

Abstract
Cas13a has already been successfully applied to virus detection. However, as a new gene interference tool, its potential in cancer treatment was not fully explored until now. This study constructed a new Cas13a expression vector, decoy minimal promoter-Cas13a-U6-guide RNA (DMP-Cas13a-U6-gRNA [DCUg]), by controlling the Cas13a and gRNA expression with a nuclear factor κB (NF-κB)-specific promoter and U6 promoter, respectively. DCUg could specifically and effectively knock down the expression of reporter genes in the 293T and HepG2 cells. DCUg could also similarly knock down the expression of endogenous oncogenes (TERT, EZH2, and RelA) at both mRNA and protein levels in a human hepatoma cell HepG2, which led to significant apoptosis and growth inhibition. In contrast, the same transfection did not affect the target gene expression, cell apoptosis, and growth of a human normal liver cell HL7702. Finally, DCUg targeting these oncogenes was packaged into adeno-associated virus (AAV) and treated four cells (HepG2, HL7702, WEHI-3, and Hepa1-6) and tumor-bearing mice. As results, the recombinant AAV significantly inhibited the growth of three cancer cells (HepG2, Hepa1-6, and WEHI-3) in vitro and the xenografted Hepa1-6 and WEHI-3 tumors in mice. This study therefore developed a new tool for the CRISPR-Cas13a-based cancer gene therapy.
Keywords: CRISPR-Cas13a, NF-κB, RNA interference, cancer, gene therapy
Graphical Abstract
This study constructed a new Cas13a expression vector, DMP-Cas13a-U6-gRNA, by controlling the Cas13a and gRNA expression with a NF-κB-specific promoter and U6 promoter, respectively. This vector provides a new tool for the CRISPR-Cas13a-based cancer gene therapy, which can specifically knock down oncogenes in cancer cells in vitro and in vivo.
Introduction
Diverse strategies for antiviral defense have been generated due to the competition for survival within microbial communities.1 Among such defense mechanisms, clustered regularly interspaced short palindromic repeat (CRISPR)-associated genes (Cas) adaptive immune pathways possessed by almost all archaea and about half of bacteria protect microbes against viruses and other foreign nucleic acids using CRISPR RNA (crRNA)-guided nucleases.2 CRISPR-Cas systems are divided into two main classes: class I, which mediates the interference with multi-effector complexes, and class II, which employs single, multi-domain effectors to mediate the interference.3 According to the genomic architecture of the CRISPR array and the signature interference effector, these two classes are further subdivided into 6 types and 33 subtypes. The class I includes types I, III, and IV and class II includes types II, V, and VI.4, 5, 6
Unlike those other Cas nucleases, the signature protein Cas13a (previously known as C2c2) of type VI-A CRISPR-Cas systems has unique features absent in other known Cas proteins, which contains two higher eukaryotes and prokaryotes nucleotide-binding domains (HEPN) responsible for separate ribonuclease activities that catalyze crRNA maturation and single-stranded RNA (ssRNA) degradation, allowing Cas13a to work as a single-component programmable RNA-guided RNA-targeting CRISPR effector.7, 8, 9 The CRISPR-Cas13a system has already been used as a rapid DNA or RNA detection technique with attomolar sensitivity and single-base mismatch specificity known as SHERLOCK (Specific High Sensitivity Enzymatic Reporter UnLOCKing).10, 11, 12, 13 This in vitro application relied on combination of collateral effect of Cas13a with isothermal amplification. The collateral effect of Cas13a refers to the engagement of activated Cas13a in “collateral” cleavage of nearby non-targeted RNAs as fluorescent RNA reporter upon recognition of its RNA target.8,14
However, Cas13a has no similar collateral effect in cells while still retaining its high-specific cleavage activity to its RNA target. Existing studies have demonstrated that LwaCas13a can be heterologously expressed in mammalian and plant cells and can be reprogrammed with guide RNAs (gRNAs) to effectively knock down either reporter or endogenous transcripts with comparable levels of knockdown efficiency as RNAi in mammalian cells and plants.3,15 However, LwaCas13a knockdown substantially reduced off-targets efficiency, providing a flexible platform for studying RNA in mammalian cells and promising therapeutic applications.11 The in vivo cleavage reactions with LwaCas13a demonstrated that the programmable RNA cleavage is most efficient with a crRNA encoding a 28-nucleotide (nt) spacer (shorter than the 29–30 nt length found in the native L. wadei CRISPR array).16 In spite of these advantages, there were only a few reports of targeting oncogenic mRNA using CRISPR-Cas13a system for cancer treatment.17, 18, 19
In this study, we constructed a new expression vector of CRISPR-Cas13a that was under the control of a nuclear factor κB (NF-κB)-specific Pol II promoter. This promoter termed as DMP (decoy minimal promoter) was constructed by fusing a NF-κB decoy sequence to a minimal promoter.20 The transcriptional activation activity of this promoter is thus mainly dependent on the NF-κB activity in cells. Because NF-κB is widely over activated in various cancers, we have demonstrated that this promoter could drive ectopic expression of an effector gene specifically in various cancer cells.21,22 In this study, we therefore aimed at specifically knocking down the expression of oncogenes with the DMP-controlled CRISPR-Cas13a system in cancer cells, in order to develop a potential CRISPR-Cas13a-based gene therapy tool of cancer. We first testified the system by knocking down the enhanced green fluorescent protein (EGFP) and mCherry in HEK293T and HepG2 cells. We then testified the system by knocking down three endogenous genes including telomerase reverse transcriptase (TERT), enhancer of zeste homolog 2 (EZH2), and RelA in HepG2 and HL7702 cells. We finally demonstrated that the system could inhibit the tumor growth in mice by packaging the system into adeno-associated virus (AAV).
Results
Antitumor Mechanism of DCUg
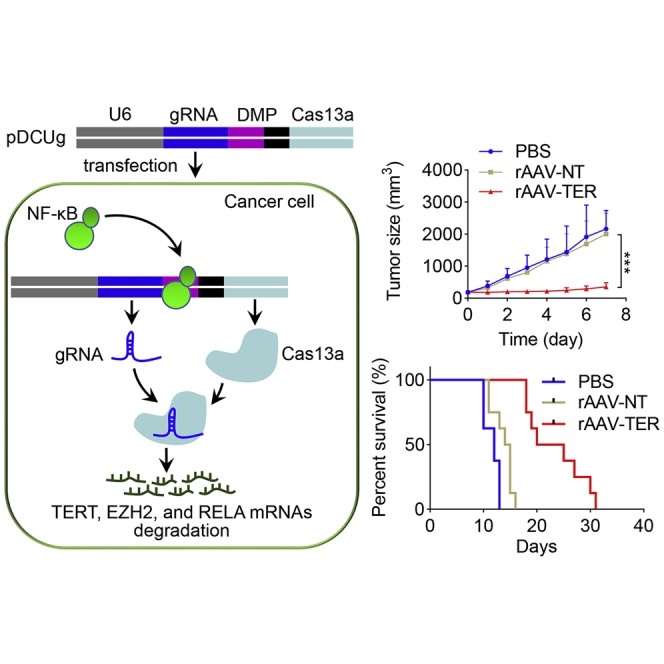
The DMP-Cas13a-U6-gRNA (DCUg) vector was constructed by controlling Cas13a and gRNA expression with a NF-κB-specific promoter, DMP, and U6 promoter, respectively. DMP consists of a NF-κB decoy and a minimal promoter. NF-κB is a sequence-specific DNA-binding transcription factor that is widely over-activated in cancer cells. However, it is not activated in normal cells. Therefore, when the DCUg vector is transfected into cancer cells, the over-activated NF-κB will bind to DMP to initiate the expression of Cas13a. The expressed Cas13a protein associates with gRNA to form Cas13a-gRNA complex, which can degrade the target mRNAs and ultimately leads to significant apoptosis of cancer cells (Figure 1). However, no Cas13a protein can be expressed in normal cells due to lack of NF-κB. In this study, we used mRNAs of TERT, EZH2, and RELA as targets of gRNA because the high expression of these genes was closely related with tumorigenesis.
Figure 1.

Schematic Illustration of Antitumor Mechanism of DMP-Cas13a-U6-gRNA (DCUg)
Gene Knockdown Effects of DCUg
In order to evaluate the in vitro knockdown efficiency and specificity of DCUg, we first designed a gRNA targeting EGFP transcript and cloned it into pDCUg. The HEK293T cells, which were demonstrated to have NF-κB activity by our previous studies,21,22 were then co-transfected by the pDCUg-EGFP, pEGFP, and pmCherry, allowing the EGFP transcript to serve as target and the pmCherry transcript as a negative control. The microscopic imaging of cells indicated that the pDCUg-EGFP transfection significantly decreased the EGFP but not mCherry fluorescence (Figure 2A; Figure S1). The results were also verified by quantitative analysis of cell fluorescence with flow cytometry (Figure 2B; Figure S2). In order to further verify the knockdown efficiency and specificity DCUg, we then designed a gRNA targeting mCherry transcript and cloned it into pDCUg. The HEK293T cells were then co-transfected by the pDCUg-mCherry, pEGFP, and pmCherry, allowing the mCherry transcript to serve as target and the EGFP transcript as a negative control. As a result, the similar results were obtained (Figures 2A and 2B; Figures S3 and S4). The pDCUg-mCherry transfection significantly decreased the mCherry but not EGFP fluorescence. In these detections, a negative pDCUg that could express a guide targeting no transcripts (NTs) in mammalian cells was used as control (pDCUg-NT), which did not affect expression of both EGFP and mCherry fluorescence. These data demonstrated the high knockdown efficiency and specificity of DCUg.
Figure 2.

Interference of Ectopic Gene Expression in HEK293T Cell with DCUg
Cells were transfected by various vectors and detected at 24 h post transfection. (A) Representative fluorescent images of cells. pDCUg-NT/EGFP/mCherry, plasmid expressing DMP-controlled Cas13a and U6-controlled gRNA targeting no, EGFP, and mCherry transcripts. (B) Flow cytometry analysis of fluorescence intensity. (C) qPCR analysis of mRNA expression. RQ, relative quantification. All values are mean ± SEM with n = 3. ∗p < 0.05; ∗∗p < 0.01.
As reporter genes, the transfection of pDCUg-EGFP and pDCUg-mCherry clearly demonstrated that the expressions of EGFP and mCherry proteins were significantly and specifically knocked down by DCUg. In order to further confirm the pDCUg knockdown, we detected the target gene expression at the mRNA level with quantitative PCR (qPCR). As expected, we found that the mRNA levels of both EGFP and mCherry were decreased (Figure 2C), in agreement with our expectation and previously reports that Cas13a-gRNA knock down its target RNA.
The same transfections were also performed with the HepG2 cells, which were also demonstrated to have NF-κB activity by our previous studies.21,22 The similar results were obtained in this cell line. DCUg significantly and specifically knocked down both EGFP and mCherry proteins (Figures 3A and 3B; Figures S5–S8). The qPCR detection revealed that mRNA levels of both EGFP and mCherry were also decreased in this cell line (Figure 3C).
Figure 3.

Interference of Ectopic Gene Expression in HepG2 Cell with DCUg
Cells were transfected by various vectors and detected at 24 h post transfection. (A) Representative fluorescent images of cells. pDCUg-NT/EGFP/mCherry, plasmid expressing DMP-controlled Cas13a and U6-controlled gRNA targeting no, EGFP, and mCherry transcripts. (B) Flow cytometry analysis of fluorescence intensity. (C) qPCR analysis of mRNA expression. All values are mean ± SEM with n = 3. ∗p < 0.05; ∗∗p < 0.01.
Antitumor Effects of DCUg In Vitro
To test whether DCUg could efficiently knock down endogenous genes, we next designed gRNAs targeting three oncogenes including TERT, EZH2, and RelA. The gRNAs were designed for both human and mouse genes (Table S1). The gRNAs targeting human genes were used to human cells and those targeting mouse genes were used to mouse cells. The effects of these guides were evaluated by transfecting the human liver cancer cell HepG2 and normal cell HL7702. Cells were also transfected by pDCUg-NT as control. Cells were imaged with microscope (Figure S9) and their apoptosis was detected by flow cytometry (Figure 4A; Figure S10). The results indicated that the HepG2 cells were significantly induced to apoptosis by all targeting gRNAs but not by the non-targeting gRNA. Moreover, the HepG2 cells were most significantly induced to apoptosis by the pDCUg-TER that could co-express gRNAs targeting TERT, EZH2, and RelA. However, the HepG2 cells were not significantly induced to apoptosis by pDCUg-NT. In contrast, the HL7702 cells were not significantly induced to apoptosis by all guides.
Figure 4.

Interference of Endogenous Gene Expression with DCUg
Cells were transfected by various vectors and detected at 24 h post transfection. (A) Flow cytometry analysis of cell apoptosis. (B) Expression level of Cas13a mRNA in HepG2 and HL7702 cells. (C) qPCR analysis of mRNA expression. (D) Western blot analysis of protein expression. The representative image and quantified optical density were shown. Lip, Lipofectamine; NT/TERT/EZH2/RelA/TER, pDCUg-NT/TERT/EZH2/RelA/TER. (E) CCK-8 assay of cell growth. Blank, cells without any treatment. Lipofectamine, cells treated with Lipofectamine only. pDCUg-NT/TERT/EZH2/RelA/TER, plasmid expressing DMP-controlled Cas13a and U6-controlled gRNA targeting no transcript (NT), TERT, EZH2, RelA, and TER transcripts. TER, TERT&EZH2&RelA. All values are mean ± SEM with n = 3, unless otherwise noted. ∗p < 0.05; ∗∗p < 0.01.
To further explore the cancer cell-specific expression of effector gene Cas13a of DCUg, we first detected Cas13a mRNA in the two cells under various treatments (Figure 4B). The results indicated that Cas13a expressed in all detected groups except lipofectamine control of HepG2 cell line; however, the Cas13a mRNA was not detected in all the treated groups of HL7702 cell line. These results indicated that DCUg could be activated in cancer cells rather than normal cells, which led to cancer cell-specific expression of effector gene.
We next detected the expression of target genes by qPCR (Figure 4C). The results demonstrated that the levels of target mRNAs were also significantly downregulated by the targeting gRNAs in the HepG2 cells but not changed in the HL7702 cells. We then detected the protein levels of TERT, EZH2, and RelA with western blot (WB) (Figure 4D). The results showed that the expressions of TERT, EZH2, and RelA proteins were significantly suppressed by DCUg in the HepG2 cells treated by the targeting gRNAs.
Given the important role of the three genes (TERT, EZH2, and RelA) in the progression of cancers, we next evaluated the antitumor effects of DCUg-mediated TERT, EZH2, and RelA mRNA interference in HepG2 and HL7702 cells. We therefore performed Cell Counting Kit-8 (CCK-8) assays to measure the cell growth rate in cells treated with DCUg (Figure 4E). The results indicated that the transfection of three targeting gRNAs significantly suppressed the growth of HepG2 cells over a 120-h time course. Moreover, this effect was strongest in the co-expression of three targeting gRNAs (pDCUg-TER). However, all transfections induced no obvious growth change in the HL7702 cells.
Antitumor Effects of DCUg In Vivo
To investigate whether there were anti-tumor effects of DCUg targeting above three oncogenic mRNAs, we packaged DCUg into AAV vectors. The anti-tumor effects of virus were first evaluated with four cell lines including HepG2, Hepa1-6, HL7702, and WEHI-3. The cells were seeded into 24-well plates (1 × 105 cells/well) and cultivated for 12 h. The cells were then transfected with the virus (5 × 1010 vg/well) by adding rAAV-MCS, rAAV-NT, rAAV-TERT, rAAV-EZH2, rAAV-RelA, and rAAV-TER in culture media, respectively. It should be noted that human cells (HepG2 and HL7702) and mouse cells (Hepa1-6 and WEHI-3) were infected with rAAVs that target human and mouse genes, respectively; both human and mouse cells can be infected with rAAV-MCS and rAAV-NT. The cells were cultured for another 72 h and imaged at time points of 24 h (Figure S11), 48 h (Figure S12), and 72 h (Figure 5A), respectively. The viability of cells was finally measured using a CCK-8 assay at 72 h post transfection. The results indicated that HepG2, Hepa1-6, and WEHI-3 cells were induced to significant apoptosis by four targeting AAVs but not by both rAAV-MCS and rAAV-NT (Figure 5B). Moreover, the rAAV-TER induced the most significant apoptosis in these three cells. Therefore, we applied the rAAV-TER to the further animal experiment. In contrast, all virus transfections induced no significant apoptosis in the HL7702 cells.
Figure 5.

Antitumor Effects of DCUg Virus In Vitro
Cells were inoculated in 24-well plate (1 × 105 cells/well) and cultured for 12 h. Cells were then treated with viruses (5 × 1010 vg/well) including rAAV-MCS, rAAV-NT, rAAV-TERT, rAAV-EZH2, rAAV-RelA, and rAAV-TER, respectively. Cells were cultured for 72 h and imaged at three time points, including 24 h, 48 h, and 72 h, respectively. The cells viability was evaluated using a CCK-8 assay at 72 h post transfection. (A) Representative images of cells at 72 h post transfection. (B) The cells viability at 72 h post transfection. All values are mean ± SEM with n = 3. ∗p < 0.05; ∗∗p < 0.01.
To evaluate whether the DCUg could be applied to treat cancers in vivo, we then performed two animal experiments. In the first animal model, ten mice were transplanted with the in vitro cultured mouse hepatocarcinoma Hepa1-6 cells. After 7 days, the tumor-bearing mice were divided into 2 groups and intravenously injected with 2 variant viruses (rAAV-TER and rAAV-NT), respectively. The measurement of tumor size revealed that the growth of tumors on the rAAV-TER-treated mice was significantly inhibited compared to the rAAV-NT group (Figures 6A and 6B). To demonstrate the in vivo tumor-specific expression of the rAAV vector further, we detected the presence of AAV viral DNA and expression of Cas13a and target genes in various tissues. Tissues, including heart, liver, spleen, lung, kidney, and tumor tissues, were collected from the two experimental groups of mice. The qPCR detection revealed that the AAV viral genomic DNA distributed in all detected tissues (Figure 6C) while Cas13a was only expressed in tumors of mice intravenously injected with rAAV-NT and rAAV-TER (Figure 6D). Moreover, the transcription of TERT, EZH2, and RelA was only downregulated in tumor (Figure 6E), indicating that cancer-specific expression of DCUg in vivo. Moreover, the qPCR detection revealed three oncogenes did not express in all normal tissues but tumor (Figure 6E). Moreover, the upregulation of these oncogenes in tumor was significantly knocked down by the rAAV-TER but not by rAAV-NT (Figure 6E), indicating the targeting of DCUg in vivo.
Figure 6.

The Antitumor Effects of the DCUg Virus in the Hepa1-6 Xenograft Mice
(A) Images of mice that were xenographed with the cancer cell Hepa1-6 transfected by virus rAAV-NT and rAAV-TER. (B) Tumor size before and after virus treatment. (C) Abundance of virus DNA in tissues. (D) Expression of Cas13a mRNA in various tissues of tumor-bearing mice intravenously injected with rAAV-NT and rAAV-TER. All other values were compared with the value of heart of rAAV-NT-treated mice. (E) Abundance of target mRNAs in tissues. All values are mean ± SEM with n = 3. ∗p < 0.05; ∗∗p < 0.01.
To further evaluate the antitumor effect, we treated a second tumor animal model that was made by subcutaneously transplanting BALB/c mice with WEHI-3 cells. The tumor-bearing mice were randomly divided into 6 treatment groups (PBS-1, PBS-2, rAAV-NT-1, rAAV-NT-2, rAAV-TER-1, and rAAV-TER-2) and intravenously administered two times with PBS (PBS-1 and PBS-2), rAAV-NT (rAAV-NT-1 and rAAV-NT-2), and rAAV-TER (rAAV-TER-1 and rAAV-TER-2), respectively. Tumor size was monitored every day. Three groups (PBS-1, rAAV-NT-1, and rAAV-TER-1) were euthanized on the seventh day post treatment for collecting tumors to detect tumor weight, virus infection, and gene expression (Figure 7A). The other three groups (PBS-2, rAAV-NT-2, and rAAV-TER-2) were fed until death to evaluate the mice survival over time (Figure 7B). The results indicated that the tumor growth was remarkably inhibited (Figures 7C to 7F) and the survival rate was significantly improved (Figure 7G) by the treatment of rAAV-TER, whereas the rAAV-NT treatment resulted in no effect in comparison with the PBS treatment (Figures 7C to 7G). Additionally, qPCR detection revealed that rAAV distributed in all detected tissues (Figure 8A), but the effector gene Cas13a was only expressed in tumor (Figure 8B). Moreover, qPCR detection also indicated that three target genes (TERT, EZH2, and RelA) were highly expressed only in tumors, which were significantly downregulated by the treatment of rAAV-TER (Figure 8C). Furthermore, the expression of two tumor growth markers, CD31 and Ki67, was also significantly downregulated by the treatment of rAAV-TER (Figure 8D). However, the expression of an apoptosis marker, Caspase-3, was significantly upregulated by the treatment of rAAV-TER (Figure 8D). In contrast, the expression of these genes was not affected by the treatment of rAAV-NT. Consistent with these results, the H&E staining of tumor sections revealed that only the treatment of rAAV-TER led to substantial necrosis (Figure 8E).
Figure 7.

The Antitumor Effect of DCUg Virus in the WEHI-3 Xenograft Mice
(A and B) Schematic illustration of the xenograft modeling and treatment (A, for groups PBS-1, rAAV-NT-1, and rAAV-TER-1; B, for groups PBS-2, rAAV-NT-2, and rAAV-TER-2). (C–E) Tumor growth curve (C), tumor weight (D), and tumor imaging (E) of mice treated with intravenously injected PBS, rAAV-NT, and rAAV-TER, respectively (groups PBS-1, rAAV-NT-1, and rAAV-TER-1). (F and G) Tumor growth curve (F) and Kaplan-Meier survival curves (G) of mice treated with intravenously injected PBS, rAAV-NT, and rAAV-TER, respectively (groups PBS-2, rAAV-NT-2, and rAAV-TER-2). All values are mean ± SEM with n = 8. ∗p < 0.05; ∗∗p < 0.01, ∗∗∗p < 0.001. s.c., subcutaneous injection; i.v., intravenous injection.
Figure 8.

The Antitumor Effect of DCUg Virus in the WEHI-3 Xenograft Mice
The tumor-bearing mice were intravenously injected with PBS, rAAV-NT, and rAAV-TER, respectively (groups PBS-1, rAAV-NT-1, and rAAV-TER-1). (A) Abundance of virus DNA in tissues. (B) Expression of Cas13a mRNA in tissues. All other values were compared with the value of heart of PBS-treated mice. (C) Abundance of target mRNAs in tissues. (D) Expression of CD31, Ki67, and Caspase-3 in tumors. (E) Images of tumors and their H&E-stained histological sections. All values are mean ± SEM with n = 3. ∗p < 0.05; ∗∗p < 0.01.
Discussion
Other than genome editing using widely known Cas9, which introduces genetic changes at the DNA level,23 Cas13a is a novel member of the family of CRISPR effectors,24 which specifically recognizes and cleaves RNA at sites guided by crRNA containing a 28 bp spacer.14,25,26 In recent years, many molecular tools utilizing CRISPR-Cas13a system to disrupt gene expression at the transcriptional level or edit gene transcripts have been successfully developed. In previous reports, it was shown that LwaCas13a protein exhibited a higher knockdown efficacy than the LshCas13a and LbuCas13a proteins.16 The CRISPR-Cas13a system has already been used to achieve robust RNA knockdown in mammalian cells by plasmid transfection.11,16,27
In our present study, we constructed a new CRISPR-Cas13a expression vector, DCUg. We expected to knock down target genes with the new CRISPR-Cas13a construct by depending on its reported mRNA-cleavage activity in mammalian cells.16 By controlling its specific expression in cancer cells with our previous developed cancer cell-specific promoter DMP, we expected to develop a new tool for the CRISPR-Cas13a-based cancer therapy. As a result, we found that the new CRISPR-Cas13a system could specifically and efficiently knock down the expression of both endogenous and exogenous target genes at both the mRNA and protein levels, which is in consistent with the previous reports.16 Moreover, under the control of a cancer cell-specific promoter, DMP, the expression of endogenous target genes could be specifically and efficiently knocked down at both the mRNA and protein levels only in cancer cells. The specific knockdown of oncogenes in cancer cells by DCUg induced apoptosis of cancer cells in vitro and in vivo. These results indicated that DCUg provides a new tool for the Cas13a-based cancer gene therapy.
Given that AAV is commonly used for in vivo gene delivery.28,29 LwaCas13a-msfGFP (4,335 bp) and U6-gRNA (326 bp) together are too big for the low packaging capacity of AAV (4.7 kb), restricting the in vivo application of LwaCas13a, especially targeting variant genes with multiple gRNAs. Therefore, a reported Cas13a-based cancer treatment used the purified LwaCas13a protein and crRNA for the efficient and specific knockdown of KRAS-G12D mRNA, finding that the co-transfection with LwaCas13a protein and crRNA significantly knocked down mutant KRAS mRNA expression and specifically distinguished KRAS-G12D mRNA from wild-type (WT) KRAS mRNA after introducing a second mismatch into crRNA.17 More importantly, LwaCas13a-mediated KRAS G12D mRNA knockdown potently induced apoptosis in vitro and caused marked tumor shrinkage in mice,17 providing a promising platform for further development of CRISPR-Cas13a in cancer treatment. However, this application did not use the mammalian cell LwaCas13a expression vectors in vivo.
In this study, we chose three well-known oncogenes as targets. Telomerase is encoded by the TERT gene, and its activity is limited in most normal somatic cells while active in 90% of tumor cells.30,31 Enhancer of EZH2, a histone-lysine N-methyltransferase enzyme encoded by EZH2 gene, is found in larger amounts in a wide range of cancers including breast, prostate, bladder, uterine, and renal cancers, as well as melanoma and lymphoma, than in healthy cells. Mutation or overexpression of EZH2 has been linked to many forms of cancers.32 EZH2 inhibits genes responsible for suppressing tumor development, and blocking EZH2 activity can slow tumor growth. The EZH2 inhibitors are currently developed as promising epigenetic therapy drugs for cancers.33,34 RelA is the most important regulator of NF-κB activity. Abnormal activation of NF-κB and its signaling pathway contribute to many human diseases, notably inflammatory diseases and cancers.35
In this study, we chose the three genes as targets of the optimized CRISPR-Cas13a system for exploring the apoptotic effect in HepG2 and HL7702 cells. Our data demonstrated that only cancer cells (HepG2 and Hepa1-6) could be significantly induced to apoptosis by targeting the three genes via DCUg. Moreover, this effect is strongest by simultaneously targeting the three transcripts (pDCUg-TER), indicating the synergistic effect on the apoptosis of cancer cells when disturbing multiple genes at the same time. Simultaneously targeting multiple genes provides DCUg a new advantage for cancer treatment over other CRISPR-Cas systems such as Cas9 that cannot make their gRNAs mature by themselves. In addition, there was no obvious change in the growth or apoptosis rates in normal liver HL7702 cells treated with the same vectors, indicating that the strategy to treat tumors with DCUg is tumor-specific in vitro. To further study the anti-tumor effect of this system in vivo, we packaged DCUg into AAV and treated two kinds of tumors in vivo via intravenous administration of rAAVs. The recombinant AAVs could efficiently deliver DCUg into cancer cells and resulted in tumor inhibition, this is beneficial for in vivo tumor gene therapy. Importantly, the in vivo cancer cell-specific activity of DCUg was strongly supported by the detection of Cas13a expression in various tissues which demonstrated that Cas13a was only expressed in tumor.
Conclusions
This study developed a new tool for cancer gene therapy that knocks down the oncogene expression specifically in cancer cells using a CRISPR-Cas13a expression vector, DCUg. This study verified the cancer cell-specific knockdown of oncogenes by knocking down three oncogenes in the cultured human cancer cell lines and xenografted tumors in mice. DCUg thus provides a useful platform for specifically knocking down various genes in cancer cells in vitro and in vivo, which should have potential application in cancer treatment.
Materials and Methods
Vector Construction
A chemically synthesized NF-κB-specific promoter, DMP, was cloned into pMD19-T simple (TAKARA) to obtain pMD19-T-DMP. The human codon optimized Cas13a coding sequence was amplified from the pC013-Twinstrep-SUMO-huLwCas13a (Addgene) by PCR and cloned into pMD19-T-DMP to obtain pMD19-T-DMP-Cas13a. The chemically synthesized U6 promoter sequence and direct repeat sequence of gRNA separated by BbsI restriction sites were cloned into pMD19-T-DMP-Cas13a to generate pDMP-Cas13a-U6-gRNA backbone (referred to as pDCUg). The particular gRNA targeting genes of interest are listed in Table S1.
gRNAs targeting NT, EGFP, mCherry, human and murine TERT, human and murine EZH2, and human and murine RelA were chosen with CHOPCHOP (http://chopchop.cbu.uib.no/). The complementary oligonucleotides containing a 28-bp gRNA target-specific region and two flanking BbsI sites (Table S2) were chemically synthesized and annealed into double-stranded oligonucleotides, which were then ligated into pDCUg. The ligation reaction (10 μL) consisted of 10 units of BbsI (NEB), 600 units of T4 DNA ligase (NEB), 1 × T4 DNA ligase buffer, 0.1 mg/mL bovine serum albumen, 1 nM double-stranded oligonucleotide, and 50 ng plasmid pDCUg. The ligation reaction was run on a PCR cycler as fellows: 10 rounds of 37°C 5 min and 16°C 10 min, 37°C 30 min, and 80°C 5 min. The generated plasmids were named as pDCUg-NT, pDCUg-EGFP, pDCUg-mCherry, pDCUg-TERT, pDCUg-EZH2, pDCUg-RelA, respectively. Two plasmids co-expressing gRNAs targeting human and murine TERT, EZH2, and RelA were also constructed, which was named as pDCUg-TER.
The pAAV-DCUg-NT/TERT/EZH2/RelA/TER vectors were constructed by cloning the DCUg-NT/TERT/EZH2/RelA/TER coding sequences into pAAV-MCS (VPK-410, Stratagene) by using MluI (upstream) and XbaI (downstream) restriction sites. The DCUg-NT/TERT/EZH2/RelA/TER coding sequences were amplified by PCR from pDCUg-NT/TERT/EZH2/RelA/TER.
The reporter constructs pEGFP and pmCherry were kept by our lab, in which the expression of EGFP and mCherry was under the control of CMV promoter.
Cell Culture and Transfection
HEK293T, HepG2, HL7702, Hepa1-6, and WEHI-3 were obtained from the cell resource center of Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences. Cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) (HEK293T, HepG2, Hepa1-6, and WEHI-3) with high glucose or Roswell Park Memorial Institute (RPMI) 1640 medium (HL7702) supplemented with 10% fetal bovine serum (VWR Seradigm), 100 units/mL penicillin (Thermo Fisher Scientific), and 100 μg/mL streptomycin (Thermo Fisher Scientific). Cells were incubated in atmosphere with 5% CO2 at 37°C. Unless otherwise noted, cells were transfected in 24-well plates using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s instructions. Cells were plated in 24-well plates 18–24 h prior to transfection to make sure 80% confluence overnight. For interfering with the ectopic gene expression, cells in each well were transfected by 500 ng of pDCUg and 150 ng of reporter construct (pEGFP/mCherry). For interfering with the endogenous gene expression, cells were transfected by 500 ng of pDCUg. Cells were imaged by a fluorescence microscope (IX51, Olympus) at a constant magnification of 100.
Flow Cytometry
For quantifying the ectopic EGFP and mCherry expression in cells, cells were collected by trypsinization at 24 h post transfection and quantified with flow cytometry (Calibur, BD, USA). For quantifying cell apoptosis, cells were collected by trypsinization at 24 h post transfection and detected with the Annexin V-fluorescein isothiocyanate (FITC) Apoptosis Detection Kit (BD, USA) according to the manufacturer’s instructions. The fluorescence intensity of cells was quantified with flow cytometry (Calibur, BD, USA). Flow cytometry data analysis and figure preparation were done using BD software.
CCK-8 Assay
Cells were plated in 96-well plates at a density of 5,000 cells/well. Each treatment was performed with six groups of cells and each group had four replicates. Cells were cultivated for 18 h (overnight) and transfected with various plasmids (100 ng plasmid/well). At variant time points (0 days, 1 day, 2 days, 3 days, 4 days, and 5 days) post transfection, 10 μL of CCK-8 solution (BS350B, Biosharp) was added to each well. After 1 h, the optical density at 450 nm was measured using a microplate reader (BioTek).
WB
Cells were seeded into 6-well plates and grown to 80% confluence overnight. Cells in each well were transfected by 1,000 ng of DCUg plasmid. At 24 h post transfection, the whole-cell and nuclear extracts were prepared using a phosphoprotein extraction kit (SA6034-100T, Signalway Antibody, USA) according to the manufacturer’s instructions. The protein lysates (20 μg/sample) were resolved by SDS-PAGE and the target proteins were detected with WB using the antibodies as follows: GAPDH mouse monoclonal antibody (97166S, CST, USA), TBP rabbit monoclonal antibody (12578S, CST, USA), TERT rabbit monoclonal antibody (ab191523, Abcam, UK), EZH2 mouse monoclonal antibody (3147S, CST, USA), and RelA mouse monoclonal antibody (6956S, CST, USA). The second antibodies were IRDye 800CW goat anti-mouse or rabbit immunoglobulin G (IgG) (Li-Cor). The blots were detected and fluorescence intensity was quantified with the Odyssey Infrared Fluorescence Imaging System (Li-Cor).
Virus Preparation
HEK293T cells were seeded into 75 cm2 flasks at a density of 5 × 106 cells per flask and cultivated for 12–20 h. Cells were then co-transfected with two helper plasmids (pHelper and pAAV-RC; Stratagene) and one of the pAAV plasmids (pAAV-MCS, pAAV-DCUg-TER, pAAV-DCUg-EZH2, pAAV-DCUg-RelA, and pAAV-DCUg-TER) using Lipofectamine 2000 according to the manufacturer’s instructions. Following transfection, cells were cultured with fresh medium for 72 h. Viruses were then collected and purified as previously described.36 Titers of AAVs were determined by qPCR using the primers AAV-F/R (Table S3). A 20 μL qPCR reaction contained 1 × SYBR Green Real-time PCR Master Mix (Roche), 0.25 μM primers, and 2 μL virus or 2-fold dilutions of a standard DNA (a 159-bp AAV genomic DNA (gDNA) fragment pre-amplified with primers AAV-F/R). The qPCR program was performed as follows: 95°C for 10 s, 45 cycles of 95°C for 15 s, and 60°C for 1 min. A melting curve was then constructed to monitor PCR amplification specificity. Data analysis was performed using Applied Biosystems StepOne v2.3. The concentration of the virus genome (vg) was calculated according to the standard curve. Quantified viruses were aliquoted and kept at −80°C for later use. The obtained viruses were referred to as rAAV-MCS, rAAV-NT, rAAV-TERT, rAAV-EZH2, rAAV-RelA, and rAAV-TER.
Virus Evaluation
Cells were seeded into a 24-well plate (1 × 105 cells/well) and cultivated for 24 h. Cells were then transfected with the virus (5 × 1010 vg/well) through cultivation in a medium containing rAAV-MCS, rAAV-NT, rAAV-TERT, rAAV-EZH2, rAAV-RelA, and rAAV-TER, respectively. Cells were cultured for another 72 h and were imaged by optical microscope (Olympus) at 24 h, 48 h, and 72 h post transfection, respectively. The cell viability was evaluated using a CCK-8 assay (BS350B, Biosharp) at 72 h post transfection.
Animal Experiment
All mice were obtained from the Changzhou Cavens Laboratory Animal (China). This study was carried out in accordance with the principles of the Basel Declaration and recommendations of Animal Care and Use guidelines of Southeast University (Nanjing, China), Experimental animal ethics committee of Southeast University. The protocol was approved by the experimental animal ethics committee of Southeast University. In the first animal model, the hepatic cancer xenograft was formed in 4-week-old female BALB/c-Foxn1nu mice with an average weight of 20 g by subcutaneously injecting with Hepa1-6 on two spots at 2 × 106 cells/place (5 mice/group). After breeding for 7–10 days, the mice were randomly divided into two experimental groups: rAAV-NT and rAAV-TER, and the tumor size was measured with a precision caliper before intravenously injected with variant AAVs at 1 × 109 vg/mouse. Tumor volume was calculated using formula V = (ab2)/2, where a is the tumor’s long axis and b is the short axis. Mice were euthanized and photographed on the 7th day post virus injection, and the tumor size was measured and calculated as described above.
In the second animal model, 4-week-old BALB/c female mice were subcutaneously transplanted with 1 × 107 WEHI-3 cells into groin. After breeding for 7 days, the tumor-bearing mice were randomly divided into 6 treatment groups (PBS-1, PBS-2, rAAV-NT-1, rAAV-NT-2, rAAV-TER-1, rAAV-TER-2, n = 8). The tumor-bearing mice were intravenously administered two times with PBS (PBS-1 and PBS-2), rAAV-NT (rAAV-NT-1 and rAAV-NT-2), and rAAV-TER (rAAV-TER-1 and rAAV-TER-2), respectively. The dosage of virus per injection was same as above. The animals of PBS-1, rAAV-NT-1, and rAAV-TER-1 were euthanized on the 7th day post treatment. The animals of PBS-2, rAAV-NT-2, and rAAV-TER-2 were fed until death. Tumor size was measured and calculated as above every day. Tumors were isolated and tumor weight was measured. Kaplan-Meier analysis was used to analyze the mice survival over time.
H&E Staining of Tumor Tissue Sections
Tumor masses were resected and fixed in 4% paraformaldehyde solution at room temperature overnight. Fixed specimens were paraffin-embedded and then cut into 5–8 μm-thick sections. The paraffin-embedded tissue sections were deparaffinized with xylene and rehydrated. The hydrated tissue sections were soaked and washed with phosphate buffer saline (PBS) three times for 5 min each time. The samples were then stained in hematoxylin staining solution (C0107, Beyotime) for 10 min and washed in running water for 10 min. Next, the samples were differentiated in 1% acid alcohol for 10 s, washed in running water for 30 min, and were then counterstained in eosin staining solution (C0109, Beyotime) for 3 min and washed in running water for 10 min. After the staining, the tissue sections were dehydrated in a gradient manner, with concentrations of 80% ethanol for 5 s, 95% ethanol for 2 min, and anhydrous ethanol for 2 min, respectively. Last, the tissue sections were sealed by a drop of neutral gum over the tissue and then covered by a coverslip. The prepared slides were then observed by a microscope (IX51, Olympus).
qPCR
Total RNA was extracted from cells at 24 h post transfection or mouse tissues using TRIzol (Invitrogen). cDNA was generated using the FastKing RT kit (TIANGEN) according to the manufacturer’s instruction. The gDNA was extracted from mouse tissues using the TIANamp Genomic DNA Kit (TIANGEN) according to the manufacturer’s instructions. The cDNA and gDNA were detected by qPCR using the Hieff qPCR SYBR Green Master Mix (Yeasen) according to the manufacturer’s instructions. GADPH was used as an internal control. qPCR programs were run with the ABI Step One Plus (Applied Biosystems). Each sample was analyzed in triplicate. Relative mRNA transcript levels and virus DNA abundance were calculated as relative quantity (RQ) according to the following equation: RQ = 2–ΔΔCt. All the qPCR primers were verified as being specific on the basis of melting curve analysis and were listed in Table S3.
Statistical Analyses
Data were showed as means values ± standard error of mean (SEM), accompanied by the number of experiments performed independently, and analyzed by t test. Differences at p < 0.05 were considered statistically significant. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Author Contributions
J.G. contributed to all cells and animal experiments. T.L. and N.L. contributed to the second animal tumor model experiments. S.Z. helped to construct vectors. J.W. and J.G. wrote the manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (61971122). The funding source did not have any role in design and conduct of the study, collection, management, analysis and interpretation of the data, or preparation, review or approval of the manuscript.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2020.09.004.
Supplemental Information
References
- 1.Dy R.L., Richter C., Salmond G.P.C., Fineran P.C. Remarkable mechanisms in microbes to resist phage infections. Annu. Rev. Virol. 2014;1:307–331. doi: 10.1146/annurev-virology-031413-085500. [DOI] [PubMed] [Google Scholar]
- 2.East-Seletsky A., O’Connell M.R., Burstein D., Knott G.J., Doudna J.A. RNA Targeting by Functionally Orthogonal Type VI-A CRISPR-Cas Enzymes. Mol. Cell. 2017;66:373–383.e3. doi: 10.1016/j.molcel.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aman R., Ali Z., Butt H., Mahas A., Aljedaani F., Khan M.Z., Ding S., Mahfouz M. RNA virus interference via CRISPR/Cas13a system in plants. Genome Biol. 2018;19:1. doi: 10.1186/s13059-017-1381-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Makarova K.S., Wolf Y.I., Alkhnbashi O.S., Costa F., Shah S.A., Saunders S.J., Barrangou R., Brouns S.J.J., Charpentier E., Haft D.H. An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol. 2015;13:722–736. doi: 10.1038/nrmicro3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shmakov S., Abudayyeh O.O., Makarova K.S., Wolf Y.I., Gootenberg J.S., Semenova E., Minakhin L., Joung J., Konermann S., Severinov K. Discovery and Functional Characterization of Diverse Class 2 CRISPR-Cas Systems. Mol. Cell. 2015;60:385–397. doi: 10.1016/j.molcel.2015.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Connell M.R. Molecular Mechanisms of RNA Targeting by Cas13-containing Type VI CRISPR-Cas Systems. J. Mol. Biol. 2019;431:66–87. doi: 10.1016/j.jmb.2018.06.029. [DOI] [PubMed] [Google Scholar]
- 7.Anantharaman V., Makarova K.S., Burroughs A.M., Koonin E.V., Aravind L. Comprehensive analysis of the HEPN superfamily: identification of novel roles in intra-genomic conflicts, defense, pathogenesis and RNA processing. Biol. Direct. 2013;8:15. doi: 10.1186/1745-6150-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abudayyeh O.O., Gootenberg J.S., Konermann S., Joung J., Slaymaker I.M., Cox D.B., Shmakov S., Makarova K.S., Semenova E., Minakhin L. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science. 2016;353:aaf5573. doi: 10.1126/science.aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mahas A., Neal Stewart C., Jr., Mahfouz M.M. Harnessing CRISPR/Cas systems for programmable transcriptional and post-transcriptional regulation. Biotechnol. Adv. 2018;36:295–310. doi: 10.1016/j.biotechadv.2017.11.008. [DOI] [PubMed] [Google Scholar]
- 10.Chotiwan N., Brewster C.D., Magalhaes T., Weger-Lucarelli J., Duggal N.K., Ruckert C., Nguyen C., Luna S.M.G., Fauver J.R., Andre B. Rapid and specific detection of Asian- and African-lineage Zika viruses. Sci. Transl. Med. 2017;9:eaag0538. doi: 10.1126/scitranslmed.aag0538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gootenberg J.S., Abudayyeh O.O., Lee J.W., Essletzbichler P., Dy A.J., Joung J., Verdine V., Donghia N., Daringer N.M., Freije C.A. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science. 2017;356:438–442. doi: 10.1126/science.aam9321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gootenberg J.S., Abudayyeh O.O., Kellner M.J., Joung J., Collins J.J., Zhang F. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science. 2018;360:439–444. doi: 10.1126/science.aaq0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Myhrvold C., Freije C.A., Gootenberg J.S., Abudayyeh O.O., Metsky H.C., Durbin A.F., Kellner M.J., Tan A.L., Paul L.M., Parham L.A. Field-deployable viral diagnostics using CRISPR-Cas13. Science. 2018;360:444–448. doi: 10.1126/science.aas8836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.East-Seletsky A., O’Connell M.R., Knight S.C., Burstein D., Cate J.H.D., Tjian R., Doudna J.A. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature. 2016;538:270–273. doi: 10.1038/nature19802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khan M.Z., Amin I., Hameed A., Mansoor S. CRISPR-Cas13a: Prospects for Plant Virus Resistance. Trends Biotechnol. 2018;36:1207–1210. doi: 10.1016/j.tibtech.2018.05.005. [DOI] [PubMed] [Google Scholar]
- 16.Abudayyeh O.O., Gootenberg J.S., Essletzbichler P., Han S., Joung J., Belanto J.J., Verdine V., Cox D.B.T., Kellner M.J., Regev A. RNA targeting with CRISPR-Cas13. Nature. 2017;550:280–284. doi: 10.1038/nature24049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao X., Liu L., Lang J., Cheng K., Wang Y., Li X., Shi J., Wang Y., Nie G. A CRISPR-Cas13a system for efficient and specific therapeutic targeting of mutant KRAS for pancreatic cancer treatment. Cancer Lett. 2018;431:171–181. doi: 10.1016/j.canlet.2018.05.042. [DOI] [PubMed] [Google Scholar]
- 18.Chen Y., Jiang H., Wang T., He D., Tian R., Cui Z., Tian X., Gao Q., Ma X., Yang J. In vitro and in vivo growth inhibition of human cervical cancer cells via human papillomavirus E6/E7 mRNAs’ cleavage by CRISPR/Cas13a system. Antiviral Res. 2020;178:104794. doi: 10.1016/j.antiviral.2020.104794. [DOI] [PubMed] [Google Scholar]
- 19.Li C., Guo L., Liu G., Guo M., Wei H., Yang Q., Wang J., Chen H. Reprogrammed CRISPR-Cas13a targeting the HPV16/18 E6 gene inhibits proliferation and induces apoptosis in E6-transformed keratinocytes. Exp. Ther. Med. 2020;19:3856–3860. doi: 10.3892/etm.2020.8631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang D., Tang H., Xu X., Dai W., Wu J., Wang J. Control the intracellular NF-κB activity by a sensor consisting of miRNA and decoy. Int. J. Biochem. Cell Biol. 2018;95:43–52. doi: 10.1016/j.biocel.2017.12.009. [DOI] [PubMed] [Google Scholar]
- 21.Wang D., Dai W., Wang J. A cell-specific nuclear factor-κB-activating gene expression strategy for delivering cancer immunotherapy. Hum. Gene Ther. 2019;30:471–484. doi: 10.1089/hum.2018.093. [DOI] [PubMed] [Google Scholar]
- 22.Dai W., Wu J., Wang D., Wang J. Cancer gene therapy by NF-κB-activated cancer cell-specific expression of CRISPR/Cas9 targeting telomeres. Gene Ther. 2020;27:266–280. doi: 10.1038/s41434-020-0128-x. [DOI] [PubMed] [Google Scholar]
- 23.Jing X., Xie B., Chen L., Zhang N., Jiang Y., Qin H., Wang H., Hao P., Yang S., Li X. Implementation of the CRISPR-Cas13a system in fission yeast and its repurposing for precise RNA editing. Nucleic Acids Res. 2018;46:e90. doi: 10.1093/nar/gky433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mohanraju P., Makarova K.S., Zetsche B., Zhang F., Koonin E.V., van der Oost J. Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science. 2016;353:aad5147. doi: 10.1126/science.aad5147. [DOI] [PubMed] [Google Scholar]
- 25.Liu L., Li X., Ma J., Li Z., You L., Wang J., Wang M., Zhang X., Wang Y. The Molecular Architecture for RNA-Guided RNA Cleavage by Cas13a. Cell. 2017;170:714–726.e10. doi: 10.1016/j.cell.2017.06.050. [DOI] [PubMed] [Google Scholar]
- 26.Liu L., Li X., Wang J., Wang M., Chen P., Yin M., Li J., Sheng G., Wang Y. Two Distant Catalytic Sites Are Responsible for C2c2 RNase Activities. Cell. 2017;168:121–134.e12. doi: 10.1016/j.cell.2016.12.031. [DOI] [PubMed] [Google Scholar]
- 27.Freije C.A., Myhrvold C., Boehm C.K., Lin A.E., Welch N.L., Carter A., Metsky H.C., Luo C.Y., Abudayyeh O.O., Gootenberg J.S. Programmable Inhibition and Detection of RNA Viruses Using Cas13. Mol. Cell. 2019;76:826–837.e11. doi: 10.1016/j.molcel.2019.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vasileva A., Jessberger R. Precise hit: adeno-associated virus in gene targeting. Nat. Rev. Microbiol. 2005;3:837–847. doi: 10.1038/nrmicro1266. [DOI] [PubMed] [Google Scholar]
- 29.Mingozzi F., High K.A. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat. Rev. Genet. 2011;12:341–355. doi: 10.1038/nrg2988. [DOI] [PubMed] [Google Scholar]
- 30.Zvereva M.I., Shcherbakova D.M., Dontsova O.A. Telomerase: structure, functions, and activity regulation. Biochemistry (Mosc.) 2010;75:1563–1583. doi: 10.1134/s0006297910130055. [DOI] [PubMed] [Google Scholar]
- 31.Podlevsky J.D., Chen J.J. It all comes together at the ends: telomerase structure, function, and biogenesis. Mutat. Res. 2012;730:3–11. doi: 10.1016/j.mrfmmm.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim K.H., Roberts C.W. Targeting EZH2 in cancer. Nat. Med. 2016;22:128–134. doi: 10.1038/nm.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang X., Yan J., Zhang M., Wang Y., Chen Y., Fu X., Wei R., Zheng X.L., Liu Z., Zhang X. Targeting Epigenetic Crosstalk as a Therapeutic Strategy for EZH2-Aberrant Solid Tumors. Cell. 2018;175:186–199.e19. doi: 10.1016/j.cell.2018.08.058. [DOI] [PubMed] [Google Scholar]
- 34.Gan L., Yang Y., Li Q., Feng Y., Liu T., Guo W. Epigenetic regulation of cancer progression by EZH2: from biological insights to therapeutic potential. Biomark. Res. 2018;6:10. doi: 10.1186/s40364-018-0122-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu X., Yarbrough W.G. Negative regulation of RelA phosphorylation: emerging players and their roles in cancer. Cytokine Growth Factor Rev. 2015;26:7–13. doi: 10.1016/j.cytogfr.2014.09.003. [DOI] [PubMed] [Google Scholar]
- 36.Chow R.D., Guzman C.D., Wang G., Schmidt F., Youngblood M.W., Ye L., Errami Y., Dong M.B., Martinez M.A., Zhang S. AAV-mediated direct in vivo CRISPR screen identifies functional suppressors in glioblastoma. Nat. Neurosci. 2017;20:1329–1341. doi: 10.1038/nn.4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.