

Summary
Trigeminal neuralgia (TN) is a common, debilitating neuropathic face pain syndrome often resistant to therapy. The familial clustering of TN cases suggests that genetic factors play a role in disease pathogenesis. However, no unbiased, large-scale genomic study of TN has been performed to date. Analysis of 290 whole exome-sequenced TN probands, including 20 multiplex kindreds and 70 parent-offspring trios, revealed enrichment of rare, damaging variants in GABA receptor-binding genes in cases. Mice engineered with a TN-associated de novo mutation (p.Cys188Trp) in the GABAA receptor Cl− channel γ-1 subunit (GABRG1) exhibited trigeminal mechanical allodynia and face pain behavior. Other TN probands harbored rare damaging variants in Na+ and Ca+ channels, including a significant variant burden in the α-1H subunit of the voltage-gated Ca2+ channel Cav3.2 (CACNA1H). These results provide exome-level insight into TN and implicate genetically encoded impairment of GABA signaling and neuronal ion transport in TN pathogenesis.
Subject Areas: Neuroscience, Structural Biology, Genomics
Graphical Abstract
Highlights
-
•
Genomic analysis of trigeminal neuralgia (TN) using exome sequencing
-
•
Rare mutations in GABA signaling and ion transport genes are enriched in TN cases
-
•
Generation of a genetic TN mouse model engineered with a patient-specific mutation
Neuroscience; Structural Biology; Genomics
Introduction
Trigeminal neuralgia (TN), or “tic doulourex,” is a severe neuropathic face pain syndrome characterized by recurrent, paroxysmal, lancinating face pain in the distribution of the trigeminal nerve that is variably triggered by sensory stimuli such as light touch or cold temperature (Maarbjerg et al., 2017). TN affects ∼3–4 per 100,000 people in the United States (Katusic et al., 1991; MacDonald et al., 2000). The pathogenesis of “classical TN (cTN)” is frequently attributed to hyperexcitability of trigeminal ganglion neurons (Burchiel, 1980a, 1980b; Burchiel and Baumann, 2004; Devor et al., 2002) secondary to morphological compression of the trigeminal nerve root entry root by the cerebral vasculature (i.e., neurovascular compression [NVC]) (Headache Classification Committee of the International Headache Society (IHS), 2013; Cruccu et al., 2016; Gardner and Miklos, 1959; Hilton et al., 1994; Rappaport et al., 1997). However, asymptomatic NVC has been noted in ∼13%–85% of asymptomatic subjects (Haines et al., 1980; Hamlyn, 1997a, 1997b; Jani et al., 2019). Other cases of TN are related to trigeminal nerve infection (e.g., herpes zoster), trauma, demyelination (as in multiple sclerosis), or compression from a space-occupying lesion in the cerebello-pontine angle and are termed “secondary TN.” However, a significant number of TN cases lack a demonstrable cause (“idiopathic TN [iTN]”) (Hughes et al., 2019; Maarbjerg et al., 2017), including some cases with bilateral symptoms (Brisman, 1987; Pollack et al., 1988).
First-line pharmacotherapy of TN includes the Na+ channel blocker carbamazepine or its analogs (Al-Quliti, 2015), followed by other Na+ channel- or GABA-modulating anticonvulsants such as gabapentin, lamotrigine, and topiramate (Ahmed et al., 2012). The efficacy of these drugs suggests that neuronal hyperexcitability and aberrant ion transport may be involved in TN pathogenesis. For patients resistant to medical therapy, alternative interventional and surgical treatments are offered. These include neurolysis of the trigeminal ganglion and the cisternal segment of the nerve, as well as open neurosurgical microvascular decompression (MVD) of the nerve. However, the few reported randomized, placebo-controlled trials with long-term follow-up have left continued uncertainty about the efficacy of these medical and surgical interventions (Zakrzewska and Akram, 2011). Gaps in our understanding of the cellular and molecular pathogenesis of TN have impeded the development of improved diagnostic, prognostic, and therapeutic measures.
The reported heritability of neuropathic pain conditions ranges from 16% to 50% (Hocking et al., 2012; Nielsen et al., 2012). Familial forms of TN are well documented, with many exhibiting autosomal dominant inheritance with incomplete penetrance (El Otmani et al., 2008; Rodriguez et al., 2019; Fleetwood et al., 2001; Smyth et al., 2003). The average age of onset in familial forms is 44.4 years (Fleetwood et al., 2001), 9 years younger than the average age of sporadic TN cases (Maarbjerg et al., 2015). A subset of familial forms demonstrates genetic anticipation, with progressively earlier disease onset across each succeeding generation (Harris, 1936; Smyth et al., 2003). These observations implicate genetic determinants in TN pathogenesis. Previous studies using candidate gene approaches have identified an association of TN with a common SNP in the Na+-dependent serotonin transporter SERT (SLC6A4) (Cui et al., 2014) and a gain-of-function de novo mutation (DNM) in the voltage-gated Na+ channel Nav1.6 (SCN8A) in a single patient with TN (Tanaka et al., 2016). However, no large, unbiased, whole-exome sequencing (WES) genomics study of TN has been performed to date.
The discovery of human genetic variants associated with TN could illuminate disease mechanisms, explain the variability of TN phenotypes and therapeutic responses, and identify potential drug targets for therapeutic intervention. Here, we present our analysis of WES of 290 TN probands, including 20 multiplex kindreds and 70 parent-offspring trios. This approach, proven to be successful for several neurodevelopmental disorders (Allen et al., 2013; Furey et al., 2018; Iossifov et al., 2012; Vissers et al., 2010), congenital heart disease (Jin et al., 2017; Zaidi et al., 2013), and other heritable conditions (Duran et al., 2019; Furey et al., 2018; Timberlake et al., 2016), enables unbiased identification of rare, damaging DNMs and copy number variations (CNVs), along with rare, inherited single-nucleotide variants and insertions and deletions (indels) that contribute to disease pathogenesis. We hypothesized that rare, damaging variants in genes encoding proteins with important roles in the development, structure, or function of neurons in the peripheral or central trigeminal pain circuitry might confer risk for the development of TN.
Results
Cohort Characteristics and Whole-Exome Sequencing
We ascertained 290 probands with TN-related disorders, including 41 from the UK Biobank (Table 1). UK Biobank patients included 34 singleton probands (85.4%) with a primary diagnosis of TN (ICD10 code: G50.0), 6 singleton probands (17.1%) with atypical facial pain (ICD10 code: G50.1), and one singleton proband with both. We recruited an additional 249 probands with either cTN, iTN type 1 (purely paroxysmal) or type 2 (with concomitant continuous pain) that included 70 parent-offspring trios; 63.9% (159/249) of probands had undergone neurosurgical intervention, including MVD (54.6%, 136/249), thermal or balloon rhizotomy (11.6%, 29/249), or gamma knife radiosurgery (14.9%, 37/249). Of the patients treated with MVD, 42.6% (58/136) did not have sustained symptom relief; 19.1% (26/136) of these patients underwent a repeat MVD for post-operative recurrence of symptoms. Interestingly, 36/249 (14.5%) cases were characterized by bilateral TN and 41/249 (16.5%) probands had a family history of TN.
Table 1.
Demographic and clinical characteristics of TN cases and controls
| TN Cases from Yale | TN Cases from UK BioBank | Autism Sibling Controls | |
|---|---|---|---|
| Sample size | 249 | 41 | 1,798 |
| Trios | 70 (28.1%) | 0 (0.0%) | 1,798 (100.0%) |
| Non-trio cases | 179 (71.9%) | 41 (100.0%) | 0 (0.0%) |
| Cases with family history of TN | 41 (16.5%) | NA | NA |
| Cases with ≥ 2 affected members sequenced | 20 (8.0%) | NA | NA |
| Gender | |||
| Male | 34 (13.7%) | 15 (36.6%) | 842 (46.8%) |
| Female | 215 (86.3%) | 26 (63.4%) | 956 (53.2%) |
| Ethnicity | |||
| European | 238 (95.6%) | 38 (92.7%) | 1,418 (78.9%) |
| African American | 0 (0.0%) | 1 (2.4%) | 77 (4.3%) |
| East Asian | 1 (0.4%) | 1 (2.4%) | 40 (2.2%) |
| South Asian | 1 (0.4%) | 0 (0.0%) | 88 (4.9%) |
| Mexican | 6 (2.4%) | 1 (2.4%) | 129 (7.2%) |
| Other | 3 (1.2%) | 0 (0.0%) | 46 (2.6%) |
| TN type | |||
| cTN-1 | 47 (18.9%) | NA | NA |
| cTN-2 | 80 (32.1%) | NA | NA |
| iTN-1 | 44 (17.7%) | NA | NA |
| iTN-2 | 78 (31.3%) | NA | NA |
| Bilateral symptoms | 36 (14.5%) | NA | NA |
| Neurosurgical intervention | 159 (63.9%) | NA | NA |
| MVD | 136 (54.6%) | NA | NA |
| With relief of symptoms | 75 (30.1%) | NA | NA |
| No relief of symptoms | 58 (23.3%) | NA | NA |
| Repeated MVD | 26 (10.4%) | NA | NA |
| Thermal or balloon rhizotomy | 29 (11.6%) | NA | NA |
| Gamma knife | 37 (14.9%) | NA | NA |
| Other | 22 (8.8%) | NA | NA |
The number of samples is shown in each category with the corresponding percentage in parentheses. Some Trios contain ≥ 2 affected members. Ethnicity is determined by principal component analysis compared to HapMap samples using EIGENSTRAT. See also Figure S4.
Variants in Genes Previously Associated with TN
To gain insight into the genetic architecture of TN, we first screened for rare, damaging heterozygous mutations (minor allele frequency [MAF] ≤ 1 × 10−3 in Bravo) (Program, 2018) in genes previously implicated in TN (Cui et al., 2014; Tanaka et al., 2016). Two probands each carried a deleterious missense (predicted as deleterious by MetaSVM or having a CADD score ≥ 30; D-mis) (Dong et al., 2015; Kircher et al., 2014) mutation in non-conserved residues of SCN8A. p.Ile1583Thr maps to the ion transport domain, and p.Arg475Gln is located immediately adjacent to the voltage-gated Na+ channel domain (Figures S1A and S1B). No rare damaging mutations were identified in SLC6A4.
Familial Forms of TN and SCN5A Variants
We next examined rare (MAF ≤ 5 × 10−4) damaging (i.e., D-mis or loss-of-function [LoF]) variants that segregated with TN in 20 multiplex families with at least two affected individuals available for WES (Figure 1). Of the 10 genes with segregating variants in at least two families (Table S1A), only SCN5A, encoding Na+ channel Nav1.5, was intolerant to both LoF (pLI ≥ 0.9) and missense variants (missense z-score ≥ 2) (Figures S1C and S1D). In iTN-1 family TRGN201, SCN5A p.Arg1826His was shared by affected siblings TRGN201-1 and TRGN201-2 (Figure S1E). p.Arg1826His maps to a conserved residue in the Nav1.5 C-terminal cytoplasmic domain and is reported in ClinVar as pathogenic for long QT syndrome (Figure S1D). Nav1.5 Arg1826His-mediated Na+ currents exhibit delayed inactivation and a 2- to 3-fold increase in late Na+ current (Ackerman et al., 2001; Wei et al., 2013). SCN5A p.Phe1293Ser was shared by cTN-1 proband TRGN141-1 and her affected maternal grandmother TRGN141-4 (Figure S1E). p.Phe1293Ser maps to SCN5A domain III and is predicted to alter Nav1.5 structure (Figure S1D). Of note, SCN5A-mutant TN patients did not report a history of cardiac arrhythmias or sudden death (Roberts, 2006).
Figure 1.

Pedigrees for 20 TN Familial Cases
20 familial cases with ≥2 members available for whole-exome sequencing (WES) are shown with sample IDs. Black circle/squares: Subjects with TN diagnosis. See also Table S1.
Copy Number Variation in Familial TN
We also identified four rare CNVs (MAF ≤ 1 × 10−3) that segregated in three multiplex TN families (Table S1B). Among them, one ∼500-kb duplication covering the serine-threonine kinase MAPK3 was shared by the proband and the affected mother, but not the unaffected father in family TRGN190. MAPK3 has been previously implicated in the pathogenesis of multiple trigeminal pain models (Alter et al., 2010; Liverman et al., 2009; Smyth et al., 2003; Sun et al., 2019). Interestingly, the TRGN190 proband with the MAPK3 duplication was diagnosed with bilateral cTN-1 at 11 years of age and exhibited bilateral NVC on brain magnetic resonance imaging (MRI). The patient was treated with bilateral MVD, followed 1 year later by repeat bilateral MVD for symptom recurrence. The proband's mother, also with an MAPK3 duplication, was diagnosed with bilateral cTN-1 at 43 years of age. Although NVC was observed on her MRI, she declined surgery. Both patients reported significant symptom relief with the voltage-gated Na+ channel blocker oxcarbazepine. The proband's sister was also diagnosed with oxcarbazepine-responsive unilateral iTN at 17 years of age, but was not available for WES.
Rare, Damaging De Novo and Inherited Variants in GABA Signaling Genes
We next examined the contribution of DNMs, including CNVs, to TN risk. Our TN cohort demonstrated a rate of 1.06 coding region DNMs per proband, following the expected Poisson distribution and closely matching the burden of DNMs in the control cohort (Tables S2A and S2B). No genes contained more than one protein-altering DNM (Table S2C). Eleven de novo CNVs were identified, including a duplication in KCNK1, encoding the inward-rectifying K+ channel TWIK1 (Table S2D). Of note, pain insensitivity in multiple African rodents has been attributed in part to the down-regulation of kcnk1 expression (Eigenbrod et al., 2019). No recurrent CNVs were observed.
We performed Gene Ontology (GO) enrichment analysis (Raudvere et al., 2019) of genes harboring damaging DNMs with high brain expression (above 75% percentile among all genes from murine RNA sequencing) (Flegel et al., 2015). Analysis showed the greatest enrichment among genes associated with the molecular function term “GABA receptor binding genes” (GO:0050811) (GABRG1, TRAK1 [Figure 2A]; enrichment = 226.6, adjusted p value = 5.9 × 10−3 [Table S3A]). Damaging DNMs in GO:0050811 genes were enriched in TN cases but not in controls (enrichment = 114.0, p value = 1.5 × 10−4) (Table S3B). Interestingly, several other notable genes with rare damaging DNMs but not included under the GO term GO:0050811 were identified that, similar to GABRG1 and TRAK1, have elevated brain expression and play important roles in GABA signaling or neurotransmission. These include ASTN2 (c.1736+2T > C) (Behesti et al., 2018), EEF2 (p.Arg839His) (Heise et al., 2017), UNC80 (p.Lys2794∗) (Philippart and Khaliq, 2018; Yeh et al., 2008), and KIF1B (p.Arg928Trp) (Aulchenko et al., 2008; Lyons et al., 2009; Zuchner et al., 2004).
Figure 2.

Damaging De Novo and Inherited Mutations in GABRG1 and TRAK1 in TN Probands
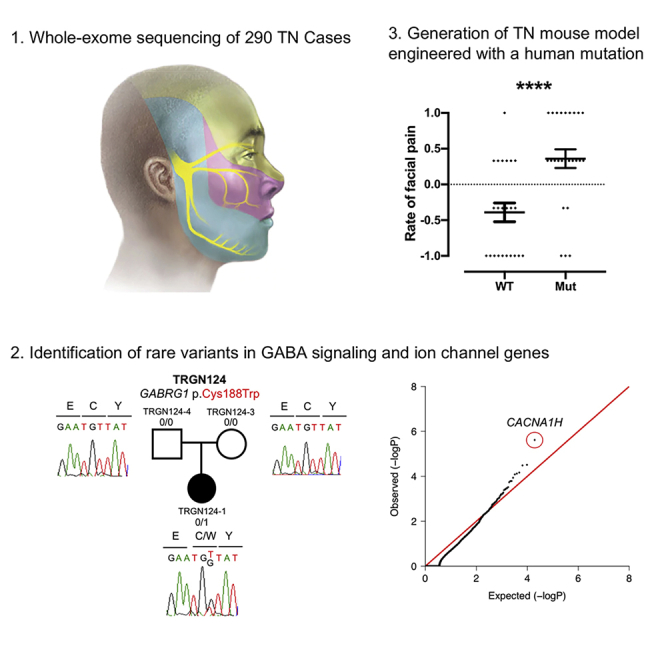
(A) De novo and inherited mutations in GABRG1 and TRAK1. Pedigrees with Sanger-validated mutant bases marked on the chromatograms.
(B) Mapping of GABRG1 and TRAK1 mutations. GABRG1 p.Cys188Trp and p.Tyr178His impact conserved residues at the neurotransmitter-gated ligand-binding domain of GABRG1. TRAK1 p.Glu798Lys affects a conserved residue of the second kinesin-binding Milton domain, whereas TRAK1 p.Arg124Gln maps to a conserved residue of the HAP1_N domain.
(C) Structural modeling of GABRG1 p.Cys188Trp and p.Tyr178His mutations. p.Cys188 disulfide bonds with Cys202. Mutation to Trp188 disrupts this conserved disulfide bond with calculated ΔΔG = 3.8 kcal/mol. In the midst of surrounding hydrophobic residues, the side chain hydroxyl of Tyr178 hydrogen bonds with the guanidinium side chain of Arg166, stabilizing interactions between their adjacent β-sheets. Disruption of this hydrogen bond by Arg mutation to His is predicted to destabilize ΔΔG by 1.7 kcal/mol.
(D) WT and mutant mice were examined for nocifensive withdrawal behavior following stimulation of the trigeminal nerve region in response to mechanical stimulation using a single von Frey filament (#7; 0.6 g). The y axis values represent average responses to three stimulations (on different days; +1 = presence of a stimulus-associated grooming response; −1 = absence of grooming behavior). The Mann-Whitney test was applied to assess statistical difference between WT and mutant mice. A Kruskal-Wallis test evaluated significant differences among all sub-groups: male and female WT and mutants. p value ∗ <0.05; ∗∗ <0.01; ∗∗∗ <1 × 10−3; ∗∗∗∗ <1 × 10−4.
(E and F) Measurement of nociceptive withdrawal threshold using the using the Simplified Up-Down method (SUDO) (Bonin et al., 2014). (E) Graph showing withdrawal threshold results using an adaptation of the method to test in the region innervated by the trigeminal nerve (see Methods; Taylor et al. Pain, 2012). (F) Nociceptive withdrawal threshold in response to mechanical stimulation of the hind paw using calibrated von Frey filaments. p value ∗∗ <0.01.
See also Figures S1 and S2; Tables S1, S3, S4, and S6; and Videos S1, S2, S3, S4, S5, S6, S7, and S8.
GABRG1 encodes the γ-1 subunit of the heteromeric ligand-gated gamma-aminobutyric acid type A receptor (GABAAR) Cl− channel. Patient TRGN124-1 with iTN-1 had a rare D-mis GABRG1 DNM (p.Cys188Trp) in a conserved residue of the GABAAR γ-1 ligand-binding domain that is predicted to disrupt a disulfide bond with Cys202 (ΔΔG = 3.8 kcal/mol) (Figures 2A–2C). Unrelated patient TRGN343-1 with iTN-2 carried a rare D-mis GABRG1 mutation p.Tyr178His (MAF = 8.2 × 10−6), which also maps to a conserved residue in the ligand-binding domain and is predicted to disrupt a hydrogen bond between adjacent β-sheets (ΔΔG = 1.7 kcal/mol) (Figures 2A–2C). Another patient with TN in the UK Biobank carried a rare stop-gain mutation in GABRG1 (p.Trp53∗) located just before the ligand-binding domain.
TRAK1 encodes a kinesin adaptor protein that regulates the anterograde axonal transport of mitochondria and GABAARs (Barel et al., 2017). Patient TRGN261-1 with iTN-2 had a rare D-mis TRAK1 DNM (p.Glu798Lys) (Figure S2A) in a conserved residue of the second kinesin-binding Milton domain of TRAK1 (Figure S2B). Unrelated patient TRGN107-1 with cTN-1 carried the unphased heterozygous D-mis TRAK1 mutation (p.Arg124Gln) with an MAF just above our threshold (1.6 × 10−5). p.Arg124Gln maps to a highly conserved residue in the TRAK1 HAP1_N domain (IPR006933) that directly participates in GABAAR trafficking in conjunction with kinesin (KIF) proteins (Gilbert et al., 2006). Recessive TRAK1 mutations cause early infantile epileptic encephalopathy (OMIM# 618201) (Barel et al., 2017), and Trak1 knockout mice exhibit severely reduced central nervous system (CNS) abundance of GABAARs (Gilbert et al., 2006).
A total of nine rare (MAF ≤1.0 × 10−5) damaging de novo, transmitted, or unphased mutations were identified in GO:0050811 GABA receptor-binding genes, yielding enrichment compared with gnomAD controls (odds ratio [OR] = 3.3, one-tailed Fisher's exact p value = 3.8 × 10−3; Tables S3C and S3D). These genes included GABAAR subunit α-5 GABRA5 and trafficking regulators PLCL1, JAKMIP1, and ARFGEF2. Extension of our search to the 21 genes encoding HUGO Gene Nomenclature Committee (HGNC)-designated GABA receptor subunits identified two additional rare, damaging heterozygous mutations in the GABAAR α-6 and ϵ subunits, GABRA6 and GABRE (Figure S1G). GABRA5 p.Glu107Gln and GABRA6 p.Glu90Ala both map to respective ligand-binding domains, whereas GABRE p.Trp300∗ affects the neurotransmitter gating domain (Figures S1H–S1J).
Mouse Model of TN Engineered with a Patient-Specific GABRG1 Mutation
As proof of principle, we generated a mouse model of one of the identified human TN-associated GABAAR DNMs (GABRG1 p.Cys188Trp), using Crispr/CAS9 mutagenesis (Figure S2). To test for trigeminal pain hypersensitivity, we quantified nocifensive behaviors using a modified version of a facial stimulation test (Bailey and Ribeiro-da-Silva, 2006). In contrast to wild-type littermates (n = 23 mice), mutant mice (n = 25) showed significant nocifensive behaviors in response to tactile stimulation of the trigeminal nerve region (Figure 2D; p value < 1 × 10−4; Mann-Whitney test; Videos S1, S2, S3, S4, S5, S6, S7, and S8 and Table S4A) in both males (n = 11 wild-type and 12 mutants) and females (n = 12 wild-type and 13 mutants; p value = 0.045 for females; p value = 9 × 10−4 for males; Kruskal-Wallis test). No significant difference in nocifensive behavior was observed between male and female mutants (p value = 0.40). To measure nociceptive withdrawal threshold, we also used a modified mechanical threshold test (Bonin et al., 2014; Taylor et al., 2012). Mutant mice (n = 11) showed a significantly lower nociceptive withdrawal threshold than wild-types (n = 9; p value = 1.0 × 10−3; Mann-Whitney test Figure 2E and Table S4B). Hind paw nociceptive withdrawal threshold was also significantly reduced in mutant mice (p value = 1.6 × 10−3; Figures 2F and Table S4B).
The white dot in the top left corner appears at the moment of the single stimulation using filament #7 (0.6 g), and the dot is displayed as long as the subsequent grooming response continues. Positive (presence of a stimulus-associated grooming response) and negative (absence of grooming behavior) responses were attributed a +1 or −1 value, respectively.
The white dot in the top left corner appears at the moment of the single stimulation using filament #7 (0.6 g), and the dot is displayed as long as the subsequent grooming response continues. Positive (presence of a stimulus-associated grooming response) and negative (absence of grooming behavior) responses were attributed a +1 or −1 value, respectively.
The white dot in the top left corner appears at the moment of the single stimulation using filament #7 (0.6 g), and the dot is displayed as long as the subsequent grooming response continues. Positive (presence of a stimulus-associated grooming response) and negative (absence of grooming behavior) responses were attributed a +1 or −1 value, respectively.
The white dot in the top left corner appears at the moment of the single stimulation using filament #7 (0.6 g), and the dot is displayed as long as the subsequent grooming response continues. Positive (presence of a stimulus-associated grooming response) and negative (absence of grooming behavior) responses were attributed a +1 or −1 value, respectively.
The white dot in the top left corner appears at the moment of the single stimulation using filament #7 (0.6 g), and the dot is displayed as long as the subsequent grooming response continues. Positive (presence of a stimulus-associated grooming response) and negative (absence of grooming behavior) responses were attributed a +1 or −1 value, respectively.
The white dot in the top left corner appears at the moment of the single stimulation using filament #7 (0.6 g), and the dot is displayed as long as the subsequent grooming response continues. Positive (presence of a stimulus-associated grooming response) and negative (absence of grooming behavior) responses were attributed a +1 or −1 value, respectively.
The white dot in the top left corner appears at the moment of the single stimulation using filament #7 (0.6 g), and the dot is displayed as long as the subsequent grooming response continues. Positive (presence of a stimulus-associated grooming response) and negative (absence of grooming behavior) responses were attributed a +1 or −1 value, respectively.
The white dot in the top left corner appears at the moment of the single stimulation using filament #7 (0.6 g), and the dot is displayed as long as the subsequent grooming response continues. Positive (presence of a stimulus-associated grooming response) and negative (absence of grooming behavior) responses were attributed a +1 or −1 value, respectively.
Significant Burden of Inherited and Unphased CACNA1H Variants
Next, we performed burden analysis of rare de novo and inherited/unphased variants in 290 TN probands, adjusting for de novo mutability using a one-tailed binomial test. Analysis of ultra-rare variants at MAF ≤ 1 × 10−5 did not identify significantly enriched genes. However, among moderately rare variants at MAF ≤ 1 × 10−4, the Cav3.2 T-type Ca2+ channel α-1H subunit (CACNA1H) reached genome-wide significant enrichment in cases (enrichment = 3.7, p value = 2.4 × 10−6; Figure 3A and Table S5A). CACNA1H contained 19 predicted damaging variants, including one LoF variant and 18 D-mis variants (Table S5B). Among these, 16 are unphased and 3 are transmitted with incomplete penetrance. The CACNA1H variants map to extracellular, intracellular, and intra-membrane regions of the ion channel (Figure 3B) and are predicted to impact Cav3.2 protein structure (Figures S3A–S3N). In one family, the CACNA1H D-mis variant p.Arg1674His was shared by proband TRGN282-1 and his sister TRGN282-2, both similarly affected by medically intractable iTN1.
Figure 3.

Gene Burden Analysis for Heterozygous Damaging Mutations and Mutation Mapping in Ca2+ Channels Encoded by CACNA1H and CACNA1F
(A) Quantile-quantile plot of observed versus expected p values for damaging (LoF and D-mis) variants with MAF ≤1 × 10−4. The genome-wide significant gene CACNA1H is circled in red. The genome significance cutoff is 2.6 × 10−6, 0.05/19,347.
(B and C) Mutation mapping of (B) CACNA1H and (C) CACNA1F. CACNA1H graph was adapted from Rzhepetskyy et al. (Rzhepetskyy et al., 2016); CACNA1F graph was modified from (Haeseleer et al., 2016).
Identification of Hemizygous Variants in CACNA1F
Lastly, we examined recessive and hemizygous genotypes. No genes harbored more than one recessive genotype in 249 TN probands. However, case-control analysis comparing rare damaging hemizygous variants (MAF ≤ 5 × 10−5) in 49 male TN probands with male controls in gnomAD identified CACNA1F, encoding the Cav3.2 T-type Ca2+ channel α-1H subunit, as the most significantly enriched gene (OR = 12.0, p value = 2.43 × 10−3; Table S5C). CACNA1F contained three rare D-mis variants (Figure 3C and Table S5D). p.Ala1335Thr and pArg1289Gly, respectively, mapped to the exofacial and endofacial surfaces of the channel. p.Ile721Val mapped to an extracellular loop of the channel. All three variants are predicted to significantly impact CACNA1F channel structure (Figures S3O–S3T).
Discussion
These results provide insight into the genomic architecture of TN. We ascertained the largest collection of familial forms of TN to date and identified several candidate genes with mutational enrichment in TN probands. Our findings implicate de novo and inherited rare, damaging variants in GABA signaling and other ion transport genes in TN pathogenesis in a subset of patients. Our creation of a knock-in mouse with the TN-associated de novo GABRG1 p.Cys188Trp mutation represents an important attempt to engineer a TN animal model with a human mutation.
Previous candidate gene sequencing approaches had identified SCN8A and SLC6A4 as potential TN-associated genes (Cui et al., 2014; Tanaka et al., 2016). In our WES study, we detected only two rare transmitted mutations in SCN8A and none in SLC6A4. Nonetheless, our data implicate other ion transport pathways in the genetic architecture of TN, including genes related to the function of the ligand-gated GABAAR Cl− channel. These findings are consistent with the expression of GABAARs along sensory axons (Bhisitkul et al., 1990; Oyelese et al., 1997), with the well-documented role of GABAAR signaling in trigeminal pathway nociception (Dieb and Hafidi, 2015; Jang et al., 2017; Kaushal et al., 2016; Martin et al., 2010; Wei et al., 2013), with the efficacy of GABA-modulating drugs in some patients with TN (Granger et al., 1995; White et al., 2000) (Table S6), and with the established importance of GABAAR disinhibition and consequent neuronal hyperexcitability in neuropathic pain (Dieb and Hafidi, 2015; Jang et al., 2017; Martin et al., 2010). These observations support our speculation that frequent co-occurrence of anxiety, depression, and other psychiatric conditions with TN, especially those in younger patients (Mousavi et al., 2016) (often unresponsive to MVD) (Hamlyn, 1997a), may reflect pleiotropy of germline variants impacting other aspects of GABA signaling.
In addition to variants in GABA signaling genes, we also identified rare dominant variants in the SCN8A-related Nav1.5 Na+ channel gene SCN5A in two multiplex TN families, a genome-wide significant enrichment of rare dominant variants in the Cav3.2 α-1H subunit CACNA1H and multiple rare X-linked hemizygous variants in the related Cav3.2 α-1F subunit CACNA1F. Not only are SCN5A mutations well known to cause cardiac rhythm disorders including long QT syndrome subtype 3, Brugada syndrome, and cardiac conduction disease (Zimmer and Surber, 2008), but they are also expressed in the brain (Kerr et al., 2004) and have been implicated in both epilepsy (Parisi et al., 2013) and schizophrenia (Roberts, 2006). CACNA1H variants have been previously implicated in congenital pain (Souza et al., 2016) and epilepsy (Eckle et al., 2014). T-type Ca2+ currents mediated by Cav3.2/α-1H are activated during GABAAR-mediated depolarizations and subsequently trigger action potential in sensory neurons (Aptel et al., 2007) and mechanosensation in nerve root ganglia (Shin et al., 2003). Increased expression of Cav3.2 in damaged dorsal root ganglion neurons contributes to the development of neuropathic pain triggered by spared nerve injury (Kang et al., 2018). The closely-related Cav3.1 has been shown to be a key element in the pathophysiology of a mouse model of trigeminal neuropathic pain (Choi et al., 2016). Mechanical thresholds of pain are significantly altered in a rat model of congenital stationary night blindness with a Cacna1f mutation (An et al., 2012). Detailed electrophysiology of these TN-associated SCN5A, CACNA1H, and CACNA1F channel variants in cell culture systems and animal models will be rich topics for future investigation.
How might these mutations and other gene variants contribute to TN pathology? One possibility is a “genetic-mechanical” model, in which a germline mutation confers increased sensitivity to the trigeminal ganglia or axons to NVC by an offending blood vessel, such as the superior cerebellar artery in the cerebello-pontine angle. This mechanism may contribute to unilaterality of symptoms in some patients with NVC and has been proposed for a gain-of-function mutation in SCN8A (Tanaka et al., 2016). A germline mutation could also predispose patients to the development of bilateral symptoms, as is seen in some patients with TN. Alternatively, a germline mutation could predispose an individual to later-onset TN arising from a second somatic mutation in the other allele of the same, or another, gene, in trigeminal ganglion neurons or other downstream brainstem or thalamo-cortical projection neurons in the trigeminal system circuitry. Such a “two-hit” model has been seen in other neurovascular cutaneous syndromes with unilateral or multifocal lesions (Brouillard et al., 2002; Pagenstecher et al., 2009; Revencu et al., 2013).
Our data suggest that rare, damaging exonic variants with large effect likely contribute to the pathogenesis of a small fraction of TN cases. Nonetheless, the investigation of such rare variants, especially DNMs, is a proven strategy to gain insight into disease pathogenesis. Therefore, continued gene discovery and functional analysis, including mechanistic work and drug screening in humanized animal models such as our GABRG1 p.Cys188Trp knock-in mouse, could further elucidate TN pathophysiology, improve diagnostics, and optimally stratify certain patients for specific treatments (e.g., a GABA-modulating drug instead of a craniotomy for MVD). Moreover, the identification of rare, damaging mutations with large effect (even in single patients) has the potential to identify unexpected targets for the development of non-addictive analgesics that could have broader relevance for other neuropathic pain syndromes (Yekkirala et al., 2017). Our findings suggest a WES approach might also be suitable to study hemifacial spasm, another cranial nerve pain syndrome with known familial occurrences that has been classically attributed to NVC and treated with neurosurgical MVD (Campbell and Keedy, 1947; Carter et al., 1990; Coad et al., 1991; Friedman et al., 1989; Haller et al., 2016; Lagalla et al., 2010; Miwa et al., 2002).
Limitations of the Study
A limitation of our study, necessary given our article's genomic focus and the magnitude of the effort, time, and expense required for patient recruitment, phenotyping, and bioinformatic analysis, is our lack of functional and mechanistic follow-up. In future work, the functional consequences of the identified mutations on channel or protein function using electrophysiological techniques in heterologous expression systems, or even better, model organisms with engineered patient mutations (starting with our GABRG1 mutant mouse), will be required. In these models, it will be important to establish the effect of mutated proteins on trigeminal pain circuitry; the expression of some our mutated proteins in second- or higher-order neurons of the trigeminal nucleus suggests that CNS defects might contribute to pathology. Our current genomics article sets a foundation for these detailed functional studies.
Another limitation of our study is its focus on the role of rare coding variants. Given the impact of common variants in multiple neuropathic pain disorders (Gormley et al., 2018; Meng et al., 2015a, 2015b), we examined the contribution to TN from common variants through a genome-wide association analysis of 236 European cases and 348,028 ethnicity-matching controls from the UK Biobank. No common variants reached genome-wide significance (Figures S4A and S4B). However, the small number of available cases limited the power of our analysis. A power calculation suggests that ∼1,600 cases are needed to achieve 80% power assuming moderate effect size (λ = 1.3) and an MAF of 0.2 for risk alleles (Figure S4C). Our analysis for these variants is thus underpowered. Future studies examining common variants will require a greater numbers of TN probands.
Although childhood and adolescent onset of TN has been reported (Bahgat et al., 2011), the high prevalence of TN and its inconsistent segregation patterns within families suggests that complex epistatic interactions, gene-environment interactions, or effects from common polygenic variants could also play important pathogenic roles in TN (Gormley et al., 2018; Zorina-Lichtenwalter et al., 2018). Addressing these questions will be important and challenging topics of future investigations.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact (kristopher.kahle@yale.edu).
Materials Availability
No plasmids were generated in this study. Mouse lines generated in this study will be deposited to the Knockout Mouse Project (KOMP) and are available upon request.
Data and Code Availability
The WES data generated during this study are available at dbGap with accession number phs000744. Original data for the mouse experiments in Figures 2D–2F in the article are available at Table S4. Our in-house Python and R pipelines are available from the corresponding author on request.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank the patients and families who participated in this research. We acknowledge Dr. Hongyu Zhao from the Department of Biostatistics, Yale School of Public Health for his mentorship on the statistical analysis and providing access to the UK Biobank Resource under Application Number 29900. We acknowledge support from the Yale-NIH Center for Mendelian Genomics (5U54HG006504) and an NIH NRCDP award to K.T.K. We acknowledge the Canadian Institute of Health Research (CIHR) Foundation grant (FDN389050) to Y.D.K. and CIHR grant (MOP-111072 and MOP-130373) to M.C. We acknowledge Dr. Kaya Bilguvar for his help on exome sequencing for the samples at Yale-NIH Center for Mendelian Genomics. W.D. was supported by American Heart Association Predoctoral Fellowship (19PRE34380842). S.C.J. was supported by the James Hudson Brown-Alexander Brown Coxe Postdoctoral Fellowship, the American Heart Association Postdoctoral Fellowship (18POST34060008), and the K99/R00 Pathway to Independence Award (K99HL143036 and R00HL143036-02). Y.D.K. was supported by a Canada Research Chair in Chronic Pain and Related Brain Disorders.
Author Contributions
Study design and conceptualization: W.D., R.P.L., and K.T.K. Cohort ascertainment, recruitment, and phenotypic characterization: A.A., S.P., J.H., A.S., J.G., A.D., C.G.F., and S.C. Exome sequencing production and validation: S.M., C.C., F.L.-G., and J.R.K. WES analysis: W.D., S.C.J., X.Z., A.H.S., J.C., and M.C.S. Statistical analysis: W.D., S.C.J., A.H.S., S.P., B.L., Q.L., G.Z., W.L., and X.L. Mouse experiments: A.C., L.-E.L., G.B., K.B., and Y.D.K. Sanger sequencing validation: C.N.-W. Biophysical simulation: B.I. and S.H. Resources: R.P.L., and K.T.K. Writing and review of manuscript: W.D., S.C.J., A.A., S.P., A.H.S., E.K., P.Q.D., A.J.K., R.F.S., J.M.S., E.N.E., C.G., J.M., M.G., J.L.G., S.D.-H., S.G.W., F.G.B., S.L.A., M.C., R.P.L., and K.T.K. Project administration: W.D., E.L., Y.D.K., R.P.L., and K.T.K. Funding acquisition and supervision: R.P.L. and K.T.K.
Declaration of Interests
The authors declare no competing interests.
Published: October 23, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101552.
Supplemental Information
(a) Genes with at least two segregated mutations in 20 TN families.
(b) Segregated CNVs in 20 TN families.
(a) De novo mutation rate closely approximates Poisson distribution in cases and controls.
(b) De novo enrichment analysis in 70 TN cases and 1,798 controls.
(c) Detailed information of de novo mutations.
(d) De novo CNVs from 70 TN trios.
(a) Significantly enriched GO terms from g:Profiler Gene Ontology (Molecular Function) analysis using 10 genes with damaging de novo mutations and with brain expression rank ≥75%.
(b) De novo enrichment analysis in GABA receptor-binding genes.
(c) Nine de novo and rare transmitted/unphased mutations in genes encoding GABA receptor-binding proteins.
(d) Case-control analysis for variants in GABA receptor-binding genes.
(a) Trigeminal repetitive stimulation.
(b) Nociceptive withdrawal threshold.
(a) Top 10 genes from gene burden analysis for damaging (LoF and D-mis) variants with MAF ≤1✕10−4. The genome significance cutoff is 2.6✕10−6, 0.05/19347. Gene with genome-wide significance is in red.
(b) Damaging heterozygous mutations in CACNA1H at MAF≤1✕10−4.
(c) Case-control analysis of rare damaging hemizygous variants (MAF≤5✕10−5) in 32 European male cases compared with gnomAD European male subjects.
(d) Damaging hemizygous mutations in CACNA1F at MAF≤5✕10−5.
References
- Ackerman M.J., Siu B.L., Sturner W.Q., Tester D.J., Valdivia C.R., Makielski J.C., Towbin J.A. Postmortem molecular analysis of SCN5A defects in sudden infant death syndrome. JAMA. 2001;286:2264–2269. doi: 10.1001/jama.286.18.2264. [DOI] [PubMed] [Google Scholar]
- Ahmed O.L., Akinyele O.A., Akindayo A.O., Bamidele K. Management of trigeminal neuralgia using amitriptyline and pregablin combination therapy. Afr. J. Biomed. Res. 2012;15:201–203. [Google Scholar]
- Al-Quliti K.W. Update on neuropathic pain treatment for trigeminal neuralgia. The pharmacological and surgical options. Neurosciences (Riyadh) 2015;20:107–114. doi: 10.17712/nsj.2015.2.20140501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen A.S., Berkovic S.F., Cossette P., Delanty N., Dlugos D., Eichler E.E., Epstein M.P., Glauser T., Goldstein D.B., Han Y. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–221. doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alter B.J., Zhao C., Karim F., Landreth G.E., Gereau R.W.t. Genetic targeting of ERK1 suggests a predominant role for ERK2 in murine pain models. J. Neurosci. 2010;30:11537–11547. doi: 10.1523/JNEUROSCI.6103-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An J., Wang L., Guo Q., Li L., Xia F., Zhang Z. Behavioral phenotypic properties of a natural occurring rat model of congenital stationary night blindness with Cacna1f mutation. J. Neurogenet. 2012;26:363–373. doi: 10.3109/01677063.2012.684416. [DOI] [PubMed] [Google Scholar]
- Aptel H., Hilaire C., Pieraut S., Boukhaddaoui H., Mallie S., Valmier J., Scamps F. The Cav3.2/alpha1H T-type Ca2+ current is a molecular determinant of excitatory effects of GABA in adult sensory neurons. Mol. Cell Neurosci. 2007;36:293–303. doi: 10.1016/j.mcn.2007.07.009. [DOI] [PubMed] [Google Scholar]
- Aulchenko Y.S., Hoppenbrouwers I.A., Ramagopalan S.V., Broer L., Jafari N., Hillert J., Link J., Lundstrom W., Greiner E., Dessa Sadovnick A. Genetic variation in the KIF1B locus influences susceptibility to multiple sclerosis. Nat. Genet. 2008;40:1402–1403. doi: 10.1038/ng.251. [DOI] [PubMed] [Google Scholar]
- Bahgat D., Ray D.K., Raslan A.M., McCartney S., Burchiel K.J. Trigeminal neuralgia in young adults. J. Neurosurg. 2011;114:1306–1311. doi: 10.3171/2010.10.JNS10781. [DOI] [PubMed] [Google Scholar]
- Bailey A.L., Ribeiro-da-Silva A. Transient loss of terminals from non-peptidergic nociceptive fibers in the substantia gelatinosa of spinal cord following chronic constriction injury of the sciatic nerve. Neuroscience. 2006;138:675–690. doi: 10.1016/j.neuroscience.2005.11.051. [DOI] [PubMed] [Google Scholar]
- Barel O., Malicdan M.C.V., Ben-Zeev B., Kandel J., Pri-Chen H., Stephen J., Castro I.G., Metz J., Atawa O., Moshkovitz S. Deleterious variants in TRAK1 disrupt mitochondrial movement and cause fatal encephalopathy. Brain. 2017;140:568–581. doi: 10.1093/brain/awx002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behesti H., Fore T.R., Wu P., Horn Z., Leppert M., Hull C., Hatten M.E. ASTN2 modulates synaptic strength by trafficking and degradation of surface proteins. Proc. Natl. Acad. Sci. U S A. 2018;115:E9717–E9726. doi: 10.1073/pnas.1809382115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhisitkul R.B., Kocsis J.D., Gordon T.R., Waxman S.G. Trophic influence of the distal nerve segment on GABAA receptor expression in axotomized adult sensory neurons. Exp. Neurol. 1990;109:273–278. doi: 10.1016/s0014-4886(05)80017-2. [DOI] [PubMed] [Google Scholar]
- Bonin R.P., Bories C., De Koninck Y. A simplified up-down method (SUDO) for measuring mechanical nociception in rodents using von Frey filaments. Mol. Pain. 2014;10:26. doi: 10.1186/1744-8069-10-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brisman R. Bilateral trigeminal neuralgia. J. Neurosurg. 1987;67:44–48. doi: 10.3171/jns.1987.67.1.0044. [DOI] [PubMed] [Google Scholar]
- Brouillard P., Boon L.M., Mulliken J.B., Enjolras O., Ghassibe M., Warman M.L., Tan O.T., Olsen B.R., Vikkula M. Mutations in a novel factor, glomulin, are responsible for glomuvenous malformations ("glomangiomas") Am. J. Hum. Genet. 2002;70:866–874. doi: 10.1086/339492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burchiel K.J. Abnormal impulse generation in focally demyelinated trigeminal roots. J. Neurosurg. 1980;53:674–683. doi: 10.3171/jns.1980.53.5.0674. [DOI] [PubMed] [Google Scholar]
- Burchiel K.J. Ectopic impulse generation in focally demyelinated trigeminal nerve. Exp. Neurol. 1980;69:423–429. doi: 10.1016/0014-4886(80)90225-3. [DOI] [PubMed] [Google Scholar]
- Burchiel K.J., Baumann T.K. Pathophysiology of trigeminal neuralgia: new evidence from a trigeminal ganglion intraoperative microneurographic recording. J. Neurosurg. 2004;101:872–873. doi: 10.3171/jns.2004.101.5.0872. [DOI] [PubMed] [Google Scholar]
- Campbell E., Keedy C. Hemifacial spasm; a note on the etiology in two cases. J. Neurosurg. 1947;4:342–347. doi: 10.3171/jns.1947.4.4.0342. [DOI] [PubMed] [Google Scholar]
- Carter J.B., Patrinely J.R., Jankovic J., McCrary J.A., 3rd, Boniuk M. Familial hemifacial spasm. Arch. Ophthalmol. 1990;108:249–250. doi: 10.1001/archopht.1990.01070040101040. [DOI] [PubMed] [Google Scholar]
- Choi S., Yu E., Hwang E., Llinas R.R. Pathophysiological implication of CaV3.1 T-type Ca2+ channels in trigeminal neuropathic pain. Proc. Natl. Acad. Sci. U S A. 2016;113:2270–2275. doi: 10.1073/pnas.1600418113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coad J.E., Wirtschafter J.D., Haines S.J., Heros R.C., Perrone T. Familial hemifacial spasm associated with arterial compression of the facial nerve. J. Neurosurg. 1991;74:290–296. doi: 10.3171/jns.1991.74.2.0290. [DOI] [PubMed] [Google Scholar]
- Cruccu G., Finnerup N.B., Jensen T.S., Scholz J., Sindou M., Svensson P., Treede R.D., Zakrzewska J.M., Nurmikko T. Trigeminal neuralgia: new classification and diagnostic grading for practice and research. Neurology. 2016;87:220–228. doi: 10.1212/WNL.0000000000002840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui W., Yu X., Zhang H. The serotonin transporter gene polymorphism is associated with the susceptibility and the pain severity in idiopathic trigeminal neuralgia patients. J. Headache Pain. 2014;15:42. doi: 10.1186/1129-2377-15-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devor M., Amir R., Rappaport Z.H. Pathophysiology of trigeminal neuralgia: the ignition hypothesis. Clin. J. Pain. 2002;18:4–13. doi: 10.1097/00002508-200201000-00002. [DOI] [PubMed] [Google Scholar]
- Dieb W., Hafidi A. Mechanism of GABA involvement in post-traumatic trigeminal neuropathic pain: activation of neuronal circuitry composed of PKCgamma interneurons and pERK1/2 expressing neurons. Eur. J. Pain. 2015;19:85–96. doi: 10.1002/ejp.525. [DOI] [PubMed] [Google Scholar]
- Dong C., Wei P., Jian X., Gibbs R., Boerwinkle E., Wang K., Liu X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015;24:2125–2137. doi: 10.1093/hmg/ddu733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran D., Zeng X., Jin S.C., Choi J., Nelson-Williams C., Yatsula B., Gaillard J., Furey C.G., Lu Q., Timberlake A.T. Mutations in chromatin modifier and ephrin signaling genes in vein of galen malformation. Neuron. 2019;101:429–443 e424. doi: 10.1016/j.neuron.2018.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckle V.S., Shcheglovitov A., Vitko I., Dey D., Yap C.C., Winckler B., Perez-Reyes E., Perez-Reyes E. Mechanisms by which a CACNA1H mutation in epilepsy patients increases seizure susceptibility. J. Physiol. 2014;592:795–809. doi: 10.1113/jphysiol.2013.264176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eigenbrod O., Debus K.Y., Reznick J., Bennett N.C., Sanchez-Carranza O., Omerbasic D., Hart D.W., Barker A.J., Zhong W., Lutermann H. Rapid molecular evolution of pain insensitivity in multiple African rodents. Science. 2019;364:852–859. doi: 10.1126/science.aau0236. [DOI] [PubMed] [Google Scholar]
- Fleetwood I.G., Innes A.M., Hansen S.R., Steinberg G.K. Familial trigeminal neuralgia. Case report and review of the literature. J. Neurosurg. 2001;95:513–517. doi: 10.3171/jns.2001.95.3.0513. [DOI] [PubMed] [Google Scholar]
- Flegel C., Schobel N., Altmuller J., Becker C., Tannapfel A., Hatt H., Gisselmann G. RNA-seq analysis of human trigeminal and dorsal root ganglia with a focus on chemoreceptors. PLoS One. 2015;10:e0128951. doi: 10.1371/journal.pone.0128951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman A., Jamrozik Z., Bojakowski J. Familial hemifacial spasm. Mov. Disord. 1989;4:213–218. doi: 10.1002/mds.870040303. [DOI] [PubMed] [Google Scholar]
- Furey C.G., Choi J., Jin S.C., Zeng X., Timberlake A.T., Nelson-Williams C., Mansuri M.S., Lu Q., Duran D., Panchagnula S. De novo mutation in genes regulating neural Stem cell fate in human congenital hydrocephalus. Neuron. 2018;99:302–314 e304. doi: 10.1016/j.neuron.2018.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner W.J., Miklos M.V. Response of trigeminal neuralgia to decompression of sensory root; discussion of cause of trigeminal neuralgia. J. Am. Med. Assoc. 1959;170:1773–1776. doi: 10.1001/jama.1959.03010150017004. [DOI] [PubMed] [Google Scholar]
- Gilbert S.L., Zhang L., Forster M.L., Anderson J.R., Iwase T., Soliven B., Donahue L.R., Sweet H.O., Bronson R.T., Davisson M.T. Trak1 mutation disrupts GABA(A) receptor homeostasis in hypertonic mice. Nat. Genet. 2006;38:245–250. doi: 10.1038/ng1715. [DOI] [PubMed] [Google Scholar]
- Gormley P., Kurki M.I., Hiekkala M.E., Veerapen K., Happola P., Mitchell A.A., Lal D., Palta P., Surakka I., Kaunisto M.A. Common variant burden contributes to the familial aggregation of migraine in 1,589 families. Neuron. 2018;99:1098. doi: 10.1016/j.neuron.2018.08.029. [DOI] [PubMed] [Google Scholar]
- Granger P., Biton B., Faure C., Vige X., Depoortere H., Graham D., Langer S.Z., Scatton B., Avenet P. Modulation of the gamma-aminobutyric acid type A receptor by the antiepileptic drugs carbamazepine and phenytoin. Mol. Pharmacol. 1995;47:1189–1196. [PubMed] [Google Scholar]
- Haeseleer F., Williams B., Lee A. Characterization of C-terminal Splice variants of Cav1.4 Ca2+ channels in human retina. J. Biol. Chem. 2016;291:15663–15673. doi: 10.1074/jbc.M116.731737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haines S.J., Jannetta P.J., Zorub D.S. Microvascular relations of the trigeminal nerve. An anatomical study with clinical correlation. J. Neurosurg. 1980;52:381–386. doi: 10.3171/jns.1980.52.3.0381. [DOI] [PubMed] [Google Scholar]
- Haller S., Etienne L., Kovari E., Varoquaux A.D., Urbach H., Becker M. Imaging of neurovascular compression syndromes: trigeminal neuralgia, hemifacial spasm, vestibular paroxysmia, and glossopharyngeal neuralgia. Am. J. Neuroradiol. 2016;37:1384–1392. doi: 10.3174/ajnr.A4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamlyn P.J. Neurovascular relationships in the posterior cranial fossa, with special reference to trigeminal neuralgia. 1. Review of the literature and development of a new method of vascular injection-filling in cadaveric controls. Clin. Anat. 1997;10:371–379. doi: 10.1002/(SICI)1098-2353(1997)10:6<371::AID-CA1>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Hamlyn P.J. Neurovascular relationships in the posterior cranial fossa, with special reference to trigeminal neuralgia. 2. Neurovascular compression of the trigeminal nerve in cadaveric controls and patients with trigeminal neuralgia: quantification and influence of method. Clin. Anat. 1997;10:380–388. doi: 10.1002/(SICI)1098-2353(1997)10:6<380::AID-CA2>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Harris W. Bilateral trigeminal tic: its association with heredity and disseminated sclerosis. Ann. Surg. 1936;103:161–172. doi: 10.1097/00000658-193602000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Headache Classification Committee of the International Headache Society (IHS) The international classification of Headache disorders, 3rd edition (beta version) Cephalalgia. 2013;33:629–808. doi: 10.1177/0333102413485658. [DOI] [PubMed] [Google Scholar]
- Heise C., Taha E., Murru L., Ponzoni L., Cattaneo A., Guarnieri F.C., Montani C., Mossa A., Vezzoli E., Ippolito G. eEF2K/eEF2 pathway controls the excitation/inhibition balance and susceptibility to epileptic Seizures. Cereb. Cortex. 2017;27:2226–2248. doi: 10.1093/cercor/bhw075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton D.A., Love S., Gradidge T., Coakham H.B. Pathological findings associated with trigeminal neuralgia caused by vascular compression. Neurosurgery. 1994;35:299–303. doi: 10.1227/00006123-199408000-00017. discussion 303. [DOI] [PubMed] [Google Scholar]
- Hocking L.J., Generation S., Morris A.D., Dominiczak A.F., Porteous D.J., Smith B.H. Heritability of chronic pain in 2195 extended families. Eur. J. Pain. 2012;16:1053–1063. doi: 10.1002/j.1532-2149.2011.00095.x. [DOI] [PubMed] [Google Scholar]
- Hughes M.A., Jani R.H., Fakhran S., Chang Y.F., Branstetter B.F., Thirumala P.D., Sekula R.F. Significance of degree of neurovascular compression in surgery for trigeminal neuralgia. J. Neurosurg. 2019;133:411–416. doi: 10.3171/2019.3.JNS183174. [DOI] [PubMed] [Google Scholar]
- Iossifov I., Ronemus M., Levy D., Wang Z., Hakker I., Rosenbaum J., Yamrom B., Lee Y.H., Narzisi G., Leotta A. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang I.J., Davies A.J., Akimoto N., Back S.K., Lee P.R., Na H.S., Furue H., Jung S.J., Kim Y.H., Oh S.B. Acute inflammation reveals GABAA receptor-mediated nociception in mouse dorsal root ganglion neurons via PGE2 receptor 4 signaling. Physiol. Rep. 2017;5:e13178. doi: 10.14814/phy2.13178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jani R.H., Hughes M.A., Gold M.S., Branstetter B.F., Ligus Z.E., Sekula R.F., Jr. Trigeminal nerve compression without trigeminal neuralgia: intraoperative vs imaging evidence. Neurosurgery. 2019;84:60–65. doi: 10.1093/neuros/nyx636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin S.C., Homsy J., Zaidi S., Lu Q., Morton S., DePalma S.R., Zeng X., Qi H., Chang W., Sierant M.C. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat. Genet. 2017;49:1593–1601. doi: 10.1038/ng.3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang X.J., Chi Y.N., Chen W., Liu F.Y., Cui S., Liao F.F., Cai J., Wan Y. Increased expression of CaV3.2 T-type calcium channels in damaged DRG neurons contributes to neuropathic pain in rats with spared nerve injury. Mol. Pain. 2018;14 doi: 10.1177/1744806918765808. 1744806918765808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katusic S., Williams D.B., Beard C.M., Bergstralh E.J., Kurland L.T. Epidemiology and clinical features of idiopathic trigeminal neuralgia and glossopharyngeal neuralgia: similarities and differences, Rochester, Minnesota, 1945-1984. Neuroepidemiology. 1991;10:276–281. doi: 10.1159/000110284. [DOI] [PubMed] [Google Scholar]
- Kaushal R., Taylor B.K., Jamal A.B., Zhang L., Ma F., Donahue R., Westlund K.N. GABA-A receptor activity in the noradrenergic locus coeruleus drives trigeminal neuropathic pain in the rat; contribution of NAalpha1 receptors in the medial prefrontal cortex. Neuroscience. 2016;334:148–159. doi: 10.1016/j.neuroscience.2016.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr N.C., Holmes F.E., Wynick D. Novel isoforms of the sodium channels Nav1.8 and Nav1.5 are produced by a conserved mechanism in mouse and rat. J. Biol. Chem. 2004;279:24826–24833. doi: 10.1074/jbc.M401281200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M., Witten D.M., Jain P., O'Roak B.J., Cooper G.M., Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagalla G., Logullo F., Di Bella P., Haghighipour R., Provinciali L. Familial hemifacial spasm and determinants of late onset. Neurol. Sci. 2010;31:17–22. doi: 10.1007/s10072-009-0153-4. [DOI] [PubMed] [Google Scholar]
- Liverman C.S., Brown J.W., Sandhir R., Klein R.M., McCarson K., Berman N.E. Oestrogen increases nociception through ERK activation in the trigeminal ganglion: evidence for a peripheral mechanism of allodynia. Cephalalgia. 2009;29:520–531. doi: 10.1111/j.1468-2982.2008.01755.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons D.A., Naylor S.G., Scholze A., Talbot W.S. Kif1b is essential for mRNA localization in oligodendrocytes and development of myelinated axons. Nat. Genet. 2009;41:854–858. doi: 10.1038/ng.376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maarbjerg S., Wolfram F., Gozalov A., Olesen J., Bendtsen L. Significance of neurovascular contact in classical trigeminal neuralgia. Brain. 2015;138:311–319. doi: 10.1093/brain/awu349. [DOI] [PubMed] [Google Scholar]
- Maarbjerg S., Di Stefano G., Bendtsen L., Cruccu G. Trigeminal neuralgia - diagnosis and treatment. Cephalalgia. 2017;37:648–657. doi: 10.1177/0333102416687280. [DOI] [PubMed] [Google Scholar]
- MacDonald B.K., Cockerell O.C., Sander J.W., Shorvon S.D. The incidence and lifetime prevalence of neurological disorders in a prospective community-based study in the UK. Brain. 2000;123(Pt 4):665–676. doi: 10.1093/brain/123.4.665. [DOI] [PubMed] [Google Scholar]
- Martin Y.B., Malmierca E., Avendano C., Nunez A. Neuronal disinhibition in the trigeminal nucleus caudalis in a model of chronic neuropathic pain. Eur. J. Neurosci. 2010;32:399–408. doi: 10.1111/j.1460-9568.2010.07302.x. [DOI] [PubMed] [Google Scholar]
- Meng W., Deshmukh H.A., Donnelly L.A., Wellcome Trust Case Control Consortium 2, Surrogate Markers for Micro and Macro-Vascular Hard Endpoints for Innovative Diabetes Tools Study Group, Torrance N., Colhoun H.M., Palmer C.N., Smith B.H. A genome-wide association study provides evidence of Sex-specific involvement of Chr1p35.1 (ZSCAN20-TLR12P) and Chr8p23.1 (HMGB1P46) with diabetic neuropathic pain. EBioMedicine. 2015;2:1386–1393. doi: 10.1016/j.ebiom.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng W., Deshmukh H.A., van Zuydam N.R., Liu Y., Donnelly L.A., Zhou K., Wellcome Trust Case Control Consortium 2, Surrogate Markers for Micro and Macro-Vascular Hard Endpoints for Innovative Diabetes Tools Study Group, Morris A.D., Holhuan H.M. A genome-wide association study suggests an association of Chr8p21.3 (GFRA2) with diabetic neuropathic pain. Eur. J. Pain. 2015;19:392–399. doi: 10.1002/ejp.560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miwa H., Mizuno Y., Kondo T. Familial hemifacial spasm: report of cases and review of literature. J. Neurol. Sci. 2002;193:97–102. doi: 10.1016/s0022-510x(01)00651-7. [DOI] [PubMed] [Google Scholar]
- Mousavi S.H., Sekula R.F., Gildengers A., Gardner P., Lunsford L.D. Concomitant depression and anxiety negatively affect pain outcomes in surgically managed young patients with trigeminal neuralgia: long-term clinical outcome. Surg. Neurol. Int. 2016;7:98. doi: 10.4103/2152-7806.194145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen C.S., Knudsen G.P., Steingrimsdottir O.A. Twin studies of pain. Clin. Genet. 2012;82:331–340. doi: 10.1111/j.1399-0004.2012.01938.x. [DOI] [PubMed] [Google Scholar]
- El Otmani H., Moutaouakil F., Fadel H., Slassi I. [Familial trigeminal neuralgia] Rev. Neurol. (Paris) 2008;164:384–387. doi: 10.1016/j.neurol.2007.10.010. [DOI] [PubMed] [Google Scholar]
- Oyelese A.A., Rizzo M.A., Waxman S.G., Kocsis J.D. Differential effects of NGF and BDNF on axotomy-induced changes in GABA(A)-receptor-mediated conductance and sodium currents in cutaneous afferent neurons. J. Neurophysiol. 1997;78:31–42. doi: 10.1152/jn.1997.78.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagenstecher A., Stahl S., Sure U., Felbor U. A two-hit mechanism causes cerebral cavernous malformations: complete inactivation of CCM1, CCM2 or CCM3 in affected endothelial cells. Hum. Mol. Genet. 2009;18:911–918. doi: 10.1093/hmg/ddn420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi P., Oliva A., Coll Vidal M., Partemi S., Campuzano O., Iglesias A., Pisani D., Pascali V.L., Paolino M.C., Villa M.P. Coexistence of epilepsy and Brugada syndrome in a family with SCN5A mutation. Epilepsy Res. 2013;105:415–418. doi: 10.1016/j.eplepsyres.2013.02.024. [DOI] [PubMed] [Google Scholar]
- Philippart F., Khaliq Z.A.-O. G(i/o) protein-coupled receptors in dopamine neurons inhibit the sodium leak channel NALCN. Elife. 2018;7:e40984. doi: 10.7554/eLife.40984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollack I.F., Jannetta P.J., Bissonette D.J. Bilateral trigeminal neuralgia: a 14-year experience with microvascular decompression. J. Neurosurg. 1988;68:559–565. doi: 10.3171/jns.1988.68.4.0559. [DOI] [PubMed] [Google Scholar]
- The NHLBI Trans-Omics for Precision Medicine (TOPMed) Whole Genome Sequencing Program BRAVO variant browser: University of Michigan and NHLBI. 2018. https://bravo.sph.umich.edu/freeze5/hg38/ Available from:
- Rappaport Z.H., Govrin-Lippmann R., Devor M. An electron-microscopic analysis of biopsy samples of the trigeminal root taken during microvascular decompressive surgery. Stereotact. Funct. Neurosurg. 1997;68:182–186. doi: 10.1159/000099920. [DOI] [PubMed] [Google Scholar]
- Raudvere U., Kolberg L., Kuzmin I., Arak T., Adler P., Peterson H., Vilo J. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update) Nucleic Acids Res. 2019;47:W191–W198. doi: 10.1093/nar/gkz369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revencu N., Boon L.M., Mendola A., Cordisco M.R., Dubois J., Clapuyt P., Hammer F., Amor D.J., Irvine A.D., Baselga E. RASA1 mutations and associated phenotypes in 68 families with capillary malformation-arteriovenous malformation. Hum. Mutat. 2013;34:1632–1641. doi: 10.1002/humu.22431. [DOI] [PubMed] [Google Scholar]
- Roberts E. GABAergic malfunction in the limbic system resulting from an aboriginal genetic defect in voltage-gated Na+-channel SCN5A is proposed to give rise to susceptibility to schizophrenia. Adv. Pharmocol. 2006;54:119–145. doi: 10.1016/s1054-3589(06)54006-2. [DOI] [PubMed] [Google Scholar]
- Rodriguez F.B., Simonet C., Cerdan D.M., Morollon N., Guerrero P., Tabernero C., Duarte J. Familial classic trigeminal neuralgia. Neurologia. 2019;34:229–233. doi: 10.1016/j.nrl.2016.12.004. [DOI] [PubMed] [Google Scholar]
- Rzhepetskyy Y., Lazniewska J., Blesneac I., Pamphlett R., Weiss N. CACNA1H missense mutations associated with amyotrophic lateral sclerosis alter Cav3.2 T-type calcium channel activity and reticular thalamic neuron firing. Channels (Austin) 2016;10:466–477. doi: 10.1080/19336950.2016.1204497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J.B., Martinez-Salgado C., Heppenstall P.A., Lewin G.R. A T-type calcium channel required for normal function of a mammalian mechanoreceptor. Nat. Neurosci. 2003;6:724–730. doi: 10.1038/nn1076. [DOI] [PubMed] [Google Scholar]
- Smyth P., Greenough G., Stommel E. Familial trigeminal neuralgia: case reports and review of the literature. Headache. 2003;43:910–915. doi: 10.1046/j.1526-4610.2003.03172.x. [DOI] [PubMed] [Google Scholar]
- Souza I.A., Gandini M.A., Wan M.M., Zamponi G.W. Two heterozygous Cav3.2 channel mutations in a pediatric chronic pain patient: recording condition-dependent biophysical effects. Pflugers Arch. 2016;468:635–642. doi: 10.1007/s00424-015-1776-3. [DOI] [PubMed] [Google Scholar]
- Sun S., Sun J., Jiang W., Wang W., Ni L. Nav1.7 via promotion of ERK in the trigeminal ganglion plays an important role in the induction of pulpitis inflammatory pain. Biomed. Res. Int. 2019;2019:6973932. doi: 10.1155/2019/6973932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka B.S., Zhao P., Dib-Hajj F.B., Morisset V., Tate S., Waxman S.G., Dib-Hajj S.D. A gain-of-function mutation in Nav1.6 in a case of trigeminal neuralgia. Mol. Med. 2016;22:338–348. doi: 10.2119/molmed.2016.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor A.M., Osikowicz M., Ribeiro-da-Silva A. Consequences of the ablation of nonpeptidergic afferents in an animal model of trigeminal neuropathic pain. Pain. 2012;153:1311–1319. doi: 10.1016/j.pain.2012.03.023. [DOI] [PubMed] [Google Scholar]
- Timberlake A.T., Choi J., Zaidi S., Lu Q., Nelson-Williams C., Brooks E.D., Bilguvar K., Tikhonova I., Mane S., Yang J.F. Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and common BMP2 alleles. Elife. 2016;5:e20125. doi: 10.7554/eLife.20125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissers L.E., de Ligt J., Gilissen C., Janssen I., Steehouwer M., de Vries P., van Lier B., Arts P., Wieskamp N., del Rosario M. A de novo paradigm for mental retardation. Nat. Genet. 2010;42:1109–1112. doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- Wei B., Kumada T., Furukawa T., Inoue K., Watanabe M., Sato K., Fukuda A. Pre- and post-synaptic switches of GABA actions associated with Cl- homeostatic changes are induced in the spinal nucleus of the trigeminal nerve in a rat model of trigeminal neuropathic pain. Neuroscience. 2013;228:334–348. doi: 10.1016/j.neuroscience.2012.10.043. [DOI] [PubMed] [Google Scholar]
- White H.S., Brown S.D., Woodhead J.H., Skeen G.A., Wolf H.H. Topiramate modulates GABA-evoked currents in murine cortical neurons by a nonbenzodiazepine mechanism. Epilepsia. 2000;41:S17–S20. [PubMed] [Google Scholar]
- Yeh E., Ng S., Zhang M., Bouhours M., Wang Y., Wang M., Hung W., Aoyagi K., Melnik-Martinez K., Li M. A putative cation channel, NCA-1, and a novel protein, UNC-80, transmit neuronal activity in C. elegans. PLoS Biol. 2008;6:e55. doi: 10.1371/journal.pbio.0060055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yekkirala A.S., Roberson D.P., Bean B.P., Woolf C.J. Breaking barriers to novel analgesic drug development. Nat. Rev. Drug Discov. 2017;16:545–564. doi: 10.1038/nrd.2017.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi S., Choi M., Wakimoto H., Ma L., Jiang J., Overton J.D., Romano-Adesman A., Bjornson R.D., Breitbart R.E., Brown K.K. De novo mutations in histone-modifying genes in congenital heart disease. Nature. 2013;498:220–223. doi: 10.1038/nature12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakrzewska J.M., Akram H. Neurosurgical interventions for the treatment of classical trigeminal neuralgia. Cochrane Database Syst. Rev. 2011 doi: 10.1002/14651858.CD007312.pub2. CD007312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer T., Surber R. SCN5A channelopathies--an update on mutations and mechanisms. Prog. Biophys. Mol. Biol. 2008;98:120–136. doi: 10.1016/j.pbiomolbio.2008.10.005. [DOI] [PubMed] [Google Scholar]
- Zorina-Lichtenwalter K., Parisien M., Diatchenko L. Genetic studies of human neuropathic pain conditions: a review. Pain. 2018;159:583–594. doi: 10.1097/j.pain.0000000000001099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuchner S., Mersiyanova I.V., Muglia M., Bissar-Tadmouri N., Rochelle J., Dadali E.L., Zappia M., Nelis E., Patitucci A., Senderek J. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004;36:449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The white dot in the top left corner appears at the moment of the single stimulation using filament #7 (0.6 g), and the dot is displayed as long as the subsequent grooming response continues. Positive (presence of a stimulus-associated grooming response) and negative (absence of grooming behavior) responses were attributed a +1 or −1 value, respectively.
The white dot in the top left corner appears at the moment of the single stimulation using filament #7 (0.6 g), and the dot is displayed as long as the subsequent grooming response continues. Positive (presence of a stimulus-associated grooming response) and negative (absence of grooming behavior) responses were attributed a +1 or −1 value, respectively.
The white dot in the top left corner appears at the moment of the single stimulation using filament #7 (0.6 g), and the dot is displayed as long as the subsequent grooming response continues. Positive (presence of a stimulus-associated grooming response) and negative (absence of grooming behavior) responses were attributed a +1 or −1 value, respectively.
The white dot in the top left corner appears at the moment of the single stimulation using filament #7 (0.6 g), and the dot is displayed as long as the subsequent grooming response continues. Positive (presence of a stimulus-associated grooming response) and negative (absence of grooming behavior) responses were attributed a +1 or −1 value, respectively.
The white dot in the top left corner appears at the moment of the single stimulation using filament #7 (0.6 g), and the dot is displayed as long as the subsequent grooming response continues. Positive (presence of a stimulus-associated grooming response) and negative (absence of grooming behavior) responses were attributed a +1 or −1 value, respectively.
The white dot in the top left corner appears at the moment of the single stimulation using filament #7 (0.6 g), and the dot is displayed as long as the subsequent grooming response continues. Positive (presence of a stimulus-associated grooming response) and negative (absence of grooming behavior) responses were attributed a +1 or −1 value, respectively.
The white dot in the top left corner appears at the moment of the single stimulation using filament #7 (0.6 g), and the dot is displayed as long as the subsequent grooming response continues. Positive (presence of a stimulus-associated grooming response) and negative (absence of grooming behavior) responses were attributed a +1 or −1 value, respectively.
The white dot in the top left corner appears at the moment of the single stimulation using filament #7 (0.6 g), and the dot is displayed as long as the subsequent grooming response continues. Positive (presence of a stimulus-associated grooming response) and negative (absence of grooming behavior) responses were attributed a +1 or −1 value, respectively.
(a) Genes with at least two segregated mutations in 20 TN families.
(b) Segregated CNVs in 20 TN families.
(a) De novo mutation rate closely approximates Poisson distribution in cases and controls.
(b) De novo enrichment analysis in 70 TN cases and 1,798 controls.
(c) Detailed information of de novo mutations.
(d) De novo CNVs from 70 TN trios.
(a) Significantly enriched GO terms from g:Profiler Gene Ontology (Molecular Function) analysis using 10 genes with damaging de novo mutations and with brain expression rank ≥75%.
(b) De novo enrichment analysis in GABA receptor-binding genes.
(c) Nine de novo and rare transmitted/unphased mutations in genes encoding GABA receptor-binding proteins.
(d) Case-control analysis for variants in GABA receptor-binding genes.
(a) Trigeminal repetitive stimulation.
(b) Nociceptive withdrawal threshold.
(a) Top 10 genes from gene burden analysis for damaging (LoF and D-mis) variants with MAF ≤1✕10−4. The genome significance cutoff is 2.6✕10−6, 0.05/19347. Gene with genome-wide significance is in red.
(b) Damaging heterozygous mutations in CACNA1H at MAF≤1✕10−4.
(c) Case-control analysis of rare damaging hemizygous variants (MAF≤5✕10−5) in 32 European male cases compared with gnomAD European male subjects.
(d) Damaging hemizygous mutations in CACNA1F at MAF≤5✕10−5.
Data Availability Statement
The WES data generated during this study are available at dbGap with accession number phs000744. Original data for the mouse experiments in Figures 2D–2F in the article are available at Table S4. Our in-house Python and R pipelines are available from the corresponding author on request.