

Abstract
Phosphorylation of sphingosine by sphingosine kinase 1 (SPHK1) produces the bioactive sphingolipid sphingosine-1-phosphate (S1P), a microvascular and immuno-modulator associated with vascular remodeling in pulmonary arterial hypertension (PAH). The low intracellular concentration of S1P is under tight spatial-temporal control. Molecular mechanisms that mediate S1P burden and S1P regulation of vascular remodeling are poorly understood. Similarities between two early response pro-inflammatory cytokine gene transcript activation profiles, S1P and Endothelial Monocyte Activating Polypeptide II (EMAP II), suggested a strategic link between their signaling pathways. We determined that EMAP II triggers a bimodal phosphorylation, transcriptional regulation and membrane translocation of SPHK1 through a common upstream process in both macrophages and pulmonary artery smooth muscle cells (PASMCs). EMAP II initiates a dual function of ERK1/2: phosphorylation of SPHK1 and regulation of the transcription factor EGR1 that induces expression of SPHK1. Activated ERK1/2 induces a bimodal phosphorylation of SPHK1 which reciprocally increases S1P levels. This identified common upstream signaling mechanism between a protein and a bioactive lipid initiates cell specific downstream signaling representing a multifactorial mechanism that contributes to inflammation and PASMC proliferation which are cardinal histopathological phenotypes of PAH.
Keywords: Pulmonary Arterial Hypertension, Bioactive lipid, sphingosine kinase 1, endothelial monocyte activating polypeptide II
Introduction
Pulmonary Arterial Hypertension (PAH) is a progressive, incurable and fatal cardiopulmonary vascular disease. Despite the recent advances in etiological and treatment landscape of PAH, it still remains difficult to be diagnosed, treated and managed [1,2]. PAH can be idiopathic or associated with other diseases resembling idiopathic PAH suggesting that there are similarities in mechanism of disease progression. Pathogenesis of PAH is multifactorial with hypoxia, viral infections, congenital heart disease, and other physiological/exogenous being triggers of PAH. Untreated PAH leads to right ventricular hypertrophy, overload and heart failure. Pulmonary vascular remodeling in concert with vascular proliferation/fibrosis and vessel obstruction are cardinal histopathological marks in PAH [3]. Recent patients and animal studies of PAH revealed that inflammation and angiogenesis are alluring contributors to vessel wall remodeling and cellular hyperproliferation, hallmarks of severe PAH [3-6].
Progression of PAH is allied with multiple cell types in the pulmonary arterial wall and pulmonary arterial circulation that contribute to the injury response. Pulmonary arterial smooth muscle cells (PASMC) and inflammatory cells are two major cellular populations that contribute to the pathogenesis of PAH. Microvascular and immuno-modulators such as Sphingosine 1-phosphate (S1P), induced upon injurious stimuli, direct vascular remodeling through a plethora of pathologic cellular processes in different cell types. Hyper-proliferation of PASMC is a vital characteristic in PAH pathogenesis leading to prominent intimal thickening and muscularization of the small arterioles [7,8]. Recent studies have provided a glimpse at S1P signaling as a regulator of PASMC proliferation while S1P elevated levels in human PAH lungs [9,10] suggests that modulation of S1P levels and understanding the signaling cascade of S1P in the development of PAH would unravel therapeutic targets to treat PAH.
Phosphorylation of sphingosine by sphingosine kinases (SPHK1 and SPHK2) generates the bioactive sphingolipid S1P, a key mediator of various biological processes such as cell proliferation and growth, apoptosis, inflammation, angiogenesis and vascular integrity.[11] S1P is present at low intracellular concentrations under tight spatial-temporal control. Upon tissue injury or stress, intracellular levels of prototypical signal molecule, S1P is induced which in turn can function as an intracellular secondary messenger or be transported out of the cells and act as an extracellular signaling molecule by binding to one of five cell-specific G-protein coupled receptors, S1P receptor 1 (S1PR1) - S1P receptor 5 (S1PR5) in autocrine or paracrine manner. ‘Inside-out’ relocation of S1P can activate several signaling pathways including Janus tyrosine kinase/signal transducer and activators of transcription (JAK/STAT) and Extracellular Regulated Kinases (ERK) [12-14]. Importantly, during tissue injury and homeostasis early tissue injury response signaling molecules such as S1P recruit phagocytes and induce secretion of proinflammatory cytokines such as IL-1β, IL6 and TNFα [15,16].
In the present study, we investigated the role of another key microvascular/ immune- early response mediator, Endothelial Monocyte Activating Polypeptide II (EMAP II) in modulating S1P homeostasis and key intermediates in the SPHK1/S1P signaling pathway. The role of EMAP II in PAH is not known, however recent studies implicate a role for EMAP II as secondary PAH was noted in Bronchopulmonary Dysplasia (BPD) like phenotypic mouse model [17]. Our recent studies identified a link between S1P and EMAP II as EMAP II mediates a distinct inflammatory gene profile in recruited macrophages through a JAK/STAT signaling pathway similar to S1P signaling axis [18,19]. EMAP II is the cleaved C-terminal peptide of the pervasively expressed 34-kDa intracellular protein Aminoacyl tRNA synthetase complex Interacting Multifunctional Protein 1 (AIMPI, also known as p43/ proEMAP II) that functions as one of three scaffold proteins for the multi-synthetase complex (MSC) that mediate protein translation [20]. Once EMAP II is released, it functions as a promiscuous pro-inflammatory signaling cytokine that predominately signals through tyrosine kinases [21-24].
We identified EMAP II as a strategic early regulator of SPHK1 mediated S1P expression where it signals in both macrophages and SMCs through ERK1/2 and transcriptional regulator Early growth response 1 (EGR1). Furthermore, we demonstrate that EMAP II mediated SPHK1/S1P signaling generates cell-type specific response such as macrophage pro-inflammatory activation and PASMC proliferation. Although recent studies target the inhibition of SPHK1 as a protective treatment for SPHK1/S1P signaling activated inflammatory and proliferative role of altered S1P homeostasis [25], our results suggest that EMAP II initiated bimodal phosphorylation of SPHK1 is an important upstream regulator of the SPHK1/S1P mediated signaling mechanism. Thus, shedding new insight into the upstream regulation of SPHK1/S1P signaling mechanism and offering hope of more specific and effective therapeutic targets in treating PAH and several pathophysiological conditions of inflammation and PASMC proliferation.
Materials and methods
Materials.
The primary antibodies STAT3, pSTAT3 (Y705), ERK1/2, pERK1/2, SPHK1, EGR1, Lamin B1 and cyclin 1 were from Cell Signaling Technology (CST) (Danvers, MA), SPHK1 and alpha-tubulin was from Abcam and phospho-SPHK1 (Ser225) was purchased from ECM Biosciences. HRP-conjugated rabbit and mouse secondary antibodies were from CST, Alexa fluorescent labeled secondary antibodies and phalloidin were purchased from Thermo Fischer Scientific (Waltham, MA). Inhibitors W 146 (iS1PR1), JTE 013 (iS1PR2), TY 52156 (iS1PR3) and PF543 (iSPHK1) were from Cayman Chemical (Ann Arbor, MI), PD 184161 (iERK) was from Santa Cruz Biotechnology (Dallas, TX) and Ruxolitinib (iJAK1//2) and Trametinib (iERK1) were purchased from LC laboratories (Woburn, MA). EMAP II neutralizing antibody (Anti-EMAP II) was used to block EMAP II activity [26].
EMAP II preparation
6x-His tagged EMAP II was prepared as previously described [27] The endotoxin levels in all preparations used for this study were below detectable limits, containing <0.1 ng/EU (GenScript).
Cell culture and treatments
RAW 264.7 (ATCC) mouse macrophages and HEK-293T (ATCC) cells were cultured in DMEM media with 10% (v/v) fetal bovine serum (FBS) and 2 mM glutamine. HEPES buffer and beta-mercaptoethanol were added to the growth medium used for RAW 264.7 macrophages. Primary human pulmonary artery smooth muscle cells (hPASMC) purchased from Lonza (Walkersville, MD) were cultured in complete growth medium containing smooth muscle growth media-2 (SmGM-2) with 10% FBS and growth factors provided as a kit by the supplier (Lonza, (Walkersville, MD). THP-1 human monocyte cells were cultured in RPMI 1640 with 10% FBS and glutamine. Cells were cultured in a humidified atmosphere with 5% CO2 at 37°C. For all studies passages 5-14 were used for RAW 264.7 and passages 5-10 were used for hPASMCs. For treatment studies, subconfluent cells plated in multi-well plates were serum starved and if required, pretreated with inhibitors before stimulation with 2 μg/mL recombinant EMAP II. All the controls were handled similar to the test samples. Cells were treated with EMAP II or vehicle, cells were rinsed at the desired time point, and lysis buffer was applied.
Mouse Lung Tissues
Animal experimental procedure was performed in accordance with the guidelines issued by the University of Notre Dame/Indiana University Institutional Animal and Use Committee. Lung tissues were harvested from postnatal day 15 C57BL/6 mice (Jackson laboratories) that were injected with either EMAP II or PBS at Day 3. Total protein was extracted and quantified. 1 mg of total protein was subjected to S1P quantification by HPLC/MS/MS.
ELISA for S1P quantification
Equal number of hPASMC cells (1,000,000) were plated and serum starved for overnight. Cells were stimulated for in a series of time points and collected the cells. Cells were pelleted out and washed with PBS. After the lysis protein concentration was measured. S1P levels were measured using S1P ELISA kit (MyBioSource, MBS069092) following manufacturer’s instructions. S1P levels were normalized against protein levels.
Analysis of S1P, Sphingosine and Ceramides by LC-MS/MS
Collected cells were rinsed with PBS, pelleted. Cell pellets and 1 mg of protein tissue extracts were frozen at −80 °C. Sphingolipids extraction and analysis was carried out at the Medical University of South Carolina (MUSC) core facility using eight-point calibration curves generated for each target analyte by High Performance Liquid Chromatography-Tandem Mass Spectrometry (HPLC/MS/MS) system operating in positive multiple reaction-monitoring (MRM) mode engaging a gradient elution. Sphingolipids were normalized to inorganic phosphate levels in PASMC which was determined from the Bligh & Dyer lipid extraction method. Biological samples were from different passages collected on different days.
Immunoblotting
After appropriate treatments, protein lysates were prepared by using RIPA buffer supplemented with phosphatase and protease inhibitors (Thermo Fisher Scientific). Alpha-tubulin was used as a loading control. For nuclear and cytoplasmic fractionation, NE-PER kit from Thermo Fischer Scientific was used. Western blots were performed according to standard methods and quantified using densitometry using Image Studio Lite software from LI-COR Biosciences.
Differentiation of THP-1 cells
Several passages after thawing of THP-1 cell line, cells were seeded in Nunc Lab-Tek™ II 4-well imaging plates and differentiated into macrophages with 100 nM Phorbol myristate acetate (InvivoGen, CA) for 72 hours.
Immunofluorescence staining
Primary hPASMCs cells or human THP-1 macrophages were seeded in Nunc Lab-Tek™ II 4-well imaging plates (Thermo Fisher Scientific) and cultured in growth medium. After the serum starvation cells were stimulated with EMAP II. Treated cells were rinsed with PBS and fixed with 4% paraformaldehyde at room temperature for 15 minutes and permeabilized with 0.1% Triton X-100 at room temperature for 10 min. After washing with PBS three times, the cells were incubated with EGR1, lamin, or pSPHK1 antibody at 4 °C overnight. The cells were then rinsed with PBS three times and subsequently incubated with respective secondary antibody conjugated with Alexa Fluor 647 and 488 or Phalloidin 488 at room temperature for 1h. The cells were rinsed with PBS three times and coverslips were mounted with SlowFade Gold Antifade Mountant with DAPI (Thermo Fischer Scientific) and the cells were examined under Olympus microscope with 40X water objective lens for EGR1 expression and with 60X for pSPHK1 water immersion.
Transfection with Small Interfering RNA
Knockdown of endogenous EGR1 was carried out by transfecting with 25 nM final concentration of ON-TARGETplus siRNAs specific for EGR1 or non-targeting control (GE Dharmacon, Lafayette, CO) using Viromer Blue (Lipocalyx) per manufacturer’s instructions. Cells were stimulated with EMAP II 48 h post-transfection. Non-targeting controls and EGR1 knocked-down samples were washed, treated and handled similar to each other.
Transfection and luciferase assay
HEK 293-T cells cultured in DMEM media containing 10% serum were reverse co-transfected with pGL4.47[luc2P/SIE/Hygro] and pGL4.74 [hRluc/TK] using Attractene Transfection Reagent (301005, Qiagen) as per manufacturer’s protocol. Pretreated with inhibitors for 1 hour as required. After the treatment, cells were incubated for 24 hours and luciferase activity was measured using Dual-Glo Luciferase Assay (E2920, Promega) per manufacturer’s guidelines.
Cell proliferation assay
Cell proliferation of hPASMCs was assessed using WST-1 cell proliferation reagent (Roche Diagnostic Corporation, IN) in a 96- well plate. Human PASMCs were seeded at 4200 cells/well density in 100 μl complete medium and serum starved for overnight. Cells were stimulated for 48 hours with 2 μg/ mL EMAP II following the pretreatment with inhibitors. WST-1 reagent (10 μL) was added into each well and incubated for 90 minutes in a humidified atmosphere with 5% CO2 at 37 °C. Absorbance was measured at 450 nm using microplate reader. Blank control well was with medium and WST-1 reagent without cultured cells. All samples were in duplicates. Cell proliferation was determined by subtracting the background reading from average of duplicate of each sample.
RNA extraction and quantification
Total RNA was extracted with TriZol using standard RNA extraction protocol and reverse transcribed with Superscript III Reverse Transcriptase (Thermo Fisher Scientific). SPHK1, Tnf, Il6, EGR1, Gapdh, Hprt1 transcripts were quantified using PrimePCR SYBR Green assays from BioRad.
Statistical analysis
The data are presented as means ± 1 standard error of mean (SEM) from at least three independent experiments. In time-course studies, all the time points were performed on the same day and repeated at least three times over different days. Statistical significance was determined with unpaired Student’s t-test or one-way or two-way ANOVA using GraphPad Prism software.
Results
EMAP II modulates S1P levels in vitro and in vivo.
EMAP II mediates a distinct inflammatory gene profile in recruited macrophages through a JAK/STAT signaling pathway similar to S1P signaling axis [18,19]. Moreover, our previous studies reported EMAP II promoted secondary PAH in BPD-like phenotypic mouse model [17]. Therefore, we investigated the potential of EMAP II to modulate S1P levels in two major cell populations in PAH, the pulmonary arterial wall component, PASMCs and one of the major inflammatory cells, macrophages. Following stimulation by EMAP II in a time course, intracellular S1P levels of smooth muscle cells (SMCs) measured by ELISA peaked at 1 hour, subsided at 2 hours peaked again at the 6 hour time point before subsiding at 24 hour exhibiting a significant bimodal pattern in S1P elevation (Figure 1A). The cellular S1P measurements in both SMCs and macrophages by HPLC-MS/MS confirmed the bimodal trend of S1P elevation at 1 hour and 6 hour (Figure 1B and 1E) and moreover, examination of the immediate precursor of S1P, sphingosine and S1P’s interconvertible lipid, ceramide by HPLC-MS/MS showed a decreasing trend over time both in SMCs and macrophages after EMAP II treatment (normalized against inorganic phosphates) (Figure 1B, 1C, 1E and 1F). Consistently, S1P (product of SPHK1): sphingosine (substrate of SPHK1) ratio in both EMAP II treated SMCs and macrophages showed a bimodal peaking trend at 1 hour and 6 hour (Figure 1B and 1E). Extracellular S1P in SMCs was elevated after 1 hour of EMAP II treatment compared to the control (Figure 1D) suggesting the possibility of S1P acting as an extracellular ligand for S1P receptors.
Figure 1. EMAP II modulates in vitro and in vivo S1P levels.

(A) ELISA-cellular C18- S1P levels normalized against 1 μg of protein, (B) HPLC/MS/MS-cellular C18- S1P, sphingosine levels and S1P: sphingosine ratio, (C) HPLC/MS/MS-cellular ceramide levels (C16-Cer, C18-Cer, C20-Cer, C22-Cer and C24-Cer) normalized against cell phosphates in hPASMC cells following EMAP II treatment for 0, 1, 2, 6 and 24 hours. (D) HPLC/MS/MS-extracellular C18-S1P in 1 mL of hPASMC growth media at 0 and 1 hour of EMAP II treatment. (E) HPLC/MS/MS- cellular C18- S1P, sphingosine levels and S1P: sphingosine ratio, (F) HPLC/MS/MS-cellular ceramide levels (C16-Cer, C18-Cer, C20-Cer, C22-Cer and C24-Cer) normalized against cell phosphates in RAW 264.7 cells following EMAP II treatment for 0, 1, 2, 6 and 24 hours. (G) HPLC/MS/MS tissue C18- S1P levels normalized against 1 mg of protein in postnatal day 15 mouse lung tissues following EMAP II treatment on postnatal days 3-15. Results are shown as means ± SEM. n=3, *p<0.05, **p<0.005 using unpaired t-test ((A) and (G)) or two-way ANOVA following Dunnett’s test ((B) **p= 0.0016, *p= 0.0314) and (E) all **p<0.005, *p=0.0487) or one-way ANOVA following Tukey’s test ((C) and (F)) compared to the control or time=0.
Previously, delivery of recombinant EMAP II subQ to neonatal mice on days of life 3-15 has shown to induce distal pulmonary artery thickening and right ventricular hypertrophy consistent with phenotypes of pulmonary hypertension [29]. Consistent with in vitro studies, EMAP II stimulation, significantly increased S1P levels in whole lungs of mice at postnatal age day 15 as assessed by HPLC/MS/MS compared to the control (Figure 1G).
EMAP II induces time dependent bimodal ERK activation and overexpression of immediate early transcription factor EGR1.
As EMAP II is a promiscuous mediator of tyrosine kinase signaling [21-24], the activation of a common tyrosine kinase-signaling factor, ERK1/2 can be a potential target of EMAP II signaling. To determine the impact and timing of EMAP II induced ERK1/2 phosphorylation, a 24-hour time course examined the impact of EMAP II on pERK1/2 in two known S1P signaling cellular targets, macrophages and SMCs. We identified a time dependent statistically significant bimodal activation of pERK1/2 (normalized to ERK1/2) in macrophages (Figure 2A and 2B) as early as 5 minutes and reached the peak at 1 hour before subsiding only to again be noted to increase at 24 hours. EMAP II induced pERK1/2 in SMCs in a similar time dependent manner (Figure 2D and 2E) where pERK1/2 reached significance as early as 3 minutes, subsided and steadily started increasing back again at 24 hours. Since EMAP II signaling is intertwined around both ERK activation (Figure 2A and 2B) and S1P signaling (Figure 1), we explored the effect of EMAP II on an immediate early transcription factor, EGR1 which has SPHK1 as a target gene [30,31] and is known to be transcriptionally mediated by ERK1/2 [32,33]. Protein levels of EGR1 in whole cell lysates from time course studies in both macrophages and SMCs demonstrated a significant upregulation of EGR1 expression at 1 to 2 hours following EMAP II treatment in macrophages and SMCs (Figure 2A, 2C, 2D and 2F).
Figure 2. EMAP II induces time dependent bi-modal ERK activation and overexpression of immediate early transcription factor EGR1.
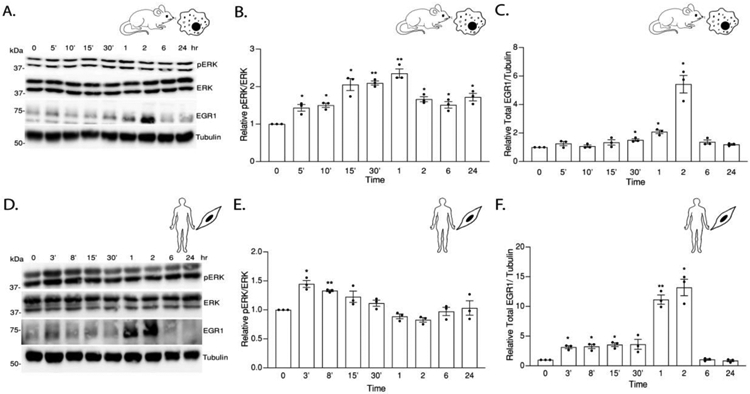
(A) Representative western blot probed for ERK and EGR1 in whole cell lysates of EMAP II treated RAW 264.7 cells had a time dependent phosphorylation of ERK and an overexpression of EGR1. (B) quantification of pERK/ERK (C) quantification of EGR1/ tubulin. (D) Representative western blot probed for ERK and EGR1 in whole cell lysates of EMAP II treated hPASMC cells had a time dependent phosphorylation of ERK and an overexpression of EGR1. (E) quantification of pERK/ERK (F) quantification of EGR1/tubulin. Results are shown as means ± SEM. n=3, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 using unpaired t-test.
EMAP II induced upregulation of nuclear EGR1 expression is mediated through ERK activation.
Previous studies have identified nuclear pERK1/2 as an important transcriptional regulator of EGR1 [34]. Utilizing nuclear and cytoplasmic fractions, the impact of EMAP II stimulation on pERK1/2 and EGR1 nuclear expression in macrophage and SMC populations was studied. Converse to pERK1/2 mediated EGR1 transcription, MEK inhibition by iERK (MEK inhibitor, 10 μM) and iERK1 (MEK inhibitor, 10 μM) blocked EMAP II induced EGR1 protein expression in both macrophages (Figure 3A and 3B) and SMCs (Figure 3D and 3E). To determine whether EGR1 transcription is mediated through ERK activation, EGR1 transcripts were quantified in macrophages and SMCs with or without iERK1 (MEK inhibitor, 10 μM) pretreatment following EMAP II stimulation and normalized against Hprt1 in macrophages (Figure 3C) and Gapdh in SMCs (Figure 3F). Results demonstrated EMAP II induced EGR1 transcription can be inhibited by blocking ERK1/2 phosphorylation. Furthermore, EMAP II significantly (~ 9-fold) upregulated nuclear expression of EGR1 in macrophages at 2 hours (Figure 4A and 4B) and in SMCs, EMAP II upregulated EGR1 expression (~ 2.5-fold) at 1 hour before subsiding (Figure 4C and 4D). Immunofluorescent analysis of SMCs stimulated with EMAP II for 1 hour demonstrated significantly elevated nuclear EGR1 expression as indicated by co-localization of EGR1 (red), DNA (blue) and lamin (green) as compared to control (Figure 4E and 4F). These results suggest that EMAP II upregulated EGR1 play a major role as a transcription factor in EMAP II mediated signaling cascade.
Figure 3. EGR1 upregulation is mediated through EMAP II induced ERK activation.
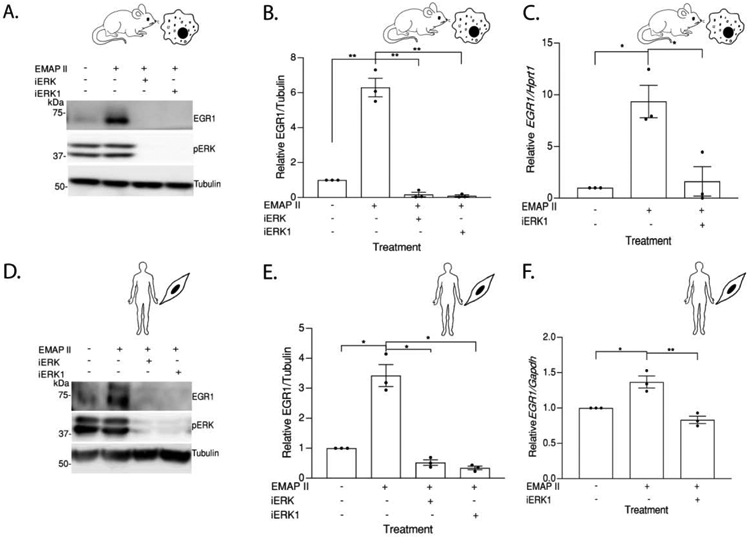
(A) Representative western blot probed for EGR1 in whole cell lysates of RAW 264.7 cells pretreated with PD184161 (iERK), Trametinib (iERK1) or vehicle exposed to EMAP II or vehicle for 2 hours (B) quantitation of EGR1/ Tubulin. (C) EGR1 expression levels normalized against Hprt1 in RAW 264.7 cells pretreated with Trametinib (iERK1) or vehicle and followed EMAP II treatment for 45 minutes. (D) Representative western blot probed for EGR1 in whole cell lysates of hPASMC cells pretreated with PD184161 (iERK), Trametinib (iERK1) or vehicle exposed to EMAP II or vehicle for 1 hour (E) quantitation of EGR1/ Tubulin. (F) EGR1 expression levels normalized against Gapdh in hPASMC cells pretreated with Trametinib (iERK1) or vehicle and followed EMAP II treatment for 45 minutes. Results are shown as means ± SEM. n=3, *p<0.05, **p<0.01 using unpaired t-test.
Figure 4. EMAP II induced upregulation of EGR1 expression is nuclear localized.

(A) Representative western blot probed for EGR1 and pERK in cytoplasmic and nuclear fractions of RAW 264.7 following EMAP II treatment for 0, 1, 2, 3 hours (B) quantification of nuclear EGR1/ lamin. (C) Representative western blot probed for EGR1 and pERK in cytoplasmic and nuclear fractions of hPASMC following EMAP II treatment for 0, 20 minutes and 1 and 2 hours (D) quantification of nuclear EGR1/ lamin. (E) Immunocytochemistry images of EGR1 expression in vehicle (i) or EMAPII (ii) treated hPASMC cells for 1 hour (red= EGR1, green= actin and blue=DNA), scale bar is 20 μm. Enlarged images of the nucleus in (ii) are on right. (F) quantification of fluorescence intensity of EGR1. Results are shown as means ± SEM. n=3, ns= non-significant, *p<0.05, **p<0.01, ***p<0.001 using unpaired t-test.
EMAP II upregulates SPHK1 transcription and translation through EGR1.
As EGR1 has been previously identified as a SPHK1 transcriptional mediator, the effect of EMAP II signaling on SPHK1 expression was measured. EMAP II significantly increased expression of SPHK1 at 6 hours in both macrophages (Figure 5A and 5B) and SMCs (Figure 5D and 5E). To determine the effect of EMAP II on SPHK1 transcription, qRT-PCR of SPHK1 transcription was performed. EMAP II induced a time-dependent increase in SPHK1 transcription that peaked at 3 hours after EMAP II stimulation in both macrophages and SMC types (Figure 5C and 5F).
Figure 5. EMAP II upregulates SPHK1 transcription and translation through EGR1.
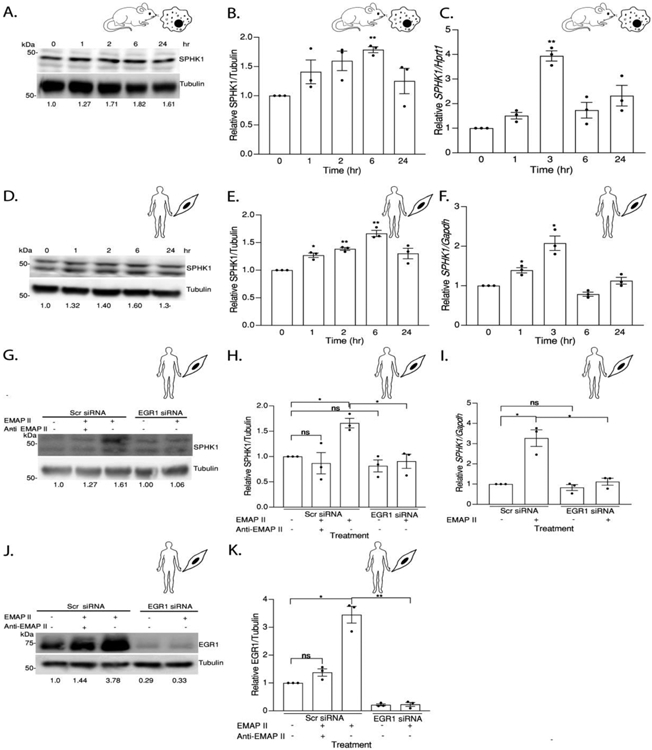
(A) Representative western blot probed for SPHK1 in whole cell lysates of RAW 264.7 cells following EMAP II treatment for 0, 1, 2, 6 and 24 hours (B) quantitation of SPHK1/ Tubulin. (C) SPHK1 expression levels normalized against Hprt1 in RAW 264.7 cells following EMAP II treatment for 0,1, 3, 6 and 24 hours. (D) Representative western blot probed for SPHK1 in whole cell lysates of hPASMC cells following treatment with EMAP II 0, 1, 2, 6, 24 hours (E) quantitation of SPHK1/ Tubulin. F) SPHK1 expression levels normalized against Gapdh in hPASMC cells following EMAP II treatment for 0, 1, 3, 6 and 24 hours. (G) Representative western blot probed for SPHK1 in whole cell lysates of hPASMC cells following siRNA mediated EGR1 silencing and EMAP II treatment for 6 hours (H) quantitation of SPHK1/ Tubulin. (I) SPHK1 expression levels normalized against Gapdh in hPASMC cells following siRNA mediated EGR1 silencing and EMAP II treatment for 3 hours. (J) Representative western blot probed for EGR1 in whole cell lysates of hPASMC cells following siRNA mediated EGR1 silencing and EMAP II treatment for 6 hours (K) quantitation of EGR1/ Tubulin. Results are shown as means ± SEM. n=3, ns= non-significant, *p<0.05, **p<0.01 using unpaired t-test.
Furthermore, knockdown of EGR1 using specific siRNA and blocking EMAP II activity by pretreating with an EMAP II neutralizing antibody significantly attenuated EMAP II induced SPHK1 protein upregulation at 6 hours (Figure 5G and 5H) and upregulation of transcript level at 3 hours (Figure 5I). Successful knockdown of EGR1 was confirmed by immunoblotting (Figure 5J and 5K).
EMAP II induces bimodal phosphorylation and plasma membrane localization of SPHK1.
Having established that SPHK1 is upregulated by EMAP II through EGR1, we sought to determine whether EMAP II affects the activation of SPHK1. Since phosphorylation of SPHK1 at Ser225 is required for SPHK1 activity [35], we initially studied the pSPHK1 protein expression after EMAP II stimulation at specific time points. In line with EMAP II induced bimodal S1P peaks (Figure 1), immunoblotting demonstrated that EMAP II induced bimodal SPHK1 phosphorylation at 1 and 6 hours in SMCs (Figure 6A and 6B). SPHK1 resides in the cytoplasm where upon phosphorylation by ERK1/2, it translocates to the plasma membrane [35]. Consistent with the immunoblotting data, immunofluorescence showed increased pSPHK1 signal (red) co-localized with actin (green) staining boundaries in SMCs stimulated with EMAP II for 15 minutes yet persisted for 60 minutes (Figure 6C). As human SMCs and mouse macrophages, human macrophages THP-1 also exhibited EMAP II induced bimodal ERK activation, EGR1 upregulation, SPHK1 overexpression and S1P generation as shown in Supplementary Figure 1. Moreover, elevated levels of pSPHK1 was observed near the THP1 cell membrane at 1 hour of EMAP II stimulation (Figure 6D) as indicated by actin staining. In addition, we observed that in SMCs after 6 hours of EMAP II treatment, pSPHK1 is concentrated around the membrane (Figure 6E) demonstrating bimodal phosphorylation of SPHK1 at 1 and 6 hours as suggested by the immunoblotting for pSPHK1.
Figure 6. EMAP II induces bi-modal phosphorylation and plasma membrane localization of SPHK1.

(A) Representative western blot probed for pSPHK1 in whole cell lysates of hPASMC cells following EMAP II treatment for 0, 1, 2, 6 and 24 hours (B) quantitation of pSPHK1/ Tubulin. (C) Representative immunofluorescence images of EMAP II treated primary hPASMC at 0, 15 min and 60 min. (D) Representative immunofluorescence images of vehicle (i) EMAP II (ii) treated THP1 human macrophages at 1 hour. (E) Representative immunofluorescence images of vehicle (i) EMAP II (ii) treated primary hPASMC at 6 hours. (red= pSPHK1, green= actin and blue= DNA and arrows indicate the membrane localization of pSPHK1), scale bar is 10 μm. Results are shown as means ± SEM. n=3, *p<0.05 using unpaired t-test.
EMAP II induced STAT3 activation in macrophages is linked with S1P signaling and ERK activation.
Our previous studies demonstrated that EMAP II induces a JAK dependent biphasic tyrosine phosphorylation of STAT3 (Y705) and increases STAT3 mediated transcriptional target genes in macrophages[19]. EMAP II induced STAT3 activation in macrophages while pretreatment with S1PR specific inhibitors [iS1PR1 (S1PR1 inhibitor, 1 μM), iS1PR2 (S1PR2 inhibitor, 1 μM), iS1PR3 (S1PR3 inhibitor, 1 μM)], SPHK1 inhibitor [iSPHK1, 100 nM)] as well as a JAK1/2 inhibitor [iJAK1/2, 1 μM] significantly inhibited EMAP II induction of pSTAT3 (Figure 7A and 7B). These findings were further supported as luminescence of STAT3 transcription reporter plasmid (Figure 7C) demonstrated that EMAP II induced STAT3 activation is linked to the S1P signaling axis. Pharmacological inhibition of SPHK1 significantly inhibited EMAP II induced STAT3 activation and luminescence of STAT3 transcription reporter plasmid (Figure 7A, 7B and 7C) suggesting that EMAP II signaling was upstream of S1PR. In SMCs, no STAT3 activation was observed (data not shown). Similar to EMAP II induction of ERK1/2 phosphorylation in endothelial cells [22], EMAP II induced ERK1/2 phosphorylation in macrophages (Figure 2) while pretreatment with a chemical inhibitor to block phosphorylation of ERK1/2 (iERK: MEK inhibitor, 10 μM) significantly inhibited EMAP II induced pSTAT3 (Figure 7D and 7E) suggesting that pSTAT3 activation may occur through EMAP II initiated ERK1/2 mediated signaling cascade.
Figure 7. EMAP II induced STAT3 activation in macrophages is linked with S1P signaling and ERK activation.
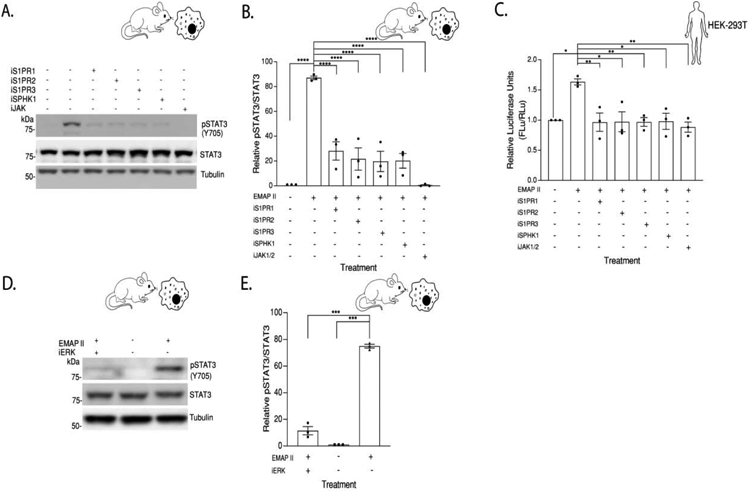
(A) Representative western blot probed for pSTAT3 in whole cell lysates of RAW 264.7 cells pretreated with W146 (iSIPR1), JTE013 (iS1PR2), TY52156 (iS1PR3), PF543 (iSPHK1), Ruxolitinib (iJak1/2) or vehicle exposed to EMAP II or vehicle for 24 hours (B) quantitation of pSTAT3/STAT3. (C) HEK293-T cells were reverse transfected with 200 ng of pGL3-STAT3 and 10 ng of pRL-null vectors, pretreated with W146 (iSIPR1), JTE013 (iS1PR2), TY52156 (iS1PR3), PF543 (iSPHK1), Ruxolitinib (iJak1/2) or vehicle exposed to EMAP II or vehicle and luciferase activity measured after 24 hours. (B) and (C) Following one-way ANOVA, Tukey’s honestly significant difference test, *p<0.05, **p<0.01, ****p<0.0001 (D) Representative western blot probed for pSTAT3 in whole cell lysates of RAW 264.7 cells pretreated with PD184161 (iERK) or vehicle exposed to EMAP II or vehicle for 24 hours. (E) quantitation of pSTAT3/STAT3, ***p<0.001 using unpaired t-test. Results are shown as means ± SEM. n=3
The downstream effects of EMAP II/S1P signaling are cell type specific.
Although EMAP II signaling activates STAT3 in macrophages as shown in Figure 7, EMAP II does not impact pSTAT3 levels in SMCs (data not shown). Moreover, EMAP II induced tumor necrosis factor (TNF) cytokine production in macrophages as demonstrated by increased Tnf transcription level (Figure 8E) while this observation was absent in SMCs (data not shown). We discovered that EMAP II modulates cyclin D1 in SMCs in an ERK1/2 dependent manner instead of TNF∝ after 48 hours of EMAP II stimulation (Figure 8A and 8B) which was confirmed by WST1 proliferation assay (Figure 8C) suggesting that EMAP II promotes cell proliferation in SMCs. Importantly, inhibition of SPHK1 ablated EMAP II promoted proliferation in SMCs (Figure 8C) without affecting the basal cell proliferation rate. Likewise, increased transcription of a major proliferative marker in SMCs [36] interleukin 6 (Il6) was found in SMCs by qRT-PCR (Figure 8D).
Figure 8. The downstream effects of EMAP II/ S1P signaling are cell type specific.
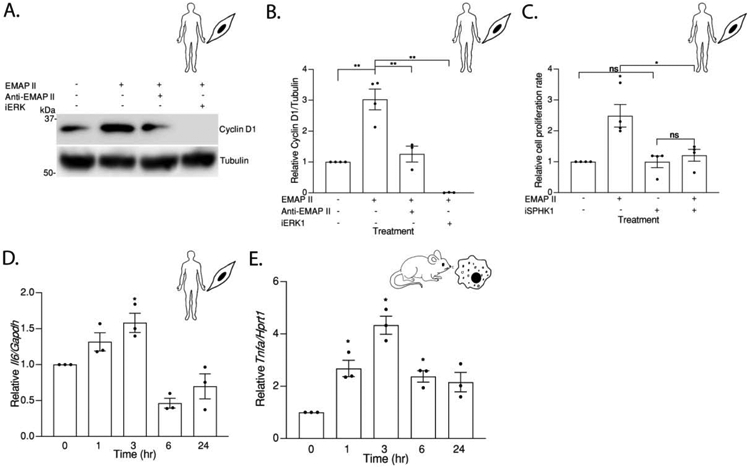
(A) Representative western blot probed for Cyclin D1 in whole cell lysates of hPASMC cells pretreated with PD184161 (iERK), EMAP II neutralizing antibody (Anti-EMAP II) or vehicle following EMAP II or vehicle treatment for 48 hours (B) quantitation of Cyclin D1/ Tubulin. (C) Cell proliferation rate in hPASMC cells pretreated with PF543 (iSPHK1) or vehicle following EMAP II treatment for 48 hours. (D) Il6 expression levels normalized against Gapdh in hPASMC cells following EMAP II treatment for 0, 1, 3, 6 and 24 hours. (E) Tnf expression levels normalized against Hprt1 in RAW 264.7 cells following EMAP II treatment for 0, 1, 3, 6 and 24 hours. Results are shown as means ± SEM. n=3-4, ns= non-significant, *p<0.05, **p<0.01 using unpaired t-test.
Discussion
The regulatory role of the bioactive lipid S1P in vasoconstriction, proliferation, fibrosis and vascular inflammation has attributed a growing recognition of S1P as a critical regulator of several cardiovascular and pulmonary pathophysiological processes including PAH[11,25,31,37]. Understanding the molecular mechanism and key regulators of enigmatic S1P could guide the discovery of potential treatments for PAH. The molecular regulation of S1P generation catalyzing kinase SPHK1 is not well described. Here, we report a novel role of EMAP II in mediating cellular responses through triggering a bimodal phosphorylation of sphingosine, generating S1P in a two-pronged manner by modulating phosphorylation, transcriptional regulation and translocation of SPHK1 through a common and coherent upstream signaling in inflammatory cells and SMCs. EMAP II initiates cell specific downstream pathophysiological functions that may contribute to the pathogenesis PAH. We identified that 1) EMAP II induces bimodal ERK1/2 activation and SPHK1 activation, 2) EMAP II induced ERK1/2 results in overexpression of transcription regulator of differentiation, inflammation, and mitogenesis protein coding gene EGR1, 3) EMAP II induced elevated nuclear expression of EGR1 regulates SPHK1 transcription, and 4) EMAP II promotes membrane localization of pSPHK1 and 0increases S1P expression (Figure 9). Thus, identifying the upstream crosstalk between S1P and EMAP II in downstream molecular and signaling consequences that stimulate macrophage pro-inflammatory mediators and SMC proliferation associated with development PAH sheds new insight into the regulation of S1P homeostasis.
Figure 9. Potential mechanism of EMAP II/ S1P signaling and modulation of S1P homeostasis.

EMAP II stimulation activates a common signaling pathway where ERK activation induces bimodal phosphorylation of SPHK1 and EGR1 overexpression. EGR1 overexpression upregulates SPHK1 transcription and translation. Increased SPHK1 and phosphorylated SPHK1 results in augmented S1P levels which generates cell type-specific downstream effects such as pro-inflammatory signals in macrophages and pro-proliferative signals in SMCs.
S1P has been implicated in several cardiopulmonary diseases. S1P levels reported to be elevated in lung tissues of patients with PAH where increased transcription of SPHK1 showed to play the critical role in disrupting the S1P homeostasis [31,38]. SPHK1 mediated increased proliferation of perivascular SMCs reported to cause PAH [39]. Moreover, a clinical study identified S1P as a stronger biomarker of severe atherosclerosis [40,41]. Recent studies reported the knockdown or pharmacological inhibition of SPHK1 attenuates bronchopulmonary dysplasia (BPD) in mouse models warranting that SPHK1/S1P would be a potential signaling pathway to be targeted in BPD [25,42]. Similarly, moonlighting roles for EMAP II in disease progression mediated by inflammation include mediating inflammation by immune cell trafficking, angiogenesis and apoptosis. Previous studies link EMAP II to endothelial cell apoptosis, a detrimental factor in atherosclerosis and increased expression of E- and P-selectins and Tnf-1 which results in monocyte and neutrophils attraction [43,44]. Moreover, EMAP II mediated macrophage recruitment and pro-inflammatory cytokine production are seen in BPD mouse models [17,45]. However, this study is the first to report the potential mechanistic contribution of EMAP II to the development of PAH.
Both immunochemistry and mass spectrometry studies showed EMAP II stimulated bimodal peaking of S1P and S1P: sphingosine ratio at 1 and 6 hours suggesting that the protein, EMAP II is a potential upstream mediator of S1P generation. Moreover, the decreasing trend of ceramide can be ascribed to increasing ceramide degradation as S1P is a derivative of ceramide hydrolysis with the ceramide-sphingosine-S1P rheostat known to control the cell fate [28]. Therefore, the depreciation of ceramides presumably, supports the observation of S1P elevation. Our recent studies reported that AIMP1 binds to phosphoinositide, a phospholipid that has a fundamental role in the cellular membrane structure and cellular signaling [46] implicating a role for EMAP II in the metabolism of membrane phospholipids. Moreover, a bimodal activation of sphingosine to S1P is further evident by immunoblotting and immunofluorescence studies of activating phosphorylation of SPHK1, the S1P conversion catalyzing enzyme. The phosphorylation of SPHK1 at Ser225 by ERK1/2 activators such as phorbol esters and PDGF increases SPHK1 enzyme activity by 14-fold and mediates the translocation of the enzyme from the cytosol to the plasma membrane bringing it into proximity to its substrates. The translocation of SPHK1 is assisted by the interaction with calmodulin-related protein calcium and integrin-binding protein 1 (CIB1) which provides a plasma membrane docking site via myristoylation where it’s retention facilitates sphingosine phosphorylation [35,47,48]. Importantly, high cytosolic Ca2+ detected upon CIB1-SPHK1 interaction [35] is consistent with previous reports that showed EMAP II treatment causes redistribution of intracellular calcium stores into the cytosol [49].
We observed a similar bimodal pattern in both ERK1/2 and SPHK1 activation, suggesting an inter-dependent relationship between ERK1/2 and SPHK1. ERK1/2 is one of the members of mitogen-activated protein kinase super family and is known to directly phosphorylate SPHK1 at Ser225 residue [35,47]. Additionally, SPHK1 carries a docking site for ERK1/2 [50]. However, there is evidence showing that ERK1/2 activated SPHK1 is capable of activating ERK1/2 back in a positive feedback mechanism [51] suggesting that the early observed (1 hr) pSPHK1 may be the activator of secondary ERK1/2 activation in EMAP II/SPHK1/S1P signaling pathway. For the first time, we report that EMAP II phosphorylates SPHK1 at Ser225 by acting as an activator of ERK1/2 which may eventually initiate the feedback loop between ERK1/2 and SPHK1.
Interestingly, we found that EMAP II induces overexpression of nuclear EGR1, another known target of ERK1/2 [32,33,35,52-54] and transcription activator of SPHK1 [30,31]. EGR1 is a 80–82-kD inducible zinc finger transcription factor that has also been known to regulate cell growth, proliferation and inflammation in lung disorders [55-57]. Identified as one of the early biomarkers of lung injury in preterm lambs, EGR1 mRNA levels were increased 120-fold within 30 minutes of injury [58]. Moreover, pharmacological inhibition of EGR1 has been reported to attenuate pulmonary vascular resistance and right ventricular hypertrophy in animal models [55]. Our experiments indicate that inhibition of EGR1 suppresses EMAP II mediated SPHK1 transcription representing a potential mechanism of regulating downstream S1P generation. Since our studies show that EMAP II signaling provokes a bimodal early upstream SPHK1 phosphorylation in addition to the later EGR1 mediated SPHK1 transcription and activation, it is questionable whether inhibition of EGR1 alone would be a promising therapeutic target for impeding EMAP II/SPHK1/S1P signaling in PAH. However, ablating ERK1/2 phosphorylation has been reported to inhibit EGR1 induced pulmonary vascular proliferation in newborn pulmonary hypertension calves models [52,55] supporting the fact that the phosphorylation of ERK1/2 is a key regulator of both SPHK1 phosphorylation and transcriptional regulation. Thus providing a critical target point in regulation of downstream sphingosine phosphorylation. In addition, we observed about 4 times induction of SPHK1 transcription in EMAP II treated macrophages in compared to the control. Nevertheless, the protein expression of SPHK1 in macrophages was only about 1.7 times. One possibility for the difference between the fold change of transcripts and protein expression levels might be due to multiple factors that affecting the protein translation including the level of mRNA and its half-life, translational efficiency, and the stability of the interested protein [59].
To demonstrate the potential of translating this novel signaling mechanism into in vivo system, EMAP II treated postnatal day 15 mice were used as in Lee et al. 2016 reported right ventricular hypertension and increased elastin deposition in neonatal mice of postnatal day 42 treated with EMAP II in 3-15 days of life that promoted BPD-like phenotype suggesting that EMAP II induces secondary PAH signs associated with BPD. Moreover, Lee et al. 2016 reported that those mice exhibit high inflammatory responses on postnatal day 15. Compared to control, EMAP II treated postnatal day 15 mice lungs showed a significant increase in the tissue S1P levels suggesting that EMAP II induced S1P could contribute to the development of PAH in BPD.
Although upstream signaling of EMAP II/ERK/EGR1/SPHK1/S1P is common in both cell types of macrophages and SMCs, our studies demonstrated cell type-specific downstream responses suggesting that downstream effects may depend on the cell type and the availability of different S1PRs. In macrophages, EMAP II signaling induces STAT3 activation via S1PRs which mediates inflammatory state as shown by prior studies [18,19] while in SMCs, EMAP II signaling results in proliferative signaling by overexpressing cell cycle regulating proteins like cyclin D1 which is required for G1/S phase transition. We report that EMAP II promotes proliferation in SMCs challenging the conventional known apoptotic function of EMAP II in endothelial cells [60,61] and is consistent with previous reports of increased peri-vascular SMC EMAP II expression in infants with BPD [62]. A recent study has shown that macrophage migration inhibitory factor upregulates cyclin D1 via ERK1/2 leading to SMC proliferation causing pulmonary hypertension in animal models [63].
As such our future studies will explore if S1P/S1PR is a potential therapeutic target to regulate these downstream effects while minimizing consequences associated with S1P signaling. Elevated early response signaling molecules recruit inflammatory cells such as neutrophils and macrophages. Macrophages are the prominent inflammatory cells in chronic inflammation due to its functions in promotion and resolution of inflammation, pathogen clearance, and tissue restoration following injury [64]. Chronic inflammation mediated by macrophages can contribute to the arterial remodeling observed PAH. The dynamic interplay between macrophages and SMCs in vascular diseases is well recognized [65]. Therefore, the source of EMAP II that acts on SMCs to promote proliferation can be expressed and secreted in recruited macrophages suggesting a novel aspect of a complex signaling mechanism to be explored. We report EMAP II as a mediator of S1P burden. However, the proteolytically cleaved protein EMAP II and the bioactive lipid S1P are signals that get triggered as early response chemokines prior to influx of inflammatory cells upon the cellular damage. Therefore, in physiological conditions although it is possible that S1P is activated prior to EMAP II, our findings suggest that there is an intimate crosstalk between two biological molecules may initiate a feedback loop mechanism elevating cellular S1P concentration.
In summary, our findings indicate that EMAP II can be a potential key mediator of the bioactive lipid S1P. EMAP II modulated S1P signaling promotes activation of key mechanistic signaling pathways that can contribute to the pro-inflammation and SMC hyperproliferation in PAH through a two-phase process. An upstream common protein initiated biphasic activation of the bioactive lipid S1P and a lipid mediated downstream cell specific signaling pathway that contributes to PAH progression. Future studies exploring this signaling mechanism in PAH are warranted to identify potential therapeutic targets aimed at treating inflammation and vascular remodeling associated with PAH.
Supplementary Material
Highlights.
Unique identification of EMAP II mediated cellular responses through triggering a bimodal phosphorylation of sphingosine generating the bioactive sphingolipid S1P in a two-pronged manner by modulating phosphorylation, transcriptional regulation and translocation of the S1P synthesis catalyzing kinase SPHK1.
Representation of a binary mechanism of mediating cell type- specific downstream S1P mediated pro-inflammatory and proliferative smooth muscle cellular responses that may contribute to pulmonary arterial hypertension.
Acknowledgement
This publication was made possible in part by National Heart, Lung, and Blood Institute (NHLBI) Grant 5R01 HL114977 (M. A. Schwarz) and R21 HO090227 (M. A. Schwarz) from NIH and; the Lilly Endowment Physician Scientist Initiative (M. A. Schwarz); the Indiana Clinical and Translational Sciences Institute funded in part by Indiana CTSI Collaboration in Biomedical/Translational Research (CBR/CTR) Pilot Program Grants; and National Institutes of Health, National Center for Advancing Translational Sciences, Clinical and Translational Sciences Award UL1TR001108. Fellowship support was provided by NIH to D. D. Lee (NHLBI Grant T32 HL091816). The authors thank Lipidomics Shared Resources, Medical University of South Carolina for HPLC/MS/MS analysis of sphingolipids.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Montani D, Günther S, Dorfmüller P, Perros F, Girerd B, Garcia G, Jaïs X, Savale L, Artaud-Macari E, Price LC, Humbert M, Simonneau G, Sitbon O, Pulmonary arterial hypertension, Orphanet J. Rare Dis. 8 (2013) 97 10.1186/1750-1172-8-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kuhr FK, Smith KA, Song MY, Levitan I, Yuan JX-J, New mechanisms of pulmonary arterial hypertension: role of Ca2+ signaling., Am. J. Physiol. Heart Circ. Physiol 302 (2012) H1546–62. 10.1152/ajpheart.00944.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Farber HW, Loscalzo J, Pulmonary Arterial Hypertension, 2004. www.nejm.org (accessed February 22, 2019). [Google Scholar]
- [4].Price LC, Wort SJ, Perros F, Dorfmüller P, Huertas A, Montani D, Cohen-Kaminsky S, Humbert M, Inflammation in Pulmonary Arterial Hypertension, Chest. 141 (2012) 210–221. 10.1378/CHEST.11-0793. [DOI] [PubMed] [Google Scholar]
- [5].Dorfmüller P, Perros F, Balabanian K, Humbert M, Brenot F, Rain B, Capron F, Galanaud P, Duroux P, Simonneau G, Emilie D, Inflammation in pulmonary arterial hypertension., Eur. Respir. J 22 (2003) 358–63. 10.1183/09031936.03.00038903. [DOI] [PubMed] [Google Scholar]
- [6].Groth A, Vrugt B, Brock M, Speich R, Ulrich S, Huber LC, Inflammatory cytokines in pulmonary hypertension., Respir. Res 15 (2014) 47 10.1186/1465-9921-15-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chan SY, Loscalzo J, Pathogenic mechanisms of pulmonary arterial hypertension, J. Mol. Cell. Cardiol 44 (2008) 14–30. 10.1016/j.yjmcc.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Noureddine H, Gary-Bobo G, Alifano M, Marcos E, Saker M, Vienney N, Amsellem V, Maitre B, Chaouat A, Chouaid C, Dubois-Rande JL, Damotte D, Adnot S, Pulmonary artery smooth muscle cell senescence is a pathogenic mechanism for pulmonary hypertension in chronic lung disease, Circ. Res 109 (2011) 543–553. 10.1161/CIRCRESAHA.111.241299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chen J, Tang H, Sysol JR, Moreno-Vinasco L, Shioura KM, Chen T, Gorshkova I, Wang L, Huang LS, Usatyuk PV, Sammani S, Zhou G, Raj JU, Garcia JGN, Berdyshev E, Yuan JXJ, Natarajan V, Machado RF, The sphingosine kinase 1/sphingosine-1-phosphate pathway in pulmonary arterial hypertension, Am. J. Respir. Crit. Care Med 190 (2014) 1032–1043. 10.1164/rccm.201401-0121OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yan X, Wang J, Zhu Y, Feng W, Zhai C, Liu L, Shi W, Wang Q, Zhang Q, Chai L, Li M, S1P induces pulmonary artery smooth muscle cell proliferation by activating calcineurin/NFAT/OPN signaling pathway, Biochem. Biophys. Res. Commun 516 (2019) 921–927. 10.1016/j.bbrc.2019.06.160. [DOI] [PubMed] [Google Scholar]
- [11].Maceyka M, Harikumar KB, Milstien S, Spiegel S, Sphingosine-1-phosphate signaling and its role in disease, Trends Cell Biol. (2012). 10.1016/j.tcb.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Mendelson K, Evans T, Hla T, Sphingosine 1-phosphate signalling, Development. (2013). 10.1242/dev.094805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wang Y, Wang D, Zhang L, Ye F, Li M, Wen K, Role of JAK-STAT pathway in reducing cardiomyocytes hypoxia/reoxygenation injury induced by S1P postconditioning, Eur. J. Pharmacol (2016). 10.1016/j.ejphar.2016.05.024. [DOI] [PubMed] [Google Scholar]
- [14].Mohammed S, Harikumar KB, Sphingosine 1-Phosphate: A Novel Target for Lung Disorders, Front. Immunol 8 (2017). 10.3389/fimmu.2017.00296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].You Y, Guo C, Zhang H, Deng S, Tang J, Xu L, Deng C, Gong F, Effect of Intranasal Instillation of Lipopolysaccharide on Lung Development and Its Related Mechanism in Newborn Mice., J. Interferon Cytokine Res (2019). 10.1089/jir.2019.0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Medina CB, Ravichandran KS, Do not let death do us part: “Find-me” signals in communication between dying cells and the phagocytes, Cell Death Differ. (2016). 10.1038/cdd.2016.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lee DD, V Lal C, Persad EA, Lowe C-W, Schwarz AM, Awasthi N, Schwarz RE, Schwarz MA, Endothelial Monocyte-Activating Polypeptide II Mediates Macrophage Migration in the Development of Hyperoxia-Induced Lung Disease of Prematurity., Am. J. Respir. Cell Mol. Biol 55 (2016) 602–612. 10.1165/rcmb.2016-0091OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Lee DD, Lal CV, Persad EA, Lowe CW, Schwarz AM, Awasthi N, Schwarz RE, Schwarz MA, Endothelial monocyte-activating polypeptide II mediates macrophage migration in the development of hyperoxia-induced lung disease of prematurity, Am. J. Respir. Cell Mol. Biol (2016). 10.1165/rcmb.2016-0091OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lee DD, Hochstetler A, Murphy C, Lowe C-W, Schwarz MA, A Distinct Transcriptional Profile in Response to Endothelial Monocyte Activating Polypeptide II is Partially Mediated by JAK-STAT3 in Murine Macrophages, Am. J. Physiol. Physiol (2019) ajpcell.00277.2018. 10.1152/ajpcell.00277.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Liu J, Schwarz MA, Identification of protease-sensitive sites in Human Endothelial-Monocyte Activating Polypeptide II protein., Exp. Cell Res 312 (2006) 2231–7. 10.1016/j.yexcr.2006.03.024. [DOI] [PubMed] [Google Scholar]
- [21].Schwarz RE, Schwarz MA, In vivo therapy of local tumor progression by targeting vascular endothelium with EMAP-II, J. Surg. Res 120 (2004) 64–72. 10.1016/j.jss.2003.10.005. [DOI] [PubMed] [Google Scholar]
- [22].Schwarz MA, Kandel J, Brett J, Li J, Hayward J, Schwarz RE, Chappey O, Wautier JL, Chabot J, Lo Gerfo P, Stern D, Endothelial-monocyte activating polypeptide II, a novel antitumor cytokine that suppresses primary and metastatic tumor growth and induces apoptosis in growing endothelial cells., J. Exp. Med 190 (1999) 341–54. 10.1084/jem.190.3.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Awasthi N, Schwarz MA, Verma V, Cappiello C, Schwarz RE, Endothelial monocyte activating polypeptide II interferes with VEGF-induced proangiogenic signaling., Lab. Invest 89 (2009) 38–46. 10.1038/labinvest.2008.106. [DOI] [PubMed] [Google Scholar]
- [24].Schwarz MA, Zheng H, Liu J, Corbett S, Schwarz RE, Endothelial-monocyte activating polypeptide II alters fibronectin based endothelial cell adhesion and matrix assembly via alpha5 beta1 integrin., Exp. Cell Res 311 (2005) 229–39. 10.1016/j.yexcr.2005.09.008. [DOI] [PubMed] [Google Scholar]
- [25].Harijith A, Pendyala S, Reddy NM, Bai T, Usatyuk PV, Berdyshev E, Gorshkova I, Huang LS, Mohan V, Garzon S, Kanteti P, Reddy SP, Raj JU, Natarajan V, Sphingosine kinase 1 deficiency confers protection against hyperoxia-induced bronchopulmonary dysplasia in a murine model: Role of s1p signaling and nox proteins, Am. J. Pathol (2013). 10.1016/j.ajpath.2013.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Schwarz MA, Zhang F, Gebb S, Starnes V, Warburton D, Endothelial Monocyte Activating Polypeptide II inhibits lung neovascularization and airway epithelial morphogenesis, Mech. Dev 95 (2000) 123–132. 10.1016/S0925-4773(00)00361-0. [DOI] [PubMed] [Google Scholar]
- [27].Schwarz MA, Zhang F, Gebb S, Starnes V, Warburton D, Endothelial Monocyte Activating Polypeptide II inhibits lung neovascularization and airway epithelial morphogenesis, Mech. Dev (2000). 10.1016/S0925-4773(00)00361-0. [DOI] [PubMed] [Google Scholar]
- [28].Loh KC, Baldwin D, Saba JD, Sphingolipid signaling and hematopoietic malignancies: to the rheostat and beyond., Anticancer. Agents Med. Chem 11 (2011) 782–93. http://www.ncbi.nlm.nih.gov/pubmed/21707493 (accessed October 10, 2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lee DD, Lal CV, Persad EA, Lowe C-W, Schwarz AM, Awasthi N, Schwarz RE, Schwarz MA, Endothelial Monocyte-Activating Polypeptide II Mediates Macrophage Migration in the Development of Hyperoxia-Induced Lung Disease of Prematurity, Am. J. Respir. Cell Mol. Biol 55 (2016) 602–612. 10.1165/rcmb.2016-0091OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sysol JR, Natarajan V, Machado RF, PDGF induces SphK1 expression via Egr-1 to promote pulmonary artery smooth muscle cell proliferation, Am. J. Physiol. Physiol (2016). 10.1152/ajpcell.00059.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Li F, Wang J, Zhu Y, Liu L, Feng W, Shi W, Wang Q, Zhang Q, Chai L, Li M, SphK1/S1P mediates PDGF-induced pulmonary arterial smooth muscle cell proliferation via miR-21/BMPRII/Id1 signaling pathway, Cell. Physiol. Biochem (2018). 10.1159/000495243. [DOI] [PubMed] [Google Scholar]
- [32].Maegawa M, Arao T, Yokote H, Matsumoto K, Kudo K, Tanaka K, Kaneda H, Fujita Y, Ito F, Nishio K, EGFR mutation up-regulates EGR1 expression through the ERK pathway., Anticancer Res. 29 (2009) 1111–7. http://www.ncbi.nlm.nih.gov/pubmed/19414352 (accessed July 10, 2019). [PubMed] [Google Scholar]
- [33].Adams KW, Kletsov S, Lamm RJ, Elman JS, Mullenbrock S, Cooper GM, Role for Egr1 in the Transcriptional Program Associated with Neuronal Differentiation of PC12 Cells, (2017). 10.1371/journal.pone.0170076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gregg J, Fraizer G, Transcriptional Regulation of EGR1 by EGF and the ERK Signaling Pathway in Prostate Cancer Cells, Genes and Cancer. 2 (2011) 900–909. 10.1177/1947601911431885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Jarman KE, Moretti PAB, Zebol JR, Pitson SM, Translocation of sphingosine kinase 1 to the plasma membrane is mediated by calcium- and integrin-binding protein 1, J. Biol. Chem (2010). 10.1074/jbc.M109.068395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yao JS, Zhai W, Fan Y, Lawton MT, Barbaro NM, Young WL, Yang G-Y, Interleukin-6 upregulates expression of KDR and stimulates proliferation of human cerebrovascular smooth muscle cells, J. Cereb. Blood Flow Metab 27 (2007) 510–520. 10.1038/sj.jcbfm.9600365. [DOI] [PubMed] [Google Scholar]
- [37].Wideman RF, Rhoads DD, Erf GF, Anthony NB, Pulmonary arterial hypertension (ascites syndrome) in broilers: A review, Poult. Sci (2013). 10.3382/ps.2012-02745. [DOI] [PubMed] [Google Scholar]
- [38].Chen J, Tang H, Sysol JR, Moreno-Vinasco L, Shioura KM, Chen T, Gorshkova I, Wang L, Huang LS, Usatyuk PV, Sammani S, Zhou G, Raj JU, Garcia JGN, Berdyshev E, Yuan JXJ, Natarajan V, Machado RF, The sphingosine kinase 1/sphingosine-1-phosphate pathway in pulmonary arterial hypertension, Am. J. Respir. Crit. Care Med 190 (2014) 1032–1043. 10.1164/rccm.201401-0121OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sysol JR, Natarajan V, Machado RF, PDGF induces SphK1 expression via Egr-1 to promote pulmonary artery smooth muscle cell proliferation, Am J Physiol Cell Physiol 310 (2016) 983–992. 10.1152/ajpcell.00059.2016.-Pulmo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Daum G, Grabski A, Reidy MA, Sphingosine 1-phosphate: A regulator of arterial lesions, Arterioscler. Thromb. Vasc. Biol 29 (2009) 1439–1443. 10.1161/ATVBAHA.108.175240. [DOI] [PubMed] [Google Scholar]
- [41].Deutschman DH, Carstens JS, Klepper RL, Smith WS, Page MT, Young TR, Gleason LA, Nakajima N, Sabbadini RA, Predicting obstructive coronary artery disease with serum sphingosine-1-phosphate, Am. Heart J 146 (2003) 62–68. 10.1016/S0002-8703(03)00118-2. [DOI] [PubMed] [Google Scholar]
- [42].Hendricks-Muñoz KD, Xu J, Voynow JA, Tracheal aspirate VEGF and sphingolipid metabolites in the preterm infant with later development of bronchopulmonary dysplasia., Pediatr. Pulmonol 53 (2018) 1046–1052. 10.1002/ppul.24022. [DOI] [PubMed] [Google Scholar]
- [43].Mogylnytska LA, [Endothelial monocyte-activating polypeptide-II: properties, functions, and pathogenetic significance]., Fiziol. Zh 61 (2015) 102–11. http://www.ncbi.nlm.nih.gov/pubmed/26040042 (accessed September 17, 2019). [DOI] [PubMed] [Google Scholar]
- [44].Chelvanambi S, Bogatcheva NV, Bednorz M, Agarwal S, Maier B, Alves NJ, Li W, Syed F, Saber MM, Dahl N, Lu H, Day RB, Smith P, Jolicoeur P, Yu Q, Dhillon NK, Weissmann N, Twigg HL, Clauss M, HIV-Nef protein persists in the lungs of aviremic patients with HIV and induces endothelial cell death, Am. J. Respir. Cell Mol. Biol 60 (2019) 357–366. 10.1165/rcmb.2018-0089OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Lal CV, Schwarz MA, Vascular mediators in chronic lung disease of infancy: Role of endothelial monocyte activating polypeptide II (EMAP II), Birth Defects Res. Part A - Clin. Mol. Teratol 100 (2014) 180–188. 10.1002/bdra.23234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lee DD, Hochstetler A, Sah E, Xu H, Lowe C-W, Santiaguel S, Thornton JL, Pajakowski A, Schwarz MA, Influence of Aminoacyl tRNA Synthetase Complex Interacting Multifunctional Protein 1 on Epithelial Differentiation and Organization During Lung Development, Am. J. Physiol. Cell. Mol. Physiol (2020). 10.1152/ajplung.00518.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Pitson SM, Moretti PAB, Zebol JR, Lynn HE, Xia P, Vadas MA, Wattenberg BW, Activation of sphingosine kinase 1 by ERK1/2-mediated phosphorylation, EMBO J. (2003). 10.1093/emboj/cdg540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Bryan L, Kordula T, Spiegel S, Milstien S, Regulation and functions of sphingosine kinases in the brain, Biochim. Biophys. Acta - Mol. Cell Biol. Lipids (2008). 10.1016/j.bbalip.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kao J, Houck K, Fan Y, Haehnel I, Libutti SK, Kayton ML, Grikscheit T, Chabot J, Nowygrod R, Greenberg S, Characterization of a novel tumor-derived cytokine. Endothelial-monocyte activating polypeptide II., J. Biol. Chem 269 (1994) 25106–19. http://www.ncbi.nlm.nih.gov/pubmed/7929199 (accessed July 18, 2019). [PubMed] [Google Scholar]
- [50].Sharrocks AD, Yang SH, Galanis A, Docking domains and substrate-specificity determination for MAP kinases, Trends Biochem. Sci (2000). 10.1016/S0968-0004(00)01627-3. [DOI] [PubMed] [Google Scholar]
- [51].Bazzazi H, Popel AS, Computational investigation of sphingosine kinase 1 (SphK1) and calcium dependent ERK1/2 activation downstream of VEGFR2 in endothelial cells, PLoS Comput. Biol (2017). 10.1371/journal.pcbi.1005332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Hartney T, Birari R, Venkataraman S, Villegas L, Martinez M, Black SM, Stenmark KR, Nozik-Grayck E, Xanthine oxidase-derived ROS upregulate Egr-1 via ERK1/2 in PA smooth muscle cells; model to test impact of extracellular ROS in chronic hypoxia, PLoS One. (2011). 10.1371/journal.pone.0027531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ghatak S, Markwald RR, Hascall VC, Dowling W, Lottes RG, Baatz JE, Beeson G, Beeson CC, Perrella MA, Thannickal VJ, Misra S, Fosang AJ, Transforming growth factor β1 (TGF β1) regulates CD44V6 expression and activity through extracellular signal-regulated kinase (ERK)-induced EGR1 in pulmonary fibrogenic fibroblasts, J. Biol. Chem (2017). 10.1074/jbc.M116.752451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Gregg J, Fraizer G, Transcriptional Regulation of EGR1 by EGF and the ERK Signaling Pathway in Prostate Cancer Cells, Genes and Cancer. (2011). 10.1177/1947601911431885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Dickinson MG, Kowalski PS, Bartelds B, Borgdorff MAJ, Van Der Feen D, Sietsma H, Molema G, Kamps JAAM, Berger RMF, Acritical role for Egr-1 during vascular remodelling in pulmonary arterial hypertension, Cardiovasc. Res (2014). 10.1093/cvr/cvu169. [DOI] [PubMed] [Google Scholar]
- [56].Extinguishing Egr-1-dependent inflammatory and thrombotic cascades after lung transplantation., FASEB J. (2001). 10.1096/fj.01-0490fje. [DOI] [PubMed] [Google Scholar]
- [57].Chu L, Wang T, Hu Y, Gu Y, Su Z, Jiang H, Activation of Egr-1 in Human Lung Epithelial Cells Exposed to Silica through MAPKs Signaling Pathways, PLoS One. (2013). 10.1371/journal.pone.0068943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Wallace MJ, Probyn ME, Zahra VA, Crossley K, Cole TJ, Davis PG, Morley CJ, Hooper SB, Early biomarkers and potential mediators of ventilation-induced lung injury in very preterm lambs, Respir. Res (2009). 10.1186/1465-9921-10-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Maier T, Güell M, Serrano L, Correlation of mRNA and protein in complex biological samples, FEBS Lett. (2009). 10.1016/j.febslet.2009.10.036. [DOI] [PubMed] [Google Scholar]
- [60].Van Horssen R, Rens JAP, Schipper D, Eggermont AMM, Hagen TLMT, EMAP-II facilitates TNF-R1 apoptotic signalling in endothelial cells and induces TRADD mobilization, Apoptosis. (2006). 10.1007/s10495-006-0284-5. [DOI] [PubMed] [Google Scholar]
- [61].Schwarz MA, Kandel J, Brett J, Li J, Hayward J, Schwarz RE, Chappey O, Wautier JL, Chabot J, Lo Gerfo P, Stern D, Endothelial-monocyte activating polypeptide II, a novel antitumor cytokine that suppresses primary and metastatic tumor growth and induces apoptosis in growing endothelial cells., J. Exp. Med 190 (1999) 341–54. 10.1084/jem.190.3.341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Quintos-Alagheband ML, White CW, Schwarz MA, Potential role for antiangiogenic proteins in the evolution of bronchopulmonary dysplasia., Antioxid. Redox Signal 6 (2004) 137–45. 10.1089/152308604771978444. [DOI] [PubMed] [Google Scholar]
- [63].Li H, Wang Y, Chen L, Han L, Li L, He H, Li Y, Huang N, Ren H, Pei F, Li G, Cheng J, Wang W, The role of MIF, cyclinD1 and ERK in the development of pulmonary hypertension in broilers, Avian Pathol. (2017). 10.1080/03079457.2016.1245409. [DOI] [PubMed] [Google Scholar]
- [64].Allison AC, Ferluga J, Prydz H, Schorlemmer HU, The role of macrophage activation in chronic inflammation., Agents Actions. 8 (1978) 27–35. 10.1007/bf01972398. [DOI] [PubMed] [Google Scholar]
- [65].Ostriker A, Horita HN, Poczobutt J, Weiser-Evans MCM, Nemenoff RA, Vascular smooth muscle cell-derived transforming growth factor-β promotes maturation of activated, neointima lesion-like macrophages, Arterioscler. Thromb. Vasc. Biol 34 (2014) 877–886. 10.1161/ATVBAHA.114.303214. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.