KM, Morgan HE, Johnson A, Nicholson RI. Structure-function analysis of a novel member of the LIV-1 subfamily of zinc transporters, ZIP14. FEBS Lett. 2005; 579: 427–432. [DOI] [PubMed] [Google Scholar]
- 15.Lakhal-Littleton S, Wolna M, Carr CA et al. Cardiac ferroportin regulates cellular iron homeostasis and is important for cardiac function. Proc Natl Acad Sci U S A. 2015; 112: 3164–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lakhal-Littleton S, Wolna M, Chung YJ et al. An essential cell-autonomous role for hepcidin in cardiac iron homeostasis. Elife. 2016; 5: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bayeva M, Khechaduri A, Puig S et al. mTOR regulates cellular iron homeostasis through tristetraprolin. Cell metabolism. 2012; 16: 645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sato T, Chang HC, Bayeva M et al. mRNA-binding protein tristetraprolin is essential for cardiac response to iron deficiency by regulating mitochondrial function. Proc Natl Acad Sci U S A. 2018; 115: E6291–E6300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anand I, Gupta P. Response by Anand and Gupta to Letter Regarding Article, “Anemia and Iron Deficiency in Heart Failure: Current Concepts and Emerging Therapies”. Circulation. 2018; 138: 2976–2977. [DOI] [PubMed] [Google Scholar]
- 20.Nanas JN, Matsouka C, Karageorgopoulos D et al. Etiology of anemia in patients with advanced heart failure. J Am Coll Cardiol. 2006; 48: 2485–2489. [DOI] [PubMed] [Google Scholar]
- 21.Eschbach JW. Erythropoietin 1991--an overview. Am J Kidney Dis. 1991; 18: 3–9. [PubMed] [Google Scholar]
- 22.Kooistra MP, Niemantsverdriet EC, van Es A, Mol-Beermann NM, Struyvenberg A, Marx JJ. Iron absorption in erythropoietin-treated haemodialysis patients: effects of iron availability, inflammation and aluminium. Nephrol Dial Transplant. 1998; 13: 82–88. [DOI] [PubMed] [Google Scholar]
- 23.McMurray JJV, Parfrey PS, Adamson JW et al. Kidney disease: Improving global outcomes (KDIGO) anemia work group. KDIGO clinical practice guideline for anemia in chronic kidney disease. Kidney International Supplements. 2012; 2: 279–335. [Google Scholar]
- 24.Anand IS, Gupta P. Anemia and iron deficiency in heart failure: current concepts and emerging therapies. Circulation. 2018; 138: 80–98. [DOI] [PubMed] [Google Scholar]
- 25.Jankowska EA, Von Haehling S, Anker SD, Macdougall IC, Ponikowski P. Iron deficiency and heart failure: diagnostic dilemmas and therapeutic perspectives. European heart journal. 2012; 34: 816–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Melenovsky V, Petrak J, Mracek T et al. Myocardial iron content and mitochondrial function in human heart failure: a direct tissue analysis. European journal of heart failure. 2017; 19: 522–530. [DOI] [PubMed] [Google Scholar]
- 27.Khechaduri A, Bayeva M, Chang H-C, Ardehali H. Heme levels are increased in human failing hearts. Journal of the American College of Cardiology. 2013; 61: 1884–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang H, Wu R, Shang M et al. Reduction in mitochondrial iron alleviates cardiac damage during injury. EMBO molecular medicine. 2016; 8: 247–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leszek P, Sochanowicz B, Szperl M et al. Myocardial iron homeostasis in advanced chronic heart failure patients. International journal of cardiology. 2012; 159: 47–52. [DOI] [PubMed] [Google Scholar]
- 30.Jankowska EA, Kasztura M, Sokolski M et al. Iron deficiency defined as depleted iron stores accompanied by unmet cellular iron requirements identifies patients at the highest risk of death after an episode of acute heart failure. European heart journal. 2014; 35: 2468–2476. [DOI] [PubMed] [Google Scholar]
- 31.Anker SD, Comin Colet J, Filippatos G et al. Ferric carboxymaltose in patients with heart failure and iron deficiency. New England Journal of Medicine. 2009; 361: 2436–2448. [DOI] [PubMed] [Google Scholar]
- 32.Ponikowski P, Van Veldhuisen DJ, Comin-Colet J et al. Beneficial effects of long-term intravenous iron therapy with ferric carboxymaltose in patients with symptomatic heart failure and iron deficiency. European heart journal. 2014; 36: 657–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Veldhuisen DJ, Ponikowski P, van der Meer P et al. Effect of ferric carboxymaltose on exercise capacity in patients with chronic heart failure and iron deficiency. Circulation. 2017; 136: 1374–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Albaramki J, Hodson EM, Craig JC, Webster AC. Parenteral versus oral iron therapy for adults and children with chronic kidney disease. Cochrane Database of Systematic Reviews. 2012; [DOI] [PubMed] [Google Scholar]
- 35.Roger SD, Tio M, Park H et al. Intravenous iron and erythropoiesis-stimulating agents in haemodialysis: A systematic review and meta-analysis. Nephrology. 2017; 22: 969–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coyne DW, Kapoian T, Suki W et al. Ferric gluconate is highly efficacious in anemic hemodialysis patients with high serum ferritin and low transferrin saturation: results of the Dialysis Patients’ Response to IV Iron with Elevated Ferritin (DRIVE) Study. Journal of the American Society of Nephrology. 2007; 18: 975–984. [DOI] [PubMed] [Google Scholar]
- 37.Avni T, Bieber A, Grossman A, Green H, Leibovici L, Gafter-Gvili A. The safety of intravenous iron preparations: systematic review and meta-analysis. Mayo clinic proceedings. 2015; Mayo clinic proceedings 90: 12–23. [DOI] [PubMed] [Google Scholar]
- 38.Agarwal R, Kusek JW, Pappas MK. A randomized trial of intravenous and oral iron in chronic kidney disease. Kidney Int. 2015; 88: 905–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Macdougall IC, Bock AH, Carrera F et al. FIND-CKD: a randomized trial of intravenous ferric carboxymaltose versus oral iron in patients with chronic kidney disease and iron deficiency anaemia. Nephrol Dial Transplant. 2014; 29: 2075–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Macdougall IC, White C, Anker SD et al. Intravenous iron in patients undergoing maintenance hemodialysis. New England Journal of Medicine. 2019; 380: 447–458. [DOI] [PubMed] [Google Scholar]
- 41.Beck-da-Silva L, Piardi D, Soder S et al. IRON-HF study: a randomized trial to assess the effects of iron in heart failure patients with anemia. International journal of cardiology. 2013; 168: 3439–3442. [DOI] [PubMed] [Google Scholar]
- 42.Lewis GD, Malhotra R, Hernandez AF et al. Effect of oral iron repletion on exercise capacity in patients with heart failure with reduced ejection fraction and iron deficiency: the IRONOUT HF randomized clinical trial. JAMA. 2017; 317: 1958–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Niehaus ED, Malhotra R, Cocca-Spofford D, Semigran M, Lewis GD. Repletion of iron stores with the use of oral iron supplementation in patients with systolic heart failure. Journal of cardiac failure. 2015; 21: 694–697. [DOI] [PubMed] [Google Scholar]
- 44.Pergola PE, Fishbane S, Ganz T. Novel Oral Iron Therapies for Iron Deficiency Anemia in Chronic Kidney Disease. Adv Chronic Kidney Dis. 2019; 26: 272–291. [DOI] [PubMed] [Google Scholar]
- 45.Fishbane S, Block GA, Loram L et al. Effects of ferric citrate in patients with nondialysis-dependent CKD and iron deficiency anemia. Journal of the American Society of Nephrology. 2017; 28: 1851–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yokoyama K, Akiba T, Fukagawa M et al. Long-term safety and efficacy of a novel iron-containing phosphate binder, JTT-751, in patients receiving hemodialysis. Journal of Renal Nutrition. 2014; 24: 261–267. [DOI] [PubMed] [Google Scholar]
- 47.Scialla JJ, Xie H, Rahman M et al. Fibroblast growth factor-23 and cardiovascular events in CKD. Journal of the American Society of Nephrology. 2014; 25: 349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pisani A, Riccio E, Sabbatini M, Andreucci M, Del Rio A, Visciano B. Effect of oral liposomal iron versus intravenous iron for treatment of iron deficiency anaemia in CKD patients: a randomized trial. Nephrology Dialysis Transplantation. 2014; 30: 645–652. [DOI] [PubMed] [Google Scholar]
- 49.Moretti D, Goede JS, Zeder C et al. Oral iron supplements increase hepcidin and decrease iron absorption from daily or twice-daily doses in iron-depleted young women. Blood. 2015; 126: 1981–1989. [DOI] [PubMed] [Google Scholar]
- 50.Stoffel NU, Cercamondi CI, Brittenham G et al. Iron absorption from oral iron supplements given on consecutive versus alternate days and as single morning doses versus twice-daily split dosing in iron-depleted women: two open-label, randomised controlled trials. The Lancet Haematology. 2017; 4: e524–e533. [DOI] [PubMed] [Google Scholar]
- 51.Kamanna VS, Ganji SH, Shelkovnikov S, Norris K, Vaziri ND. Iron sucrose promotes endothelial injury and dysfunction and monocyte adhesion/infiltration. Am J Nephrol. 2012; 35: 114–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lim CS, Vaziri ND. The effects of iron dextran on the oxidative stress in cardiovascular tissues of rats with chronic renal failure. Kidney international. 2004; [DOI] [PubMed] [Google Scholar]
- 53.Zheng H, Huang X, Zhang Q, Katz SD. Iron sucrose augments homocysteine-induced endothelial dysfunction in normal subjects. Kidney international. 2006; 69: 679–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Salahudeen AK, Oliver B, Bower JD, II LJR. Increase in plasma esterified F2-isoprostanes following intravenous iron infusion in patients on hemodialysis. Kidney international. 2001; [DOI] [PubMed] [Google Scholar]
- 55.Wolf M, Rubin J, Achebe M et al. Effects of Iron Isomaltoside vs Ferric Carboxymaltose on Hypophosphatemia in Iron-Deficiency Anemia: Two Randomized Clinical Trials. JAMA. 2020; 323: 432–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Musgrove J, Wolf M. Regulation and Effects of FGF23 in Chronic Kidney Disease. Annu Rev Physiol. 2020; 82: 365–390. [DOI] [PubMed] [Google Scholar]