

Abstract
The repurposing of FDA approved drugs is presently receiving attention for COVID-19 drug discovery. Previous studies revealed the binding potential of several FDA-approved drugs towards specific targets of SARS-CoV-2; however, limited studies are focused on the structural and molecular basis of interaction of these drugs towards multiple targets of SARS-CoV-2. The present study aimed to predict the binding potential of six FDA drugs towards fifteen protein targets of SARS-CoV-2 and propose the structural and molecular basis of the interaction by molecular docking and dynamic simulation. Based on the literature survey, fifteen potential targets of SARS-CoV-2, and six FDA drugs (Chloroquine, Hydroxychloroquine, Favipiravir, Lopinavir, Remdesivir, and Ritonavir) were selected. The binding potential of individual drug towards the selected targets was predicted by molecular docking in comparison with the binding of the same drugs with their usual targets. The stabilities of the best-docked conformations were confirmed by molecular dynamic simulation and energy calculations. Among the selected drugs, Ritonavir and Lopinavir showed better binding towards the prioritized targets with minimum binding energy (kcal/mol), cluster-RMS, number of interacting residues, and stabilizing forces when compared with the binding of Chloroquine, Favipiravir, and Hydroxychloroquine, later drugs demonstrated better binding when compared to the binding with their usual targets. Remdesvir showed better binding to the prioritized targets in comparison with the binding of Chloroquine, Favipiravir, and Hydroxychloroquine, but showed lesser binding potential when compared to the interaction between Ritonavir and Lopinavir and the prioritized targets. The structural and molecular basis of interactions suggest that the FDA drugs can be repurposed towards multiple targets of SARS-CoV-2, and the present computational models provide insights on the scope of repurposed drugs against COVID-19.
Keywords: Repurposed drugs, COVID-19, SARS-CoV-2, Potential molecular targets, Computational models, Structural and molecular mechanism
Highlights
-
•
Molecular mechanism of the binding of six drugs to multiple targets of SARS-CoV-2.
-
•
Highlight the scope of repurposing of six drugs towards 15 targets of SARS-CoV-2.
-
•
Ritonavir and Lopinavir possessed significant binding potential towards multiple targets.
-
•
MD studies showed that the repurposing of these drugs to selected targets provide future insights.
1. Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has been a threat across the world. The epidemic caused by SARS-CoV and Middle East respiratory syndrome coronavirus (MERS-CoV) was observed in 2003 and 2012, respectively [1]. Coronavirus causes respiratory tract infection with symptoms such as fever, dry cough, fatigue, chills, headache, sore throat, and loss of appetite and it can result in severe pneumonia in immunocompromised patients [2]. The infections caused by SARS-CoV-2 are called Coronavirus disease-2019 (COVID-19) and first reported in the city of Wuhan, China [3].
Coronaviruses are a large family of viruses that possess a single, positive-sense RNA strand [4]. The genome size of SARS-CoV-2 is approximately ~30 kb which codes for the entire proteome. The major proteins involved in the pathogenesis of SARS-CoV-2 are spike glycoprotein, RNA dependent RNA polymerase (RdRp), membrane proteins, small envelope proteins, and non-structural proteins. These proteins promote replication, transmission, host cell interaction of the virus, and they can act as major virulent factors [5].
Viral infection has caused both epidemiological and economical damages. COVID-19 was declared a pandemic by the World Health Organization (WHO) on March 11, 2020 because viral infections rapidly emerged across the world within a short period. According to WHO, globally, there have been 35, 659, 007 confirmed cases of COVID-19, including 1,044,269 deaths, reported as of October 07, 2020 [6]. Currently, there are no approved drugs available for COVID-19, thus the situation demands the identification of novel therapeutic agents. Several candidate vaccines are in their pre-clinical and clinical stage of development. But the availability of such vaccines requires time and effort. Thus, a rapid drug screening approach is highly preferred [7].
Identification of potential molecular targets is essential for drug repurposing [8]. Chloroquine, Hydroxychloroquine, and Remdesivir are three FDA approved drugs that were suggested for the treatment of COVID-19. Chloroquine previously used to treat malaria, and reports from China suggested that the drug reduced fever and increased the rate recovery in more than a hundred patients and recommended for a larger population [9]. Hydroxychloroquine, a derivative of Chloroquine was showed its in-vitro activity [10,11], and clinical trials are in progress. Chloroquine and Hydroxychloroquine interfere with the glycosylation of angiotensin-converting enzyme 2 (ACE2) and inhibit the biosynthesis of sialic acid. Remdesivir initially developed to treat Ebola viral infection, and this drug is presently suggested as a drug of choice against coronaviral infection [12]. The anti-viral activity of Remdesivir is due to the inhibition of RdRp [13]. Several clinical trials were conducted to study the efficacy and anti-viral potential of Remdesivir [14]. Favipiravir, Lopinavir, and Ritonavir are the other drugs currently suggested for the treatment of coronaviral infections [11]. Favipiravir showed a therapeutic response by viral clearance and disease progression [15]. Lopinavir and Ritonavir were used to treat HIV infections, and recent studies suggested that these drugs can be used to treat coronaviral infections [16], the clinical trials of their combinations are also encouraged [17].
Drug repurposing is a promising approach in which the drug already used to treat an infection can be re-used against a new disease as they underwent clinical trials approved by the FDA [8]. Drug-drug interaction (DDI) and drug-target interaction (DTI) are the fundamental aspects of drug repurposing. Though there are clinical trials in progress, presently, no potential drugs are approved towards COVID-19. Thus, the drug repurposing approach can contribute insight into the drug discovery pipeline of COVID-19. There are studies available on the computational interaction modeling of few FDA approved drugs against some of the putative targets; limited studies are available on the interaction modeling of FDA approved drugs and multiple targets of SARS-CoV-2. Hence, predicting the binding potential of repurposed drugs towards multiple protein targets of SARS-CoV-2 possesses scope and applications. Therefore, the present study is aimed to predict the binding potential of six FDA approved drugs towards fifteen protein targets of SARS-CoV-2 in comparison with the binding of these drugs towards their usual targets by molecular docking and dynamic simulation studies and propose the structural and molecular mechanism of the interactions.
2. Methodology
2.1. Identification of the molecular targets of SARS-CoV-2
2.1.1. Drugs and potential targets
The FDA approved drugs such as Chloroquine (Drug Bank ID: DB00608), Hydroxychloroquine (Drug Bank ID: DB01611), Favipiravir (Drug Bank ID: DB12466), Lopinavir (Drug Bank ID: DB01601), Remdesivir (Drug Bank ID: DB14761), and Ritonavir (Drug Bank ID: DB00503) were selected for this study, and three-dimensional (3D) structures of the targets were retrieved from Drug Bank [33]. Based on the literature survey, the proteins, which played a major role in the pathogenicity of SARS-CoV-2 were selected as molecular targets. The 3D structures of fifteen proteins were retrieved from RCSB-PDB [18]. These proteins were the spike (S) glycoprotein, the open state spike ectodomain (PDB: 6VYB; electron microscopy structure, resolution 3.2 Å) [19], the closed state spike glycoprotein (PDB: 6VXX; electron microscopy structure, resolution 2.8 Å) [20], the pre-fusion spike glycoprotein with a single receptor-binding domain (PDB: 6VSB; electron microscopy, resolution 3.46 Å) [20], the post-fusion core of S2 subunit (PDB: 6LXT; X-ray crystal structure, resolution 2.9 Å) [21], HR2 domain of S2 subunit (PDB: 6LVN; X-ray crystal structure, resolution 2.47 Å) [22], RNA dependent RNA polymerase (PDB: 6M71; electron microscopy structure, resolution 2.9 Å) [23] with nsp 12 (chain A), nsp8 (chain B, D) and nsp7 (chain C), papain-like protease (PLpro) (PDB: 6W9C; X-ray crystal structure, resolution 2.7 Å) [24, main protease (Mpro) with chain A of nsp 5 (PDB: 6Y2E; X-ray crystal structure, resolution 1.75 Å) [25], The nucleocapsid (N) proteins: N-terminal RNA binding domain (PDB: 6M3M; X-ray crystal structure, resolution 2.7 Å) [26] and C-terminal dimerization domain of nucleocapsid phosphoprotein (PDB: 6WJI; X-ray crystal structure, resolution at 2.05 Å) [27], Nsp 15 endoribonuclease (PDB: 6VWW; X-ray crystal structure, resolution 2.20 Å) [28], ADP ribose phosphatase (PDB:6VXS; X-ray crystal structure, resolution 2.03 Å) [29], RNA binding protein-nsp9 (PDB: 6W4B; X-ray crystal structure, resolution 2.95 Å) [30], nsp16 and nsp10 complex structures (PDB: 6W75; X-ray crystal structure, resolution 1.95 Å) [31] and orf7a encoded with the accessory protein (PDB: 6W37; X-ray crystal structure, resolution 2.9 Å) [32] were retrieved as prospective molecular targets.
2.2. Molecular docking studies
The 3D structures of each of the protein target retrieved from PDB, and the structures prepared by removing water molecules and bound ligands in the crystal structure of the target, adding polar hydrogen atoms, merging non-polar bonds, and computing Gasteiger charge. Similarly, the 3D structures of each ligand were retrieved from the Drug Bank, and the ligand structure prepared by detecting the root atom, setting the torsion, and the number of torsions. The target and drug files were saved as pdbqt format. The binding pocket of the receptors predicted by CastP server and the residue coordinates for x, y, and z dimensions were assigned by AutoGrid. The binding of the selected drugs against the prioritized targets was predicted by molecular docking by AutoDock Vina [34]. The best-docked conformations in each set were analyzed based on the number of hydrogen bonds, binding energy (kcal/mol), upper and lower bound root mean square deviation (RMSD), number of interacting residues, and forces, which stabilized the receptor-ligand complex.
To compare the binding efficacy of the FDA drugs towards selected targets, the binding of these drugs to their normal targets is also predicted by molecular docking. The receptors and ligands for docking studies were prepared as per the method described previously. The binding sites of the usual targets for the drug were also predicted by the CastP server, and the grid box was assigned for the coordinate residues (x, y, z dimensions) at the binding pocket. The binding interaction between Chloroquine and Glutathione S-transferase (usual target of the drug) (PDB: 1OKT, resolution 1.9 Å) [35], Hydroxychloroquine and human angiotensin-converting enzyme (ACE) (PDB: 1R42; resolution 2.2 Å) [36], Favipiravir and RNA-dependent RNA polymerase (RdRp) (PDB: 6QNW, resolution 3.31 Å) [37], Lopinavir and HIV-1 protease (PDB: 5V4Y; 1.9 Å) [38], Remdesivir and main protease (PDB: 1Q2W; resolution 1.86 Å) [39], and Ritonavir, and human cytochrome P4503A4 (PDB: 3NXU; resolution 2.0 Å) [40] were predicted by molecular docking by AutoDock Vina. The best-docked complexes in each set were analyzed based on the binding energy (kcal/mol), the number of hydrogen bonds, upper and lower bound RMSD, and the number of interacting residues. These computational models were used as the control/reference sets for the comparison between binding potential (binding energy) of the FDA-approved drugs and selected targets in this study.
2.3. Molecular dynamic simulation studies of the best-docked complexes
Among all the six sets of docked results, the stabilities of the best-docked conformations of the drug and target were confirmed by molecular dynamics (MD) simulation at 100 ns The MD simulation studies were performed by the Desmond module of Schrödinger's suite [41]. The protein and ligand in the best-docked complex were pre-processed and simulated at 300 K using the ensemble constant number of particles, pressure, and temperature (NPT) for 100 ns
The trajectories of RMSD and RMSF of the proteins and ligand interactions were obtained by the following formula:
where, N is the number of atoms; tref is the reference time, r' is the position of the selected atoms in frame x after superimposing on the reference frame, frame x recorded at time tx, T is the trajectory time over which the RMSF was calculated and r is the position of an atom.
The simulation trajectories of protein-ligand RMSD, RMSF, associated contacts, ligand properties, and torsion profiles and protein structural information was analyzed using a simulation interaction diagram tool.
2.4. Calculation of molecular mechanics Poisson-Boltzmann surface area (MMPBSA)
The binding energies of the best protein-drug complexes were calculated by GROMACS [42,43] tool at 1 ns after simulating the complexes at100 ns [44]. CGenFF (force field generator) is used to assign charges to the drug molecule to generate an index file. The CHARMM36 force field was used for the complex system. A dodecahedron solvation box was generated with a three-site water model (TIP3P) solvent. The system charges were neutralized by adding ions. The potential energy calculation of the receptor-drug complex was carried out for 1 ns The number of particles, volume, and temperature (NVT) ensemble was equilibrated before the simulation at 1 ns, and the interaction trajectories generated were used as input. The potential energies of the best complexes calculated by MMPBSA in kcal/mol.
3. Results and discussion
3.1. Putative drug targets
The salient features of the drug targets selected in the study are shown in Table 1 . Fifteen prioritized protein targets of SARS-CoV-2 included spike glycoprotein (S), RNA dependent RNA polymerase (RdRp) with non-structural proteins nsp7, nsp8, and nsp 12, papain-like protease (PL-pro), main protease (Mpro), nucleocapsid protein (N), heptad repeat of domain 2 (HR2), ADP ribose phosphatase (nsp3), RNA binding protein (nsp9), endoribonuclease (nsp15), accessory protein (orf7a) and other non-structural proteins such as nsp10 and nsp1.
Table 1.
Probable drug targets of SARS-CoV-2 that possess experimentally solved 3D structures retrieved from Protein Data Bank.
| PDB ID | Name of the protein | Chain | Resolution | R-Value Free | R-Value Work | Experimental method | References |
|---|---|---|---|---|---|---|---|
| 6M3M | SARS-CoV-2 nucleocapsid protein N-terminal RNA binding domain | A, B, C, D | 2.7 Å | 0.293 | 0.258 | X-ray diffraction | [26] |
| 6VYB | SARS-CoV-2 spike ectodomain structure (open state) | A, B, C | 3.2 Å | Aggregation state: Particle | Reconstruction method: Single particle | Electron Microscopy | [19] |
| 6VXX | Structure of the SARS-CoV-2 spike glycoprotein (closed state) | A, B, C | 2.8 Å | Aggregation state: Particle | Reconstruction method: Single particle | Electron Microscopy | [20] |
| 6M71 | SARS-Cov-2 RNA-dependent RNA polymerase in complex with cofactors | A, B. C, D | 2.9 Å | Aggregation state: Particle | Reconstruction method: Single particle | Electron Microscopy | [23] |
| 6LVN | Structure of the 2019-nCoV HR2 Domain | A, B. C, D | 2.47 Å | 0.258 | 0.215 | X-ray diffraction | [22] |
| 6LXT | Post fusion core of 2019-nCoV S2 subunit | A, B, C, D, E, F | 2.90 Å | 0.290 | 0.259 | X-ray diffraction | [21] |
| 6VSB | Prefusion 2019-nCoV spike glycoprotein with a single receptor-binding domain up | A, B, C | 3.46 Å | Aggregation state: Particle | Reconstruction method: Single particle | Electron Microscopy | [20] |
| 6VWW | NSP15 Endoribonuclease from SARS CoV-2. | A, B | 2.20 Å | 0.178 | 0.158 | X-ray diffraction | [28] |
| 6VXS | ADP ribose phosphatase of NSP3 from SARS CoV-2 | A, B | 2.03 Å | 0.234 | 0.186 | X-ray diffraction | [28] |
| 6W4B | Nsp9 RNA binding protein of SARS CoV-2 | A, B | 2.95 Å | 0.276 | 0.240 | X-ray diffraction | [30] |
| 6W9C | Papain-like protease of SARS CoV-2 | A, B, C | 2.70 Å | 0.309 | 0.235 | X-ray diffraction | [24] |
| 6W37 | SARS-CoV-2 ORF7A encoded accessory protein | A | 2.90 Å | 0.268 | 0.236 | X-ray diffraction | [32] |
| 6W75 | NSP10 – NSP16 Complex from SARS-CoV-2 | A, B, C, D | 1.95 Å | 0.174 | 0.157 | X-ray diffraction | [31] |
| 6WJI | C-terminal Dimerization Domain of Nucleocapsid Phosphoprotein from SARS-CoV-2 | A, B, C, D, E, F | 2.05 Å | 0.228 | 0.187 | X-ray diffraction | [27] |
| 6Y2E | Free enzyme of the SARS-CoV-2 (2019-nCoV) main protease | A | 1.75 Å | 0.222 | 0.171 | X-ray diffraction | [25] |
The 3D structures of these proteins are available in their native form. Spike glycoprotein S (PDB: 6VYB, 6VXX, 6LXT, 6VSB) is responsible for the transmission of the virus into the host by mediating its entry, which leads to the infection [45]. The open, closed, pre-fusion, and post-fusion conformations were selected as putative targets because they contributed to the interactions between the drug and the target. Three motifs that recognize human angiotensin-converting enzyme 2 (hACE2), responsible for host entry are present in the closed conformation of spike glycoprotein. The open state conformation is responsible for a good transmissibility rate. The post-fusion conformation consists of two heptads repeats stabilized by hydrogen bonds between them, and in the prefusion conformation, S2 (subunit 2 of S) consists of the fusion machinery, which is stabilized by S1 (subunit 1 of spike protein) [19]. Heptad repeat 2 (HR2)- domain (PDB: 6LVN) involved in the fusion of spike glycoprotein trimers (viral membrane fusion), was considered a major target [22]. RNA-dependent RNA polymerase, RdRp (PDB: 6M71) is a nucleoprotein responsible for viral replication and transcription, which contains chains A, B, and C, which code for nsp7, nsp8, and nsp 2 respectively [23]. Papain-like protease (PL-pro/nsp3; PDB: 6W9C) and main protease (Mpro) (PDB: 6Y2E) involved in the cleavage of N- and C-terminals of replicase polyproteins [24,25]. The C-terminal dimerization domain (PDB: 6WJI) and N- terminal RNA binding domain (PDB: 6M3M) of nucleocapsid are involved in viral replication [26,27]. Endoribonuclease (nsp 15) (PDB: 6VWW) cleaves RNA strand after transcription and form functional RNA [28]. RNA binding protein (PDB: 6W4B) and ADP ribose phosphatase, nsp3 (PDB: 6VXS) are also involved in viral replication [29,30]. Accessory protein, orf7a (PDB: 6W37), and non-structural proteins nsp10 and nsp16 (PDB: 6W75) is essential for the viral mRNA methylation, which contributes to the induction of apoptosis [31,32]. Based on the functional role of these proteins in the virulent mechanism of the virus, these proteins are considered as putative molecular targets in the present study.
3.2. Interaction modeling of the selected drugs and prospective targets of SARS-CoV-2
The binding potential of six FDA approved drugs towards fifteen prioritized targets were predicted by molecular docking. The description of the molecular interaction of the drugs and the targets predicted by molecular docking are shown in Table 2 . The binding potential of each drug against fifteen selected targets is predicted based on the binding energy (kcal/mol), cluster RMS, number of interacting residues, hydrogen bonds, and other weak interactions.
Table 2.
The binding potential of the selected drugs towards the probable drug targets of SARS-CoV-2 obtained by molecular docking studies by AutoDock Vina.
| Protein name | PDB ID | Name of the drug with DrugBank ID | 2D structure of the drug | Binding energy (kcal/mol) | RMSD (Å) | Interacting residues | Hydrogen bonds |
|---|---|---|---|---|---|---|---|
| ADP ribose phosphatase of NSP3 | 6VXS | Chloroquine (DB00608) | 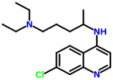 |
−5.3 | 0.0 | Leu126, Phe156 | 0 |
| C-terminal Dimerization Domain of Nucleocapsid Phosphoprotein | 6WJI | −6.6 | 0.0 | Val270, Phe274, Arg277, Trp301 | 1: Arg277 | ||
| HR2 Domain | 6LVN | −4.7 | 0.0 | Arg18, Asn20, Glu21, Lys24 | 0 | ||
| Main protease | 6Y2E | −5.2 | 0.0 | Met49, Phe140, Leu141, Asn142, His163, Met165, Glu166, Gln189 | 0 | ||
| NSP10 | 6W75·B | −4.9 | 0.0 | Gln4289, Thr4292, Cys4294, Lys4296, Asn4358 | 1: Asn4358 | ||
| NSP15 Endoribonuclease | 6VWW | −5.6 | 0.0 | Asn29, Asn30 | 0 | ||
| NSP16 | 6W75.A | −7.0 | 0.0 | Leu6819. Leu6820, Tyr7020, Val7021 | 0 | ||
| Nsp9 RNA binding protein | 6W4B | −4.9 | 0.0 | Gly39, Arg40, Phe57, Ser60, Ile66 | 0 | ||
| Nucleocapsid protein N-terminal RNA binding domain | 6M3M | −5.1 | 0.0 | Thr77, Ala156, Ile158, Val159 | 1: Thr77 | ||
| ORF7A encoded accessory protein | 6W37 | −5.0 | 0.0 | Tyr25, Phe31 | 0 | ||
| Papain-like protease | 6W9C | −4.8 | 0.0 | Lys105, Trp106, His272 | 0 | ||
| Post fusion core of S2 subunit | 6LXT | −5.0 | 0.0 | Asn914, Leu 916, Tyr917 | 0 | ||
| Prefusion of the spike glycoprotein with a single receptor-binding domain | 6VSB | −5.2 | 0.0 | Pro683, Thr732, His1058 | 0 | ||
| RNA-dependent RNA polymerase NSP12 | 6M71.A | −5.5 | 0.0 | Tyr32, Tyr129, His133, Lys780 | 0 | ||
| RNA-dependent RNA polymerase NSP7 | 6M71.C | −4.9 | 0.0 | Thr9, Phe49, Met52, Val53 | 0 | ||
| RNA-dependent RNA polymerase NSP8 | 6M71.B.D | −4.4 | 0.0 | Tyr135, Tyr138, Lys139, Asp143, Ser170 | 0 | ||
| Spike ectodomain structure (open state) | 6VYB | −5.1 | 0.0 | Met740, Tyr741, Asn8586, Leu966, Val976, Asn978 | 0 | ||
| Spike glycoprotein (closed state) | 6VXX | −5.2 | 0.0 | Tyr38, Asp40, Phe43, Glu224, Pro225 | 0 | ||
| ADP ribose phosphatase of NSP3 | 6VXS | Favipiravir (DB12466) | 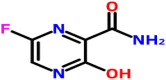 |
−4.5 | 0.0 | Lys31, Pro32, His86, Ala89, Lys90 | 2: intra hydrogen bonds within the drug molecule |
| C-terminal Dimerization Domain of Nucleocapsid Phosphoprotein | 6WJI | −4.6 | 0.0 | Val270, Arg277 | 1: Val270 | ||
| HR2 Domain | 6LVN | −4.0 | 0.0 | Glu21, Lys24 | 1: Glu21 | ||
| Main protease | 6Y2E | −4.7 | 0.0 | Glu14, Gly15, Met17, Ala70 | 0 | ||
| NSP10 | 6W75·B | −4.6 | 0.0 | Thr4292, Cys4294, Asn4358 | 1: Cys4294 | ||
| NSP15 Endoribonuclease | 6VWW | −4.7 | 0.0 | Gln202, Glu203, Phe204, Ala256, Phe259 | 1: Ala256 | ||
| NSP16 | 6W75.A | −5.0 | 0.0 | Thr6934, Phe6947, Phe6948 | 0 | ||
| Nsp9 RNA binding protein | 6W4B | −4.6 | 0.0 | Ser14, Asn26, Asp27, Ala29, Asp48 | 1: Ser14 | ||
| Nucleocapsid protein N-terminal RNA binding domain | 6M3M | −4.3 | 0.0 | Gly115, Ala120, Gly121, Tyr124, Asn141, Pro143 | 1: Tyr124 | ||
| ORF7A encoded accessory protein | 6W37 | −4.3 | 0.0 | Glu18, Ser22, Gly23, Thr24, Tyr25, Phe31 | 1: Ser22 | ||
| Papain-like protease | 6W9C | −4.7 | 0.0 | Asp76, Arg82, Tyr154, Asn156 | 0 | ||
| Post fusion core of S2 subunit | 6LXT | −4.5 | 0.0 | Ser967, Gly971, Leu1166 | 1: Leu1166 | ||
| Prefusion of the spike glycoprotein with a single receptor-binding domain | 6VSB | −4.6 | 0.0 | Tyr38, Asp40, Gly283 | 1: Asp40 | ||
| RNA-dependent RNA polymerase NSP12 | 6M71.A | −4.9 | 0.0 | Trp800 | 1: Trp800 | ||
| RNA-dependent RNA polymerase NSP7 | 6M71.C | −4.1 | 0.0 | Asp38, Ala42, Lys43, Asp44 | 0 | ||
| RNA-dependent RNA polymerase NSP8 | 6M71.B.D | −4.4 | 0.0 | Tyr135, Ser177 | 2: Tyr135, Ser177 | ||
| Spike ectodomain structure (open state) | 6VYB | −4.8 | 0.0 | Met740, Tyr741, Gly744, Leu977, Arg1000 | 0 | ||
| Spike glycoprotein (closed state) | 6VXX | −5.1 | 0.0 | Tyr741, Gly744, Leu966, Leu977, Arg1000 | 1: Gly744 | ||
| ADP ribose phosphatase of NSP3 | 6VXS | Hydroxychloroquine (DB01611) | 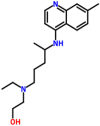 |
−5.8 | 0.0 | Ile23, Ala38, Leu126, Ser128, Ala129, Ile131, Phe132, Phe156 | 0 |
| C-terminal Dimerization Domain of Nucleocapsid Phosphoprotein | 6WJI | −5.8 | 0.0 | Arg259, Val270, Phe274, Arg277, Phe286, Ile304 | 1: Arg277 | ||
| HR2 Domain | 6LVN | −4.7 | 0.0 | Lys14, Asp17, Glu21 | 0 | ||
| Main protease | 6Y2E | −5.9 | 0.0 | Gln110, Thr111, Val202, Thr292, Phe294 | 0 | ||
| NSP10 | 6W75·B | −5.3 | 0.0 | Tyr4280, Tyr4283, Ile4291, Thr4292,Arg4331, Leu4328 | 0 | ||
| NSP15 Endoribonuclease | 6VWW | −5.9 | 0.0 | Asn29, Asn30 | 1: Asn30 | ||
| NSP16 | 6W75.A | −6.3 | 0.0 | Arg6817, Met6818, Leu6819, Leu6820, Tyr7020, Val7021 | 0 | ||
| Nsp9 RNA binding protein | 6W4B | −5.1 | 0.0 | Arg40, Val42, Ile66, Phe67, Thr68 | 0 | ||
| Nucleocapsid protein N-terminal RNA binding domain | 6M3M | −5.0 | 0.0 | Gln84, Thr136, Thr166 | 1: Thr166 | ||
| ORF7A encoded accessory protein | 6W37 | −4.8 | 0.0 | Tyr25, Phe31 | 0 | ||
| Papain-like protease | 6W9C | −5.0 | 0.0 | Tyr213, Glu214, Lys217, Tyr305, Tyr310, Thr313 | 1: Thr313 | ||
| Post fusion core of S2 subunit | 6LXT | −4.7 | 0.0 | Gly932, Lys933, Gln935, Asp936, Ser939, Lys1191, Asn1192 | 1: Asn1192 | ||
| Prefusion of the spike glycoprotein with a single receptor-binding domain | 6VSB | −5.2 | 0.0 | Ser730, thr778, Pro863, Thr866, Asp867, Glu868, his1058 | 0 | ||
| RNA-dependent RNA polymerase NSP12 | 6M71.A | −4.9 | 0.0 | Tyr273, Leu329, Val330, Ala379, Ala382 | 0 | ||
| RNA-dependent RNA polymerase NSP7 | 6M71.C | −4.6 | 0.0 | Thr9, Thr46, Phe47, Met52 | 1: Thr46 | ||
| RNA-dependent RNA polymerase NSP8 | 6M71.B.D | −4.7 | 0.0 | Phe92, Leu95, Ile107, Ala110 | 0 | ||
| Spike ectodomain structure (open state) | 6VYB | −5.0 | 0.0 | Leu517, Asn544, Leu546, Gln564, Phe565 | 0 | ||
| Spike glycoprotein (closed state) | 6VXX | −5.7 | 0.0 | Trp104, Val126, Phe192, Phe194, Val 227, Asp228 | 0 | ||
| ADP ribose phosphatase of NSP3 | 6VXS | Lopinavir (DB01601) | 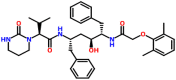 |
−8.5 | 0.0 | Ile23, Gly48, Ala52, Lys55, Leu126, Phe156 | 0 |
| C-terminal Dimerization Domain of Nucleocapsid Phosphoprotein | 6WJI | −11.0 | 0.0 | Arg259, Gln260, Ala264, Phe274, Arg277, Trp301, Phe314, Leu331, Tyr333 | 0 | ||
| HR2 Domain | 6LVN | −7.8 | 0.0 | Lys14, Arg18, Lys24, Asn27, Ile31 | 0 | ||
| Main protease | 6Y2E | −8.6 | 0.0 | Gln107, Gln110, Thr111, Ile200, Glu240, His246, Thr292, Phe294 | 0 | ||
| NSP10 | 6W75·B | −8.2 | 0.0 | Ala4279, Ile4291, Thr4292, Asn4293, Leu4328, Arg4331, Cys4332, His4333 | 0 | ||
| NSP15 Endoribonuclease | 6VWW | −9.6 | 0.0 | Ile27, Ile28, Asn29, Asn30, Val52 | 0 | ||
| NSP16 | 6W75.A | −9.9 | 0.0 | Arg6817, Leu6819, Tyr7020, Val7021 | 0 | ||
| Nsp9 RNA binding protein | 6W4B | −7.8 | 0.0 | Gly39, Arg40, Phe41, Val42, Phe57, Ser60, Ile66 | 0 | ||
| Nucleocapsid protein N-terminal RNA binding domain | 6M3M | −7.5 | 0.0 | Trp53, Asn76, Ile147, Asn151, Asn155, Ile158 | 0 | ||
| ORF7A encoded accessory protein | 6W37 | −7.0 | 0.0 | Thr24, Tyr25, Pro30, Phe31 | 1: Phe31 | ||
| Papain-like protease | 6W9C | −8.1 | 0.0 | Lys217, Lys218, Tyr305, Asn308, Tyr310 | 0 | ||
| Post fusion core of S2 subunit | 6LXT | −8.1 | 0.0 | Lys921, Asn928, Gln935, Leu1200 | 0 | ||
| Prefusion of the spike glycoprotein with a single receptor-binding domain | 6VSB | −8.2 | 0.0 | Arg815, Phe823, Leu828, Asp867, Glu868 | 0 | ||
| RNA-dependent RNA polymerase NSP12 | 6M71.A | −8.1 | 0.0 | Pro412, Phe415, Tyr420, Leu437, Phe440 | 0 | ||
| RNA-dependent RNA polymerase NSP7 | 6M71.C | −7.3 | 0.0 | Thr9, Val16, Val53, Leu56, Ile68 | 0 | ||
| RNA-dependent RNA polymerase NSP8 | 6M71.B.D | −8.2 | 0.0 | Leu122, Leu128, Val130, Val131, Tyr149 | 0 | ||
| Spike ectodomain structure (open state) | 6VYB | −8.3 | 0.0 | Asp294, Pro295, Leu296, Phe318, Val610, Ile651 | 0 | ||
| Spike glycoprotein (closed state) | 6VXX | −8.7 | 0.0 | Arg355, Asp428, Phe429, Thr430, Lys462, Pro463, Phe464, Ser514, Glu516 | 0 | ||
| ADP ribose phosphatase of NSP3 | 6VXS | Remdesivir (DB14761) | 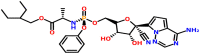 |
−7.6 | 0.0 | Asn40, Val41, Tyr42, Gly48, Lys102, Gly130, Ile131, Phe132 | 1: Asn40 |
| C-terminal Dimerization Domain of Nucleocapsid Phosphoprotein | 6WJI | −8.2 | 0.0 | Val270, Phe274, Arg277, Phe286, Leu291, Trp301, Ile304, Ala308, Tyr333 | 1: Arg277 | ||
| HR2 Domain | 6LVN | −6.4 | 0.0 | Lys14, Asp17, Arg18, Asn20, Glu21 | 0 | ||
| Main protease | 6Y2E | −7.7 | 0.0 | Gln110, Asp153, Thr292, Phe294 | 0 | ||
| NSP10 | 6W75·B | −7.1 | 0.0 | Gln4306, Ile4308, Phe4342, Asp4344, Val4369, Thr4371, Gly4374 | 1: Val4369 | ||
| NSP15 Endoribonuclease | 6VWW | −8.0 | 0.0 | Val156, Lys159, Thr193, Tyr194, Phe195, Gln197 | 2: Thr193, Gln197 | ||
| NSP16 | 6W75.A | −8.7 | 0.0 | Leu6820, Lys6822, Glu6940, Asn6941, His6972, Tyr7020 | 0 | ||
| Nsp9 RNA binding protein | 6W4B | −6.4 | 0.0 | Met13, Arg40, Val42, Phe57, Ser60, Ile66, Thr68 | 0 | ||
| Nucleocapsid protein N-terminal RNA binding domain | 6M3M | −6.4 | 0.0 | Tyr124, Asn127, Gly138, Asn141, Trp133 | 2: Tyr124, Trp133 | ||
| ORF7A encoded accessory protein | 6W37 | 6.4 | 0.0 | Tyr25, Phe31, Pro33 | 2: Tyr25, Phe31 | ||
| Papain-like protease | 6W9C | −7.4 | 0.0 | Thr74, Thr75, Arg82, Asn128, Tyr154, Gln174, His175, Asp179, Val202 | 2: Thr74, Thr75 | ||
| Post fusion core of S2 subunit | 6LXT | −7.1 | 0.0 | Asn928, Ser929, Gly932, Leu1197, Asp1199, Leu1200 | 1: Leu1200 | ||
| Prefusion of the spike glycoprotein with a single receptor-binding domain | 6VSB | −6.9 | 0.0 | Asn907, Thr912, Glu1092, Gly1093, Phe1089, Arg1107, Phe1121 | 1: Arg1107 | ||
| RNA-dependent RNA polymerase NSP12 | 6M71.A | −7.5 | 0.0 | Arg249, Trp268, Thr319, Pro322, Pro323, Leu460, Pro461 | 0 | ||
| RNA-dependent RNA polymerase NSP7 | 6M71.C | −6.9 | 0.0 | Ser25, Leu28, Val53, Ser54, Ser57, Val58, Ser61 | 0 | ||
| RNA-dependent RNA polymerase NSP8 | 6M71.B.D | −7.3 | 0.0 | Leu98, Pro121, Ala125, Ala126, Leu128, Tyr149, Trp154 | 0 | ||
| Spike ectodomain structure (open state) | 6VYB | −6.9 | 0.0 | Thr33, Phe59, Asp294, Leu296, Val608 | 0 | ||
| Spike glycoprotein (closed state) | 6VXX | −7.0 | 0.0 | Val341, Ala344, Ala348, Ser349, Tyr351, Asn354, Lys356 | 1: Asn354 | ||
| ADP ribose phosphatase of NSP3 | 6VXS | Ritonavir (DB00503) | 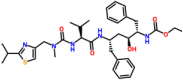 |
−6.7 | 0.0 | Ile23, Val49, Leu126, Gly130, Ile131, Pro136, Phe156, Asp157, Leu160, | 0 |
| C-terminal Dimerization Domain of Nucleocapsid Phosphoprotein | 6WJI | −8.4 | 0.0 | Arg259, Val270, Phe274, Arg277, Thr282, Leu291, Trp301, Phe314, Tyr333 | 1: Arg277 | ||
| HR2 Domain | 6LVN | −5.9 | 0.0 | ASP17, ASN20, GLU21, LYS24 | 0 | ||
| Main protease | 6Y2E | −7.1 | 0.0 | Gln107, Gln110, Val202, Asn203, His246, Ile249, Pro293 | 0 | ||
| NSP10 | 6W75·B | −6.9 | 0.0 | Ile4308, Asn4338, Asp4344, Thr4364, Val4369, Thr4371, Trp4376 | 1: Asp4344 | ||
| NSP15 Endoribonuclease | 6VWW | −7.2 | 0.0 | Ile27, Ile28, Asn29, Asn30, Pro51, Val52 | 0 | ||
| NSP16 | 6W75.A | −7.7 | 0.0 | Gln6804, Gln6850, Asn6853, Thr6854, Ser7041, Lys7047, Leu7050, Lys7051, Arg7053 | 0 | ||
| Nsp9 RNA binding protein | 6W4B | −6.3 | 0.0 | Arg40, Phe41, Val42, Phe57, Ile66, Thr68, Ile92 | 1: Val42 | ||
| Nucleocapsid protein N-terminal RNA binding domain | 6M3M | −5.5 | 0.0 | Gln161, Leu162, Gln164, Gly165, Thr166, Thr167, Leu168, Tyr173 | 1: Gly165 | ||
| ORF7A encoded accessory protein | 6W37 | −5.3 | 0.0 | Ser22, Gly23, Tyr25, Glu26, Gly27, Phe31, Pro33, Phe50, | 1: Tyr25 | ||
| Papain-like protease | 6W9C | −6.3 | 0.0 | Phe69, His73, Thr74, Thr75, Asp76, Tyr171, Gln174, His175, Ala176, Leu 178,Asp179 | 1: His175 | ||
| Post fusion core of S2 subunit | 6LXT | −7.5 | 0.0 | Asn919, Gln920, Lys921, Asn928, Ile1198, Leu1200, Gln1201 | 0 | ||
| Prefusion of the spike glycoprotein with a single receptor-binding domain | 6VSB | −6.0 | 0.0 | Thr33, Phe59, Val289, Leu293, Asp294, Lys300, Asn606 | 0 | ||
| RNA-dependent RNA polymerase NSP12 | 6M71.A | −7.3 | 0.0 | Tyr32, Lys47, Lys80, Tyr129, Asn138, Cys139, Thr141, Thr710 | 1: Tyr129 | ||
| RNA-dependent RNA polymerase NSP7 | 6M71.C | −6.3 | 0.0 | Thr9, Leu13, Phe49, Met52, Val53, Leu56, Val66, Ile68 | 0 | ||
| RNA-dependent RNA polymerase NSP8 | 6M71.B.D | −6.5 | 0.0 | Asn100, Pro121, Ala125, Ala126, Lys127, Leu128, Met129, Val131, Tyr149, Trp154, | 4: Asn100, Ala126, Lys127, Met129 | ||
| Spike ectodomain structure (open state) | 6VYB | −6.9 | 0.0 | Thr732, Phe823, Val860, Pro863, Asp867, Ile870, Pro1057, His1058 | 0 | ||
| Spike glycoprotein (closed state) | 6VXX | −7.2 | 0.0 | Ser730, Thr732, Phe782, Phe823, Val860, Pro863, Asp867, Ile870, Asn955, His1058 | 1: Asp867 |
The molecular docking studies revealed that when Chloroquine (4-N-(7-chloroquinolin-4-yl)-1-N, 1-N-diethylpentane-1, 4-diamine) docked against the spike glycoprotein in the open state conformation (PDB: 6VYB), the interaction occurred with the binding energy of −5.1 kcal/mol and Met740, Tyr741, Asn8586, Leu966, Val976, and Asn978 were identified to be the interacting residues (Fig. 1 a). The binding of the drug with closed state conformation (PDB: 6VXX) showed binding energy of −5.2 kcal/mol, and Tyr38, Asp40, Phe43, Glu224, and Pro225 identified as the interacting residues at the binding sites (Fig. 1b). When the drug docked against the post-fusion conformation of the spike glycoprotein (PDB: 6LXT), the complex showed binding energy of −5.0 kcal/mol, and Asn914, Leu 916, and Tyr917 identified as the interacting residues (Fig. 1c). When the pre-fusion conformation of the spike glycoprotein (PDB: 6VSB) interacted with the drug, the interaction stabilized by the binding energy of −5.2 kcal/mol. Pro683, Thr732, and His1058 identified to be the interacting residues (Fig. 1d).
Fig. 1.

The binding mode of Chloroquine with the probable protein targets of SARS-CoV-2 predicted by molecular docking using AutoDock Vina and visualized using MGL tools. The interacting residues and the binding energy are labeled where the residues and ligands are shown in stick figures. The best-docked conformations of Chloroquine and (a) spike glycoprotein in the open conformation (b) spike glycoprotein in the closed conformation (c) spike glycoprotein in the post-fusion conformation (d) spike glycoprotein in the pre-fusion conformation (e) HR2 domain of S2 subunit (f) non-structural protein7 of RNA dependent RNA polymerase (g) non-structural protein8 RNA dependent RNA polymerase (h) non-structural protein12 RNA dependent RNA polymerase (i) papain-like protease (j) main protease (k) C-terminal RNA binding domain (l) N-terminal RNA binding domain (m) endoribonuclease of non-structural protein15 (n) ADP ribose phosphatase of non-structural protein3 (o) RNA the binding protein of non-structural protein9 (p) non-structural protein10 (q) non-structural protein16 (r) orf7a accessory protein.
The drug showed better binding with pre-fusion and closed state conformation when compared to post-fusion and open state conformation of the spike glycoprotein. The interaction of the drug with the HR2 domain (PDB: 6LVN) showed binding energy of −4.7 kcal/mol, and Arg18, Asn20, Glu21, and Lys24 identified to be the interacting residues (Fig. 1e). The interaction of the drug with RdRp (nsp7 (PDB: 6M71_C), nsp8 (PDB: 6M71_B, D), and nsp 12 (PDB: 6M71_A)) predicted by molecular docking, and the complex showed the binding energies of −4.9, −4.4, and −5.5 kcal/mol, respectively. The drug interacted with nsp7 at Thr9, Phe49, Met52, and Val53 (Fig. 1f), nsp8 at Tyr135, Tyr138, Lys139, Asp143, and Ser170 (Fig. 1g), and nsp12 at Tyr32, Tyr129, His133, and Lys780 (Fig. 1h). The interaction of the drug with papain-like protease (PLpro) (PDB: 6W9C) and main protease (Mpro) (PDB: 6Y2E) were stabilized by binding energies of −4.8 and −5.2 kcal/mol, respectively. The drug interacted with PLpro at Lys105, Trp106, and His272 (Fig. 1i), and the drug interacted with Mpro at Met49, Phe140, Leu141, Asn142, His163, Met165, Glu166, and Gln189 (Fig. 1j). The interaction of the drug and C-terminal (PDB: 6WJI) and N-terminal RNA binding domain (PDB: 6M3M) of nucleocapsid phosphoprotein showed binding energies of −6.6 and −5.1 kcal/mol, respectively. The C-terminal domain of nucleocapsid phosphoprotein interacted with Chloroquine at Val270, Phe274, Arg277, and Trp301, and the interaction stabilized by a hydrogen bond between the ligand and the residue Arg277 (Fig. 1k). Similarly, the nucleocapsid phosphoprotein at the N-terminal domain interacted with the residues such as Thr55, Ala156, Ile158, and Val159 (Fig. 1l). When the drug interacted with nucleoproteins such as endoribonuclease of nsp15 (PDB: 6VWW), ADP ribose phosphatase of nsp3 (PDB: 6VXS), and RNA binding protein of nsp9 (PDB: 6W4B), the complexes were showed the binding energies of −5.6, −5.3 and −4.9 kcal/mol, respectively. Endoribonuclease interacted with the drugs via Asn29 and Asn30 (Fig. 1m). ADP ribose phosphatase interacted with the drugs at Leu126 and Phe156 (Fig. 1n), and RNA binding protein interacted with the drug via Gly39, Arg40, Phe57, Ser60, and Ile66 (Fig. 1o). When non-structural proteins nsp10 (PDB: 6W75_B) and nsp16 (PDB: 6W75_A) docked against Chloroquine, the complexes were showed binding energies of −4.9 and −7.0 kcal/mol respectively. Nsp10 interacted with the ligands by the residues such as Gln4289, Thr4292, Cys4294, and Lys4296, and the interaction stabilized by a hydrogen bond (Fig. 1p). Nsp16 interacted with the drug by residues such as Leu6819, Leu6820, Tyr7020, and Val7021 (Fig. 1q). The accessory protein, orf7a (PDB: 6W37) interacted with the drug, and the complex was showed binding energy of −5.0 kcal/mol, and the residues interacted were found to be Tyr25 and Phe31 (Fig. 1r). The molecular docking studied revealed that Chloroquine showed better binding energy (−7.0 kcal/mol) to the targets in comparison with the other drugs. Chloroquine is 4-aminoquinoline and known for its anti-malarial and anti-inflammatory properties [35]. Recently, Chloroquine was used to treat SARS-CoV-2 infections. This study showed the probable structural biology mechanism of the interactions between Chloroquine and potential targets of SARS-CoV-2 by molecular docking studies.
The interaction of Favipiravir (5-fluoro-2-oxo-1H-pyrazine-3-carboxamide) and the prioritized targets of SARS-CoV-2 predicted by molecular docking studies. When Favipiravir docked with the spike glycoprotein in the open state (PDB: 6VYB) and closed state conformation (PDB: 6VXX), the docked complexes were showed binding energies of −5.1 and −4.8 kcal/mol, respectively. Met740, Tyr741, Gly744, Leu977, and Arg1000 identified as the residues interacted with glycoprotein in its open conformation (Fig. 2 a). The amino acid residues Tyr741, Gly744, Leu966, Leu977, and Arg1000 involved in the binding of glycoprotein in the closed state to the drug (Fig. 2b). The binding pocket in both the conformations found to be almost the same. The complexes formed with the closed state conformation stabilized by a hydrogen bond at Gly744. The interaction of post- and pre-fusion conformations of the spike glycoprotein and the drugs were showed the binding energies of −4.5 and −4.6 kcal/mol, respectively. The post-fusion spike protein (PDB: 6LXT), and the drug interacted via Ser967, Gly971, and Leu1166 (Fig. 2c). The pre-fusion spike protein (PDB: 6VSB) interacted with the drugs at Tyr38, Asp40, Gly283, and the interaction stabilized by a hydrogen bond at Asp40 (Fig. 2d). The drug interacted with the HR2 domain (PDB: 6LVN) at Glu21 and Lys24 and the binding energy was estimated to be −4.0 kcal/mol (Fig. 2e). The interaction of the drug with RdRp in which the individual chain such as nsp7 (PDB: 6M71_C), nsp8 (PDB: 6M71_B & D), and nsp 12 (PDB: 6M71_A) were showed binding energies of −4.4, −4.8, and −4.1 kcal/mol respectively. The residues Asp38, Ala42, Lys43, and Asp44 found to be the interacting residues of nsp7 (Fig. 2f). Tyr135 and Ser177 interacted with nsp8 by a hydrogen bond (Fig. 2g), and Trp800 interacted with nsp12 with the formation of a hydrogen bond (Fig. 2h). Favipiravir interacted with PLpro at Asp76, Arg82, Tyr154, Asn156, and the binding energy estimated to be −4.7 kcal/mol (Fig. 2i). Similarly, the drug showed the binding with Mpro at Glu14, Gly15, Met17, and Ala70, and the binding energy was estimated to be −4.7 kcal/mol (Fig. 2j). The C-terminal domain of nucleocapsid protein (PDB: 6WJI) interacted with the drug (binding energy of −4.6 kcal/mol) at Val270 and Arg277 by forming a hydrogen bond at Val270 (Fig. 2k). The N-terminal domain of nucleocapsid phosphoprotein (PDB: 6M3M) interacted at Gly115, Ala120, Gly121, Tyr124, Asn141, and Pro143 (binding energy −4.3 kcal/mol), and the interaction was stabilized by a hydrogen bond with Tyr124 (Fig. 2l). Endoribonuclease (PDB: 6VWW), ADP ribose phosphatase (PDB: 6VXS), and RNA binding proteins (PDB: 6W4B) interacted with the drug and showed binding energies of −4.7, −4.5, and −4.6 kcal/mol respectively. The endoribonuclease (nsp15) interacted with the drug at Gln202, Glu203, Phe204, Ala256, and Phe259, and the interaction stabilized by a hydrogen bond at Ala256 (Fig. 2m). Similarly, ADP ribose phosphatase (nsp3) interacted with the drug at Lys31, Pro32, His86, Ala89, and Lys90 (Fig. 2n). RNA binding protein (nsp9) interacted with the drug at Ser14, Asn26, Asp27, Ala29, Asp48, and the interaction was stabilized by a hydrogen bond at Ser14 (Fig. 2o). The non-structural proteins such as nsp10 (PDB: 6W75_B) and nsp16 (PDB: 6W75_A) interacted with the drug with binding energies of −4.6 and −5.0 kcal/mol, respectively. Nsp10 interacted with the drug at Thr4292, Cys4294, and Asn4358. The interaction stabilized by a hydrogen bond at Cys4294 (Fig. 2p). Nsp16 interacted with the drug at Thr6934, Phe6947, and Phe6948 (Fig. 2q). Accessory protein, orf7a (PD: 6W37), interacted with the drug at Glu18, Ser22, Gly23, Thr24, Tyr25, and Phe31, and the binding energy was estimated to be −4.3 kcal/mol. The interaction stabilized by a hydrogen bond at Ser22 (Fig. 2r). The molecular docking studies suggested that Favipiravir showed better interaction with the closed conformation of the spike glycoprotein (binding energy-5.1 kcal/mol) in comparison to the interactions of the drug with other prioritized targets. Favipiravir is a pyrazine carboxamide derivative with activity against RNA viruses, approved as the drug of choice against the influenza virus. It is known to be effective against RNA viruses and probably acts as an eye-opener for the therapeutic development against SARS-CoV-2.
Fig. 2.

The binding mode of Favipiravir with the probable protein targets of SARS-CoV-2 predicted by molecular docking using AutoDock Vina and visualized using MGL tools. The interacting residues and the binding energy is labeled where the residues and ligands are shown in stick figures. The best-docked conformations of Favipiravir and (a) spike glycoprotein in the open conformation (b) spike glycoprotein in the closed conformation (c) spike glycoprotein in the post-fusion conformation (d) spike glycoprotein in the pre-fusion conformation (e) HR2 domain of S2 subunit (f) non-structural protein7 of RNA dependent RNA polymerase (g) non-structural protein8 RNA dependent RNA polymerase (h) non-structural protein12 RNA dependent RNA polymerase (i) papain-like protease (j) main protease (k) C-terminal RNA binding domain (l) N-terminal RNA binding domain (m) endoribonuclease of non-structural protein15 (n) ADP ribose phosphatase of non-structural protein3 (o) RNA binding protein of non-structural protein9 (p) non-structural protein10 (q) non-structural protein16 (r) orf7a accessory protein.
When Hydroxychloroquine (2-[4-[(7-chloroquinolin-4-yl) amino] pentyl-ethylamino] ethanol) docked against the open state of the spike glycoprotein (PDB: 6VYB), the complex was stabilized with a binding energy of −5.0 kcal/mol. The interacting residues present in the binding cavity were Leu517, Asn544, Leu546, Gln564, and Phe565 (Fig. 3 a). Spike glycoprotein in the closed state (PDB: 6VXX) interacted with the drug that demonstrated binding energy of −5.7 kcal/mol. The residues Trp104, Val126, Phe192, Phe194, Val227and Asp228 identified to be the residues present in the binding cavity (Fig. 3b). When the drug interacted with post-fusion (PDB: 6LXT) and pre-fusion (PDB: 6VSB) conformations of spike glycoprotein, the complexes were demonstrated the binding energies of −4.7 kcal/mol and −5.2 kcal/mol, respectively. The residues Gly932, Lys933, Gln935, Asp936, Ser939, Lys1191, and Asn1192 identified as the binding residues of the post-fusion conformation (Fig. 3c), Ser730, Thr778, Pro863, Thr866, Asp867, Glu868, and His1058 identified as the interacting residues in the pre-fusion conformation (Fig. 3d). When the drug interacted with the HR2 domain, the complex revealed that the binding energy of −4.7 kcal/mol (PDB: 6LVN), and the interacting residues identified to be Lys14, Asp17, and Glu21 (Fig. 3e). RdRp consists of nsp7 (PDB: 6M71_C), nsp8 (PDB: 6M71_B & D), and nsp 12 (PDB: 6M71_A) docked with the drug, complex revealed the binding energies of −4.6, 4.7, and −4.9 kcal/mol, respectively. Thr9, Thr46, Phe47, and Met52 involved in the interaction with nsp7, in which Thr46 formed a hydrogen bond (Fig. 3f). The residues Phe92, Leu95, Ile107, and Ala110 were interacted with nsp8 (Fig. 3g), whereas, Tyr273, Leu329, Val330, Ala379, and Ala382 were acted as the interacting residues of nsp12 (Fig. 3h). Papain-like protease (PLpro) (PDB: 6W9C) interacted with the drug demonstrated the binding energy of −5.0 kcal/mol in which Tyr213, Glu214, Lys217, Tyr305, Tyr310, and Thr313 identified to be the interacting residues. Thr313 produced H-bond with the drug (Fig. 3i). Similarly, when the main protease (Mpro) (PDB: 6Y2E) docked against the drug, the complex demonstrated a binding energy of −5.9 kcal/mol, in which Gln110, Thr111, Val202, Thr292, and Phe294 were acted as the amino acid residues (Fig. 3j). The docking of Hydroxychloroquine against the C-terminal dimerization domain of nucleocapsid protein (PDB: 6WJI) were displayed the binding energy of −5.8 kcal/mol. The interacting residues identified were Arg259, Val270, Phe274, Arg277, Phe286, and Ile304, in which Arg277 formed a hydrogen bond with the ligand (Fig. 3k). When the N-terminal domain of the nucleocapsid phosphoprotein (PDB: 6M3M) docked with the ligand, the complex showed binding energy of −5.0 kcal/mol and Gln84, Thr136, and Thr166 identified to be the interacting residues. Thr166 formed a hydrogen bond with the ligand (Fig. 3l). Endoribonuclease (nsp15) (PDB: 6VWW) interacted with the ligand with the binding energy of −5.9 kcal/mol. Asn29 and Asn30 identified to be the interacting residues in which Asn30 formed a hydrogen bond with the drug (Fig. 3m). When ADP ribose phosphatase (nsp3) (PDB: 6VXS) and RNA binding protein (nsp9) (PDB: 6W4B) interacted with the drug, the docked complexes were demonstrated the binding energies of −5.8 and −5.1 kcal/mol, respectively. The drug interacted with ADP ribose phosphatase at Ile23, Ala38, Leu126, Ser128, Ala129, Ile131, Phe132, and Phe156 (Fig. 3n), and the drug interacted with RNA binding protein at Arg40, Val42, Ile66, Phe67, and Thr68 (Fig. 3o). Nsp10 (PDB: 6W75_B) and nsp16 (PDB: 6W75_A) docked against the drug revealed that the binding energies of −5.3 kcal/mol and −6.3 kcal/mol, respectively. Tyr4280, Tyr4283, Ile4291, Thr4292, Arg4331, and Leu4328 were identified to be the interacting residues in nsp10 (Fig. 3p), and Arg6817, Met6818, Leu6819, Leu6820, Tyr7020, and Val7021 were identified as the residues involved in binding with nsp16 (Fig. 3q). The accessory protein (orf7a) (PDB: 6W37) interacted with the drug by the binding energy of −4.8 kcal/mol. Tyr25 and Phe31 were the residues present in the binding cavity (Fig. 3r). From the molecular docking studies, it was evident that Hydroxychloroquine showed the best interaction to the non-structural protein-nsp16 with a binding energy of −6.3 kcal/mol in comparison with other targets. There are studies reported that Hydroxychloroquine, an analog of Chloroquine, probably possess anti-viral and anti-inflammatory activities against coronavirus. Further, the drug is considered less toxic and safer than Chloroquine.
Fig. 3.

The binding mode of Hydroxychloroquine with the probable protein targets of SARS-CoV-2 predicted by molecular docking using AutoDock Vina and visualized using MGL tools. The interacting residues and the binding energy are labeled where the residues and ligands are shown in stick figures. The best-docked conformations of Hydroxychloroquine and (a) spike glycoprotein in the open conformation (b) spike glycoprotein in the closed conformation (c) spike glycoprotein in the post-fusion conformation (d) spike glycoprotein in the pre-fusion conformation (e) HR2 domain of S2 subunit (f) non-structural protein7 of RNA dependent RNA polymerase (g) non-structural protein8 RNA dependent RNA polymerase (h) non-structural protein12 RNA dependent RNA polymerase (i) papain-like protease (j) main protease (k) C-terminal RNA binding domain (l) N-terminal RNA binding domain (m) endoribonuclease of non-structural protein15 (n) ADP ribose phosphatase of non-structural protein3 (o) RNA binding protein of non-structural protein9 (p) non-structural protein10 (q) non-structural protein16 (r) orf7a accessory protein.
The interaction of Lopinavir ((2S)–N-[(2S,4S, 5S)-5-[[2-(2,6-dimethylphenoxy) acetyl] amino]-4-hydroxy-1,6-diphenylhexan-2-yl]-3-methyl-2-(2-oxo-1,3-diazinan-1-yl) butanamide) and the prioritized targets of SARS-CoV-2 were studied by molecular docking. When Lopinavir docked with the open state conformation and the closed state conformation of spike glycoprotein, the complexes were showed binding energies of −8.3 and −8.7 kcal/mol, respectively. The drug interacted with the open state conformation of spike glycoprotein at Asp294, Pro295, Leu296, Phe318, Val610, and Ile651 (Fig. 4 a). The residues Arg355, Asp428, Phe429, Thr430, Lys462, Pro463, Phe464, Ser514, and Glu516 identified to be the major binding residues present in the closed state conformation (Fig. 4b). The binding of Lopinavir against the post-fusion state and the pre-fusion state confirmations of spike glycoprotein were exhibited the binding energies of −8.1 and −8.2 kcal/mol, respectively. The major residues involved in the interaction of the post-fusion conformation of spike glycoprotein and the drug found to be Lys921, Asn928, Gln935, and Leu1200 (Fig. 4c). The residues involved in the interaction of the pre-fusion state of the spike glycoprotein and drug identified to be Arg815, Phe823, Leu828, Asp867, and Glu868 (Fig. 4d). The HR2 domain interacted with the drug (binding energy −7.8 kcal/mol) in which Lys14, Arg18, Lys24, Asn27, and Ile31 identified to be the major residues present in the binding cavity (Fig. 4e). The RdRp consists of nsp7, nsp8, and nsp 12 docked against Lopinavir, the complexes were showed binding energies of −7.3, −8.2, and −8.1 kcal/mol, respectively. The drug interacted with nsp7 at the position Thr9, Val16, Val53, Leu56, and Ile68 (Fig. 4f). The drug interacted with nsp8 at Leu122, Leu128, Val130, Val131, and Tyr149 (Fig. 4g), and the drug binds with nsp12 at Pro412, Phe415, Tyr420, Leu437, and Phe440 (Fig. 4h). The interaction of PLpro and the drug showed binding energy of −8.1 kcal/mol, and Lys217, Lys218, Tyr305, Asn308, and Tyr310 identified to be the interacting residues (Fig. 4i). When Lopinavir interacted with Mpro, the complex showed the binding energy of −8.6 kcal/mol in which Gln107, Gln110, Thr111, Ile200, Glu240, His246, Thr292, and Phe294 identified to be the major interacting residues (Fig. 4j). When Lopinavir docked to the C-terminal domain of nucleocapsid protein, the complex exhibited the binding energy of −11.0 kcal/mol, and Arg259, Gln260, Ala264, Phe274, Arg277, Trp301, Phe314, Leu331, and Tyr333 involved in the binding (Fig. 4k). N-terminal domain of the nucleocapsid phosphoprotein interacted with the drug, the complex showed the binding energy of −7.5 kcal/mol. The major residues interacted with the drug were Trp53, Asn76, Ile147, Asn151, Asn155, and Ile158 (Fig. 4l). When endoribonuclease (nsp15), ADP ribose phosphatase (nsp3) (PDB: 6VXS), and RNA binding proteins (nsp9) docked with the drug, the complexes revealed the binding energies of −9.6, −8.5, and −7.8 kcal/mol, respectively. The drug interacted with nsp15 at the position Ile27, Ile28, Asn29, Asn30, and Val52 (Fig. 4m), and the drug interacted with nsp3 at Ile23, Gly48, Ala52, Lys55, Leu126, and Phe156 (Fig. 4n). The major residues of nsp9 participated in the binding were Gly39, Arg40, Phe41, Val42, Phe57, Ser60, and Ile66 (Fig. 4o). When the non-structural proteins such as nsp10 and nsp16 docked with Lopinavir, the complexes were depicted the binding energies of −8.2 and −9.9 kcal/mol, respectively. The residues Ala4279, Ile4291, Thr4292, Asn4293, Leu4328, Arg4331, Cys4332, and His4333 identified as the major residues present at the binding site of nsp10 (Fig. 4p). Similarly, Arg6817, Leu6819, Tyr7020, and Val7021 identified as the main residues involved in the binding of the drug (Fig. 4q). When Orf7a docked with the drug showed the binding energy of −7.0 kcal/mol in which Thr24, Tyr25, Pro30, and Phe31 were identified to be the major interacting residues. Phe31 formed a hydrogen bond with the drug (Fig. 4r). From the interaction modeling by molecular docking, it was evident that the best binding affinity observed for the complexes of the C-terminal domain of nucleocapsid phosphoprotein and Lopinavir (−11.0 kcal/mol) in comparison with the other docked structures. Further, Lopinavir demonstrated better binding abilities to several targets in comparison with the docking results of other drugs. Lopinavir is used as an antiviral agent against coronavirus and showed reduced symptoms when used in combination with Ritonavir. Thus, there is a scope for the repurposing of Lopinavir towards the targets highlighted in this study.
Fig. 4.

The binding mode of Lopinavir with the probable protein targets of SARS-CoV-2 predicted by molecular docking using AutoDock Vina and visualized using MGL tools. The interacting residues and the binding energy are labeled where the residues and ligands are shown in stick figures. The best-docked conformations of Lopinavir and (a) spike glycoprotein in the open state conformation (b) spike glycoprotein in the closed state conformation (c) spike glycoprotein in the post-fusion conformation (d) spike glycoprotein in the pre-fusion conformation (e) HR2 domain of S2 subunit (f) non-structural protein7 of RNA dependent RNA polymerase (g) non-structural protein8 RNA dependent RNA polymerase (h) non-structural protein12 RNA dependent RNA polymerase (i) papain-like protease (j) main protease (k) C-terminal RNA binding domain (l) N-terminal RNA binding domain (m) endoribonuclease of non-structural protein15 (n) ADP ribose phosphatase of non-structural protein3 (o) RNA binding protein of non-structural protein9 (p) non-structural protein10 (q) non-structural protein16 (r) orf7a accessory protein.
Remdesivir (2-ethylbutyl (2S)-2-{[(S)-{[(2R,3S,4R, 5R)-5-{4-aminopyrrolo [2,1-f][1,2,4]triazin-7-yl}-5-cyano-3,4-dihydroxyoxolan-2-yl]methoxy}(phenoxy)phosphoryl]amino}propanoate) was docked against spike proteins; the open state conformation showed the binding energy of −6.9 kcal/mol and the major residues involved in the binding was found to be Thr33, Phe59, Asp294, Leu296 and Val608 (Fig. 5 a). The closed conformation of the target and the drug interacted with Val341, Ala344, Ala348, Ser349, Tyr351, and Lys356 in which a hydrogen bond formed by Asn354 (binding energy −7.0 kcal/mol) (Fig. 5b). The interaction of post-fusion conformation of spike glycoprotein and the drug showed the binding energy of −7.1 kcal/mol in which Asn928, Ser929, Gly932, Leu1197, and Asp1199 identified to be the major interacting residues. The binding stabilized by a hydrogen bond formed at Leu1200 (Fig. 5c). Remdesivir interacted with pre-fusion conformation at Asn907, Thr912, Glu1092, Gly1093, Phe1089, and Phe1121. Arg1107 formed a hydrogen bond with the drug (binding energy −6.9 kcal/mol) (Fig. 5d). When the drug docked to the HR-2 domain of the S2 subunit, the complex showed the binding energy of −6.4 kcal/mol, and Lys14, Asp17, Arg18, Asn20, and Glu21 found to be the major interacting residues (Fig. 5e). The interaction modeling of RdRp with nsp7, nsp8, and nsp 12 showed the binding energies of −6.9 kcal/mol (Ser25, Leu28, Val53, Ser54, Ser57, Val58, and Ser61 acted to be the major binding residues) (Figs. 5f), −7.3 kcal/mol (Leu98, Pro121, Ala125, Ala126, Leu128, Tyr149, and Trp154 found to be the main interacting residues) (Fig. 5g), and −7.5 kcal/mol (Arg249, Trp268, Thr319, Pro322, Pro323, Leu460, and Pro461 identified to be the major binding residues) (Fig. 5h), respectively. When Remdesivir docked with PLpro and Mpro, the complexes were showed binding energies of −7.4 and −7.7 kcal/mol, respectively. Arg82, Asn128, Tyr154, Gln174, His175, Asp179, and Val202 (hydrogen bond formation at Thr74 and Thr75) identified as the major residues at the binding sites of PLpro (Fig. 5i), and Gln110, Asp153, Thr292, and Phe294 identified to be the major residues identified in the binding site of Mpro (Fig. 5j). When the drug docked against the C-terminal dimerization and N-terminal RNA binding domains of nucleocapsid proteins, the complexes were showed the binding energies of −8.2 and −6.4 kcal/mol, respectively. The drug interacted with Val270, Phe274, Phe286, Leu291, Trp301, Ile304, and Ala308, Tyr333 of the C-terminal domain, and formed a hydrogen bond at Arg277 (Fig. 5k). The drug interacted with the N-terminal domain of the target in the position Asn127, Gly138, Asn141, and hydrogen bonds formed at Tyr124 and Trp133 (Figure 5l). The non-structural proteins nsp15 endoribonuclease showed interactions with the residues such as Val156, Lys159, Thr193, Tyr194, Phe195, and Gln197 in which Thr193 and Gln197 involved in the hydrogen bond formation (binding energy −8.0 kcal/mol) (Fig. 5m). Nsp3-ADP ribose phosphatase interacted with the drug at Val41, Tyr42, Gly48, Lys102, Gly130, Ile131, and Phe132 and formed a hydrogen bond at Asn40 (binding energy −7.6 kcal/mol) (Fig. 5n). Nsp9 RNA binding protein interacted with the drug with the binding energy of −6.4 kcal/mol, and Met13, Arg40, Val42, Phe57, Ser60, Ile66, and Thr68 identified as the major binding residues (Fig. 5o). The drug interacted with the non-structural proteins such as nsp10 (chain B) and nsp16 (chain A) and showed the binding energies of 7.1 kcal/mol and 8.7 kcal/mol, respectively. The major binding position in nsp10 observed to be Gln4306, Ile4308, Phe4342, Asp4344, Thr4371, Gly4374, and a hydrogen bond formed at Val4369 (Fig. 5p). The drug interacted with nsp16 at Leu6820, Lys6822, Glu6940, Asn6941, His6972, and Tyr7020 (Fig. 5q). The drug and orf7a interacted at Tyr25, Phe31 Pro33, and the interaction stabilized by hydrogen bonds with the binding energy of −6.4 kcal/mol (Fig. 5r). The molecular docking studies showed that Remdesivir has a profound binding with several prioritized targets. The drug showed better interaction to nsp16 (binding energy of −8.7 kcal/mol) when compared to the binding of the drug with other molecular targets, repurposing of Remdesivir against nsp16 has an application against COVID-19 drug discovery. Remdesivir (GS-5734), an adenosine triphosphate analog, recently suggested using against COVID-19 [46]. The predicted computational model probably offers the molecular mechanism of binding of this drug with the targets and provides insight into drug repurposing against SARS-CoV2.
Fig. 5.

The binding mode of Remdesivir with the probable protein targets of SARS-CoV-2 predicted by molecular docking using AutoDock Vina and visualized using MGL tools. The interacting residues and the binding energy are labeled where the residues and ligands are shown in stick figures. The best-docked conformations of Remdesivir and (a) spike glycoprotein in the open conformation (b) spike glycoprotein in the closed conformation (c) spike glycoprotein in the post-fusion conformation (d) spike glycoprotein in the pre-fusion conformation (e) HR2 domain of S2 subunit (f) non-structural protein7 of RNA dependent RNA polymerase (g) non-structural protein8 RNA dependent RNA polymerase (h) non-structural protein12 RNA dependent RNA polymerase (i) papain-like protease (j) main protease (k) C-terminal RNA binding domain (l) N-terminal RNA binding domain (m) endoribonuclease of non-structural protein15 (n) ADP ribose phosphatase of non-structural protein3 (o) RNA binding protein of non-structural protein9 (p) non-structural protein10 (q) non-structural protein16 (r) orf7a accessory protein.
The docking studies suggested that the interaction of the open form of the ectodomain of glycoprotein to Ritonavir showed the binding energy of −6.9 kcal/mol. The major interacting residues observed to be Thr732, Phe823, Val860, Pro863, Asp867, Ile870, Pro1057, and His1058 (Fig. 6 a). The closed state of glycoprotein and drug complex were stabilized with the binding energy of −7.2 kcal/mol, and the major interacting residues identified to be Ser730, Thr732, Phe782, Phe823, Val860, Pro863, Asp867, Ile870, Asn955, and His1058 in which Asp867 formed a hydrogen bond with the drug (Fig. 6b). The interaction of drug and post-fusion and pre-fusion state of the target demonstrated the binding energies of −7.5 and −6.0 kcal/mol, respectively. Asn919, Gln920, Lys921, Asn928, Ile1198, Leu1200, and Gln1201 were the major residues present at the binding of cavity post-fusion state (Fig. 6c). Thr33, Phe59, Val289, Leu293, Asp294, Lys300, and Asn606 were the major residues present at the binding site of the pre-fusion state (Fig. 6d). HR-2 domain of the S2 subunit, when docked with the drug, showed the binding energy of −5.9 kcal/mol and Asp17, Asn20, Glu21, and Lys24 identified to be the major interacting residues (Fig. 6e). Nsp7, nsp8, and nsp 12 of RdRp demonstrated the binding energies of −6.3, −6.5, and −7.3 kcal/mol, respectively, when the drug interacted with the target. The drug interacted with nsp7 at the position Thr9, Leu13, Phe49, Met52, Val53, Leu56, Val66, and Ile68 (Fig. 6f). The drug also interacted with nsp8 at the position Asn100, Pro121, Ala125, Ala126, Lys127, Leu128, Met129, Val131, Tyr149, and Trp154 in which Asn100, Ala126, Lys127, and Met129 formed hydrogen bonds with the ligand (Fig. 6g). The drug binds with Nsp12 at Tyr32, Lys47, Lys80, Tyr129, Asn138, Cys139, Thr141, and Thr710, in which Tyr129 formed a hydrogen bond with the drug (Fig. 6h). The PLpro and Mpro demonstrated the binding energies of −6.3 and −7.1 kcal/mol, respectively when the drug interacted with Ritonavir. Ritonavir interacted with PLpro at Phe69, His73, Thr74, Thr75, Asp76, Tyr171, Gln174, Ala176, and Leu 178, in which, Asp179 formed a hydrogen bond (Fig. 6i) and the drug interacted with Mpro in the position Gln107, Gln110, Val202, Asn203, His246, Ile249, and Pro293 (Fig. 6j). The molecular interaction between the drug-C-terminal dimerization domain of nucleocapsid proteins and the drug-N-terminal RNA binding domain of nucleocapsid were demonstrated the binding energies of −8.4 and −5.5 kcal/mol, respectively. Arg259, Val270, Phe274, Thr282, Leu291, Trp301, Phe314, and Tyr333 acted as the major binding residues of the C-terminal domain with a hydrogen bond at Arg277 (Fig. 6k). Gln161, Leu162, Gln164, Gly165, Thr166, Thr167, Leu168, and Tyr173 of the N-terminal domain of the target acted as the major binding residues in which Gln161 formed a hydrogen bond with the drug (Fig. 6l). Endoribonuclease nsp15, nsp3- ADP ribose phosphatase, and nsp9 of RNA binding protein were interacted with the drug, and the complexes were exhibited the binding energies of −7.2, −6.7, and −6.3 kcal/mol, respectively. The major interacting residues with the ligand observed in the nsp15 were Ile27, Ile28, Asn29, Asn30, Pro51, and Val52 (Fig. 6m), the interacting residues present in the nsp3 identified to be Ile23, Val49, Leu126, Gly130, Ile131, Pro136, Phe156, Asp157, and Leu160 (Fig. 6n) and the major interacting residues present in the nsp9 observed to be Arg40, Phe41, Phe57, Ile66, Thr68, and Ile92 with hydrogen bond at Val 92 (Fig. 6o). When nsp10 (chain B) and nsp16 (chain A) interacted with the drug, the complexes showed binding energies of −6.9 kcal/mol and −7.7 kcal/mol, respectively. Ile4308, Asn4338, Thr4364, Val4369, Thr4371, and Trp4376 were the major interacting residues observed in the nsp10, and Asp4344 formed a hydrogen bond with the drug (Fig. 6p). The residues Gln6804, Gln6850, Asn6853, Thr6854, Ser7041, Lys7047, Leu7050, Lys7051, and Arg7053 acts as major residues present at the binding pocket of nsp16 (Fig. 6q). The orf7a interacted with the drug at Ser22, Gly23, Glu26, Gly27, Phe31, Pro33, and Phe50 by a hydrogen bond at Tyr25 (binding energy of −5.3 kcal/mol) (Fig. 6r). From the molecular docking studies, it was clear that Ritonavir showed potential binding against the C-terminal domain of nucleocapsid phosphoprotein (binding energy −8.4 kcal/mol) in comparison with the binding of other targets. Further, Ritonavir showed profound binding with several prioritized targets. Thus, the study suggested that the repurposing of Ritonavir against several molecular targets of COVID-19 probably offers scope and application. Studies suggested that Ritonavir, a drug in combination with Lopinavir, is being investigated against COVID-19 as therapeutic agent [27], the effectiveness of the drug needs to study.
Fig. 6.

The binding mode of Ritonavir with the probable protein targets of SARS-CoV-2 predicted by molecular docking using AutoDock Vina and visualized using MGL tools. The interacting residues and the binding energy are labeled where the residues and ligands are shown in stick figures. The best-docked conformations of Ritonavir and (a) spike glycoprotein in the open conformation (b) spike glycoprotein in the closed conformation (c) spike glycoprotein in the post-fusion conformation (d) spike glycoprotein in the pre-fusion conformation (e) HR2 domain of S2 subunit (f) non-structural protein7 of RNA dependent RNA polymerase (g) non-structural protein8 RNA dependent RNA polymerase (h) non-structural protein12 RNA dependent RNA polymerase (i) papain-like protease (j) main protease (k) C-terminal RNA binding domain (l) N-terminal RNA binding domain (m) endoribonuclease of non-structural protein15 (n) ADP ribose phosphatase of non-structural protein3 (o) RNA binding protein of non-structural protein9 (p) non-structural protein10 (q) non-structural protein16 (r) orf7a accessory protein.
3.3. Binding of drugs and their usual targets
The binding energy of the interaction between FDA approved drugs and their usual molecular targets predicted by molecular docking studies illustrated in Table 3 . When Chloroquine docked against Glutathione - S - transferase (PDB: 1OKT), it showed that the interaction stabilized with the binding energy of −3.8 kcal/mol with Glu90, Leu 91, and Phe 94 as interacting residues (Fig. 7 a). Favipiravir docked against RdRp catalytic subunit (PDB: 6QNW), and the complex showed the binding energy of −4.7 kcal/mol. The interacting residues identified to be Lys308, Trp309, Asn310, Glu311, Asp445, and the interaction stabilized by two hydrogen bonds with Lys308 and Asn310 (Fig. 7b). When Hydroxychloroquine docked against ACE2 (PDB: 1R42), it showed the binding energy of −1.0 kcal/mol with Gln287 as the interacting residue (Fig. 7c). When Lopinavir interacted with protease (PDB: 5V4Y), the binding energy of −7.2 kcal/mol was observed, and Leu5, Leu23, Asp25, Thr26, Gly27, and Phe99 identified to be the interacting residues (Fig. 7d). Remdesivir interacted with the protease of SARS coronavirus (PDB: 1Q2W), and the complex exhibited the binding energy of −10.7 kcal/mol. Gly2, Lys5, Arg4, Gln127, Lys137, Ser284, Glu290, and Phe291 found to be the interacting residues. The docked complex stabilized by the formation of hydrogen bonds at Lys5 and Gln127 (Fig. 7e). Ritonavir interacted with human cytochrome P4503A4 (PDB: 3NXU) with the binding energy of −10.3 kcal/mol. Arg105, Phe108, Ser119, Arg205, Leu210, Leu211, Phe213, Phe215, Ile301, Phe304, Glu308, Thr309, Ile369, Ala370, Met371, Arg372, Gly481, Leu482, and Leu483 were identified as the interacting residues, and the binding stabilized by a hydrogen bond at Met371 (Fig. 7f). The binding energy (kcal/mol), number of interacting residues, and weak interactions involved in the binding of drugs and their usual targets were considered as the reference/control for the comparison of the binding of the drugs and prioritized targets of SARS-CoV2 in this study.
Table 3.
The binding potential of the selected drugs towards their usual targets obtained by molecular docking studies by AutoDock Vina.
| Target organism | Name of the target | PDB ID | Name of the drug with DrugBank ID | Binding energy (kcal/mol) | RMSD (Å) | Interacting residues | Hydrogen bonds |
|---|---|---|---|---|---|---|---|
| Plasmodium falciparum | Glutathione-S-transferase | 1OKT.A | Chloroquine (DB00608) | −3.8 | 0.0 | Glu90, Leu 91,Phe 94 | 0 |
| Influenza A virus (A/Northern Territory/60/1968(H3N2)) | RNA-directed RNA polymerase catalytic subunit | 6QNW.B | Favipiravir (DB12466) | −4.7 | 0.0 | Lys308, Trp309, Asn310, Glu311, Asp445 | 2: Lys308, Asn310 |
| Homo sapiens | Angiotensin converting enzyme 2 | 1R42 | Hydroxychloroquine (DB01611) | −1.0 | 0.0 | Gln287 | 0 |
| Human immunodeficiency virus 1 | Protease | 5V4Y | Lopinavir (DB01601) | −7.2 | 0.0 | Leu5, Leu23, Asp25, Thr26, Gly27, Phe99 | 0 |
| SARS - related coronavirus | Protease | 1Q2W | Remdesivir (DB14761) | −10.7 | 0.0 | Gly2, Lys5, Arg4, Gln127, Lys137, Ser284, Glu290, Phe291 | 2: Lys5, Gln127 |
| Homo sapiens | Human cytochrome P4503A4 | 3NXU. A | Ritonavir (DB00503) | −10.3 | 0.0 | Arg105, Phe108, Ser119, Arg205, Leu210, Leu211, Phe213, Phe215, Ile301, Phe304, Glu308, Thr309, Ile369, Ala370, Met371, Arg372, Gly481, Leu482, Leu483 | 1: Met371 |
Fig. 7.

The binding potential of the selected drugs towards their usual targets predicted by molecular docking studies using AutoDock Vina. The interaction of (a) Chloroquine and Glutathione transferase of Plasmodium falciparum (b) Favipiravir and RNA-directed RNA polymerase catalytic subunit of influenza virus (c) hydroxychloroquine and human angiotensin-converting enzyme 2 (hACE2) (d) Lopinavir and HIV–I protease (e) Remdesivir and SARS protease (f) Ritonavir and human cytochrome P4503A4.
In comparison to the binding affinities of the currently prescribed drugs with their respective targets, the selected drugs showed better binding energy towards most of the prioritized targets in the current study. This study observed that the interaction with Lopinavir and Ritonavir towards the prioritized targets showed the binding energy better than −6.0 kcal/mol towards all the probable targets of SARS-CoV-2 in comparison with the binding of these drugs to their usual targets. The interaction of Lopinavir, Chloroquine, and Hydroxychloroquine also showed better binding to the prioritized targets when compared to their standard target. However, in the case of Remdesvir, the control showed better binding energy in comparison with the interaction of this drug with selected targets. Thus, the molecular docking studies suggested that repurposing of these drugs have application towards the potential targets of SARS-CoV-2 and the study helps in understanding the interaction mechanism of the suggested drugs towards the probable drug targets of SARS CoV-2. Thus, the current study certainly provides the foundation on the structural and molecular mechanisms of interaction of the repurposed FDA drugs and prioritized targets.
3.4. Molecular dynamic simulation studies
The best-docked complexes that showed the minimum binding energy (kcal/mol) and maximum stabilizing interacting residues obtained by molecular docking studies simulated using the Desmond module of the Schrodinger suite to confirm the dynamics and stability of the interactions and binding conformations between the targets and drugs. The best-docked complexes selected for simulation were Chloroquine - nsp 16, Favipiravir - closed state conformation of spike glycoprotein, Hydroxychloroquine - nsp16, Lopinavir - C-terminal domain of nucleocapsid phosphoprotein, Remdesivir - nsp16, and Ritonavir - C-terminal domain of nucleocapsid phosphoprotein. Various parameters of simulation trajectories like protein-ligand RMSD, RMSF, and other binding features are illustrated in the following sections.
Molecular docking studies suggested that Chloroquine showed the best binding potential towards the non-structural protein (nsp) 16 (PDB: 6W75_A) when compared to the other SARS-CoV-2 protein targets. Therefore, this docked complex simulated for 100 ns, and its trajectories depicted in Fig. 8 . The RMSD deviation of the protein in the complex showed in Fig. 8a. The RMSD of the protein deviated between 1.2 and 3.6 Å during the simulation period of 100 ns The RMSD initially increased up to 2 Å in the first 35ns and later stabilized. This deviation might be due to the interaction of the drug and the protein. The protein RMSF ranged between 0.6 Å and 4.0 Å during the simulation (Fig. 8b). The RMSF at the N -terminal region deviated with an exponential level indicating high fluctuation. The protein RMSF found to be 4.2 Å, which ranged at the residue region 25–50 and 75–90 during the simulation period due to drug-protein interaction. The ligand RMSF ranged between 3.0 and 6.0 Å is depicted in Fig. 8c. The ligand RMS fluctuation might be due to the drug atoms that underwent a conformational change when it interacted with protein. The interactions like hydrophobic interactions, water bridges, and hydrogen bonds formed during the protein-ligand interactions are shown in Fig. 8d. The plot indicated that several weak interactions formed throughout the simulation, which included the hydrophobic interactions and hydrogen bond formation with several residues. The ligand-protein contacts that occurred during more than 10% of the simulation period are shown in Fig. 8e, which indicated that the hydrophobic interactions formed at Leu6820, Tyr7020, and Trp6974 acted as main weak interactions.
Fig. 8.

The stability of docked complex of chloroquine – nsp 16 analyzed by molecular dynamic simulation (MD) studies (a) Protein-ligand RMSD: Protein RMSD (Å) on the y-axis and simulation time on the x-axis (b) Protein RMSF: RMSF (Å) on the y-axis and atom index on the x-axis (c) Ligand RMSF: RMSF (Å) on the y-axis and atom index on the x-axis (d) The protein-ligand contacts: blue indicates water bridges, grey indicates hydrophobic interactions, pink indicates ionic bonds and green indicates hydrogen bonds (e) Major interactions between nsp16 and chloroquine during MD simulation (f) Protein-ligand contacts: Timeline representation of interactions. The total number of specific contacts and the major residues of the protein interacted with the ligand.
The protein-ligand contact observed during the simulation is shown in Fig. 8f. The top region of the figure showed that the interaction contacts observed during the 100ns simulation time, and the bottom part showed that the residue interacted with Chloroquine during the simulation period. The residues interacted are Leu6820, Trp6974, Tyr7020, His 7023, and Ala 7024. The major structural features and conformational changes of nsp 16, Chloroquine, and their complex during MD simulation are shown in supplementary materials (Fig. S1a – S2d). The MD simulation also showed that the complex of Chloroquine-nsp16 found to be stable throughout the simulation.
The structural stability and molecular dynamic mechanism of the interaction of Favipiravir to the closed conformation of the SARS-CoV-2 spike glycoprotein (PDB: 6VXX) were studied by MD simulation at 100 ns The interaction trajectories of the complex are shown in Fig. 9 . The RMSD fluctuations of the protein during the interaction are displayed in Fig. 9a. The RMSD values ranged from 2.5 to 20.0 Å in the first 40ns simulation, and later stabilized with conformational changes of the protein. The RMSF of the protein was in the range of 2.0–14.0 Å (Fig. 9b). An increased RMSF of >14.0 Å was found in the residue regions 375 to 400, and RMSF values of the residues ranged between 4.0 and 6.0 Å. Similarly, the ligand RMS fluctuated between 5.0 and 7.0 Å due to their interaction with the protein. The major forces that stabilized the protein-drug interactions are illustrated in Fig. 9d.
Fig. 9.

The stability of docked complex of favipiravir – spike protein analyzed by molecular dynamic simulation (MD) studies (a) Protein-ligand RMSD: Protein RMSD (Å) on the y-axis and simulation time on the x-axis (b) Protein RMSF: RMSF (Å) on the y-axis and atom index on the x-axis (c) Ligand RMSF: RMSF (Å) on the y-axis and atom index on the x-axis (d) The protein-ligand contacts: blue indicates water bridges, grey indicates hydrophobic interactions, pink indicates ionic bonds and green indicates hydrogen bonds (e) Major interactions between favipiravir and spike protein during MD simulation (f) Protein-ligand contacts: Timeline representation of interactions. The total number of specific contacts and the major residues of the protein interacted with the ligand.
There were water bridges, and hydrogen bonds have stabilized the interaction, but no ionic interaction. The binding of the protein-drug at 10% of the simulation time is illustrated in Fig. 9e. The residues Met740 and Phe 855 involved in hydrophobic interactions, and Ser45, Ser 46, and Asn 856, involved in polar bond formation. Asp745 is involved in a negative electrostatic bond formation with the ligand. The interacting residues and residue contacts with the drug during the simulation are shown in Fig. 9f. The residue contact during the simulation ranged from 2 to 9 in which Ser45, Met740, and Asp745 interacted consistently throughout the simulation. The major structural features and conformational changes of the closed conformation of glycoprotein and Favipiravir and the protein-drug complex during MD simulation are shown in the supplementary materials (Fig. S3a – S4d). From the MD simulation, it is clear that Favipiravir demonstrated stable binding to the closed conformation of glycoprotein with several weak interactions, and the protein-drug complex was found to be stable during the MD simulation.
The stability of the docked complex of Hydroxychloroquine and nsp 16 (PDB: 6W75_A), which showed the best interaction in the molecular docking simulated at100 ns The MD simulation trajectories of the drug-receptor complex during MD simulation are shown in Fig. 10 . The RMSD trajectory of the protein deviated between 1.6 and 3.6 Å (Fig. 10a). The deviation probably due to the interaction of the drug with the target and changed the structural conformation. The RMSF of the protein ranged between 0.8 Å- 3.2 Å and showed fluctuation at the C- and N-terminal regions of the target (Fig. 10b). Ligand RMSF fluctuated from 7.5 to 10.0 Å during the interaction (Fig. 10c). The interacting forces like hydrogen bonds, hydrophobic interactions, and water bridges formed between the target and the drug shown in Fig. 10d. The residues such as Asp7018, Trp6988, and Met6815 were formed hydrogen bonds with the drug. Met6815, Trp6988, and Met 7022 were involved in the formation of hydrophobic interactions in which Lys 6814 and Arg6817 showed positively charged interactions, and Asp7018 showed negatively charged interactions (Fig. 10e). The protein-ligand binding during the MD simulation is shown in Fig. 10f. The study suggested that Met6815, Arg6817, Trp6988, and Asp7018, were observed to be the interacting residues. The structural features and conformational changes in the interaction between Hydroxychloroquine and nsp16 and the protein-drug complex during MD simulation are shown in supplementary materials (Fig. S5a – S6d). From MD simulation studies, it is clear that Hydroxychloroquine and nsp16 showed stable binding throughout the simulation, and the interactions stabilized by several weak interactions.
Fig. 10.

The stability of docked complex of Hydroxychloroquine – nsp 16 analyzed by molecular dynamic simulation (MD) studies (a) Protein-ligand RMSD: Protein RMSD (Å) on the y-axis and simulation time on the x-axis (b) Protein RMSF: RMSF (Å) on the y-axis and atom index on the x-axis (c) Ligand RMSF: RMSF (Å) on the y-axis and atom index on the x-axis (d) The protein-ligand contacts: blue indicates water bridges, grey indicates hydrophobic interactions, pink indicates ionic bonds and green indicates hydrogen bonds (e) Major interactions between Hydroxychloroquine and nsp16 protein during MD simulation (f) Protein-ligand contacts: Timeline representation of interactions. The total number of specific contacts and the major residues of the protein interacted with the ligand.
The stability of the docked complex of Lopinavir and C-terminal domain of nucleocapsid phosphoprotein (PDB: 6WJI) were confirmed by MD simulation as the interaction of these complex showed the best binding during docking studies when compared to the binding of Lopinavir and other prioritized targets. The docked complex simulated at 100ns and the binding trajectories obtained during MD simulation are shown in Fig. 11 . The RMSD of the protein-ligand interaction is shown in Fig. 11a. The RMSD of the protein deviated from 4.8 to 7.2 Å during the simulation. The RMSD initially increased to 4.8 Å and later stabilized. The RMS deviation was probably due to the interaction between protein and the drug. The protein RMSD ranged from 0.75 Å to 4.0 Å during the simulation (Fig. 11b). The RMSF values of residues 15–30 and 62–76 varied during the simulation period, probably due to the conformational change caused due to the protein-ligand interaction. The ligand RMSF ranged between 1.0 and 3.0 Å is depicted in Fig. 11c. The RMSF values varied due to the binding of the receptor and ligand. The forces of interaction like hydrophobic interactions, water bridges, and hydrogen bond formed between the protein and the drug are shown in Fig. 11d. Hydrophobic interactions and hydrogen bonds are the main forces. The ligand-protein contacts that evident during the simulation are shown in Fig. 11e. The residues Ala264, Phe274, Trp301, Ile304, Phe 307, Pro 309, Phe314, Leu331, and Tyr333 were formed hydrophobic interactions, and Gln260 formed polar contact. The protein-ligand contacts observed during the simulation are shown in Fig. 11f. From the studies, it is clear that several amino acid residues that were involved in the stabilization of the protein and ligand complex. The structural and conformational changes due to the interaction of the Lopinavir and C-terminal domain of nucleocapsid phosphoprotein and their complex during MD simulation are shown in supplementary materials (Fig. S7a – S8d). From MD simulation, it is clear that the interaction between Lopinavir and C-terminal domain of nucleocapsid phosphoprotein showed stability throughout the simulation, and Lopinavir probably acts as an effective inhibitor against the target protein.
Fig. 11.

The stability of docked complex of Lopinavir – C-terminal domain of nucleocapsid protein analyzed by molecular dynamic simulation (MD) studies (a) Protein-ligand RMSD: Protein RMSD (Å) on the y-axis and simulation time on the x-axis (b) Protein RMSF: RMSF (Å) on the y-axis and atom index on the x-axis (c) Ligand RMSF: RMSF (Å) on the y-axis and atom index on the x-axis (d) The protein-ligand contacts: blue indicates water bridges, grey indicates hydrophobic interactions, pink indicates ionic bonds, and green indicates hydrogen bonds (e) Major interactions between lopinavir – C-terminal domain of nucleocapsid protein during MD simulation (f) Protein-ligand contacts: Timeline representation of interactions. The total number of specific contacts and the major residues of the protein interacted with the ligand.
The molecular docking studies suggested that Remdesivir showed the best binding potential towards nsp 16 in comparison with the other targets, thus, the stability of the complex was further validated by MD simulation at 100 ns The interaction trajectories obtained by MD simulation are shown in Fig. 12 . The protein RMSD showed a fluctuation from 1.3 to 3.1 Å, which was probably due to the drug molecule interacted with the target and caused conformational changes. The RMSD values of the protein ranged from 2.4 Å to 3.0 Å. The protein fluctuated at C- and N-terminals with RMSF above 5.4 Å. The other residues also showed fluctuation, RMSF ranged between 1.2 and 3.6 Å, is shown in Fig. 12b. The ligand RMSF fluctuated between the range 0.8–4.3 Å, probably due to the interactions between the receptor and the ligand (Fig. 12c). Hydrogen bonds, hydrophobic interactions, water bridges, and ionic bonds formed due to protein-drug interaction are shown in Fig. 12d. The residues such as Asn 6841, Asp6873, Asp6899, Asp6928, and Tyr6930 were formed hydrogen bonds with the drug. Hydrophobic (Tyr6930), polar (Asn 6899), and positively charged interactions (Asp6873, Asp6897, and Asp6928) were stabilized the protein-drug complex during MD simulation (Fig. 12e). The protein-drug binding in the MD simulation is shown in Fig. 12f. The residues such as Asp6873, Asp6897, Asp6899, Asp6928, and Tyr6930 responsible for binding and caused conformational changes. The structural and conformational changes of the binding between Remdesivir and nsp16 and their complex during the MD simulation are shown in supplementary materials (Fig. S9a – S10d). The simulation studies demonstrated that the docked complex of Remdesivir and nsp16 was stable during the MD simulation, and the interaction modeling provides the structural and molecular mechanisms of the binding of the drug to the selected targets.
Fig. 12.

The stability of docked complex of Remdesivir – nsp 16 analyzed by molecular dynamic simulation (MD) studies (a) Protein-ligand RMSD: Protein RMSD (Å) on the y-axis and simulation time on the x-axis (b) Protein RMSF: RMSF (Å) on the y-axis and atom index on the x-axis (c) Ligand RMSF: RMSF (Å) on the y-axis and atom index on the x-axis (d) The protein-ligand contacts: blue indicates water bridges, grey indicates hydrophobic interactions, pink indicates ionic bonds and green indicates hydrogen bonds (e) Major interactions between Remdesivir – nsp16 during MD simulation (f) Protein-ligand contacts: A timeline representation of interactions. The total number of specific contacts and the major residues of the protein interacted with the ligand.
The stability of the complex of the Ritonavir and C-terminal domain of nucleocapsid phosphoprotein (PDB: 6WJI) was studied by MD simulation at 100 ns The molecular interaction trajectories of the drug-protein complex are shown in Fig. 13 . The RMS fluctuation of the protein during the interaction observed in MD simulation is shown in Fig. 13a. The RMSD was found to be 2.4–4.8 Å and was probably due to the conformational changes of the protein during the binding. RMSF of the protein is estimated to be 0.8–6.2 Å (Fig. 13b). The ligand RMS fluctuations was probably due to the binding between the protein and drug (13c). The residue contacts of the interaction between protein and drug are shown in Fig. 13d. There were no prominent ionic interactions produced, water bridges and hydrogen bonds stabilized the receptor-ligand complex. The residues like Phe274, Phe286, Trp301, Phe314, and Tyr333 formed hydrophobic interactions, Thr282, Gln 283, and Asn 285 formed polar bonds, and Arg277 formed positive electrostatic interaction with the drug (Fig. 13e). The residue contacts that occurred during the simulation is represented in Fig. 13f. Several weak forces were involved in the stabilization of the receptor-ligand complex, in which Arg277 and Phe286 showed binding with the ligand throughout the simulation. The structural and conformational changes in the interaction between the Ritonavir and C-terminal domain of nucleocapsid phosphoprotein and their complex during MD simulation are shown in supplementary materials (Fig. S9a – S10d). Thus, simulation studies suggested that the complex of Ritonavir and C-terminal domain of nucleocapsid phosphoprotein showed stability throughout the interaction, and the present study showed the structural and molecular insight on the binding between Ritonavir and C-terminal domain of nucleocapsid phosphoprotein.
Fig. 13.

The stability of the docked complex of Ritonavir – C-terminal domain of nucleocapsid protein analyzed by molecular dynamic simulation (MD) studies (a) Protein-ligand RMSD: Protein RMSD (Å) on the y-axis and simulation time on the x-axis (b) Protein RMSF: RMSF (Å) on the y-axis and atom index on the x-axis (c) Ligand RMSF: RMSF (Å) on the y-axis and atom index on the x-axis (d) The protein-ligand contacts: blue indicates water bridges, grey indicates hydrophobic interactions, pink indicates ionic bonds and green indicates hydrogen bonds (e) Major interactions between ritonavir – C-terminal domain of nucleocapsid protein during MD simulation (f) Protein-ligand contacts: Timeline representation of interactions. The total number of specific contacts and the major residues of the protein interacted with the ligand.
3.5. MMPBSA calculation
The binding potential and stabilities of the selected docked conformations were further validated by the energy calculations using MMPBSA by employing the GROMACS force field. The binding energies of the docked complexes of Chloroquine and nsp 16, Favipiravir and the closed conformation of the spike glycoprotein, Hydroxychloroquine, and nsp16, Lopinavir and the C-terminal domain of nucleocapsid phosphoprotein, Remdesivir, and nsp16, and Ritonavir and C-terminal domain of nucleocapsid phosphoprotein were calculated to be −36.75 kcal/mol, −24.68 kcal/mol, −41.98 kcal/mol, −88.37 kcal/mol, −65.91 kcal/mol and −106.48 kcal/mol respectively. Thus, the calculated binding energy predicted by the MMPBSA approach confirmed the potential interaction of drugs towards the prioritized targets of SARS-CoV-2 as suggested by molecular docking and MD simulation studies. The energy minimization studies showed that binding of Ritonavir and the C-terminal domain of nucleocapsid phosphoprotein depicted better binding energy in comparison with the interaction between other drugs and the selected targets calculated by MMPBSA. However, all the interaction energies found to be negative and the sum of the interaction energies calculated by the MMPBSA showed the overall lower negative energy values in all the studied complexes that indicated the thermodynamic stability of the complexes studied.
Several recent studies have emphasized the application of computational biology for structure-based virtual screening to screen therapeutic agents and provide insights towards COVID-19. The protein targets of SARS-CoV-2 included were spike glycoproteins, RdRp, endoribonuclease, nucleocapsid proteins, envelope protein, membrane protein, and non-structural protein and provides new structural insights for drug repurposing of drugs against COVID-19 [46]. A study suggested that Chloroquine probably used as one of the effective therapeutic agents against SARS-CoV-2 based on host-directed therapy, although there are several studies required to confirm their efficiency [47]. Chloroquine and Hydroxychloroquine are being used in many countries to treat the infection as these antivirals are known to have inhibited viral entry and endocytosis; however, they have not shown 100% effective responses [48,49]. Favipiravir inhibit RdRp of influenza viruses, and recently suggested for the treatment of mild COVID-19 cases, the study conducted in combination with other drugs [50]. Ritonavir and Lopinavir were targeted against HIV–I protease and showed binding towards 3-chymotrypsin-like protease (3CLpro) [51]. Remdesivir initially developed against the Ebola virus, and it is known to inhibit various types of coronaviruses and is recently suggested for the treatment against COVID-19 [52]. The interaction of Remdesivir and RdRp modeled by molecular docking studies suggested that the drug can use against SARS-CoV-2 [53]. Previous studies have shown that the binding energy of Ritonavir and Remdesivir towards the main protease (Mpro) of SARS-CoV-2 estimated by MMGBSA were-72.02 and −65.19 kcal/mol, respectively [54]. The current study provides the interaction models of six drugs with each of the fifteen predicted targets of SARS-CoV-2 by molecular docking studies. Most of the studies reported on the binding potential of drugs towards limited targets or specific targets of SARS-CoV-2, however, there are limited studies prioritized the interaction prediction of FDA approved drugs towards the multiple prospective targets of SARS-CoV-2. Thus, the present study certainly provides structural and molecular insights on the binding potential of these drugs towards the multiple targets and enlighten the concept of drug repurposing approaches towards the several potential targets of SARS-CoV-2.
The present study provided structural and molecular insight into the interaction between six FDA approved drugs and the prioritized targets of SARS-CoV-2. The binding potential of six FDA approved drugs towards fifteen prospective targets were predicted by molecular docking studies. The potential docked complexes were simulated for 100ns to understand the dynamics and mechanistic aspects of the interaction and validated by energy calculations. The study suggested that Lopinavir, Remdesivir, and Ritonavir showed better binding energy towards the prioritized proteins in comparison to the binding energies of the docked complexes of Chloroquine, Hydroxychloroquine, and Favipiravir and the selected targets. However, Chloroquine, Hydroxychloroquine, and Favipiravir showed better binding with the prioritized targets of SARS-CoV-2 when compared to the binding energy of their usual targets. Thus, these drugs can use as putative inhibitors against the selected targets. The present study opens opportunities for future investigation and provides structural and molecular insights for the interaction studies that can use for the development of drugs against SARS-CoV-2, and drug repurposing concept can apply for COVID-19 drug screening.
4. Conclusion
The drug screening against COVID-19 is a worldwide concern, and there are several attempts and studies in progress. Several studies are available on the prediction of the binding of drugs towards the specific or single target of SARS-CoV-2, the binding potential of the drugs towards multi-target or the prospective molecular targets of SARS-CoV-2 are limited. With the advent of computer-aided virtual screening, the time required for a drug to reach into the market has significantly reduced. Several in silico studies reported the mechanism of binding of various lead molecules against putative targets of SARS-CoV-2. Recent studies suggested that drug repurposing against newly identified targets probably used as promising therapeutic strategies in the absence of licensed drugs. Thus, the present study aimed to predict the binding potential of six FDA approved drugs towards fifteen putative targets of SARS-CoV-2 in comparison with the binding of the drugs towards their usual targets. The study aimed to predict structural and molecular mechanisms of the binding of these drugs towards the prioritized targets of SARS-CoV-2. The study identified fifteen probable drug targets that included spike proteins, proteins such as RdRp and nucleocapsid phosphoprotein, membrane and envelope proteins, and other non-structural proteins. Chloroquine, Favipiravir, Hydroxychloroquine, Lopinavir, Remdesivir, and Ritonavir were the FDA approved drugs used for interaction modeling. When the binding potential of these drugs against prospective molecular targets were predicted by molecular docking and molecular dynamic simulation studies, Ritonavir, Lopinavir, and Remdesivir were demonstrated potential binding to most of the drug targets when compared to the interaction of their usual targets. Chloroquine, Hydroxychloroquine, and Favipiravir showed lower binding potential towards the prioritized targets in comparison with the binding of the other drugs and their targets. However, these drugs showed better binding to the selected drug targets when compared to their usual protein targets. Thus, the current study suggested that the prioritized FDA approved drugs probably used as potential inhibitors towards multiple targets of SARS-CoV-2 and repurposing of the drugs towards putative molecular targets of SARS-CoV-2 provides insight into drug development against COVID-19.
Author contributions
SS and DG designed the hypothesis, framed and standardized the methodology and performed major parts o the work, SC, PK were performed the computer aided virtual screening, AU, AGM and performed the molecular dynamic simulation studies and energy calculations and analysis. SS finally reviewed, revised and edited the manuscript. All authors carefully read and approved the final version.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Declaration of competing interest
The authors declare that they have no competing interests.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.compbiomed.2020.104054.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Pal M., Berhanu G., Desalegn C., Kandi V. Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2): an update. Cureus. 2020;12(3) doi: 10.7759/cureus.7423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shah B., Modi P., Sagar S.R. In silico studies on therapeutic agents for COVID-19: drug repurposing approach. Life Sci. 2020;252:117652. doi: 10.1016/j.lfs.2020.117652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grifoni A., Sidney J., Zhang Y., Scheuermann R.H., Peters B., Sette A. A sequence homology and bioinformatic approach can predict candidate targets for immune responses to SARS-CoV-2. Cell Host Microbe. 2020;27(4):671–680. doi: 10.1016/j.chom.2020.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buonaguro L., Tagliamonte M., Tornesello M.L., Buonaguro F.M. SARS-CoV-2 RNA polymerase as target for antiviral therapy. J. Transl. Med. 2020;18(1):185. doi: 10.1186/s12967-020-02355-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Naqvi A.A.T., Fatima K., Mohammad T T. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: structural genomics approach. Biochim. Biophys. Acta (BBA) - Mol. Basis Dis. 2020;1866(10):165878. doi: 10.1016/j.bbadis.2020.165878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.World Health Organization WHO coronavirus disease (COVID-19), 10:38am CEST. 4 September 2020. https://covid19.who.int/ Dashboard.
- 7.Glebov O.O. Understanding SARS-CoV-2 endocytosis for COVID-19 drug repurposing. FEBS J. 2020 doi: 10.1111/febs.15369. Advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang J. Fast identification of possible drug treatment of coronavirus disease-19 (COVID-19) through computational drug repurposing study. J. Chem. Inf. Model. 2020 doi: 10.1021/acs.jcim.0c00179. Advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao J., Tian Z., Yang X. Breakthrough: chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. Bioscience Trends. 2020;14(1):72–73. doi: 10.5582/bst.2020.01047. [DOI] [PubMed] [Google Scholar]
- 10.Liu J., Cao R., Xu M., Wang X., Zhang H., Hu H., Li Y., Hu Z., Zhong W., Wang M. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discovery. 2020;6:16. doi: 10.1038/s41421-020-0156-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yao T.T., Qian J.D., Zhu W.Y., Wang Y., Wang G.Q. A systematic review of lopinavir therapy for SARS coronavirus and MERS coronavirus-A possible reference for coronavirus disease-19 treatment option. J. Med. Virol. 2020;92(6):556–563. doi: 10.1002/jmv.25729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pardo J., Shukla A.M., Chamarthi G., Gupte A. The journey of remdesivir: from Ebola to COVID-19. Drugs Context. 2020;9(14):2020–2024. doi: 10.7573/dic.2020-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gordon C.J., Tchesnokov E.P., Woolner E. Remdesivir is a direct-acting antiviral that inhibits RNA-dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency. J. Biol. Chem. 2020;295(20):6785–6797. doi: 10.1074/jbc.RA120.013679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holshue M.L., DeBolt C., Lindquist S., Lofy K.H., Wiesman J., Bruce H., Spitters C., Ericson K., Wilkerson S., Tural A., Diaz G., Cohn A., Fox L., Patel A., Gerber S.I., Kim L., Tong S., Lu X., Lindstrom S., Pallansch M.A. Washington state 2019-nCoV case investigation team. First case of 2019 novel coronavirus in the United States. N. Engl. J. Med. 2020;382(10):929–936. doi: 10.1056/NEJMoa2001191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cai Q., Yang M., Liu D., Chen J., Shu D., Xia J., Liao X., Gu Y., Cai Q., Yang Y., Shen C., Li X., Peng L., Huang D., Zhang J., Zhang S., Wang F., Liu J., Chen L., Chen S., Liu L. Engineering; Beijing, China: 2020. Experimental Treatment with Favipiravir for COVID-19: an Open-Label Control Study. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choy K.T., Wong A.Y., Kaewpreedee P., Sia S.F., Chen D., Hui K., Chu D., Chan M., Cheung P.P., Huang X., Peiris M., Remdesivir H. L. Yen. lopinavir, emetine, and homoharringtonine inhibit SARS-CoV-2 replication in vitro. Antivir. Res. 2020;178:104786. doi: 10.1016/j.antiviral.2020.104786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Havlichek D., Jr. A trial of lopinavir-ritonavir in covid-19. N. Engl. J. Med. 2020;382(21):e68. doi: 10.1056/NEJMc2008043. [DOI] [PubMed] [Google Scholar]
- 18.Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The protein Data Bank. Nucleic Acids Res. 2020;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Walls A.C., Park Y.J., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181(2):281–292. doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.L., Abiona O., Graham B.S., McLellan J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science (New York, N.Y.). 2020;367(6483):1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xia S., Liu M., Wang C., Xu W., Lan Q., Feng S., Qi F., Bao L., Du L., Liu S., Qin C., Sun F., Shi Z., Zhu Y., Jiang S., Lu L. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020;30(4):343–355. doi: 10.1038/s41422-020-0305-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu Y., Sun F. 2020. Crystal Structure of HR2 Domain of 2019-nCoV S2 Subunit. [DOI] [Google Scholar]
- 23.Gao Y., Yan L., Huang Y., Liu F., Zhao Y., Cao L., Wang T., Sun Q., Ming Z., Zhang L., Ge J., Zheng L., Zhang Y., Wang H., Zhu Y., Zhu C., Hu T., Hua T., Zhang B., Yang X., Rao Z. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science. 2020;368(6492):779–782. doi: 10.1126/science.abb7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Osipiuk J., Jedrzejczak R., Tesar C., Endres M., Stols L., Babnigg G., Kim Y., Michalska K., Joachimiak A., Center for . 2020. Structural Genomics of Infectious Diseases (CSGID). The Crystal Structure of Papain-like Protease of SARS CoV-2. [DOI] [Google Scholar]
- 25.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science (New York, N.Y.). 2020;368(6489):409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen S., Kang S. 2020. Structural Insights of SARS-CoV-2 Nucleocapsid Protein RNA Binding Domain Reveal Potential Unique Drug Targeting Sites. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Minasov G., Shuvalova L., Wiersum G., Satchell K.J.F. 2020. 2.05 Angstrom Resolution Crystal Structure of C-Terminal Dimerization Domain of Nucleocapsid Phosphoprotein from SARS-CoV-2. [DOI] [Google Scholar]
- 28.Kim Y., Jedrzejczak R., Maltseva N., Endres M., Mececar A., Michalska K., Joachimiak A., Center for Structural Genomics of Infectious Diseases (CSGID) 2020. Crystal Structure of ADP Ribose Phosphatase of NSP3 from SARS CoV-2. [DOI] [Google Scholar]
- 29.Kim Y., Jedrzejczak R., Maltseva N.I., Endres M., Godzik A., Michalska K., Joachimiak A. 2020. Crystal Structure of NSP15 Endoribonuclease from SARS CoV-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tan K., Kim Y., Jedrzejczak R., Maltseva N., Endres M., Michalska K. 2020. A. Joachimiak & Center for Structural Genomics of Infectious Diseases (CSGID). The Crystal Structure of Nsp9 Replicase Protein of COVID-19. [DOI] [Google Scholar]
- 31.Minasov G., Shuvalova L., Rosas-Lemus M., Kiryukhina O., Wiersum G., Godzik A., Jaroszewski L., Stogios P.J., Skarina T., Satchell K.J.F. 2020. 1.95 Angstrom Resolution Crystal Structure of NSP10 - NSP16 Complex from SARS-CoV-2. [DOI] [Google Scholar]
- 32.Nelson C.A., Fremont D.H. 2020. Structure of the SARS-CoV-2 ORF7a Encoded Accessory Protein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wishart D.S., Feunang Y.D., Guo A.C., Lo E.J., Marcu A., Grant J.R., Sajed T., Johnson D., Li C., Sayeeda Z., Assempour N., Iynkkaran I., Liu Y., Maciejewski A., Gale N., Wilson A., Chin L., Cummings R., Le D., Pon A., Wilson M. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46:D1074–D1082. doi: 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trott O., Olson A.J., AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fritz-Wolf K., Becker A., Rahlfs S., Harwaldt P., Schirmer R.H., Kabsch W., Becker K. X-ray structure of glutathione S-transferase from the malarial parasite Plasmodium falciparum. PNAS U.S.A. 2020;100(24):13821–13826. doi: 10.1073/pnas.2333763100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Towler P., Staker B., Prasad S.G., Menon S., Tang J., Parsons T., Ryan D., Fisher M., Williams D., Dales N.A., Patane M.A., Pantoliano M.W. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J. Biol. Chem. 2020;279(17):17996–18007. doi: 10.1074/jbc.M311191200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fan H., Walker A.P., Carrique L., Keown J.R., Serna Martin I., Karia D., Sharps J., Hengrung N., Pardon E., Steyaert J., Grimes J.M., Fodor E. Structures of influenza A virus RNA polymerase offer insight into viral genome replication. Nature. 2020;573(7773):287–290. doi: 10.1038/s41586-019-1530-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Amano M., Miguel Salcedo-Gómez P., Yedidi R.S., Delino N.S., Nakata H., Venkateswara Rao K., Ghosh A.K., Mitsuya H. GRL-09510, a unique P2-crown-tetrahydrofuranylurethane -containing HIV-1 protease inhibitor, maintains its favorable antiviral activity against highly-drug-resistant HIV-1 variants in vitro. Sci. Rep. 2020;7(1):12235. doi: 10.1038/s41598-017-12052-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bonanno J.B., Fowler R., Gupta S., Hendle J., Lorimer D., Romero R., Sauder M., Wei C.L., Liu E.T., Burley S.K., Harris T. NY Times; 2003. X-ray Crystal Structure of the SARS Coronavirus Main Protease. C2-C2. [Google Scholar]
- 40.Sevrioukova I.F., Poulos T.L. Structure and mechanism of the complex between cytochrome P4503A4 and ritonavir. PNAS U.S.A. 2010;107(43):18422–18427. doi: 10.1073/pnas.1010693107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schrödinger Release 2019-3: Desmond Molecular Dynamics System. D. E. Shaw Research; New York, NY: 2019. Maestro-Desmond Interoperability Tools, Schrödinger. New York, NY, 2019. [Google Scholar]
- 42.Baker N.A., Sept D., Joseph S., Holst M.J., McCammon J.A. Electrostatics of nanosystems: application to microtubules and the ribosome. PNAS U.S.A. 2001;98(18):10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kumari R., Kumar R., Open Source Drug Discovery Consortium, Lynn A. g_mmpbsa–a GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014;54(7):1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- 44.Pronk S., Páll S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M.R., Smith J.C., Kasson P.M., van der Spoel D., Hess B., Lindahl E. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29(7):845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guo Y.R., Cao Q.D., Hong Z.S., Tan Y.Y., Chen S.D., Jin H.J., Tan K.S., Wang D.Y., Yan Y. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak -an update on the status. Military Medical Research. 2020;7(1):11. doi: 10.1186/s40779-020-00240-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu C., Liu Y., Yang Y., Zhang P., Zhong W., Wang Y., Wang Q., Xu Y., Li M., Li X., Zheng M., Chen L., Li H. Acta Pharmaceutica Sinica. B,. Advance online publication; 2020. Analysis of Therapeutic Targets for SARS-CoV-2 and Discovery of Potential Drugs by Computational Methods. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li X., Zhang C., Liu L., Gu M. Existing bitter medicines for fighting 2019-nCoV-associated infectious diseases. Faseb. J. 2020;f34(5):6008–6016. doi: 10.1096/fj.202000502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jie Z., He H., Xi H., Zhi Z. Expert consensus on chloroquine phosphate for the treatment of novel coronavirus pneumonia. Chin. J. Tuberc. Respir. Dis. 2020;43(3):185–188. doi: 10.3760/cma.j.issn.1001-0939.2020.03.009. [DOI] [PubMed] [Google Scholar]
- 49.Yao X., Ye F., Zhang M., Cui C., Huang B., Niu P., Liu X., Zhao L., Dong E., Song C., Zhan S., Lu R., Li H., Tan W., Liu D. Advance Online Publication; 2020. In Vitro Antiviral Activity and Projection of Optimized Dosing Design of Hydroxychloroquine for the Treatment of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), Clinical Infectious Diseases. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Saber-Ayad M., Saleh M.A., Abu-Gharbieh E. The rationale for potential pharmacotherapy of COVID-19. Pharmaceuticals. 2020;13(5):E96. doi: 10.3390/ph13050096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nutho B., Mahalapbutr P., Hengphasatporn K., Pattaranggoon N.C., Simanon N., Shigeta Y., Hannongbua S., Rungrotmongkol T. Why are lopinavir and ritonavir effective against the newly emerged coronavirus 2019? Atomistic insights into the inhibitory mechanisms. Biochemistry. 2020;59(18):1769–1779. doi: 10.1021/acs.biochem.0c00160. [DOI] [PubMed] [Google Scholar]
- 52.Martinez M.A. Compounds with therapeutic potential against novel respiratory 2019 coronavirus. Antimicrob. Agents Chemother. 2020;64(5) doi: 10.1128/AAC.00399-20. e00399-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Elfiky A.A. SARS-CoV-2 RNA dependent RNA polymerase (RdRp) targeting: an in-silico perspective. J. Biomol. Struct. Dynam. 2020:1–9. doi: 10.1080/07391102.2020.1761882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Al-Khafaji K., Al-Duhaidahawi D., Taskin Tok T. Using integrated computational approaches to identify safe and rapid treatment for SARS-CoV-2. J. Biomol. Struct. Dynam. 2020:1–9. doi: 10.1080/07391102.2020.1764392. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.