

Abstract
The concepts of gene therapy were initially introduced during the 1960s. Since the early 1990s, more than 1900 clinical trials have been conducted for the treatment of genetic diseases and cancers mainly using viral vectors. Although a variety of methods have also been performed for the treatment of malignant gliomas, it has been difficult to target invasive glioma cells. To overcome this problem, immortalized neural stem cell (NSC) and a nonlytic, amphotropic retroviral replicating vector (RRV) have attracted attention for gene delivery to invasive glioma. Recently, genome editing technology targeting insertions at site-specific locations has advanced; in particular, the clustered regularly interspaced palindromic repeats/CRISPR-associated-9 (CRISPR/Cas9) has been developed. Since 2015, more than 30 clinical trials have been conducted using genome editing technologies, and the results have shown the potential to achieve positive patient outcomes. Gene therapy using CRISPR technologies for the treatment of a wide range of diseases is expected to continuously advance well into the future.
Keywords: gene therapy, genome editing, ZFN, TALEN, CRISPR/Cas9
Introduction
Gene therapy is a therapeutic strategy using genetic engineering techniques to treat various diseases.1,2) In the early 1960s, gene therapy first progressed with the development of recombinant DNA (rDNA) technology,1) and was further developed using various genetic engineering tools, such as viral vectors.3–5) More than 1900 clinical trials have been conducted with gene therapeutic approaches since the early 1990s. In these procedures, DNA is randomly inserted into the host genome using conventional genetic engineering tools. In the 2000s, genome editing tootls, including zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and the recently established clustered regularly interspaced palindromic repeats/CRISPR-associated-9 (CRISPR/Cas9) technologies, were developed, which induce genome modifications at specific target sites.5) Genome editing tools are efficient for intentional genetic engineering, which has led to the development of novel treatment strategies for a wide range of diseases, such as genetic diseases and cancers. Therefore, gene therapy has again became a major focus of medical research. However, because gene therapy involves changing the genetic background, it raises important ethical concerns. In this article, we review the brief history of gene therapy and the development of genetic engineering technologies.
History of Genetic Engineering Technologies
Ethical issues
In 1968, the initial proof-of-concept of virus- mediated gene transfer was made by Rogers et al.6) who showed that foreign genetic material could be transferred into cells by viruses. In the first human gene therapy experiment, Shope papilloma virus was transduced into two patients with genetic arginase deficiency, because Rogers et al. hypothesized that the Shope papilloma virus genome contained a gene that encodes arginase. However, this gene therapy produced little improvement in the arginase levels in the patients.7) Sequencing of the Shope papilloma virus genome revealed that the virus genome did not contain an arginase gene.7)
This experiment prompted public concerns about the risks and ethical issues of gene therapy. In 1972, Friedman et al.8) proposed ethical standards for the clinical application of gene therapy to prevent premature application in human. However, in 1980, genetic engineering was unethically performed in patients with thalassemia without the approval of the institutional review board.9) The patients’ bone marrow cells were harvested and returned into their bone marrow after transduction with the plasmid DNA containing an integrated b-globin gene.9) This treatment showed no effects, and the experiments were regarded as morally dubious. The gene therapy report of the President's Commission in the United States, Splicing Life, emphasized the distinction between somatic and germline genome editing in humans, and between medical treatment and non-medical enhancement.10) An altered gene inserted into sperm or egg cells (germ cells) would lead to changes not only in the individual receiving the treatment but also in their future offspring. Interventions aimed at enhancing “normal” people also are problematic because they might lead to attempts to make “perfect” human beings.
Beginning of gene therapy using viral vector
In 1980, only nonviral methods, such as microinjection and calcium-phosphate precipitation, were used for gene delivery. Nonviral methods showed some advantages compared with viral methods, such as large-scale production and low host immunogenicity. However, nonviral methods yielded lower levels of transfection and gene expression, resulting in limited therapeutic efficacy.11) In 1989, the rDNA Advisory Committee of the National Institutes of Health proposed the first guidelines for the clinical trials of gene therapy. In 1990, retroviral infection, which is highly dependent on host cell cycle status, was first performed for the transduction of the neomycin resistance marker gene into tumor-infiltrating lymphocytes that were obtained from patients with metastatic melanoma.3,4) Then, the lymphocytes were cultured in vitro and returned to the patients’ bodies.3,4) The first Food and Drug Administration (FDA)- approved gene therapy using a retroviral vector was performed by Anderson et al. in 1990; the adenosine deaminase (ADA) gene was transduced into the white blood cells of a patient with ADA deficiency, resulting in temporary improvements in her immunity.2,12)
First severe complications
A recombinant adenoviral (AV) vector was developed after advances in the use of the retroviral vector. In 1999, a clinical trial was performed for ornithine transcarbamylase (OTC) deficiency. A ubiquitous DNA AV vector (Ad5) containing the OTC gene was delivered into the patient. Four days after administration, the patient died from multiple organ failure that was caused by a cytokine storm.13,14) In 1999, of the 20 patients enrolled in two trials for severe combined immunodeficiency (SCID)-X1, T-cell leukemia was observed in five patients at 2–5.5 years after the treatment. Hematopoietic stem cells with a conventional, amphotropic, murine leukemia virus-based vector and a gibbon-ape leukemia virus-pseudotyped retrovirus were used for gene transduction in those trials.15,16) Although four patients fully recovered after the treatment, one patient died15,16) because oncogene activation was mediated by viral insertion.15,16)
Development of viral vectors
Viral vectors continued to be crucial components in the manufacture of cell and gene therapy. Adeno- associated viral (AAV) vectors were applied for many genetic diseases including Leber’s Congenital Amaurosis (LCA), and reverse lipoprotein lipase deficiency (LPLD). In 2008, remarkable success was reported for LCA type II in phase I/II clinical trials.17) LCA is a rare hereditary retinal degeneration disorder caused by mutations in the RPE65 gene (Retinoid Isomerohydrolase RPE65), which is highly expressed in the retinal pigment epithelium and encodes retinoid isomerase.17) These trials confirm that RPE65 could be delivered into retinal pigment epithelial cells using recombinant AAV2/2 vectors, resulting in clinical benefits without adverse events.17) Recently, the FDA approved voretigene neparvovec-rzyl (Luxturna, Spark Therapeutics, Philadelphia, PA, USA) for patients with LCA type II. Alipogene tiparvovec Glybera (uniQure, Lexington, MA, USA) is the first gene-therapy-based drug to reverse LPLD to be approved in Europe in 2012. The AAV1 vector delivers an intact LPL gene to the muscle cells.18) To date, more than 200 clinical trials have been performed using AAV vectors for several genetic diseases, including spinal muscular atrophy,19) retinal dystrophy,20) and hemophilia.21)
Retrovirus is still one of the mainstays of gene therapeutic approaches. Strimvelis (GlaxoSmithKline, London, UK) is an FDA-approved drug consisting of an autologous CD34 (+)-enriched cell population that includes a gammaretrovirus containing the ADA gene that was used as the first ex-vivo stem cell gene therapy in patients with SCID because of ADA deficiency.22) Subsequently, retroviral vectors were often used for other genetic diseases, including X-SCID.23)
Lentivirus belongs to a family of viruses that are responsible for diseases, such as aquired immunodeficiency syndrome caused by the human immunodeficiency virus (HIV) that causes infection by inserting DNA into the genome of their host cells.24) The lentivirus can infect non-dividing cells; therefore, it has a wider range of potential applications. Successful treatment of the patients with X-linked adrenoleukodystrophy was demonstrated using a lentiviral vector with the deficient peroxisomal adenosine triphosphate–binding cassette D1.25) Despite the use of a lentiviral vector with an internal viral long terminal repeat, no oncogene activation was observed.25)
A timeline showing the history of scientific progress in gene therapy is highlighted in Table 1.
Table 1. History of gene therapy.
| Year | History of GT |
|---|---|
| 1962 | Successful transformation of human cells with genomic DNA was achieved. |
| 1970 | Treatment strategy using viral vectors was developed. |
| 1972 | The concept of GT was established. Technologies using recombinant DNA were developed. |
| 1974 | Advisory committee was established for recombinant DNA |
| 1980 | Unapproved GT was performed. |
| 1981 | Retroviral vector was developed. |
| 1983 | Non-replicating retroviral vector was developed. |
| 1986 | Guideline of GT was established. |
| 1989 | A marker gene was first transduced into patient TILs using a retroviral vector. |
| History of GT for hereditary disease | History of GT for malignant tumor | ||
|---|---|---|---|
| 1990 | GT was performed for patients with ADA deficiency. | 1992 | GT was first applied for malignant tumors (glioblastoma). |
| 1999 | A patient with OTC deficiency died after receiving GT. | 2006 | TCR therapy was effective for the patients with melanoma. |
| 2002 | Leukemia was observed in patients with X-SCID after GT. | 2013 | CAR-T showed excellent clinical efficacy for hematological malignancies. |
| 2008-2011 | The effectiveness of GT was reported for patients with LCA, ALD, and Hemophilia B. | 2015 | Imlygic was approved for melanoma GT. |
| 2012 | A GT-based drug (Glybera) was first approved for LPL deficiency. | 2017 | Two CD19-targeting CAR-T cell products, Kymriah and Yescarta, were approved for B-ALL and DLBCL, respectively. Luxturna was approved for the patients with LCA. |
| 2016 | Strimvelis was approved for treatment ADA deficiency. | ||
| 2019 | Zolgensma was approved for SMA. | ||
ADA: adenosine deaminase, ALD: adrenoleukodystrophy, B-ALL: B cell acute lymphoblastic leukemia, CAR: chimeric antigen receptor-modified, DLBCL: diffuse large B-cell lymphoma, GT: gene therapy, LCA: Leber’s congenital amaurosis, LPL: lipoprotein lipase deficiency, OTC: ornithine transcarbamylase, SCID: severe combined immunodeficiency, SMA: spinal muscular atrophy, TCR: T cell receptor, TIL: tumor infiltrating lymphocyte
Gene Therapeutic Strategies for Brain Tumor
A variety of studies were performed to apply gene therapy to malignant tumors. The concept of gene therapy for tumors is different from that for genetic diseases, in which new genes are added to a patient's cells to replace missing or malfunctioning genes. In malignant tumors, the breakthrough in gene therapeutic strategy involved designing suicide gene therapy,26) which was first applied for malignant glioma in 1992.26,27) The first clinical study was performed on 15 patients with malignant gliomas by Ram et al (phase I/II).27) Stereotactic intratumoral injections of murine fibroblasts producing a replication-deficient retrovirus vector with a suicide gene (herpes simplex virus-thymidine kinase [HSV-TK]) achieved anti-tumor activity in four patients through bystander killing effects.27) Subsequently, various types of therapeutic genes have been used to treat malignant glioma. Suicide genes (cytosine deaminase [CD]), genes for immunomodulatory cytokines (interferon [IFN]-β, interleukin [IL]-12, granulocyte- macrophage colony-stimulating factor [GM-CSF]), and genes for reprogramming (p53, and phosphatase and tensin homolog deleted from chromosome [PTEN]) have been applied to the treatment of malignant glioma using viral vectors.28,29)
Recently, a nonlytic, amphotropic retroviral replicating vector (RRV) and immortalized human neural stem cell (NSC) line were used for gene delivery to invasive glioma.30–32) In 2012, a nonlytic, amphotropic RRV called Toca 511 was developed for the delivery of a suicide gene (CD) to tumors.32) A tumor-selective Toca 511 combined with a prodrug (Toca FC) was evaluated in patients with recurrent high-grade glioma in phase I clinical trial.30) The complete response rate was 11.3% in 53 patients.30) In addition, the sub-analysis of this clinical trial revealed that the objective response was 21.7% in the 23-patient phase III eligible subgroup.33) However, in the recent phase III trial, treatment with Toca 511 and Toca FC did not improve overall survival compared with standard therapy in patients with recurrent high-grade glioma. A further combinational treatment strategy using programmed cell-death ligand 1 (PD-L1) checkpoint blockade delivered by TOCA-511 was evaluated in experimental models, which may lead to future clinical application.34) Since 2010, intracranial administration of allogeneic NSCs containing CD gene (HB1.F3. CD) has been performed by a team at City of Hope. Autopsy specimens indicate the HB1.F3. CD migrates toward invaded tumor areas, suggesting a high tumor-trophic migratory capacity of NSCs.31) No severe toxicities were observed in the trial. Generally, it is difficult to obtain NSCs derived from human embryonic or fetal tissue. The use of human embryos for research on embryonic stem cells is ethically controversial because it involves the destruction of human embryos, and the use of fetal tissue associated with abortion also raises ethical considerations.35) Recently, the tumor-trophic migratory activity of NSCs derived from human-induced pluripotent stem cells (hiPSCs) was shown using organotypic brain slice culture.36) Moreover, hiPSC-derived NSCs with the HSV-TK suicide gene system demonstrated considerable therapeutic potential for the treatment of experimental glioma models.36) Furthermore, iPSCs have the ability to overcome ethical and practical issues of NSCs in clinical application.
New Genetic Engineering Technologies for Gene Therapy
Genetic engineering technologies using viral vectors to randomly insert therapeutic genes into a host genome raised concerns about insertional mutagenesis and oncogene activation. Therefore, new technology to intentionally insert genes at site-specific locations was needed. Genome editing is a genetic engineering method that uses nucleases or molecular scissors to intentionally introduce alterations into the genome of living organisms.6) As of 2015, three types of engineered nucleases have been used: ZFNs, TALENs, and CRISPR/Cas (Table 2).6)
Table 2. Characteristics of genome editing technologies.
| ZFNs | TALENs | CRISPR/Cas9 | |
|---|---|---|---|
| Length of recognized DNA target | 9–18bp | 30–40bp | 22bp + PAM sequence |
| DNA recognition | Multimeric protein-DNA interaction | Protein-DNA interaction | RNA-DNA interaction |
| Nuclease design | Difficult | Feasible | Easy |
| Cost | High | Moderate | Low |
| Success rate of nuclease design | Low | High | High |
| Potential off-target effects | Yes | Yes | Yes |
| Specificity | Moderate | High | Moderate |
| Sensitivity to DNA methylation | Not known | Sensitive to CpG methylation | Not sensitive to CpG methylation |
CRISPR/Cas9: clustered regularly interspaced short palindromic repeats/CRISPR-associated proteins 9, PAM: protospacer adjacent motif, TALENs: transcription activator-like effector nucleases, ZFNs: zinc finger nucleases
Genome editing tools
ZFNs are fusions of the nonspecific DNA cleavage domain of the Fok I restriction endonuclease and zinc-finger proteins that lead to DNA double-strand breaks (DSBs). Zinc-finger domains recognize a trinucleotide DNA sequence (Fig. 1). However, design and selection of zinc-finber arrays is difficult and time-consuming.37)
Fig. 1.
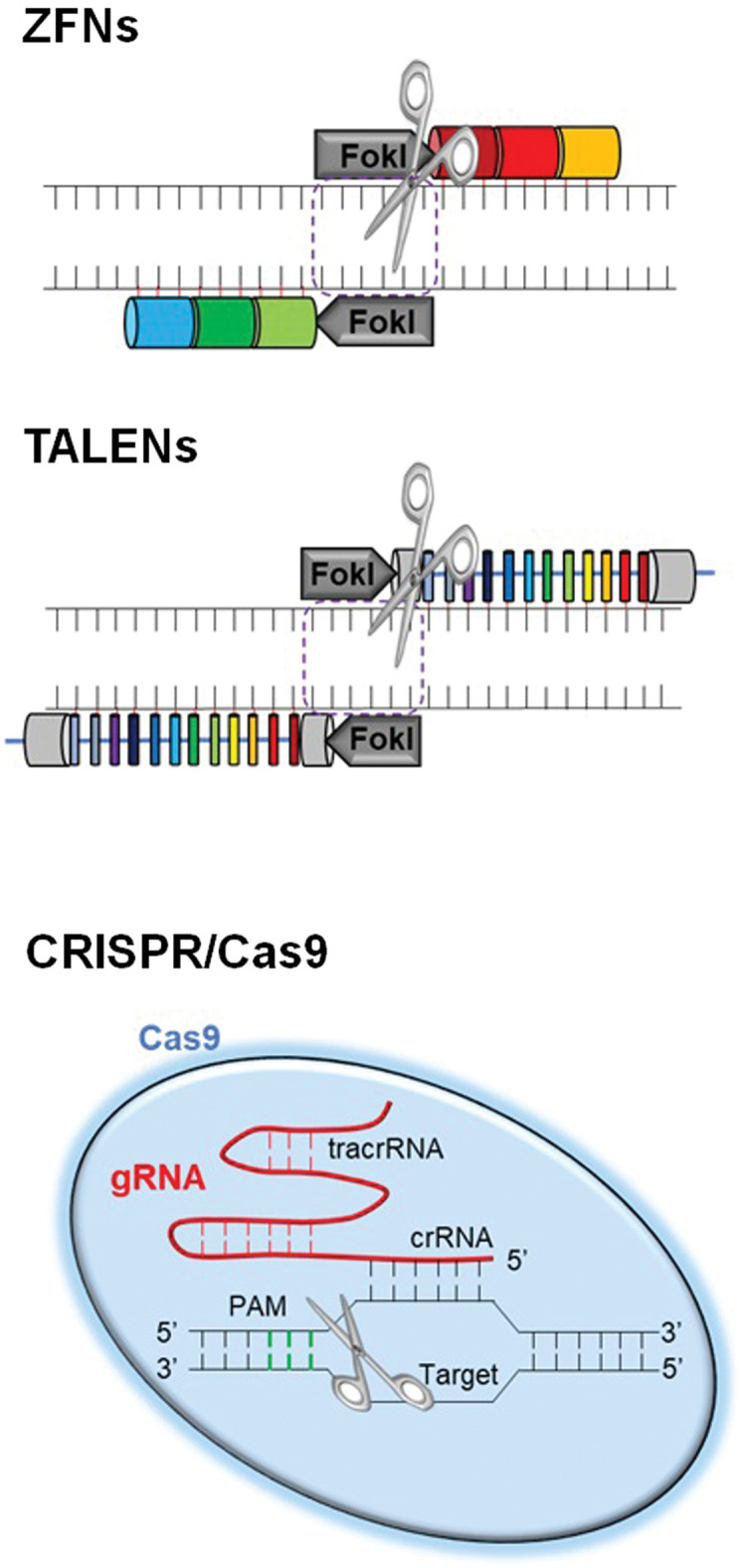
Genome editing tools. Three types of genome editing tools including ZFNs, TALENs, and CRISPR/Cas9 are shown. ZFNs are hybrid proteins using zinc-finger arrays and the catalytic domain of FokI endonuclease. TALENs are hybrid proteins containing the TAL effector backbone and the catalytic domain of FokI endonuclease. The CRISPR/Cas9 system is composed of Cas9 endonuclease and sgRNA. Cas9: CRISPR-associated-9, CRISPR: clustered regularly interspaced palindromic repeats, sgRNA: single-guide RNA, TALENs: transcription activator-like effector nucleases, ZFNs: zinc-finger nucleases.
TALENs are fusions of the Fok I cleavage domain and DNA-binding domains derived from TALE proteins. TALEs have multiple 33–35 amino acid repeat domains that recognizes a single base pair, leading to the targeted DSBs, similar to ZFNs (Fig. 1).38)
The CRISPR/Cas9 system consists of Cas9 nuclease and two RNAs (CRISPR RNA [crRNA] and trans- activating CRISPR RNA [tracrRNA]).39) The crRNA/tracrRNA complex (gRNA) induces the Cas9 nuclease and cleaves DNA upstream of a protospacer-adjacent motif (PAM, 5’-NGG-3’ for S. pyogenes) (Fig. 1).40) Currently, Cas9 from S. pyogenes (SpCas9) is the most popular tool for genome editing.40)
Critical issues in geneome editing
Several studies have demonstrated the off-target effects of Cas9/gRNA complexes.41) It is important to select unique target sites without closely homologous sequences, resulting in minimum off-target effects.42) Additionally, other CRISPR/Cas9 gene editing tools were developed to mitigate off-target effects, including gRNA modifications (slightly truncated gRNAs with shorter regions of target complementarity <20 nucleotides)43) and SpCas9 variants, such as Cas9 paired nickases (a Cas9 nickase mutant or dimeric Cas9 proteins combined with pairs of gRNAs).44) The type I CRISPR-mediated distinct DNA cleavage (CRISPR/Cas3 system) was developed recently in Japan to decrease the risk of off-target effets. Cas3 triggered long-range deletions upstream of the PAM (5'-ARG).45)
A confirmatory screening of off-target effects is necessary for ensuring the safe application of genome editing technologies.46) Although off-target mutations in the genome, including the noncoding region, can be evaluated using whole genome sequencing, this method is expensive and time-consuming. With the development of unbiased genome-wide cell-based methods, GUIDE-seq (genome-wide, unbiased identification of DSBs enabled by sequencing)47) and BLESS (direct in situ breaks labeling, enrichment on streptavidin; next-generation sequencing)48) were developed to detect off-target cleavage sites, and these methods do not require high sequencing read counts.
Applications of Genome Editing Technologies
Gene therapy has in- vivo and ex- vivo strategies. For the in- vivo strategy, vectors containing therapeutic genes are directly delivered into the patients, and genetic modification occurs in situ. For the ex- vivo strategy, the harvested cells are modified by the appropriate gene delivery tools in vitro (e.g., recombinant viruses and genome editing technologies). The modified cells are then delivered back to the patient via autologous or allogeneic transplantation after the evaluation of off-target effects (Fig. 2).
Fig. 2.

In- vivo and ex- vivo strategies of gene therapy. In- vivo and ex- vivo gene transfer strategies are shown. For in- vivo gene transfer, genetic materials containing therapeutic genes, such as viral vectors, nanoparticles, and ribosomes, are delivered directly to the patient, and genetic modification occurs in situ. For ex- vivo gene transfer, the harvested cells are modified by the appropriate gene delivery tools in vitro (e.g., recombinant viruses genome editing technologies). The modified cells are then delivered back to the patient via autologous or allogeneic transplantation after the evaluation of off-target effects.
HIV-resistant T cells were established by ZFN- mediated disruption of the C-C chemokine receptor (CCR) 5 coreceptor for HIV-I, which is being evaluated as an ex- vivo modification in early-stage clinical trials.49,50) Disruption of CCR5 using ZFNs was the first-in-human application of a genome editing tool. Regarding hematologic disorders, since 2016, clinical trials have attempted the knock-in of the factor IX gene using AAV/ZFN-mediated genome editing approach for patients with hemophilia B.51)
In addition to these promising ongoing clinical trials for genetic diseases, CRISPR/Cas9 and TALEN technologies have improved the effect of cancer immunotherapy using genome-engineered T cells. Engineered T cells express synthetic receptors (chimeric antigen receptors, CARs) that can recognize epitopes on tumor cells. The FDA approved two CD19-targeting CAR-T-cell products for B-cell acute lymphoblastic leukemia and diffuse large B-cell lymphoma.52,53) Engineered CARs target many other antigens of blood cancers, including CD30 in Hodgkin's lymphoma as well as CD33, CD123, and FLT3 of acute myeloid leukemia.54) Recent research has shown that Cas9-mediated PD-1 disruption in the CAR-T cells improved the anti-tumor effect observed in in- vitro and in- vivo experimental models, leading to the performance of a clinical trial.55,56) All other ongoing clinical trials using genome-editing technologies are highlighted in Table 3.
Table 3. Recent clinical trials using genome editing technologies.
| Trial number (Phase) | Disease | Target gene | Technology | Vector | Start date |
|---|---|---|---|---|---|
| NCT04244656 (I) | Multiple myeloma | BCMA | CRISPR/Cas9 | CAR-T | Jan. 2020 |
| NCT04037566 (I) | Leukemia or Lymphoma | XYF19 | CRISPR/Cas9 | CAR-T | Aug. 2019 |
| NCT04035434 (I/II) | B-Cell malignancies | N/A | CRISPR/Cas9 | CAR-T | Jul. 2019 |
| NCT03728322 (I) | β-thalassemia | HBB | CRISPR/Cas9 | iHSCs | Jan. 2019 |
| NCT03745287 (I/II) | Sickle cell disease | BCL11A | CRISPR/Cas9 | CD34+ hematopoietic stem cells | Nov. 2018 |
| NCT03747965 (I) | Multiple solid tumors | PD-1 | CRISPR/Cas9 | CAR-T | Nov. 2018 |
| NCT03655678 (I/II) | β-thalassemia | BCL11A | CRISPR/Cas9 | CD34+ hematopoietic stem cells | Sep. 2018 |
| NCT03399448 (I) | Multiple myeloma, Melanoma, Synovial sarcoma, Myxoid/Round cell Liposarcoma | PD-1, TCR | CRISPR/Cas9 | T cell | Sep. 2018 |
| NCT03538613 (I/II) | Gastrointestinal epithelial cancer | CISH | CRISPR/Cas9 | CAR-T | May. 2018 |
| NCT03398967 (I/II) | B cell lymphoma | N/A | CRISPR/Cas9 | CAR-T | Jan. 2018 |
| NCT03545815 (I) | Multiple solid tumors | PD-1 and TCR | CRISPR/Cas9 | CAR-T | Jan. 2018 |
| NCT03057912 (I) | HPV-related cervical Intraepithelial neoplasia I | HPV 16 and 18 E7 oncogene | CRISPR/Cas9, TALEN | Plasmid | Jan. 2018 |
| NCT03166878 (I/II) | B cell lymphoma | TCR and B2M | CRISPR/Cas9 | CAR-T | Jun. 2017 |
| NCT03164135 (N/A) | HIV | CCR5 | CRISPR/Cas9 | CD34+ cell | May. 2017 |
| NCT03164135 (N/A) | HIV | CCR5 | CRISPR/Cas9 | Hematopoietic stem cells | May. 2017 |
| NCT03081715 (N/A) | Esophageal cancer | PD-1 | CRISPR/Cas9 | T cell | Mar. 2017 |
| NCT03044743 (I/II) | Epstein-Barr virus associated malignancies | PD-1 | CRISPR/Cas9 | T cell | Apr. 2017 |
| NCT02867345 (I) | Prostate cancer | PD-1 | CRISPR/Cas9 | T cell | Nov. 2016 |
| NCT02867332 (I) | Renal cell carcinoma | PD-1 | CRISPR/Cas9 | T cell | Nov. 2016 |
| NCT02863913 (I) | Bladder cancer | PD-1 | CRISPR/Cas9 | T cell | Sep. 2016 |
| NCT02793856 (I) | Non-small cell lung cancer | PD-1 | CRISPR/Cas9 | T cell | Aug. 2016 |
| NCT04150497 (I) | B-cell acute lymphoblastic leukemia | N/A | TALEN | CAR-T | Oct. 2019 |
| NCT03226470 (I) | Cervical precancerous lesions | HPV16 E6 and E7 DNA | TALEN | T27 and T512 | Jan. 2018 |
| NCT03190278 (I) | Acute myeloid leukemia | N/A | TALEN | CAR-T | Jun. 2017 |
| NCT03653247 (I/II) | Sickle cell disease | BCL11A | ZFN | Hematopoietic stem cell | Jun. 2019 |
| NCT00842634 (I) | HIV | CCR5 | ZFN | T cell | Jan. 2019 |
| NCT03432364 (I/II) | β-thalassemia | BCL11A | ZFN | Hematopoietic stem cells | Mar. 2018 |
| NCT03041324 (I/II) | MPS II | ALB | ZFN | AAV | May. 2017 |
| NCT02702115 (I/II) | MPS I | ALB | ZFN | AAV | May. 2017 |
| NCT02800369 (I) | Cervical precancerous lesions | HPV16 and 18 E7 oncogene | ZFN | N/A | Dec. 2016 |
| NCT02695160 (I) | Hemophilia B | ALB | ZFN | AAV | Nov. 2016 |
| NCT02500849 (I) | HIV | CCR5 | ZFN | Hematopoietic stem cells | Jul. 2015 |
| NCT01252641 (I/II) | HIV | CCR5 | ZFN | T cell | Nov. 2010 |
AAV: adeno-associated virus, CAR: chimeric antigen receptor, CRISPR/Cas9: clustered regularly interspaced short palindromic repeats/CRISPR-associated 9 proteins, HIV: human immunodeficiency virus, HPV: human papillovirus, MPS: mucopolysaccharidosis, N/A: not available, PD-1: programmed cell death-1, TALEN: transcription activator-like effector nucleases, ZFN: zinc finger nucleases
Future Direction
Gene therapy has advanced treatments for patients with congenital diseases and cancers throughout recent decades by optimizating various types of vectors and the introduction of new techniques including genome editing tools. The CRISPR/Cas9 system is considered one of the most powerful tools for genetic engineering because of its high efficiency, low cost, and ease of use. CRISPR technologies have progressed and are expected to continuously advance. Although there are still many challenging obstacles to overcome to achieve safe clinical application, these methods provide the possibility of treatment for a wide variety of human diseases.
Acknowledgement
We thank Lisa Kreiner, PhD, from Edanz Group (https://en-author-services.edanzgroup.com/) for editing a draft of this manuscript.
Footnotes
Conflicts of Interest Disclosure
The authors declare no conflicts of interest associated with this manuscript. This work was supported in part by grants from the Japan Society for the Promotion of Science (JSPS) (18K19622 to M.T.). All authors have registered online Self-reported COI Disclosure Statement Forms through the website for JNS members.
References
- 1).Szybalska EH, Szybalski W: Genetics of human cess line. IV. DNA-mediated heritable transformation of a biochemical trait. Proc Natl Acad Sci USA 48: 2026–2034, 1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2).Anderson WF: Prospects for human gene therapy. Science 226: 401–409, 1984 [DOI] [PubMed] [Google Scholar]
- 3).Rosenberg SA: Immunotherapy and gene therapy of cancer. Cancer Res 51: 5074s–5079s, 1991 [PubMed] [Google Scholar]
- 4).Rosenberg SA, Aebersold P, Cornetta K, et al. : Gene transfer into humans–immunotherapy of patients with advanced melanoma, using tumor-infiltrating lymphocytes modified by retroviral gene transduction. N Engl J Med 323: 570–578, 1990 [DOI] [PubMed] [Google Scholar]
- 5).Gaj T, Gersbach CA, Barbas CF: ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31: 397–405, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Rogers S, Pfuderer P: Use of viruses as carriers of added genetic information. Nature 219: 749–751, 1968 [DOI] [PubMed] [Google Scholar]
- 7).Rogers S, Lowenthal A, Terheggen HG, Columbo JP: Induction of arginase activity with the Shope papilloma virus in tissue culture cells from an argininemic patient. J Exp Med 137: 1091–1096, 1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8).Friedmann T, Roblin R: Gene therapy for human genetic disease? Science 175: 949–955, 1972 [DOI] [PubMed] [Google Scholar]
- 9).Johnson RS: Gene transfer experiment in humans meets with scant approval. JAMA 244: 2139–2140, 1980 [PubMed] [Google Scholar]
- 10).President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research : Splicing life: a report on the social and ethical issues of genetic engineering with human beings. US Government Printing Office, Washington, 1982. [Google Scholar]
- 11).Jin L, Zeng X, Liu M, Deng Y, He N: Current progress in gene delivery technology based on chemical methods and nano-carriers. Theranostics 4: 240–255, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12).Blaese RM, Culver KW, Miller AD, et al. : T lymphocyte-directed gene therapy for ADA- SCID: initial trial results after 4 years. Science 270: 475–480, 1995 [DOI] [PubMed] [Google Scholar]
- 13).Raper SE, Chirmule N, Lee FS, et al. : Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol Genet Metab 80: 148–158, 2003 [DOI] [PubMed] [Google Scholar]
- 14).Wilson JM: Lessons learned from the gene therapy trial for ornithine transcarbamylase deficiency. Mol Genet Metab 96: 151–157, 2009 [DOI] [PubMed] [Google Scholar]
- 15).Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G, et al. : Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science 288: 669–672, 2000 [DOI] [PubMed] [Google Scholar]
- 16).Hacein-Bey-Abina S, von Kalle C, Schmidt M, et al. : A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N Engl J Med 348: 255–256, 2003 [DOI] [PubMed] [Google Scholar]
- 17).Maguire AM, Simonelli F, Pierce EA, et al. : Safety and efficacy of gene transfer for Leber's congenital amaurosis. N Engl J Med 358: 2240–2248, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18).Bryant LM, Christopher DM, Giles AR, et al. : Lessons learned from the clinical development and market authorization of Glybera. Hum Gene Ther Clin Dev 24: 55–64, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19).Mendell JR, Al-Zaidy S, Shell R, et al. : Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med 377: 1713–1722, 2017 [DOI] [PubMed] [Google Scholar]
- 20).Bainbridge JW, Mehat MS, Sundaram V, et al. : Long-term effect of gene therapy on Leber's congenital amaurosis. N Engl J Med 372: 1887–1897, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21).Nathwani AC, Reiss UM, Tuddenham EG, et al. : Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med 371: 1994–2004, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22).Cicalese MP, Ferrua F, Castagnaro L, et al. : Update on the safety and efficacy of retroviral gene therapy for immunodeficiency due to adenosine deaminase deficiency. Blood 128: 45–54, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23).Hacein-Bey-Abina S, Pai SY, Gaspar HB, et al. : A modified g-retrovirus vector for X-linked severe combined immunodeficiency. N Engl J Med 371: 1407–1417, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24).Cockrell AS, Kafri T: Gene delivery by lentivirus vectors. Mol Biotechnol 36: 184–204, 2007 [DOI] [PubMed] [Google Scholar]
- 25).Mamcarz E, Zhou S, Lockey T, et al. : Lentiviral gene therapy combined with low-dose busulfan in infants with SCID-X1. N Engl J Med 380: 1525–1534, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26).Tamura R, Miyoshi H, Yoshida K, Okano H, Toda M: Recent progress in the research of suicide gene therapy for malignant glioma. Neurosurg Rev 28: 1–21, 2019 [DOI] [PubMed] [Google Scholar]
- 27).Ram Z, Culver KW, Oshiro EM, et al. : Therapy of malignant brain tumors by intratumoral implantation of retroviral vector-producing cells. Nat Med 3: 1354–1361, 1997 [DOI] [PubMed] [Google Scholar]
- 28).Toda M, Martuza RL, Rabkin SD: Tumor growth inhibition by intratumoral inoculation of defective herpes simplex virus vectors expressing granulocyte- macrophage colony-stimulating factor. Mol Ther 2: 324–329, 2000 [DOI] [PubMed] [Google Scholar]
- 29).Parker JN, Gillespie GY, Love CE, Randall S, Whitley RJ, Markert JM: Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc Natl Acad Sci USA 97: 2208–2213, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30).Cloughesy TF, Landolfi J, Hogan DJ, et al. : Phase 1 trial of vocimagene amiretrorepvec and 5-fluorocytosine for recurrent high-grade glioma. Sci Transl Med 8: 341ra75, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31).Portnow J, Synold TW, Badie B, et al. : Neural stem cell-based anticancer gene therapy: a first-in-human study in recurrent high-grade glioma patients. Clin Cancer Res 23: 2951–2960, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32).Ostertag D, Amundson KK, Lopez Espinoza F, et al. : Brain tumor eradication and prolonged survival from intratumoral conversion of 5-fluorocytosine to 5-fluorouracil using a nonlytic retroviral replicating vector. Neuro-oncology 14: 145–159, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33).Cloughesy TF, Landolfi J, Vogelbaum MA, et al. : Durable complete responses in some recurrent high-grade glioma patients treated with Toca 511 + Toca FC. Neuro-oncology 20: 1383–1392, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34).Mitchell LA, Yagiz K, Hofacre A, et al. : PD-L1 checkpoint blockade delivered by retroviral replicating vector confers anti-tumor efficacy in murine tumor models. Oncotarget 10: 2252–2269, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35).Lo B, Parham L: Ethical issues in stem cell research. Endocr Rev 30: 204–213, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36).Tamura R, Miyoshi H, Morimoto Y, et al. : Gene therapy using neural stem/progenitor cells derived from human induced pluripotent stem cells: visualization of migration and bystander killing effect. Hum Gene Ther 31: 352–366, 2020 [DOI] [PubMed] [Google Scholar]
- 37).Kim YG, Cha J, Chandrasegaran S: Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci USA 93: 1156–1160, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38).Christian M, Cermak T, Doyle EL, et al. : Targeting DNA double-strand breaks with TAL effector nucleases. Genetics 186: 757–761, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39).Cong L, Ran FA, Cox D, et al. : Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40).Yamano T, Nishimasu H, Zetsche B, et al. : Crystal structure of Cpf1 in complex with guide RNA and target DNA. Cell 165: 949–962, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41).Kempton HR, Qi LS: When genome editing goes off-target. Science 364: 234–236, 2019 [DOI] [PubMed] [Google Scholar]
- 42).Wang Q, Ui-Tei K: Computational prediction of CRISPR/Cas9 target sites reveals potential off-target risks in human and mouse. Methods Mol Biol 1630: 43–53, 2017 [DOI] [PubMed] [Google Scholar]
- 43).Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK: Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol 32: 279–284, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44).Ran FA, Hsu PD, Lin CY, et al. : Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154: 1380–1389, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45).Morisaka H, Yoshimi K, Okuzaki Y, et al. : CRISPR-Cas3 induces broad and unidirectional genome editing in human cells. Nat Commun 10: 5302, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46).Zischewski J, Fischer R, Bortesi L: Detection of on-target and off-target mutations generated by CRISPR/Cas9 and other sequence-specific nucleases. Biotechnol Adv 35: 95–104, 2017 [DOI] [PubMed] [Google Scholar]
- 47).Tsai SQ, Zheng Z, Nguyen NT, et al. : GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol 33: 187–197, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48).Crosetto N, Mitra A, Silva MJ, et al. : Nucleotide- resolution DNA double-strand break mapping by next- generation sequencing. Nat Methods 10: 361–365, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49).Tebas P, Stein D, Binder-Scholl G, et al. : Antiviral effects of autologous CD4 T cells genetically modified with a conditionally replicating lentiviral vector expressing long antisense to HIV. Blood 121: 1524–1533, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50).Wang CX, Cannon PM: The clinical applications of genome editing in HIV. Blood 127: 2546–2552, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51).Sharma R, Anguela XM, Doyon Y, et al. : In vivo genome editing of the albumin locus as a platform for protein replacement therapy. Blood 126: 1777–1784, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52).Fournier C, Martin F, Zitvogel L, Kroemer G, Galluzzi L, Apetoh L: Trial watch: adoptively transferred cells for anticancer immunotherapy. Oncoimmunology 6: e1363139, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53).Zhang H, Ye ZL, Yuan ZG, Luo ZQ, Jin HJ, Qian QJ: New strategies for the treatment of solid tumors with CAR-T cells. Int J Biol Sci 12: 718–729, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54).Schultz L, Mackall C: Driving CAR T cell translation forward. Sci Transl Med 11: pii:eaaw2127, 2019 [DOI] [PubMed] [Google Scholar]
- 55).Hegde UP, Mukherji B: Current status of chimeric antigen receptor engineered T cell-based and immune checkpoint blockade-based cancer immunotherapies. Cancer Immunol Immunother 66: 1113–1121, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56).Rupp LJ, Schumann K, Roybal KT, et al. : CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci Rep 7: 737, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]