

Abstract
Triacylglycerol (TG) is a class of lipids that is responsible for energy storage and cell metabolism in biological systems; it is found in relatively high abundances in biological fluids such as human plasma. Due to structural complexity, analyzing TGs using shotgun lipidomic approaches is challenging because of the presence of multiple fatty acyl compositional isomers. In this work, reversed-phase liquid chromatography (RPLC) was used for separation of TG species due to the capability of separating lipids based on fatty acyl chain lengths and degrees of unsaturation. Although long retention times allow separating TGs with different locations of unsaturation, RPLC does not provide structurally informative information for the location of carbon-carbon double-bonds (C=Cs) without using synthesized standards that correspond to each species analyzed. The Paternò–Büchi (PB) reaction was employed online to confidently characterize the location of C=Cs within lipid species via photo-initiated modification of the alkene group with acetone, which was later subjected to electrospray ionization (ESI) and tandem mass spectrometry (MS/MS) to form signature fragmentation peaks. This online RPLC-PB-MS/MS system was able to distinguish fatty acyl level and C=C level isomeric species. The systems allowed for the identification of 46 TG molecular species in human plasma with confident C=C location assignment in fatty acyls at a limit of identification of 50 nM.
Graphical Abstract
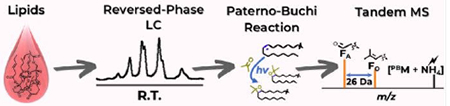
The developed online RPLC-PB-MS/MS system allows large scale analysis of isomeric triacylglycerol lipids differing in C=C locations.
Introduction
Prokaryotic and eukaryotic cells are composed of a substantial number of triacylglycerols (TGs). A major role of TGs is energy storage as they are the key component of adipose cells; on the other hand, these neutral lipids are important for the delivery of fatty acids to cellular processes. 1 TG composition within human plasma is as a result of both internal biosynthesis and dietary intake, which results in increased complexity in the TG content within a lipidome. Fish oils and milk fat, which both contribute to the Standard American Diet,2 have the most complex composition of TGs.3 TG molecular species have shown promise in acting as predictors to obesity and type II diabetes (T2D), while also strongly correlating with body-mass index (BMI),4–6 For example, Ståhlman and co-workers found that there is an increase in vaccenic acid within human plasma TG species in T2D patients versus a control group.7 Both vaccenic and oleic acid, two fatty acids only differing in the location of unsaturation, can be present in TG fatty acyl composition alongside many other carbon-carbon double bond (C=C) isomers. Being able to distinguish isomers will help improve the understanding of the roles of individual lipid species.
TG structures are composed of three fatty acids esterified onto a glycerol backbone. Each fatty acyl chain has varying lengths, degrees of unsaturation, and location of unsaturation that are important for its overall physical properties. Mass spectrometry (MS) is an important tool for the characterization of TG molecular species. Shotgun analysis is the injection of a crude sample extract for direct analysis without prior separation and has the advantages of speed and high sensitivity. Methods such as multiple reaction monitoring (MRM) utilizes a shotgun approach for rapidly profiling of large sample sets (1000 samples in less than 5 days).8 The matrix effect and species overlapping is prevalent in shotgun analysis leading to limited capability of TG characterization due to ion suppression and complex mass spectra. Chromatography coupled with mass spectrometry is a means of overcoming these challenges for the improved resolution of molecular species.
In the past, gas chromatography was used for the separation of intact TG but was inefficient at providing characterization due to sample degradation of some species resulting from its low volatility and thermal instability.9 Another separation technique is liquid chromatography (LC) which has multiple modes such as normal phase and reversed-phase (RP) LC. Molecular species separation is possible with RPLC which allows for the separation of isomeric and isobaric lipid species.10–13 RPLC is often coupled with electrospray ionization tandem mass spectrometry (ESI-MS/MS). The separation of C=C location isomers is possible with long separation time.3 However, it is difficult to determine differences in unsaturation site in a high throughput manner due to complexity in TG fatty acyl composition and lack of synthesized standards for retention time comparison.14 Although many methods for structural elucidation at C=C location exist, there is a lack of methods available for localization of C=Cs in a high-throughput manner. Electron impact excitation of ions from organics (EIEIO) coupled with differential mobility spectrometry (DMS) revealed key structural information of TGs in Portuguese olive oil by reacting the species with a beam of electrons creating informative fragment ions.15. Ozone-induced dissociation (OzID) uses gas-phase ion-molecule reaction between unsaturated lipids and ozone to localize the C=C.16–18 The Paternò–Büchi (PB) reaction is a C=C derivatization method; an oxetane ring is formed from [2+2] cycloaddition of a carbonyl group to an alkene. The PB reaction has been applied to unsaturated lipids to give C=C location upon low-energy collision-induced dissociation (CID). Activation of the product results in fragmentation at the oxetane ring leading to C=C location specific peaks. The PB reaction has been successfully applied to TGs using benzophenone and undergoing RPLC-MS/MS for analysis19, in direct analysis of biological tissues,20 and in shotgun analysis via charge-tagging PB reactions.21
Our group has shown the use of online hydrophilic interaction liquid chromatography (HILIC) PB-MS/MS for identification of polar lipids; however TG has little retention and separation under that condition.22 Recently, our group demonstrated that enhanced phospholipid isomer analysis can be achieved via coupling RPLC online with acetone PB reaction, suggesting that RPLC is compatible with online acetone PB reaction. In fact, the traditional method for TG species separation is RPLC. This method has allowed for separation of synthetic and naturally occurring TG molecular species.10,14,23–26 In order to obtain more accurately defined molecular species, TG standards are required for calibration of the separation and longer elution times are necessary for separation of species at C=C location. These parameters make it difficult to analyze C=C location in a high-throughput manner.3 Herein, we show the conditions for TG analysis for structural characterization at C=C location level without the need for standards of each molecular species C=C position.
This work shows the pairing of the PB reaction with RPLC for the detailed characterization of TG species. This online RPLC-PB-MS/MS system allowed for TG separation and characterization at C=C position. The lipid sample was first separated by RPLC, then subjected to a microreactor to undergo the online PB reaction and subsequent MS/MS for detailed structural characterization. The PB derivatized sample was probed for structural characterization and a table of identified species was built. The method allowed for distinction of C=C positional isomers within a RPLC-MS/MS workflow. Herein we show the potential to reduce the time require to distinguish C=C isomer because the species do not need to be completely separated but instead can be identified using MS/MS spectra.
EXPERIMENTAL SECTION
Nomenclature
Lipid structural identification followed the shorthand notation guidelines provided by LIPID MAPS.27 TG standards have specified fatty acyl carbon number, degrees of unsaturation, sn-positions, C=C position, and stereo-orientation. For example, TG(16:0/18:1(9Z)/18:2(9Z, 12Z) represents a TG with C16:0, C18:1, and 08:2 at sn-1, sn-2 and sn-3, respectively, where the numbers before “:” identify the carbon chain lengths and those afterward represent the degrees of unsaturation. The known sn-positions were indicated using “/” where the first number represents the sn-1 chain, the second number represents the sn-2 chain, and the third number represents the sn-3 chain. “9Z” nomenclature indicates that the C=C is located between the 9th and 10th carbon from the alpha carbon of the fatty acyl ester and alkene bond stereo configuration were cis. The notation containing “_” was used when the sn-position is unidentified, i.e. TG (16:0_18:1(Δ9)_18:1(Δ9), where “Δ9” was used when stereo-configuration of the C=C was ambiguous. The PB product fragment ions were denoted using FA and FO corresponding to species containing an aldehyde and olefin, respectively.
Chemicals
Triolein (TG (18:1(9Z)/18:1(9Z)/18:1(9Z)), triα-linolenic acid (TG (18:3(9Z, 12Z, 15Z)/18:3(9Z, 12Z, 15Z)/ 18:3(9Z, 12Z, 15Z))), and 1-Palmitoyl-2-oleoyl-3-linoleoyl-rac-glycerol (TG (16:0/18:1(9Z)/18:2(9Z, 12Z))) standards species were purchased from Sigma Aldrich. SPLASH Lipidomix Mass Spectrometry Standard was purchased from Avanti Polar Lipids (Alabaster, AL, US). Pooled human plasma (anticoagulant: Li Heparin) was purchased from Innovative Research (Novi, MI, US). Acetone, methanol, ethyl acetate, acetonitrile, 2-propanol, and ammonium acetate were purchased from Fisher Chemical (Pittsburgh, PA, US). Isooctane and chloroform were purchased from Beijing Tong Guang Fine Chemicals Company (Chaoyang Dist., Beijing, China). A purification system at 0.03 μS cm (model: Micropure UV; Thermo Scientific; San Jose, CA, USA) was used to obtain the deionized water.
Sample preparation
The human plasma lipid extraction was based on the Folch protocol followed by a silica solid phase extraction (SPE) column method 28 for glycerol lipid isolation. In summary, 50 μL of human plasma as combined with 1.5 mL of chloroform/methanol/water (v/v/v, 2/1/0.75), vortexed and centrifuged at 10,000 RPM for 10 minutes. The chloroform layer was collected and dried down using nitrogen gas. The sample was reconstituted in 0.5 mL of isooctane/ethyl acetate (80:1, v/v) and applied to a silica SPE column pretreated with 1 mL of isooctane/ethyl acetate (80:1, v/v). Three fractions of isooctane/ethyl acetate solutions (80:1, 20:1, 75:25, v/v, 5 mL total volume) were sequentially applied to the SPE column and individually collected. The second fractions contained TGs, respectively. The eluant was dried and the samples were stored in a −80°C freezer until analysis.
LC-PB-MS(MS) Experiments
Glycerolipid samples were dissolved in methanol and placed in an autosampler, which injected 2 μL of solution into an ExionLC AC system (Sciex, Concord, Ontario, Canada) equipped with an Ascentis Express C8-HPLC column (150 x 3.0 mm, 2.7 μm, Sigma-Aldrich (St. Louis, MO, US). The separation was performed isocratically at 90% mobile phase A for 45 minutes with a temperature of 55° C using mobile containing predominately acetone (A: 70/29/1, v/v/v, acetone/acetonitrile/2-propanol; B: 10 mM aqueous ammonium acetate, flowrate 0.3 mL/min). A homebuilt microreactor designed similarly to the previously described19 conducted the online PB reaction and was connected to the electrospray ionization (ESI) source for analysis using a X500R Q-TOF (Sciex, Concord, Ontario, Canada). Instrument control and data processing were performed using Analyst software equipped with the QTRAP 4500 (Sciex). All data interpretation was handled manually using an excel formula sheet created in-house. MS parameters were as follows: ESI voltage: 5000 V; ion source gas: 33 psi; curtain gas: 30 psi; declustering potential: 80 V; accumulation time 0.25 s for MS experiments and 0.1 s for MS/MS experiments; temperature 450° C; collision energy: 35 eV (beam-type CID). Approximately 5-40 scans were averaged for each spectrum.
Results and Discussions
RPLC-PB-MS/MS of Synthetic Glycerol Lipids.
Synthetic standards, TG (18:1(9Z)/18:1(9Z)/18:1(9Z)), TG (18:3(9Z, 12Z, 15Z)/18:3(9Z, 12Z, 15Z)/18:3(9Z, 12Z, 15Z)), and TG (16:0/18:1(9Z)/18:2(9Z, 12Z)) were used to establish the online acetone RPLC-PB-MS/MS protocol. In order to facilitate the PB reaction, solvent conditions for reaction optimization is necessary for efficient C=C localization. Typical solvent conditions for RPLC-separation of TGs may include high concentrations of 2-propanol, acetonitrile and/or methanol. 2-Propanol and methanol have shown to slow down the PB reaction between a lipid olefin group and acetone resulting in a decreased product yield.29 Acetone containing solvent were used as an eluent and reaction reagent in this workflow. A representation of the online RPLC-PB-MS/MS setup is shown in Scheme 1. A RPLC-column was used for the distinct separation of TG species based on equivalent carbon numbers (ECNs).23 ECN is given by the number of carbon atoms in the fatty acyl chains minus twice the number of C=Cs (CN – 2 DBN). The separated lipids flowed through a microreactor undergoing the PB reaction modifying the TG with an acetone. The reaction time was 18 seconds based on the flowrate 0.3 mL/min used for LC separation and optimized reaction conditions. The PB modified species were then subjected to ESI and underwent tandem MS for structural analysis. The resulting spectrum presented ions that differ by 26 Da and allowed for the distinct determination of molecular species at C=C position.
Scheme 1.
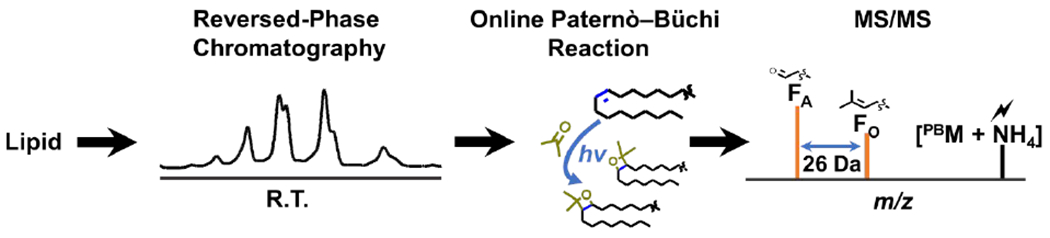
The RPLC-PB-MS/MS workflow for the analysis of unsaturated TG species. Lipid samples are injected into the LC system for reversed-phase separation, post-column acetone PB reaction, and MS/MS to obtain fragment ions specific to C=C location.
As shown in Figure 1a, TG (54:9) eludes from the column first followed by TG (52:3) and species TG (54:3), respectively, as expected due to their ECNs 36, 46, 48, respectively. The spectrum highlights that the solvent conditions show to be reliable for separation of species. The PB reaction was applied post-column and this allowed for the PB products (monitored as [PBM + NH4]+, blue trace) to share similar RT as the unmodified species ([M + NH4]+, black trace) as shown in Figure 1a. The data in Figure 1a suggest that the PB reaction product and the unmodified species can be directly linked by m/z values and RT, thus facilitating targeted analysis in subsequent PB-MS/MS.
Figure 1.
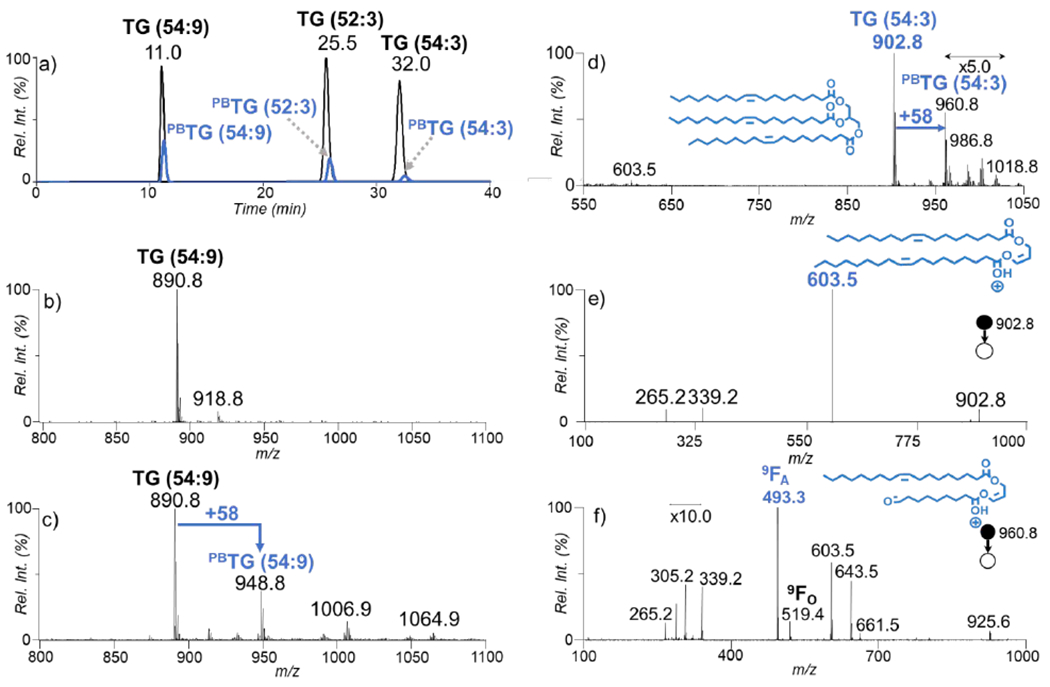
a) The overlaying XICs of ions ([M+NH4]+, black trace) corresponding to TG (54:9), TG (52:3), and TG (54:3) before and after the PB reaction ([PBM+NH4]+, blue trace, zoomed-in by 10 times), (b) An averaged mass spectrum for the ions present at RT = 11.0 minutes before the PB reaction, c) The post PB reaction spectrum of the ions present at 11.0 minutes, d) An averaged mass spectrum for the ions present at 32.0 minutes after the PB reaction, e) The CID spectrum of the ions population at m/z 902.8 f) The CID spectrum the ions population at m/z 960.8 from panel d).
The incorporation of ammonium acetate to the mobile phase allowed for intact and modified TG species to be readily adducted by a single ammonium and produce relatively abundant ion signal. Figure 1b shows the ion signal of TG (54:9) at m/z 890.8 ([M + NH4]+) without undergoing the PB reaction. The post PB reaction spectra for the same species is shown in Figure 1c exposing that the PB product peak at m/z 948.8 is 58 Da higher in mass than the intact species at corresponding to the acetone addition. TG (54:9) perceives to have a higher product yield than the other two species based on the relative intensity of XIC of the PB product at time 11 minutes. This is expected to be because of the presence of more double bonds creating a higher reaction efficiency. The PB reaction also initiated the addition of more than a single acetone onto the TG species. Due to the presence of nine double bonds, also present is the doubly and triply acetone-derivatized species in smaller abundances at m/z 1006.9 and 1064.9, respectively. In the PB reactions, acetone addition did not show selectivity toward a specific location of a C=C in fatty acyl. The post PB reaction spectrum of TG (54:3) is shown in Figure 1d, in which the PB product appears at m/z 960.8 ([PBM + NH4]+). There is also the presence of sequential acetone addition products at m/z 1018.8 as a minor product. It is worth noting that we chose to optimize reaction conditions for the single addition of acetone because the MS/MS spectrum of which allows for high sensitivity and straightforward identification of the C=C diagnostic ions.
For MS2 CID of unmodified TG (54:3) ([M+NH4]+), the combined neutral losses of ammonia and fatty acyl as a carboxylic acid, acylium ion plus 74 Da, and acylium ion are commonly observed, which provide basic information for fatty acyl composition analysis.1 Using TG (54:3) as an example (Figure 1e), the peaks at m/z 603.5, m/z 265.2, and m/z 339.2 correspond to loss of FA C18:1 (−281 Da), acylium ion of FA C18:1, and acylium ion plus 74 Da, respectively. The lack of any other fragment ions corresponding to a fatty acyl chains loss other than ions at m/z 603.5 verifies that the standard consists of three C18:1 fatty acyl chains, thus identifying the species as TG (18:1/18:1/18:1).
In order to pinpoint the location of C=C in unsaturated fatty acyls, such as in C18:1 of TG (54:3), MS2 CID was applied to the PB product at m/z 960.8 ([PBM+NH4]+). The fragmentation of the PB product shows a single set of C=C diagnostic ions that are 26 Da apart at m/z 493.3 (9FA) and 519.4 (9FO), allowing confident C=C location assignment for C18:1. The diagnostic peaks may be formed as a sequential fragmentation after neutral loss of a fatty acyl. It is not evident that there is a secondary fragmentation pathway of the PB products before the fatty acyl chain loss forming the diacylglycerol (DG)-like species. There is the presence of the fatty acyl chain loss from the PB reacted species at m/z 661.5 and its water loss at m/z 643.5. The C18:1 acylium ion and acylium + 74 Da are still present for the PB derivatized species. There is also a population of ions at m/z 305.2, which may be as a result to the water loss of a PB derivatized acylium ion. The signature 26 Da separation of the PB product is shown and can be used to verify the C=C locations at the Δ9 position upon the C18:1 fatty acyl chains identifying the species as TG (18:1(Δ9)/18:1(Δ9)/18:1(Δ9)).
When using the solvent conditions for RPLC-MS/MS to determine the species identity at fatty acyl chain level, TG (18:1/18:1/18:1) can be distinguished at a detection limit of 10 pM based on the presence of the C18:1 loss (m/z 603.5) three times the noise level in the CID spectrum. When applying the PB reaction the ability to determine TG (18:1(Δ9)/18:1(Δ9)/18:1(Δ9)) has a limit of detection of 50 nM based on the C=C diagnostic ion (m/z 493.3 and m/z 519.4) signal-to-noise ratio (S/N) being at least 3. The localization of C=Cs using the PB reaction depends on fragmentation of at the C=C after photoaddition, which results in two diagnostic ions. One of the diagnostic ions (m/z 519.4) forms less than 30% relative signal when compared with the corresponding diagnostic peak (m/z 493) leading to a reduced limit of detection. This limit of detection is 1 order of magnitude less than that of detecting the C=C location using shotgun analysis of PC.29 The hinderance of the LOD may be due to the difference in ionization efficiency combined with smaller abundance of the diagnostic ions.
Analysis of TGs in Human Plasma.
Obtaining abundant lipidome coverage is difficult without modification or separation prior to MS detection. The signal for TG is suppressed in the presence of polar lipids that readily ionize due to basic sites or fixed charges. TGs are neutral species that require an adduct for ionization, which are susceptible to matrix effect in LC-MS/MS analysis.30 A silica SPE column prior to RPLC separation was used for the removal of polar lipids from the extracts. Separation techniques that have been traditionally used for C=C localization are gas chromatography and silver ion high performance LC.31 Higher resolution separation of TGs has been reported using non-aqueous RPLC,23,32,33 however, for efficient incorporation of the PB reaction an aqueous mobile phase is preferred. Addition of water to the mobile phase system leads to the elution times that are greater than one hour for TG species. We chose C8 column for this study because it led to faster separation of TG at reasonable separation capability. Herein we show the separation of TG species in human plasma under 45 minutes followed by the C=C localization using RPLC.
Figure 2a shows the separation of human plasma TG species by ECN. RPLC is used here for separating species within a modest time frame compared to other LC methods geared at full separation. The peak shapes for a given ECN is non-gaussian due to the presence of multiple species eluting at slightly different times within the span of about 4 minutes. The species with an ECN of 48 are highlighted in Figure 2b where the XIC of m/z 824.8, m/z 850.8, m/z 876.8, and m/z 902.8 are shown which correspond to ammonium adducted TG (48:0), TG (50:1), TG (52:2), and TG (54:3), respectively. It is worth noting that the species elute in ascending mass order which is relative to the number of C=Cs present with the ions at m/z 824.8 having 0 and m/z 902.8 having 3 degrees of unsaturation. The TGs identified at subclass level in this work are consistent with the interlaboratory exercise composed by LipidMAPS.34 Although improved separation can be achieved in future work, this method proves to be efficient at partially separating TG species and the goal herein is not complete separation, but instead modest separation that allows for efficient lipid coverage and characterization at C=C level.
Figure 2.
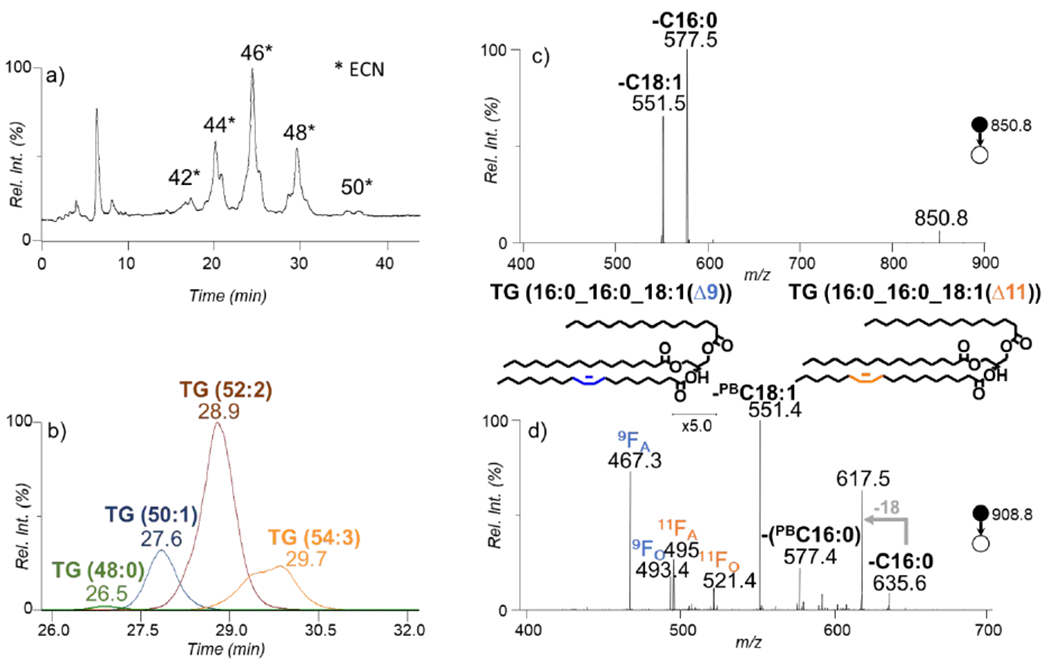
a) Total ion chromatograph (TIC) of the reversed phase separation of pooled human plasma in positive ionization mode. b) The overlaying XIC of the corresponding to the species with an ECN of 48 from panel a) (m/z 824.8, m/z 850.8, m/z 876.8, m/z 902.8). c) The MS2 CID spectrum of the ions at m/z 850.8 shown as the blue trace at time 27.6 minutes in panel b). d) The CID spectrum of the mass corresponding to the PB product of ions at m/z 850.8 at m/z 908.8.
Further characterization was activating via CID the ions at m/z 850 that correspond to TG (50:1). The CID spectrum in Figure 2c shows two abundant peaks at m/z 551.4 and m/z 577.4 which represent the neutral loss of Cl8:1 and C16:0, respectively, from the glycerol backbone. The presence of the mentioned acyl chain losses helped identify the species at fatty acyl chain level as TG (16:0_16:0_18:1). The PB reaction was applied to the sample and the post reaction CID spectrum of TG (16:0_16:0_18:1) is shown in Figure 2d. After fragmentation, the PB modified C18:1 (PBC18:1) and C16:0 are lost forming a DG-like species. The reacted species was also fragmented at the oxetane ring forming two pairs of diagnostic species. The pairs correspond to diagnostic species being formed after the loss of a C16:0 forming fragmentation ions at m/z 467.3/493.4 and m/z 495.4/521.4 congruent with an C18:1(Δ9) and an C18:1(Δ11), respectively. The species are identified as TG (16:0_16:0_18:1(Δ9)) and TG (16:0_16:0_18:1(Δ11)). This is an example of how confident C=C location isomers can be distinguished using this method at modest RT without the requirement of a standard sample for RT comparison.
Another challenge with human plasma analysis is the presence of multiple molecular TG species of closely spaced masses making spectra complex. A zoomed-in spectrum (m/z 895 – m/z 907) of the averaged total ion chromatograph is shown in Figure 3a showing that there can be much isotopic interference hindering the ability to identify species and suppression of low abundant species. The XIC for m/z 904.8 in Figure 3b emphasizes that the [M + 2]+ isotope of the ions at m/z 902.8 significantly contributes to the signal at m/z 904.8. The CID of the full XIC spectrum shows that there are interference peaks and species at m/z 904.8 cannot be clearly determined at fatty acyl chain level which is a previously reported phenomenon.35 Figure 3c shows the CID spectrum for the ions present at retention time 35.8 min in Figure 3b. The presence of the ions at m/z 603.5 and m/z 605.5, due to the losses of C18:0 and C18:1, respectively, are consistent with the presence of TG (18:0_18:1_18:1). Fragmentation of the PB modified TG (18:0_18:1_18:1) yields even greater information showcasing the power of obtaining a more in-depth characterization of the species as presented in Figure 3d. Four sets of diagnostic ions are yielded upon CID of the PB product corresponding to the C18:1(Δ9) and C18:1(Δ11). It is possible to identify the species as TG (18:0_18:1(Δ9)_18:1(Δ9)), (TG (18:0_18:1(Δ11)_18:1(Δ11)) and/or TG(18:0_18:1(Δ9)_18:1(Δ11)). Ammonium-adducted TG species readily lose all three of the fatty acyl chains thus in the case of TG (18:0_18:1_18:1) the diagnostic peaks can be produced after the loss of 18:1 or 18:0. There are two sets of diagnostic ions per C=C location for C18:1(Δ9), resulting from a combined loss of C18:0 (m/z 493.4/519.5) or C18:1 (m/z 495.4/521.5). The other two sets of ions, m/z 521.5/547.4 and m/z 523.5/549.4, correspond to similar fragmentation channels, however resulting from cleavage of the double bond in C18:1(Δ11). The 9FO (521.447) ions resulting from sequential loss of C18:1 and the 11FA (521.416) ions resulting from sequential loss of C18:0 have the same nominal mass. By using high resolution MS, the isobars were resolved at 6 ppm thus the MS/MS spectra can be used for confident determination of isobaric overlap. However, a limitation of our method is that when molecular species contain two of the same unsaturated fatty acyl chains it cannot always be determined whether the C=C location isomers exist within the same molecular species or as different molecules.
Figure 3.
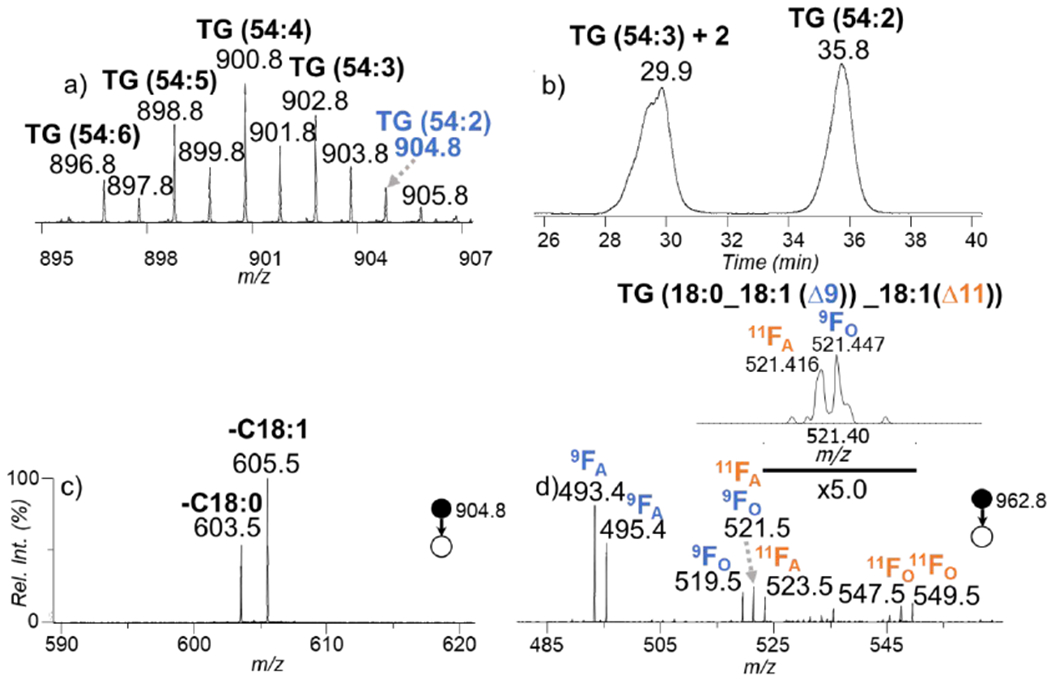
a) The zoomed-in MS spectrum (m/z 895 – m/z 907) of TG from pooled human plasma in positive ionization mode. b) XIC of m/z 904.8. c) The CID spectrum of the ions at m/z 904 eluted at 35.8 minutes from b). d) The CID spectrum of the ions at m/z 962.8 eluted at 35.8 minutes. Inset in d is the zoomed in spectrum from panel d) showing the resolution of 9FO and 11FA using high resolution MS.
The current RPLC-PB-MS/MS is capable of separating unmodified and the PB modified species of the same nominal mass, thus reducing chemical interference when compared to shotgun PB-MS/MS methods. Figure 4a displays the overlaying XICs of ions at m/z 822.7 (blue trace) and m/z 880.7 (orange trace) for the TG extract from human plasma with the online PB reaction applied to the sample. The latter species has the characteristic mass increase of 58 Da relative to the former one, which could be attributed to forming the PB modified lipids. XIC of m/z 822.7 shows an apparent peak at retention time 22.4 minutes, identified as intact TG (14:0_16:0_18:1) with a minor signal for TG (14:0_16:1_18:0) (Supplementary Figure S1a). XIC of the PB products for the ions at m/z 822.7 is expected to appear at the same retention time as the intact lipid. However, besides the expected PB products eluting at 22.4 min, the XIC of m/z 880.7 contains several abundant species eluting at longer retention times. The most abundant species elutes at a retention time of 33.7 minutes and corresponds to the [M+2]+ isotope of the TG (52:1) (monoisotopic ions at m/z 878.7). The second abundant species at retention time 39.9 is intact TG (52:0), later identified as TG (18:0_18:0_16:0) (Supplementary Figure S1b). Due to good separations from these more abundant isobaric interferences, PB-MS/MS of TG (14:0_16:0_18:1) (Figure 4b) shows a clean spectrum ideal for C=C location determination. For instance, there are two sets of diagnostic ions at m/z 439.3/465.3 and m/z 467.3/493.4, representing the presence of C18:1(Δ9). Another set of diagnostic ions are present at m/z 495/521 corresponding to C18:1(Δ11). The species present are TG (18:1(Δ9)_16:0_14:0) and a minor abundance of TG (18:1(Δ11)_16:0_14:0). This example showcases the ability to identify the species of interest without the concern of pre-existing species and this allows for fatty acyl chain and C=C location information to be obtained within the same run.
Figure 4.
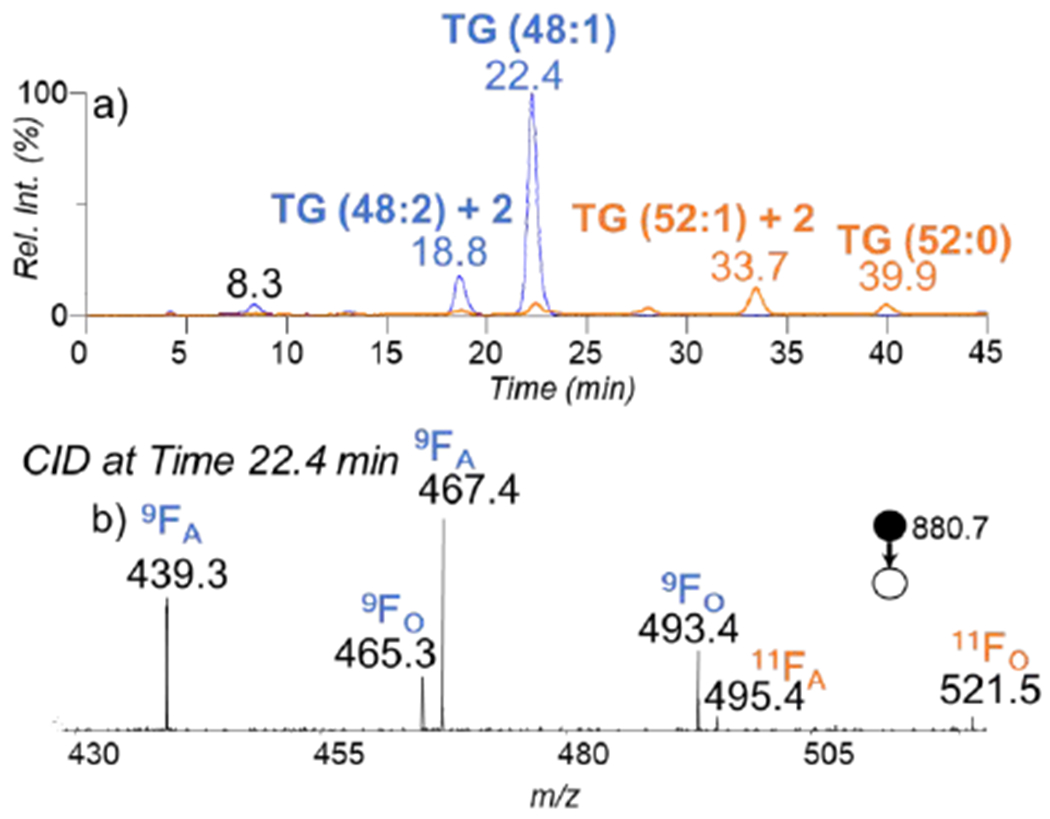
a) The overlaying XIC of m/z 822.7 and m/z 880.7 traced in blue and orange, respectively. b) The CID spectrum of the PB product of TG (14:0_16:0_18:1) (m/z 880.7), eluted at 22.4 minutes.
Separating fatty acyl compositional isomers is beneficial for confident characterization of TG species because it is possible to have as many as 9 fatty acyl chain fragments in a single CID spectrum.1 Further modifications such as the PB reaction make the MS/MS spectra even more complex and it makes identification a challenge. The major component of the species that have an ECN of 44 (Figure 2a) is TG (52:4) at m/z 872.7. The CID spectrum of the ions at m/z 872.7 loses C16:1, C18:1, C18:2, C18:4, C16:0, and C20:4 however the presences of the fragments for different fatty acyl chain losses depend on the retention time range being averaged suggesting that this group consist of multiple isomeric molecular species and are retained at different retention times. Figure 5a shows the overlaying XICs for the loss of C18:2, C18:3 and C20:4 after the CID of the ions with a m/z of 872.7. The fatty chain losses are not fully resolved because some species are composed of the same fatty acyls and better separation would be necessary for full resolving power. The average of scans at different fatty acyl chain peaks were used for spectrum collection and show that there were at least 4 different fatty acyl chain isomers present TG (16:1_18:1_18:2), TG (16:0_18:2_18:2), TG (16:0_18:1_18:3), and TG (16:0_16:0_20:4). The PB reaction was applied to the sample and fragmentation of the product resulted in the presence of diagnostic peaks which can be separated for identification of each species at C=C location.
Figure 5.
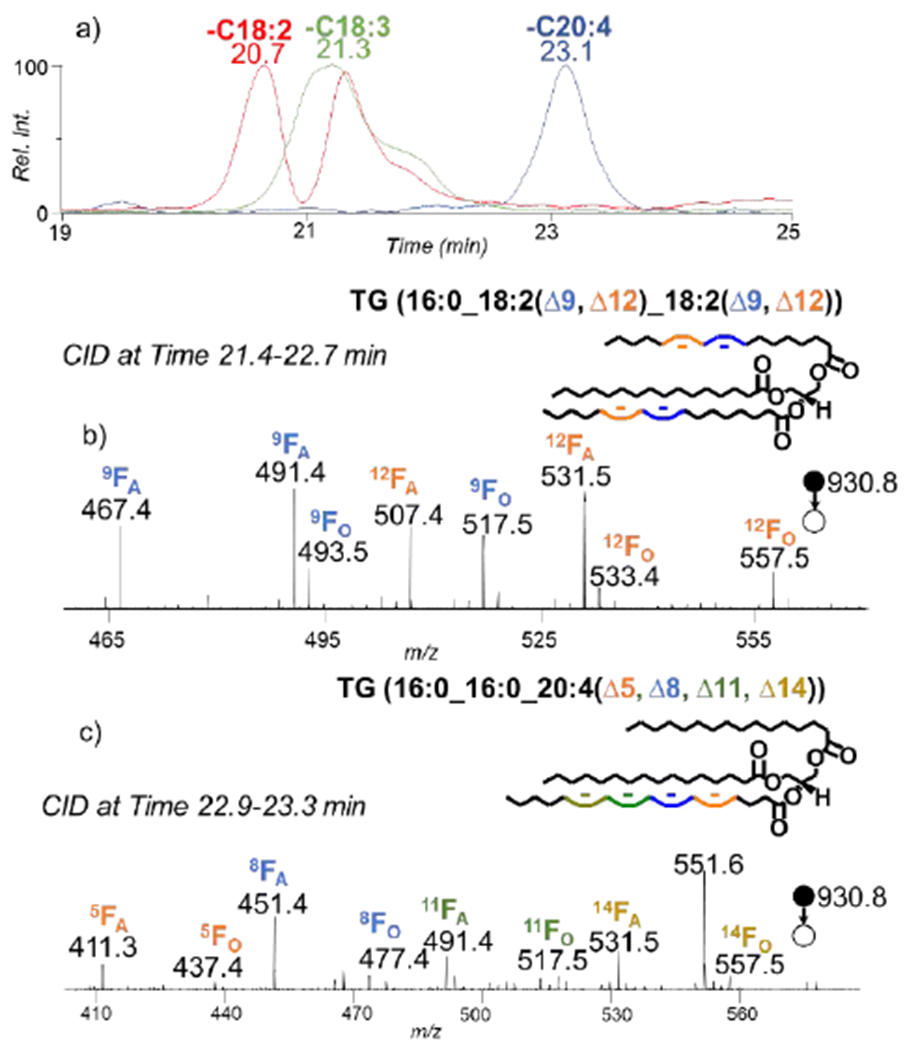
a) The overlaying XIC of the masses corresponding to the losses of the fatty acyl chains 18:2, 18:3, and 20:4. b) The CID spectrum of the ions at m/z 930.8 (the PB product of 872.7) eluted from time 21.4-22.7 minutes, c) The CID spectrum of the ions at m/z 930.8 (the PB product of 872.7) eluted from time 22.9-23.3 minutes.
The first species to elude within this subclass is TG (16:1_18:1_18:2). The two pairs of diagnostic ions at m/z 465.3/491.4 and m/z 493.4/519.4 in the PB-MS/MS spectrum (Supplementary Figure S2a) correspond to the Δ9 position on the C18:2. There is another set of pairs at m/z 505.4/531.4 and m/z 533.4/559.5 corresponding to the Δ12 position on the C18:2 identifying TG (16:1_18:1_18:2(Δ9, Δ12)). The diagnostic ions for C18:1(Δ9), C16:1(Δ9), and the Δ9 of C18:2 have isomeric overlap, which makes it challenging to determine which unsaturation site leads to the signal. TG (16:0_18:2_18:2) is the second molecular species that eludes. Figure 4b shows two sets of distinct diagnostic ions per C=C position help with identifying the species as TG (16:0_18:2(Δ9, Δ12)_18:2(Δ9, Δ12)). The other two molecular species were identified as TG (16:0_18:1_18:3) and TG (16:0_16:0_20:4). In Supplementary Figure S2b, there are two sets of diagnostic ions for C18:3(Δ6, Δ9, Δ12) and there are two pairs of diagnostic ions for C18:1(Δ9) identifying the species as TG(16:0_18:1(Δ9)_18:3(Δ6, Δ9, Δ12). The last species to elude was TG (16:0_16:0_20:4 and 1 set of diagnostic ions are formed as shown in Figure 4c because only one of the fatty acyl chains is unsaturated so only the loss of C16:0 will produce diagnostic ions identifying TG (16:0_16:0_20:4(Δ5, Δ8, Δ11, Δ14)). This shows that subclasses of TG the complexity of TG composition with a sample.
A total of 61 TGs at subclass level were detected in the human plasma sample as shown in Figure 6. Over 80 TG molecular species were identified at fatty acyl chain composition in human plasma using RPLC-MS/MS (Supplementary Table S1), 15 of which were saturated and 67 contained unsaturated acyl chains. Forty-six TG molecular species were identified at C=C location level, which originated from 24 subclasses and 37 fatty acyl chain groups. The results are slightly greater than the 42 subclasses identified in an interlaboratory comparisons of the human plasma lipidome.34 The differences in the reports may be due to quality control measures taken by the interlaboratory study leaving some identified species out of the consensus. The species that were analyzed using RPLC-PB-MS/MS for C=C localization are presented in Supplementary Table S2. The chart in Figure 6 also depicts a limitation of the current study where more than 40% of unsaturated TGs at fatty acyl composition level were not characterized for C=C location. This shows a contrast to acetone PB-MS/MS for phospholipids, where more than 90% of unsaturated lipids at fatty acyl level has been identified for C=C location.22 This limitation mainly stems from the ambiguity in assigning C=C locations to a specific unsaturated fatty acyl chain when a TG contains two or three unsaturated fatty acyls chains. In this study, the ambiguous species were not included in the table or pie chart summaries. Even so, this method has the potential for further application such as biomarker detection in human plasma. Using this method, we found 16 molecular species which contain C18:1 meaning that there is a possibility to probe 16 species for oleic (C18:1 (Δ9)) vs. vaccenic acid (C18:1 (Δ11)) composition which has been the aim in literature for biomarker detection.4,7
Figure 6.
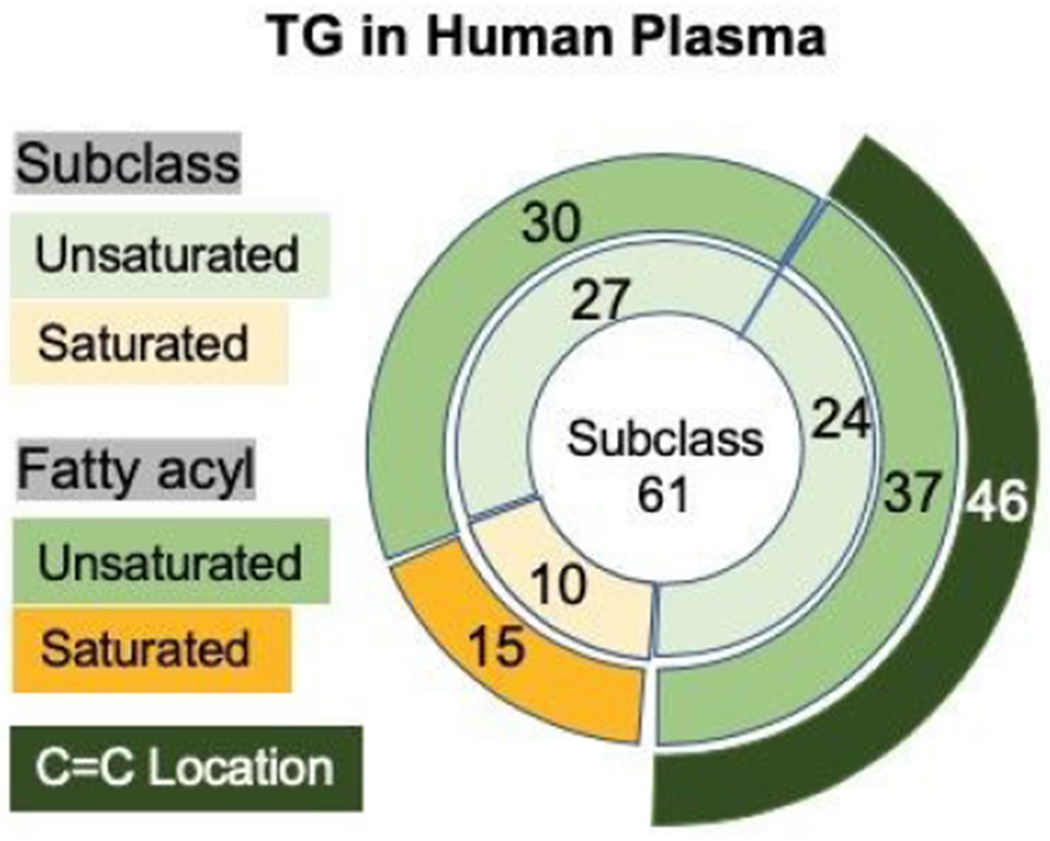
A pie chart depicting the quality of TGs characters at the subclass, fatty acyl chain, and C=C location level.
Conclusion
In this work, the online PB reaction with RPLC-MS/MS was introduced which allowed for the separation of species based on their ECN. Sum compositional isomers were partially separated and over 80 lipids were identified at fatty acyl chain level. Upon application of the PB reaction, tandem mass spectra allowed for the confident identification of C=C position and the determination isomers that differ in unsaturation site. The molecular species were better defined upon application of the PB reaction. TGs with 1 or 2 C=Cs have been shown to be the most promising for biomarker detection and this method could be useful for further characterization of unsaturation information. Future work could go into overcoming the challenge of overlapping fragment ions within a species in the post PB reaction CID spectrum. Another possible promise is the use of a sodium or lithium adduct instead of ammonium followed by MS3 experiments such that the adducted PB product forms the modified fatty acyl chains and in MS3 spectrum reveals individual fatty acyl chain C=C information.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by National Institutes of Health (R01GM118484 to Y. X.).
Footnotes
Publisher's Disclaimer: Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. We will replace this Accepted Manuscript with the edited and formatted Advance Article as soon as it is available.
The authors declare no competing financial interest.
REFERENCES
- 1.Murphy RC, in Tandem Mass Spectrometry of Lipids: Molecular Analysis of Complex Lipids, Royal Society of Chemistry, Cambridge, 2014, pp. 105–129. [Google Scholar]
- 2.Grotto D and Zied E, Nutrition in Clinical Practice, 2010, 25, 603–612. [DOI] [PubMed] [Google Scholar]
- 3.Řezanka T, Pádrová K and Sigler K, Analytical Biochemistry, 2017, 524, 3–12. [DOI] [PubMed] [Google Scholar]
- 4.Balgoma D, Pettersson C and Hedeland M, Trends in Endocrinology & Metabolism, 2019, 30, 283–285. [DOI] [PubMed] [Google Scholar]
- 5.Quehenberger O, The New England Journal of Medicine, 2011, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suvitaival T, Bondia-Pons I, Yetukuri L, Pöhö P, Nolan JJ, Hyötyläinen T, Kuusisto J and Orešič M, Metabolism, 2018, 78, 1–12. [DOI] [PubMed] [Google Scholar]
- 7.Ståhlman M, Pham HT, Adiels M, Mitchell TW, Blanksby SJ, Fagerberg B, Ekroos K and Borén J, Diabetologia, 2012, 55, 1156–1166. [DOI] [PubMed] [Google Scholar]
- 8.Yannell KE, Ferreira CR, Tichy SE and Cooks RG, The Analyst, 2018, 143, 5014–5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marěs P, Progress in Lipid Research, 1988,27, 107–133. [DOI] [PubMed] [Google Scholar]
- 10.Malone M and Evans JJ, Lipids, 2004, 39, 273–284. [DOI] [PubMed] [Google Scholar]
- 11.Bird SS, Marur VR, Sniatynski MJ, Greenberg ΗK and Kristal BS, Analytical Chemistry, 2011, 83, 6648–6657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagy K, Sandoz L, Destaillats F and Schafer O, Journal of Lipid Research, 2013, 54, 290–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cvačka J, Hovorka O, Jiroš P, Kindi J, Stránský K and Valterová I, Journal of Chromatography A, 2006, 1101, 226–237. [DOI] [PubMed] [Google Scholar]
- 14.Murphy RC, TrAC Trends in Analytical Chemistry, 2018, 107, 91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baba T, Campbell JL, Le Blanc JCY and Baker PRS, Journal of Lipid Research, 2016, 57, 2015–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thomas MC, Mitchell TW, Harman DG, Deeley JM, Nealon JR and Blanksby SJ, Analytical Chemistry, 2008, 80, 303–311. [DOI] [PubMed] [Google Scholar]
- 17.Marshall DL, Pham ΗT, Bhujel M, Chin JSR, Yew JY, Mori K, Mitchell TW and Blanksby SJ, Analytical Chemistry, 2016, 88, 2685–2692. [DOI] [PubMed] [Google Scholar]
- 18.Marshall DL, Criscuolo A, Young RSE, Poad BLJ, Zeller M, Reid GE, Mitchell TW and Blanksby SJ, Journal of The American Society for Mass Spectrometry, 2019, 30, 1621–1630. [DOI] [PubMed] [Google Scholar]
- 19.Xu T, Pi Z, Song F, Liu S and Liu Z, Analytica Chimica Acta, 2018, 1028, 32–44. [DOI] [PubMed] [Google Scholar]
- 20.Deng J, Yang Y, Liu Y, Fang L, Lin L and Luan T, Analytical Chemistry, 2019, 91, 4592–4599. [DOI] [PubMed] [Google Scholar]
- 21.Esch P and Heiles S, Journal of The American Society for Mass Spectrometry,, DOI: 10.1007/s13361-018-2023-x. [DOI] [PubMed] [Google Scholar]
- 22.Zhang W, Zhang D, Chen Q, Wu J, Ouyang Z and Xia Y, Nature Communications, DOI: 10.1038/s41467-018-07963-8. [DOI] [Google Scholar]
- 23.Hvattum E, Rapid Communications in Mass Spectrometry, 2001, 15, 187–190. [DOI] [PubMed] [Google Scholar]
- 24.Momchilova S, Tsuji K, Itabashi Y, Nikolova-Damyanova B and Kuksis A, Journal of Separation Science, 2004, 27, 1033–1036. [DOI] [PubMed] [Google Scholar]
- 25.Holčapek M, VelÃ-nská H, LÃ-sa M and Česla P, Journal of Separation Science, 2009, 32, 3672–3680. [DOI] [PubMed] [Google Scholar]
- 26.Kauhanen D, Sysi-Aho M, Koistinen KM, Laaksonen R, Sinisalo J and Ekroos K, Analytical andBioanalytical Chemistry, 2016, 408, 3475–3483. [DOI] [PubMed] [Google Scholar]
- 27.Liebisch G, Vizcaíno JA, Köfeler H, Trötzmüller M, Griffiths WJ, Schmitz G, Spener F and Wakelam MJO, Journal of Lipid Research, 2013, 54, 1523–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ingalls ST, Kriaris MS, Xu Y, DeWulf DW, Tsemg K-Y and Hoppel CL, Journal of Chromatography B: Biomedical Sciences and Applications, 1993, 619, 9–19. [DOI] [PubMed] [Google Scholar]
- 29.Stinson CA and Xia Y, The Analyst, 2016, 141, 3696–3704. [DOI] [PubMed] [Google Scholar]
- 30.Pucci V, Di Palma S, Alfieri A, Bonelli F and Monteagudo E, Journal of Pharmaceutical and Biomedical Analysis, 2009, 50, 867–871. [DOI] [PubMed] [Google Scholar]
- 31.Hancock SE, Poad BLJ, Batarseh A, Abbott SK and Mitchell TW, Analytical Biochemistry, 2017, 524, 45–55. [DOI] [PubMed] [Google Scholar]
- 32.Nagai T, Gotoh N, Mizobe H, Yoshinaga K, Kojima K, Matsumoto Y and Wada S, Journal of Oleo Science, 2011, 60, 345–350. [DOI] [PubMed] [Google Scholar]
- 33.Nagai T, Watanabe N, Yoshinaga K, Mizobe H, Kojima K, Kuroda I, Odanaka Y, Saito T, Beppu F and Gotoh N, Journal of Oleo Science., 2015, 64, 943–952. [DOI] [PubMed] [Google Scholar]
- 34.Bowden JA, Heckert A, Ulmer CZ, Jones CM, Koelmel JP, Abdullah L, Ahonen L, Alnouti Y, Armando AM, Asara JM, Bamba T, Barr JR, Bergquist J, Borchers CH, Brandsma J, Breitkopf SB, Cajka T, Cazenave-Gassiot A, Checa A, Cinel MA, Colas RA, Cremers S, Dennis EA, Evans JE, Fauland A, Fiehn O, Gardner MS, Garrett TJ, Gotlinger KH, Han J, Huang Y, Neo AH, Hyötyläinen T, Izumi Y, Jiang H, Jiang H, Jiang J, Kachman M, Kiyonami R, Klavins K, Klose C, Köfeler HC, Kolmert J, Koal T, Koster G, Kuklenyik Z, Kurland IJ, Leadley M, Lin K, Maddipati KR, McDougall D, Meikle PJ, Mellett NA, Monnin C, Moseley MA, Nandakumar R, Oresic M, Patterson R, Peake D, Pierce JS, Post M, Postle AD, Pugh R, Qiu Y, Quehenberger O, Ramrup P, Rees J, Rembiesa B, Reynaud D, Roth MR, Sales S, Schuhmann K, Schwartzman ML, Serhan CN, Shevchenko A, Somerville SE, St. John-Williams L, Surma MA, Takeda H, Thakare R, Thompson JW, Torta F, Triebl A, Trötzmüller M, Ubhayasekera SJK, Vuckovic D, Weir JM, Welti R, Wenk MR, Wheelock CE, Yao L, Yuan M, Zhao XH and Zhou S, Journal of Lipid Research, 2017, 58, 2275–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holčapek M, Liebisch G and Ekroos K, Analytical Chemistry, 2018, 90, 4249–4257. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.