

Key Points
Real-world patient outcomes after CAR T therapy closely resemble data from landmark phase 2 trials.
The benefit of CAR T over alternate therapies is less clear after adjusting for baseline risk factors.
Abstract
The prognosis of patients with relapsed or refractory (R/R) diffuse large B-cell lymphoma (DLBCL) is poor. Chimeric antigen receptor (CAR) T-cell therapy has been approved for R/R DLBCL after 2 prior lines of therapy based on data from single-arm phase 2 trials, with complete responses (CRs) in 40% to 60% of patients. However, a direct comparison with other treatments is not available and, moreover, its true efficacy in real-world patients is unknown. In this single center, retrospective, observational study of 215 patients, we compared outcomes in patients treated with CAR T-cell therapy (n = 69) with a historical population treated with alternate therapies (n = 146). Patients treated with CAR T cell vs alternate therapies demonstrated a CR rate of 52% vs 22% (P < .001), median progression-free survival (PFS) of 5.2 vs 2.3 months (P = .01), and median overall survival (OS) of 19.3 vs 6.5 months (P = .006), and this advantage appeared to persist irrespective of the number of lines of prior therapy. After adjusting for unfavorable pretreatment disease characteristics, superior overall response rate in the CAR T cohort remained significant; however, differences in PFS and OS between cohorts did not. In addition, patients who responded to alternate therapies demonstrated prolonged remissions comparable to those who responded to CAR T therapy. We contend that in select clinical scenarios alternate therapies may be as efficacious as CAR T therapy; thus, additional study is warranted, ideally with randomized prospective trials.
Visual Abstract

Introduction
Relapsed or refractory (R/R) diffuse large B-cell lymphoma (DLBCL) is associated with a generally poor prognosis. Patients with R/R disease after frontline anthracycline-based chemotherapy have a 3-year event-free survival of ∼30%.1 Even in fit patients with chemosensitive disease to salvage therapy, ∼50% will ultimately progress after autologous hematopoietic cell transplant (AHCT) and experience a median overall survival (OS) of 10 months.2,3 Those who have disease refractory to primary or salvage chemotherapy or relapse in ≤12 months after AHCT have especially poor outcomes, with an overall response rate (ORR) of 26% to the next line of therapy and median OS of 6.3 months.4 Data for specific therapies in the third or later lines are limited without a standard of care for these patients.
Chimeric antigen receptor (CAR) T-cell therapy has been approved by the US Food and Drug Administration and other regulatory agencies for patients with R/R DLBCL (including high-grade B-cell lymphoma, primary mediastinal B-cell lymphoma, and transformed follicular lymphoma) after 2 prior lines of therapy based on data from single-arm phase 2 trials. These studies demonstrated impressive 40% to 60% complete responses (CR) rates.5,6 However, in the ZUMA-1 trial, approximately 60% of responding patients ultimately progressed, and median progression-free survival (PFS) was only 5.9 months.5 Furthermore, the median duration of response for those with incomplete responses was only 1.9 months. In the JULIET trial, median PFS and OS were approximately 3 and 12 months, respectively.6
Although lacking the scientific rigor of prospective studies, real-world evidence can provide valuable insight into various treatment outcome parameters and hypothesis generation. Real-world evidence also has the potential to capture a more generalizable patient population, including patients otherwise unfit for enrollment in clinical trials. Data from observational studies of patients receiving commercial CAR T therapy for DLBCL recently presented at he American Society of Hematology (ASH) Annual Meeting the annual meeting of the American Society of Hematology have shown response rates comparable to the pivotal trials for both axicabtagene ciloleucel and tisagenlecleucel. However, follow-up in these studies is limited, and long-term outcomes are unknown.7,8 In addition, cross-study comparisons are limited by the heterogeneity of patient populations, variable institutional practices, and differences in study methodology. We sought to compare outcomes between CAR T and alternate (non-CAR T) therapies in patients with DLBCL who had R/R disease after at least 2 prior lines of systemic treatment in this single institution, retrospective study.
Methods
This was a single center retrospective study of adult patients (age ≥18 years) diagnosed with R/R DLBCL and treated with CAR T or alternate (non-CAR T) therapies at Memorial Sloan Kettering Cancer Center (MSKCC). The study was approved by the institutional review board and conducted in accordance with the Declaration of Helsinki.
Eligible patients were identified by performing an institutional database query, and data were collected exclusively from electronic medical records. To be eligible for inclusion into the CAR T cohort, patients must have received either tisagenlecleucel or axicabtagene ciloleucel on-label between February 2018 and September 2019 at MSKCC. De novo DLBCL (including T-cell/histiocyte-rich large B-cell lymphoma [THRLBCL]), transformed indolent lymphoma, primary mediastinal B-cell lymphoma (PMBCL), and high-grade B-cell lymphoma with translocations involving MYC and BCL2 or BCL6 (HGBL) histology were allowed. All patients who received at least 2 prior lines of systemic therapy were included for the primary analysis. Patients who received bridging therapy were included, but this was not considered a separate line of therapy. Last, patients were required to have had long enough follow-up to reach the first response assessment time point, which was generally performed at 30 days after infusion. To be eligible for inclusion in the alternate therapy cohort, patients had to have had 2 prior lines of aggressive lymphoma-directed systemic therapy and long enough follow-up to reach the first response assessment to third-line therapy. All types of systemic third-line therapies were included, including experimental therapies. Patients with de novo DLBCL, transformed indolent lymphoma, PMBCL, THRLBCL, and HGBL histology were included. Patients with central nervous system (CNS) disease at time of third-line therapy were included if there was concurrent systemic disease as well (consistent with our practices for CAR T eligibility). Future enrollment in a CAR T protocol was not grounds for exclusion, although data were censored at the time of infusion. All patients who went on to receive commercial CAR T were considered duplicate patients and only analyzed in the CAR T cohort.
The primary study objective was to compare outcomes of CAR T therapy (after >2 prior lines) and alternate (or non-CAR T) therapies (after only 2 prior lines) with outcomes of interest including response rate (CR and ORR), PFS, and OS. Second, a subgroup analysis for each treatment cohort was performed, evaluating PFS among patients that achieved either a CR or partial response (PR) as best objective response. A subgroup analysis was also performed on patients in both cohorts with elevated lactate dehydrogenase (LDH; >upper limit of normal) and with bulky (>10 cm) disease. Third, an additional analysis compared outcomes between treatment groups when matched by number of prior lines of therapy (2, 3, or 4 or more prior lines). Disease response assessment of CR, PR, stable disease, and progressive disease was made using the Lugano criteria.9 ORR was defined as the sum of CR and PR rates. PFS was defined as time from start of treatment until time of aggressive lymphoma progression or relapse or death from any cause. OS was defined as duration of time from start of treatment until time to death of any cause. The start of treatment in the CAR T cohort was defined as date of cell infusion.
Patient and disease characteristics at time of diagnosis and time of treatment were compared between the CAR T and alternate cohorts using the χ2 test of independence and Fisher’s exact tests for categorical variables and the Wilcoxon rank-sum test for continuous variables. Disease responses were compared across the 2 cohorts using the Fisher’s exact test. Kaplan-Meier methods were used for analysis of PFS and OS and to calculate 6- and 12-month event rates along with 95% confidence intervals. Differences in PFS and OS between the 2 cohorts were compared using the long-rank test. Univariable Cox regression models were performed for high-risk prognostic factors, and significant covariates at the 0.05 level (including treatment cohort) were included in the multivariable survival model. Odds ratios were calculated to determine the association between high-risk prognostic factors and ORR. Significant covariates (including treatment cohort) from the univariate ORR model were included in the multivariable ORR analysis. The same statistical methods were used for the subgroup analyses. All computations were performed in R v3.6.1 (Vienna, Austria). P < .05 was considered statistically significant.
Results
For the primary objective, 215 total patients were included. The alternate treatment cohort consisted of 146 patients, and the CAR T cohort consisted of 69 patients. For the alternate cohort, 186 of 2551 patients treated for DLBCL since 2001 underwent at least 3 lines of therapy and had available outcome data at the third line. Of these patients, 12 were excluded because they were treated with indolent lymphoma-directed therapy (eg, single-agent rituximab) as a prior line, 23 were excluded for CNS-only disease, and 5 went on to receive commercial CAR T therapy and were thus analyzed in the other cohort. Of the 72 patients referred to MSKCC for commercial CAR T therapy beginning February 2018, only 3 patients were excluded from the CAR T cohort because they were treated off-label as part of a research protocol (Figure 1).
Figure 1.
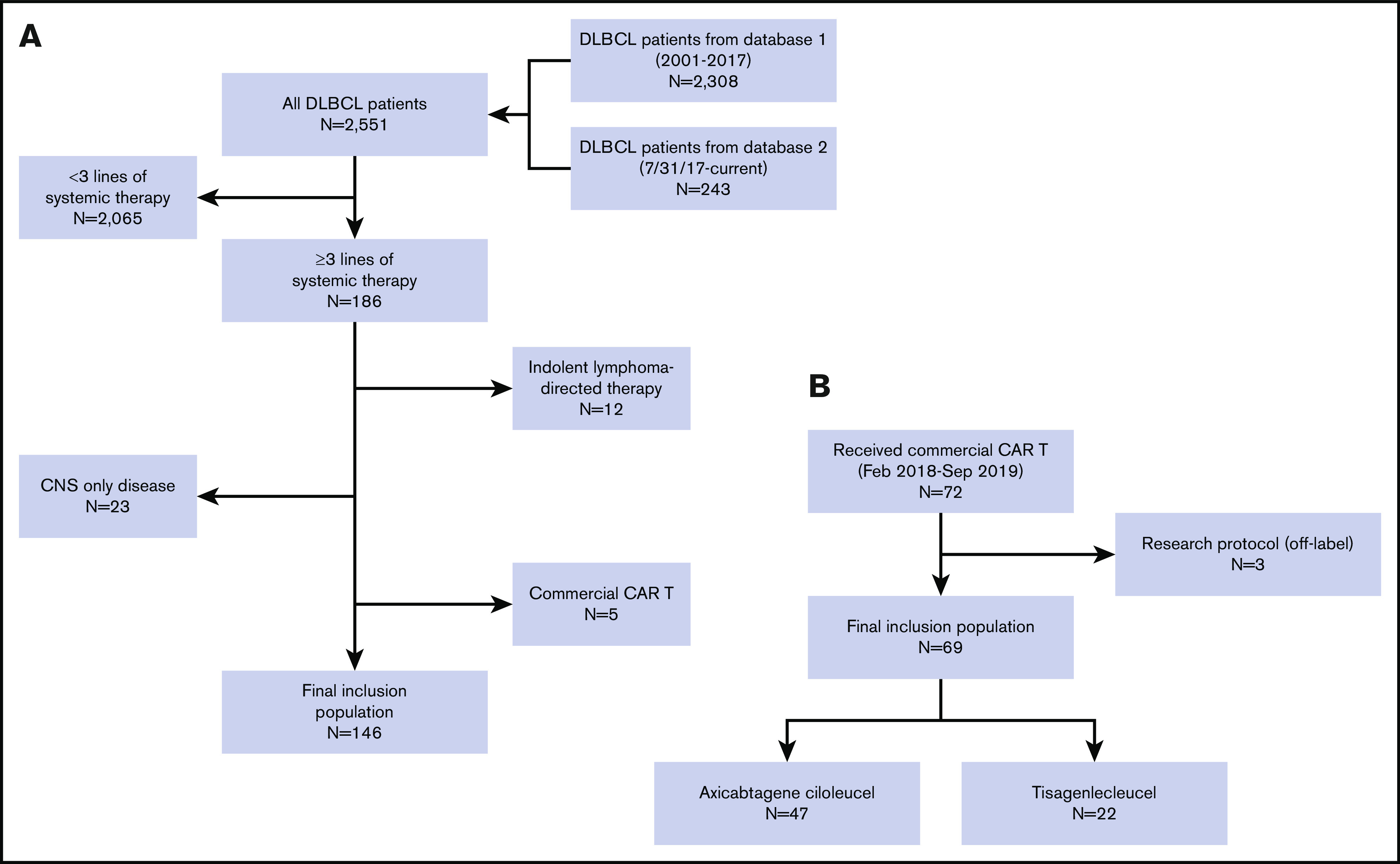
Consort. (A) Cohort 1: alternate therapy group. Of 2551 total patients diagnosed with DLBCL and treated at MSKCC, 186 patients had at least 3 lines of therapy and available outcome data at the third line. Of these, 12 were excluded because they were treated with indolent lymphoma-directed therapy (eg, rituximab) as a prior line, 23 were excluded for CNS-only disease, and 5 who ultimately received commercial CAR T therapy were duplicate patients included in the other cohort. The final inclusion population consisted of 146 patients. (B) Cohort 2: CAR T therapy group. Of 72 patients who were referred to MSKCC for commercial CAR T therapy beginning in February 2018, 3 were excluded because they were treated off-label as part of a research protocol. The final inclusion population consisted of 69 patients, of which 47 received axicabtagene ciloleucel and 22 tisagenlecleucel.
Patient and disease characteristics at time of diagnosis are listed in supplemental Table 1. There were 7 patients with PMBCL and 4 with THRLBCL in the alternate cohort and 1 with THRLBCL in the CAR T cohort. There was a higher frequency of transformed low-grade lymphoma in the CAR T compared with the alternate cohort (P = .005). In the CAR T cohort, 51% had activated B-cell phenotype, 20% were double expressor, 41% had positive MYC expression, and 12% were HGBL (supplemental Table 2). Molecular and histologic subtypes within the alternate cohort were infrequently reported; thus, these data were not analyzed for this study. Disease characteristics at time of treatment with alternate or CAR T treatment are listed in Table 1. The median age was 66 and 63 years in the alternate and CAR T cohorts, respectively (P = .5). ECOG was >1 in 13% of patients treated with CAR T therapy compared with 8.5% in the alternate therapy cohort (P = .4). LDH was more frequently elevated in the alternate cohort compared with the CAR T cohort (66% vs 45%; P = .007). Patients treated with alternate therapies were more likely to have refractory disease (stable disease or progressive disease to last prior therapy or relapse < 12 months after AHCT) compared with those treated with CAR T (79% vs 33%; P < .001). Seven patients treated with alternate therapy had known prior or active CNS disease compared with 1 in the CAR T group. The number of patients with prior AHCT was similar for the alternate and CAR T cohorts (14% vs 20%; P = .2). Median number of prior lines of therapy in the CAR T cohort was 3 (range, 2-7). The most frequently used third-line therapies in the alternate cohort were platinum-based regimens (29%), investigational agents (26%), etoposide-based regimens (8.9%), and anthracycline-based regimens (8.9%). Ten patients (6.8%) received consolidation with allogeneic hematopoietic cell transplantation and 12 with AHCT (8.2%) (Table 2). In the CAR T cohort, 47 received axicabtagene ciloleucel and 22 received tisagenlecleucel (supplemental Table 3).
Table 1.
Disease characteristics at time of treatment
| Characteristics | Alternate | CAR T | P |
|---|---|---|---|
| Total, n | 146 | 69 | |
| Age | |||
| Median (range), y | 66 (27-91) | 63 (19-85) | .5 |
| >60 y, n (%) | 90 (62) | 43 (62) | >.9 |
| ECOG | .4 | ||
| 0-1, n (%) | 130 (92) | 60 (87) | |
| ≥2, n (%) | 12 (8.5) | 9 (13) | |
| Unknown, n | 4 | 0 | |
| Bulk >10 cm, n (%) | 23 (16) | 12 (17) | >.9 |
| Unknown, n | 3 | 0 | |
| Number of EN sites, n (%) | .5 | ||
| 0-1 | 97 (66) | 42 (61) | |
| >1 | 49 (34) | 27 (39) | |
| Elevated LDH, n (%) | 86 (66) | 31 (45) | .007 |
| Unknown, n | 15 | 0 | |
| Stage | >.9 | ||
| Limited, n (%) | 24 (16) | 11 (16) | |
| Advanced, n (%) | 122 (84) | 58 (84) | |
| BM involvement, n (%) | 5 (3.6) | 10 (21) | <.001 |
| Unknown, n | 9 | 22 | |
| Refractory disease | <.001 | ||
| No, n (%) | 31 (21) | 46 (67) | |
| Yes, n (%) | 114 (79) | 23 (33) | |
| Missing, n | 1 | 0 | |
| Prior AHCT, n (%) | 20 (14) | 14 (20) | .2 |
| Prior allogeneic-HCT, n (%) | 3 (2) | 4 (6) | .2 |
P values in bold are statistically significant.
BM, bone marrow; ECOG, Eastern cooperative oncology group; EN; extranodal.
Table 2.
Third-line therapies used in alternate cohort
| Third-line regimens | n (%) |
|---|---|
| Platinum-based combination (eg, DHAP) | 43 (29) |
| Clinical trial | 38 (26) |
| Etoposide-based (eg, CEPP) | 13 (8.9) |
| Anthracycline-based combination (eg, CHOP) | 13 (8.9) |
| Bendamustine-rituximab | 9 (6.2) |
| Lenalidomide-rituximab | 8 (5.4) |
| Gemcitabine-based | 6 (4.1) |
| Ibrutinib | 5 (3.4) |
| IVAC | 3 (2.0) |
| Rituximab monotherapy | 2 (1.3) |
| Other | 6 (4.1) |
| Consolidation after third line | |
| Allogeneic transplant | 10 (6.8) |
| Autologous transplant | 12 (8.2) |
CEPP, cyclophosphamide/etoposide/procarbazine/prednisone; CHOP, cyclophosphamide/doxorubicin/vincristine/prednisone; DHAP; dexamethasone/cytarabine/cisplatin; IVAC, ifosfamide/etoposide/cytarabine.
The median follow-up for the CAR T and alternate cohorts was 14.6 (range, 1.2-18.9) and 30.6 months (range, 2.1-162), respectively. The ORR in the CAR T and alternate cohorts was 72% vs 32% (P < .001), and the CR rate was 52% vs 22%, respectively (P < .001). The median PFS and OS in the CAR T cohort were 5.2 and 15.8 months, respectively, contrasted with 2.3 and 6.5 months, respectively, in the alternate cohort (Table 3; Figure 2). In univariate analysis, pretreatment ECOG >1, elevated LDH, tumor bulk (>10 cm), >1 extranodal site, and refractory disease were adverse factors associated with both shorter PFS and OS (all P < .05). Use of non-CAR T therapy was also associated with worse PFS and OS. In multivariate analysis, only pretreatment LDH and tumor bulk remained significant for both PFS and OS, whereas ECOG remained significant for PFS, and extranodal sites were significant for OS. Type of treatment was no longer significant for PFS or OS in this model. In a subgroup of 117 patients with elevated LDH, the median PFS in the CAR T and alternate groups was 2.8 and 1.7 months, respectively (P = .1), and median OS in the CAR T and alternate groups was 7.7 and 4.2 months, respectively (P = .1). In 35 patients with bulky disease, the median PFS in the CAR T and alternate groups was 1.2 and 1.8 months, respectively (P = .2), and median OS in the CAR T and alternate groups was 3.7 and 5.2 months, respectively (P = .3) (supplemental Figure 1). In a univariate analysis for ORR, elevated pretreatment LDH, 2 or more extranodal sites, and refractory disease were associated with decreased likelihood of response, as was alternate treatment. Adjusting for these variables in a multivariable model, only elevated LDH and alternate treatment remained significant (Table 4).
Table 3.
Outcomes by treatment cohort
| Outcomes | Alternate | CAR T | P |
|---|---|---|---|
| Total, n | 146 | 69 | |
| CR rate, % | 22 | 52 | <.001 |
| ORR, % | 32 | 72 | <.001 |
| 6-mo OS, % (95% CI) | 55 (47-64) | 71 (61-82) | |
| 12-mo OS, % (95% CI) | 39 (31-48) | 64 (54-77) | |
| Median OS, mo | 6.5 | 19.3 | .006 |
| 6-mo PFS, % (95% CI) | 29 (23-38) | 49 (39-63) | |
| 12-mo PFS, % (95% CI) | 25 (19-33) | 44 (33-58) | |
| Median PFS, mo | 2.3 | 5.2 | .01 |
P values in bold are statistically significant.
CI, confidence interval.
Figure 2.
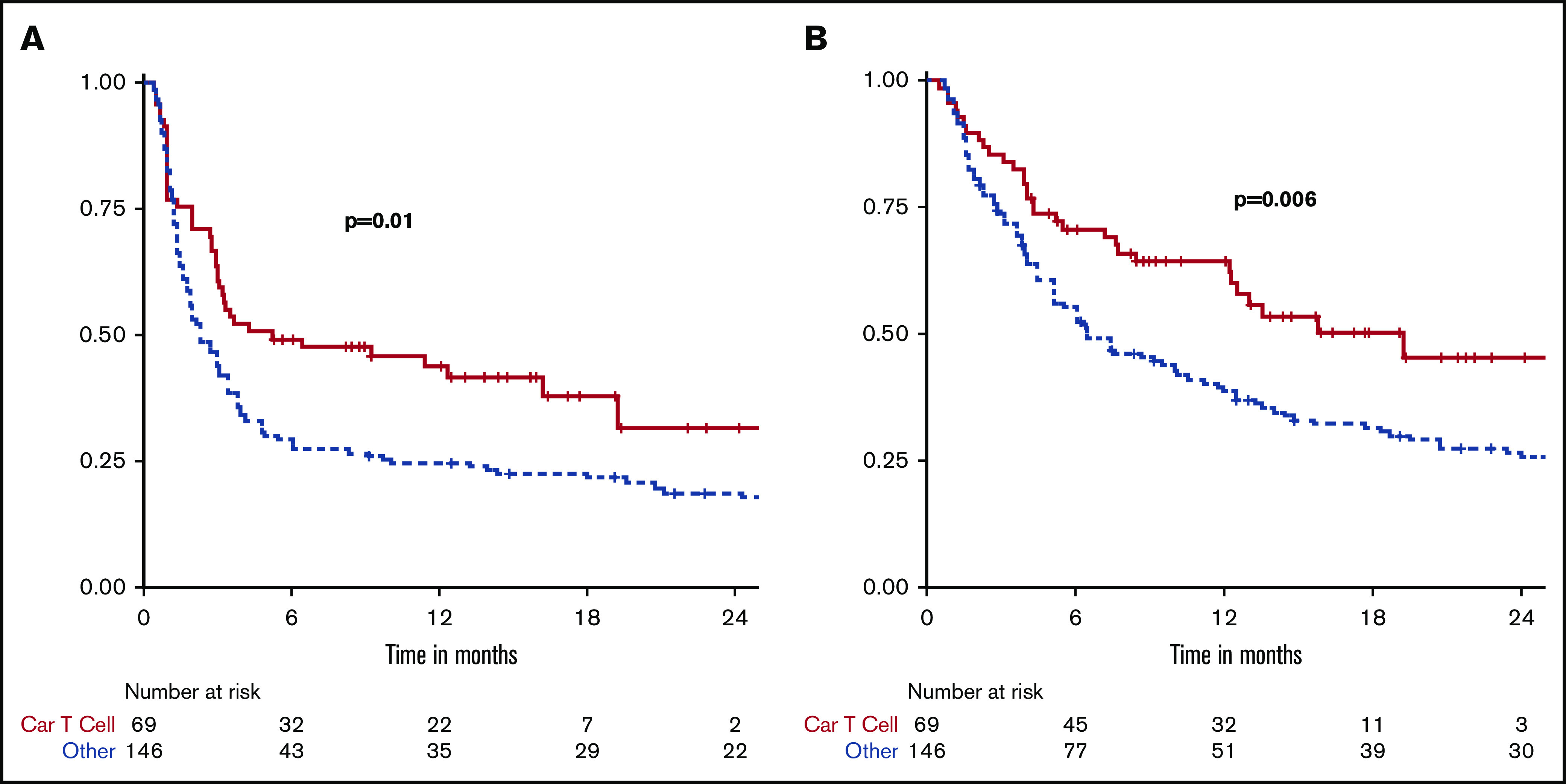
Kaplan-Meier curves of all 215 patients comparing outcomes by treatment cohort. (A) PFS. (B) OS.
Table 4.
Factors prognostic of PFS, OS, and ORR on multivariable analysis
| Prognostic factors | PFS | OS | ORR | |||
|---|---|---|---|---|---|---|
| HR (95% CI) | P | HR (95% CI) | P | OR (95% CI) | P | |
| Elevated LDH | 1.7 (1.2-2.7) | .009 | 2.1 (1.4-3.2) | <.001 | 0.3 (0.2-0.6) | <.001 |
| Transformed | 0.9 (0.6-1.3) | .5 | 0.8 (0.5-1.1) | .2 | 1.2 (0.6-2.5) | .6 |
| Bulk >10cm | 1.8 (1.2-2.9) | .01 | 1.7 (1.1-2.7) | .03 | — | — |
| >1 EN site | 1.2 (0.8-1.8) | .3 | 1.6 (1.1-2.3) | .02 | 0.6 (0.3-1.2) | .1 |
| ECOG >1 | 2.1 (1.2-3.7) | .01 | 1.7 (1.0-3.1) | .06 | — | — |
| BM positive | 0.7 (0.3-1.6) | .4 | — | — | — | — |
| Refractory | 1.4 (0.9-2.3) | .1 | 1.5 (1.0-2.3) | .07 | 0.6 (0.3-1.2) | .1 |
| CAR T | Ref | Ref | Ref | |||
| Alternate | 1.1 (0.7-1.8) | .7 | 1.3 (0.8-2.1) | .2 | 0.2 (0.1-0.4) | <.001 |
P values in bold are statistically significant. Dashes indicate analysis was not performed on these factors as they were not significant in the univariable analysis.
EN, extranodal; HR, hazard ratio; LDH, lactate dehydrogenase.
For patients who only received 2 prior lines of treatment, ORR with CAR T (n = 32) compared with alternate therapy (n = 146) was 75% vs 32% (P < .001) and median PFS was 6.4 vs 2.3 months (P = .04). After 3 prior lines of treatment, ORR with CAR T (n = 23) contrasted to alternate therapy (n = 59) was 74% vs 16% (P < .001), and median PFS was 12.3 vs 1.7 months (P < .001). After 4 or more prior lines, ORR with CAR T (n = 14) compared with alternate therapy (n = 26) was 64% vs 8%, and median PFS was 3.0 vs 1.4 months (P = .008). In this group, number of prior therapies ranged from 4 to 7, with a median of 4 and 5 in the alternate and CAR T groups, respectively (Table 5; Figure 3).
Table 5.
Outcomes by treatment cohort and number of prior lines of therapy
| Outcomes | Alternate | CAR T | P |
|---|---|---|---|
| 2 prior lines | |||
| Total, n | 146 | 32 | |
| CR rate, % | 22 | 56 | <.001 |
| ORR, % | 32 | 75 | <.001 |
| Median OS, mo | 6.5 | 19.3 | .02 |
| Median PFS, mo | 2.3 | 6.4 | .04 |
| 3 prior lines | |||
| Total, n | 59 | 23 | |
| CR rate, % | 8.6 | 52 | <.001 |
| ORR, % | 16 | 74 | <.001 |
| Median OS, mo | 4.7 | Not reached | .001 |
| Median PFS, mo | 1.7 | 12.3 | <.001 |
| 4 prior lines | |||
| Total, n | 26 | 14 | |
| CR rate, % | 8 | 43 | .02 |
| ORR, % | 8 | 64 | <.001 |
| Median OS, mo | 2.9 | 7.0 | .03 |
| Median PFS, mo | 1.4 | 3.0 | .008 |
P values in bold are statistically significant.
Figure 3.
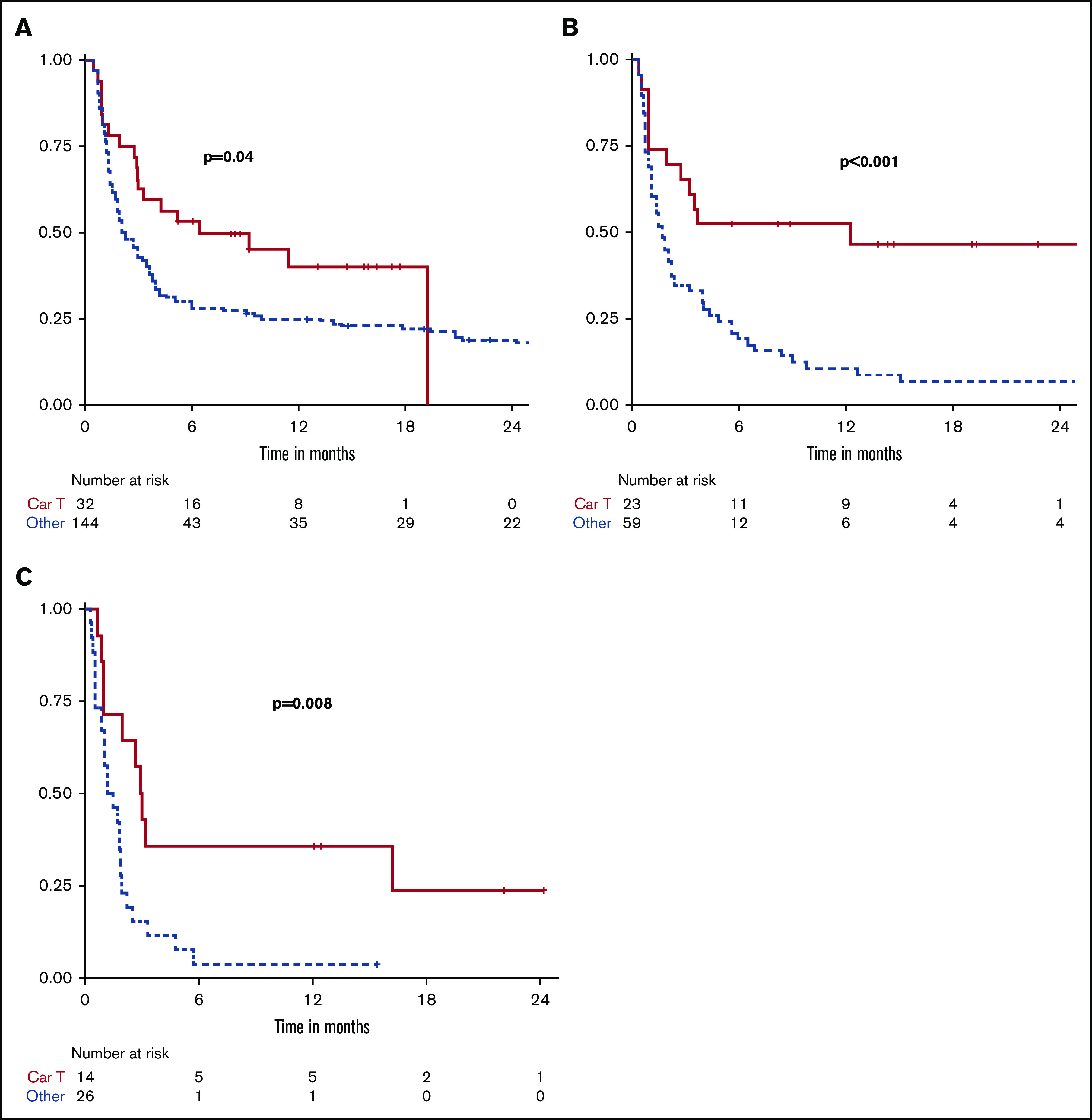
Kaplan-Meier curves comparing PFS between treatments when classified by number of prior lines of therapy. Two prior lines (A), 3 prior lines (B), and 4 or more prior lines (C).
Among the 36 patients within the CAR T cohort who had a CR, the 12-month PFS was 70%, and the median was not reached. Of the patients in the CAR T cohort with PR as their best response, 12-month PFS was 18%, and the median was 2.2 months. Of the 32 patients in the alternate treatment cohort who had a CR, the 12-month PFS was 81%, and the median PFS was 65 months. Of the 14 who had PR, there was a 50% 12-month PFS with a median of 9.9 months (supplemental Table 4; Figure 4). In this group of responders to alternate therapy, of the 21 patients who proceeded to consolidation with either autologous (n = 11) or an allogeneic hematopoietic cell transplant (HCT; n = 10), 3 (14%) relapsed. Of the 25 patients who did not undergo HCT, 14 (56%) ultimately relapsed.
Figure 4.
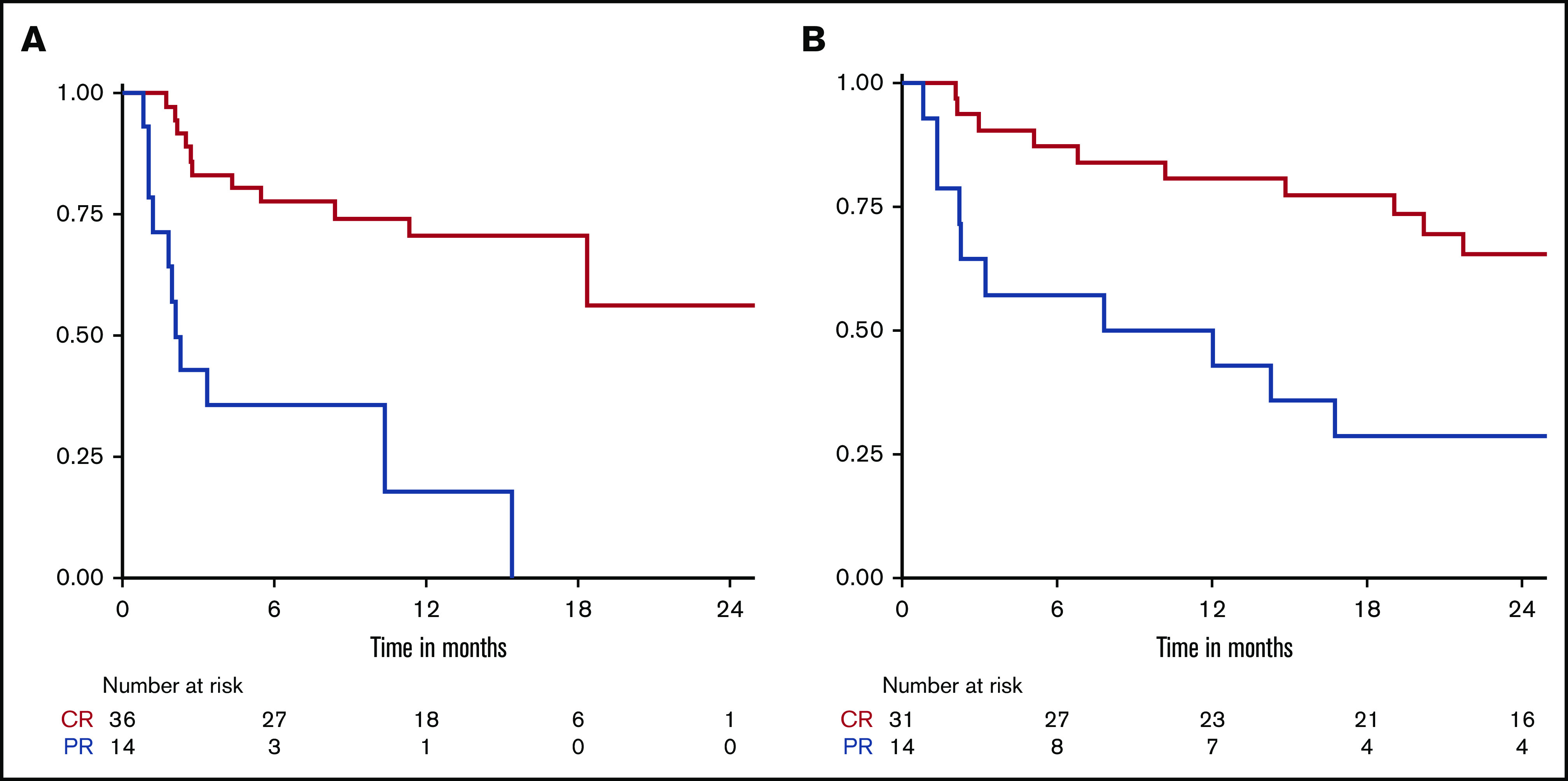
Kaplan-Meier curves comparing PFS between patients who achieved CR vs PR. (A) CAR T cohort. (B) Alternate treatment cohort.
Discussion
The introduction of CAR T therapy has rapidly shifted the landscape of treatment of aggressive lymphoid malignancies. Since the initial approval of CD19-targeted CAR T cells for acute lymphoblastic leukemia in August 2017, there have been 2 additional US Food and Drug Administration approvals for second-generation CAR T therapies within the span of 1 year, with several more likely on the horizon. Despite encouraging response rates in clinical trial populations, a minority of patients have long-term durable responses. Furthermore, data demonstrating the true efficacy and broad feasibility of CAR T therapy in real-world populations are limited. In addition, cross-study comparisons between prospective clinical trials and observational studies (eg, SCHOLAR-1), which typically consist of trial-ineligible patients, should be made cautiously.10 In the current study, we describe our single center experience treating patients with commercial CAR T therapy for R/R DLBCL. We report response rates, PFS, and OS comparable to the pivotal trials and markedly better than outcomes seen in patients treated with alternate therapies before the CAR T era. However, when adjusting for baseline unfavorable disease biology, the seemingly inferior outcomes associated with use of alternate therapies are less clear. Moreover, this CAR T cohort selectively excludes patients that are shunted to alternate therapy given rapid disease kinetics prohibiting logistical hurdles of apheresis and CAR T-cell manufacturing.
Characteristics and outcomes of patients who received commercial CAR T therapy at our center resemble data from other real-world studies.7,8,11-13 The Center for International Blood & Marrow Transplant Research (CIBMTR) postmarketing observational study for axicabtagene ciloleucel demonstrated a CR rate of 52%. In this study of 295 patients, the population was slightly younger (median age of 61) and fitter (95% with ECOG 0-1) than ours; however, 66% had chemotherapy-resistant disease before infusion (compared with 33% in ours).7 In the CIBMTR postmarketing study for tisagenlecleucel, of 47 evaluable patients, the CR rate was 38%, and 30% of patients died of disease progression at a median follow-up of about 6 months. In this population, median age was 65, but only 3 patients had ECOG > 1.8 Finally, a real-world analysis of 91 patients with DLBCL who received commercial CAR T therapy in the United Kingdom demonstrated notably poorer responses and rates of durable remissions compared with the pivotal trials. With median follow-up of 4.8 months, median event-free survival was only 3.1 months overall. For patients receiving axicabtagene ciloleucel, the CR rate was 21% and about 60% experienced progression. For tisagenlecleucel, the CR rate was 17% and about 70% progressed. In this patient population, median age was lower at 56 years; however, 88% were refractory to the most recent prior treatment.11
Baseline characteristics of patients included in our study population were fairly skewed for higher-risk disease (eg, high International Prognostic Index) as expected. At time of treatment with third-line alternate therapy or with CAR T cells (third line or greater), the cohorts did not differ significantly in age or ECOG status. However, patients who received CAR T therapy tended to have lower LDH and a lower frequency of refractory disease to most recent prior therapy, indicating these patients may have had more favorable lymphoma biology. Of note, in the CAR T group, the percentage of patients with poor-risk histologies (eg, double hit or double expressor) was representative of DLBCL in the general population. In the alternate cohort, when examining the types of treatment received by patients at their third line, a large number of patients were fit enough to receive platinum regimens, undergo HCT, or were eligible for a clinical trial.
In our study, response rates to CAR T therapy were almost identical to that of the pivotal clinical trials and those reported by the CIBMTR in the real-world observational cohorts. Interestingly, the response rates in the alternate therapy group were better than those reported in the SCHOLAR-1 study (22% CR rate vs 7%). However, patient selection for SCHOLAR-1 was selectively enriched with very high-risk patients, only including those with primary or secondary refractory disease or experiencing relapse in under 12 months. Still, OS was comparable between our population and SCHOLAR-1 with a median OS of 6 months (vs 6.3 months) and 25% OS (vs 30%) in 1 year. When outcomes between the 2 cohorts in our study were compared directly, response rate, PFS, and OS were significantly better in the CAR T therapy group, albeit with shorter follow-up in this group. When analyzing more homogeneous populations by subclassifying each cohort by number of prior treatment lines, the benefit of CAR T therapy appears to persist, irrespective of the number of lines of prior therapy. Also as expected, we observed a steady decline in both response rate and PFS with increasing subsequent lines of therapy in both cohorts.
Despite the encouraging response rates, PFS, and OS of the CAR T cohort, greater than two-thirds ultimately progressed after CAR T cells. Moreover, after adjusting for poor prognostic variables in both cohorts, including elevated LDH, bulky disease, extranodal sites, elevated ECOG status, and refractory disease, the superiority of CAR T therapy was less pronounced for PFS and OS. Indeed, patients who received alternate therapy had a significantly increased incidence of elevated LDH and refractory disease compared with those patients who were referred for CAR T cells. Furthermore, the subgroup analysis performed on patients with elevated LDH and bulky disease demonstrated that CAR T therapy did not significantly improve PFS and OS (supplemental Figure 1). The superior response rate seen with CAR T therapy, however, appears unaffected by these aggressive disease features. In the multivariate analysis, results for ORR adjusted for the same prognostic covariates demonstrated the difference in treatment cohort remained statistically significant. Finally, in an analysis that specifically evaluated outcomes in responding patients, the rate of relapse or progression was higher in patients that received CAR T cells compared with alternate therapies. This is especially true for patients only achieving a PR as best response to CAR T cells (not reached vs 58% 12-month PFS). Although relatively small in number, the patients who did experience a response to alternate therapies had encouraging long-term outcomes, especially those with a CR. Although many of these patients proceeded to either autologous or allogeneic HCT, those that did not still experienced relatively favorable outcomes.
There are several limitations to the current study. First, as a single center study conducted at a large academic center, the patient population is not fully representative of the broader community. Therefore, our data should be validated across other institutions before drawing generalizable conclusions. Second, the alternate cohort patients were treated over a long period of time (2001-2018), which may be associated with significant differences in management practices and patient outcomes irrespective of the type of the therapy received. However, this would be expected to bias against this cohort compared with the CAR T patients who received their care more recently. Patient selection bias is a common limitation to retrospective studies; however, this is controlled to some extent by multivariate analysis, which adjusts for baseline variables of patients. Also, many additional confounding variables likely exist that are not identified. Additionally, heterogeneity exists between the cohorts. For example, patients in the alternate cohort all received 2 prior lines compared with those in the CAR T cohort, who have received between 2 and 7 prior lines. However, outcomes are typically worse after successive lines of therapy, and when analyzing outcomes classified by number of prior lines, the results are consistent with that from the primary analysis. In fact, we see greater separation of the curves between the 2 cohorts with subsequent lines of prior therapy.
In summary, patients who received CAR T cells appear to have superior response rates, PFS, and OS compared with a historical group of patients receiving alternate or non-CAR T therapy. When adjusting for unfavorable pretreatment factors, although response rate remains significantly better, overall PFS and OS do not. This suggests that patients treated with CAR T cells may have less aggressive disease biology and growth kinetics that permit up to 1 month or more for the manufacturing process to be complete. Our data also show that patients with objective responses to alternate third-line therapy may have long-term remissions, although a significant number were exposed to the extra burdens of HCT. Therefore, if patients are treated at a center where CAR T therapy is an option, it may be a useful tool for cases of significant disease burden that necessitate treatment with a high likelihood of response. Despite the potential for unique toxicities, the experiences at our institution have underscored that CAR T therapy is generally well tolerated by a broad range of patients, including those traditionally considered unfit because of advanced age.14,15 However, there continues to be an important role for use of alternate therapy in the appropriate setting for patients with R/R disease. For instance, if CAR T therapy is unavailable at a given location, patients may still benefit from immediately available conventional or targeted therapy, and if a good response is attained, they may not need to be referred to a CAR T center and could be considered, if eligible, for autologous or allogeneic HCT. Alternate off-the-shelf therapies may also be necessary in cases of rapid disease kinetics. Finally, a significant percentage of patients with R/R disease will still have prohibitively poor performance status, making them ineligible for CAR T cells. Future CAR T studies should focus on identifying the most appropriate sequencing with other therapies (including HCT) and patient selection variables. Prospective (ideally) randomized studies that compare CAR T cells to conventional therapies head-to-head are also warranted. Of note, the phase 3 trials BELINDA (NCT03570892), ZUMA-7 (NCT03391466), and TRANSFORM (NCT03575351) are currently ongoing, and we eagerly await these results. There continues to be a need for novel next-generation CAR T therapies that lead to more durable responses.
In conclusion, the CD19-targeted CAR T products axicabtagene ciloleucel and tisagenlecleucel remain an important treatment option for patients with R/R DLBCL. However, we contend that alternate therapies may be as efficacious as CAR T therapy in select clinical scenarios. Therefore, additional study continues to be warranted, ideally with randomized prospective trials.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
This research was supported in part by National Institutes of Health (NIH), National Cancer Institute Cancer Center Support Grant P30 CA008748. M.P. was supported by an American-Italian Cancer Foundation Post-Doctoral Research Fellowship and by AIL Milano e Porvincia Onlus.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
For information on data, please contact the corresponding author at sauterc@mskcc.org.
Authorship
Contribution: D. Sermer contributed to study formulation/design, data collection, data analysis, and manuscript preparation; J.F. and S.D. contributed to data analysis and manuscript preparation; M.-A.P., M.P., A.A., and M.S. contributed to data collection; C.S. contributed to study formulation/design and manuscript preparation; and all other authors made substantial contributions to all aspects of the preparation of this manuscript.
Conflict-of-interest disclosure: C.B. received grant funding from Janssen, Novartis, Epizyme, Xynomics, Bayer, BMS; served as a consultant for Life Sci, GLG, Celgene, Seattle Genetics, Xynomics; and holds honorarium from Dava Oncology. C.H. received honorarium from Invivoscribe Inc. G.S. received research funding from Janssen and Amgen. M.-A.P. reports honoraria from AbbVie, Bellicum, Celgene, Bristol-Myers Squibb, Incyte, Kite/Gilead, Merck, Novartis, Nektar Therapeutics, Omeros, and Takeda; serves on DSMBs for Cidara Therapeutics, Servier and Medigene and the scientific advisory boards of MolMed and NexImmune; received research support for clinical trials from Incyte, Kite/Gilead and Miltenyi Biotec; and serves in a volunteer capacity as a member of the Board of Directors of American Society for Transplantation and Cellular Therapy (ASTCT) and Be The Match (National Marrow Donor Program, NMDP), as well as on the CIBMTR Cellular Immunotherapy Data Resource (CIDR) Committee. M.S. has served as a consultant for McKinsey & Company, Angiocrine Bioscience, Inc., Omeros Corporation; received research funding from Angiocrine Bioscience, Inc., and has served on an ad hoc advisory board for Kite–A Gilead Company. P.D. serves on the advisory board for Kite/Gilead. P.H. receives research support from Portola, Novartis/GSK, Molecular Templates, and Janssen Pharmaceuticals and served as consultant for Karyopharm, Juno, Portola, Celgene, and AstraZeneca. S.H. received research funding from ADCT therapeutics, Aileron, Forty-Seven, Verastem, Kyowa Hakko Kirin, Millennium Pharmaceuticals Inc, Celgene, Trillium, and Daiichii Sankyo and consults for Astex, Affimed, Merck Sharp and Dome, Kyowa Hakko Kirin Pharma, Corvus Pharmaceuticals Inc., Celgene, Portola Pharmaceuticals, Takeda Millennium, Innate Pharma, Verastem, Miragen Therapeutics Inc, Seattle Genetics, and ADCT. A.K. receives research funding from AbbVie Pharmaceuticals, Adaptive Biotechnologies, Pharmacyclics, and Seattle Genetics and serves on advisory board for Celgene and Astra Zeneca. A.N. receives honoraria from Janssen, Pharmacyclics, and Prime Oncology; consults for Medscape; serves on advisory board for Janssen; serves on the speakers bureau for Prime Oncology; and receives research funding for Rafael Pharma and Pharmacyclics. A.M. receives research support from Seattle Genetics, Merck, Bristol-Myers Squibb, and Incyte and receives honorarium from Kyowa Hakko Kirin Pharma, Miragen Therapeutics, Takeda Pharmaceuticals, ADC Therapeutics, Seattle Genetics, Cell Medica, Bristol-Myers Squibb, and Erytech Pharma. D. Straus consults for InPractice Elselvier and Seattle Genetics and is on speakers bureau for Medical Crossfire. A.Z. consults for Genentech/Roche, Gilead, Celgene, Janssen, Amgen, Novartis, Adaptive Biotechnology, and Verastem; serves on advisory boards for MorphoSys, Gilead, Genentech, AbbVie, AstraZeneca, and Pharmacyclics; and receives research funding from MEI Pharmaceuticals, Roche, Gilead, and Beigene. A.Y. receives research support from Janssen, Curis, Merck, BMS, Syndax, and Roche and honorarium from Janssen, AbbVie, Merck, Curis, Epizyme, Roche, and Takeda and consults for Biopath, Xynomics, Epizyme, Roche, Celgene, and HCM. C.S. receives research support from Juno Therapeutics, Celgene, Precision Biosciences, and Sanofi-Genzyme and consults for Juno Therapeutics, Sanofi-Genzyme, Spectrum Pharmaceuticals, Novartis, Genmab, Precision Biosciences, Kite, Celgene, Gamida Cell, and GSK. The remaining authors declare no competing financial interests.
Correspondence: Craig Sauter, Memorial Sloan Kettering Cancer Center, 1275 York Ave, Box 276, New York, NY 10065; e-mail: sauterc@mskcc.org.
References
- 1.Gisselbrecht C, Glass B, Mounier N, et al. . Salvage regimens with autologous transplantation for relapsed large B-cell lymphoma in the rituximab era. J Clin Oncol. 2010;28(27):4184-4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gisselbrecht C, Schmitz N, Mounier N, et al. . Rituximab maintenance therapy after autologous stem-cell transplantation in patients with relapsed CD20(+) diffuse large B-cell lymphoma: final analysis of the collaborative trial in relapsed aggressive lymphoma. J Clin Oncol. 2012;30(36):4462-4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Epperla N, Badar T, Szabo A, et al. . Postrelapse survival in diffuse large B-cell lymphoma after therapy failure following autologous transplantation. Blood Adv. 2019;3(11):1661-1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crump M, Neelapu SS, Farooq U, et al. . Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study [published correction in Blood. 2018;131(5):587-588]. Blood. 2017;130(16):1800-1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Locke FL, Ghobadi A, Jacobson CA, et al. . Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 2019;20(1):31-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schuster SJ, Bishop MR, Tam CS, et al. ; JULIET Investigators . Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019;380(1):45-56. [DOI] [PubMed] [Google Scholar]
- 7.Pasquini MC, Locke FL, Herrera AF, et al. . Post-marketing use outcomes of an anti-CD19 chimeric antigen receptor (CAR) T cell therapy, axicabtagene ciloleucel (Axi-Cel), for the treatment of large B Cell lymphoma (LBCL) in the United States (US) [abstract]. Blood. 2019;134(suppl 1):Abstract 764. [Google Scholar]
- 8.Jaglowski S, Hu ZH, Zhang Y, et al. . Tisagenlecleucel chimeric antigen receptor (CAR) T-cell therapy for adults with diffuse large B-cell lymphoma (DLBCL): real world experience from the Center for International Blood & Marrow Transplant Research (CIBMTR) Cellular Therapy (CT) Registry [abstract]. Blood. 2019;134(suppl 1):Abstract 766. [Google Scholar]
- 9.Cheson BD, Fisher RI, Barrington SF, et al. ; United Kingdom National Cancer Research Institute . Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol. 2014;32(27):3059-3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rutherford SC, Leonard JP. Lymphoma “benchmark” or “bench-smudge”? Blood. 2017;130(16):1778-1779. [DOI] [PubMed] [Google Scholar]
- 11.Kuhnl A, Roddie C, Martinez-Cibrian N, et al. . Real-world data of high-grade lymphoma patients treated with CD19 CAR T in England [abstract]. Blood. 2019;134(suppl 1):Abstract 767. [Google Scholar]
- 12.Nastoupil LJ, Jain MD, Spiegel JY, et al. . Axicabtagene ciloleucel (axi-cel) CD19 chimeric antigen receptor (CAR) T-cell therapy for relapsed/refractory large B-cell lymphoma: real world experience [abstract]. Blood. 2018;132(suppl 1):Abstract 91. [Google Scholar]
- 13.Jacobson CA, Hunter B, Armand P, et al. . Axicabtagene ciloleucel in the real world: outcomes and predictors of response, resistance, and toxicity [abstract]. Blood. 2018;132(suppl 1):Abstract 92. [Google Scholar]
- 14.Pennisi M, Jain T, Santomasso BD, et al. . Comparing CAR T-cell toxicity grading systems: application of the ASTCT grading system and implications for management. Blood Adv. 2020;4(4):676-686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin RJ, Lobaugh SM, Pennisi M, et al. . Impact and safety of chimeric antigen receptor T cell therapy in older, vulnerable patients with relapsed/refractory large B-cell lymphoma [published online ahead of print 20 February 2020]. Haematologica. doi:10.3324/haematol.2019.243246. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.