

Key Points
Circulating cytotoxic CD69−TTE cells mediating antimyeloma responses and noncytotoxic BM-resident CD69+TTE cells exist in NDMM patients.
A balance between CD69−TTE and CD69+TTE cells may regulate antimyeloma responses and contribute to clinical heterogeneity in NDMM patients.
Abstract
CD8+CD57+ terminal effector T (TTE) cells are a component of marrow-infiltrating lymphocytes and may contribute to the altered immune responses in multiple myeloma (MM) patients. We analyzed TTE cells in the bone marrow (BM) and peripheral blood (PB) of age-matched controls and patients with monoclonal gammopathy of undetermined significance (MGUS), smoldering MM (SMM), and newly diagnosed (ND) MM using flow cytometry, mass cytometry, and FlowSOM clustering. TTE cells are heterogeneous in all subjects, with BM containing both CD69− and CD69+ subsets, while only CD69− cells are found in PB. Within the BM-TTE compartment, CD69− and CD69+ cells are found in comparable proportions in controls, while CD69− cells are dominant in MGUS and SMM and predominantly either CD69− or CD69+ cells in NDMM. A positive relationship between CD69+TTE and CD69−TTE cells is observed in the BM of controls, lost in MGUS, and converted to an inverse relationship in NDMM. CD69−TTE cells include multiple oligoclonal expansions of T-cell receptor/Vβ families shared between BM and PB of NDMM. Oligoclonal expanded CD69−TTE cells from the PB include myeloma-reactive cells capable of killing autologous CD38hi plasma cells in vitro, involving degranulation and high expression of perforin and granzyme. In contrast to CD69−TTE cells, oligoclonal expansions are not evident within CD69+TTE cells, which possess low perforin and granzyme expression and high inhibitory checkpoint expression and resemble T resident memory cells. Both CD69−TTE and CD69+TTE cells from the BM of NDMM produce large amounts of the inflammatory cytokines interferon-γ and tumor necrosis factor α. The balance between CD69− and CD69+ cells within the BM-TTE compartment may regulate immune responses in NDMM and contribute to the clinical heterogeneity of the disease.
Visual Abstract

Introduction
Multiple myeloma (MM) is a plasma cell (PC) neoplasm that is preceded by the premalignant condition monoclonal gammopathy of undetermined significance (MGUS) or asymptomatic, smoldering MM (SMM). In MM, malignant PCs in the bone marrow (BM) are accessible to T cells (marrow-infiltrating lymphocytes [MILs]) entering the BM by blood circulation. This proximity between PCs and T cells may facilitate autologous T-cell–mediated immune responses against malignant PCs. Direct evidence of autologous T-cell–mediated antimyeloma responses has been demonstrated in the Vk*MYC mouse model,1,2 while a number of clinical studies provide indirect evidence to support autologous antimyeloma responses in humans.3-6
CD57 has been most widely explored as a marker of senescent CD8+T cells.7 Persistent immune stimulation is believed to induce the conversion of memory T (TM) cells from CD28+CD57− cells to senescent CD28−CD57+ cells characterized by limited proliferative capacity.8 Acquisition of CD57 is thought to reflect a shift toward highly cytotoxic terminally differentiated effector T (TTE) cells, with increased perforin and granzyme production.9
We have previously reported on the existence of oligoclonal expansions of TTE cells, identified by expression of T-cell receptor (TCR) variable β (TCR-Vβ) families, in the peripheral blood (PB) of the majority of MM patients7 and related their presence to a favorable prognosis.10 Such oligoclonal expanded TTE cells have lower expression of the inhibitory checkpoint CD279 (PD-1), suggesting that these cells may not be an optimal target for checkpoint blockade immunotherapy.11,12 It has also been suggested that TTE cells within MILs may impair immune responses to myeloma due to their senescent status.13
Expansion of oligoclonal TTE cells in MM patients may result from persistent stimulation of CD8+T cells by myeloma-associated antigens6,14 in the absence of effective clearance of malignant clones. Recently, it has also been reported that progression from MGUS to MM involves attrition of the BM-resident T-cell compartment15 and the appearance of exhausted T cells.16 We considered that cytotoxic TTE cells, being a constituent of MILs, may undergo changes that can help explain the altered immune responses observed in MM patients and provide novel ground for future immunotherapeutic approaches.17
In this study, we analyzed CD8+CD57+TTE cells in the BM and PB of age-matched controls and patients with MGUS, SMM, and newly diagnosed (ND) MM using fluorescence flow cytometry, mass cytometry,18 and unsupervised FlowSOM clustering algorithm analyses.19 We found that TTE cells in all subjects can be subdivided by expression of CD69. CD69−TTE cells circulate between PB and BM, while CD69+TTE are restricted to the BM and have many characteristics in common with T resident memory (TRM) cells. Within the BM-TTE compartment, CD69− and CD69+ cells are found in comparable proportions in controls, while CD69− cells are dominant in MGUS and SMM and predominantly either CD69− or CD69+ cells in NDMM. Within the BM-TTE compartment, a positive relationship between CD69+ and CD69− cells is observed in controls, lost in MGUS, and converted to an inverse relationship in NDMM. We also demonstrated that the previously described oligoclonal expansions7 are found within the CD69−TTE cells in BM and PB, but not in CD69+TTE cells, and that oligoclonal expanded cells from the PB of NDMM patients are capable of eliminating autologous CD38hiPCs in vitro involving degranulation and high expression of perforin and granzyme B.
Materials and methods
Patients and controls
MGUS, SMM, and NDMM patients, diagnosed using the criteria established by the International Myeloma Working Group,20 were recruited through the Department of Hematology, Royal Prince Alfred Hospital (RPAH). Age-matched controls included healthy blood donors and patients without diagnosed malignancy or active infection, undergoing hip arthroplasty at the Department of Orthopedic Surgery, RPAH. Patients and controls characteristics are shown in Table 1. Where possible, paired BM and PB samples were collected and, dependent on sample availability, analyzed by mass cytometry, fluorescence flow cytometry, or both. The study was approved by the institutional human ethics committee. All patients signed informed consent before sample collection in accordance with the amended Declaration of Helsinki.
Table 1.
General and clinical characteristics of patients and controls
| NDMM (n = 36) | SMM (n = 11) | MGUS (n = 24) | Controls (n = 26) | |
|---|---|---|---|---|
| Age, median (range), y | 69 (38-90) | 73 (50-85) | 64 (42-89) | 60 (35-84) |
| Sex, male, n (%) | 20 (55) | 5 (45) | 7 (29) | 12 (46) |
| ISS stage,* n (%) | ||||
| ISS1 | 11 (34) | 3 (37.5) | NA | NA |
| ISS2 | 10 (32) | 3 (37.5) | NA | NA |
| ISS3 | 11 (36) | 2 (25) | NA | NA |
| Isotype, n (%) | ||||
| IgG | 21 (60) | NA | NA | NA |
| IgA | 8 (23) | NA | NA | NA |
| Light chain | 6 (17) | NA | NA | NA |
| Oligosecretory | 1 (3) | NA | NA | NA |
| Cytogenetics, n (%) | 33 (92) | NA | NA | NA |
| 17p deletion | 4 (12) | NA | NA | NA |
| −1p and/or +1q | 13 (40) | NA | NA | NA |
| FISH,† n (%) | 20 (55) | NA | NA | NA |
| t(4;14) | 2 (11) | NA | NA | NA |
| t(14;16) | 0 (0) | NA | NA | NA |
| LDH,‡ median (range), U/L | 157 (89-407) | NA | NA | NA |
| Above normal, n (%) | 5 (18) | NA | NA | NA |
FISH, fluorescence in situ hybridization; IgA, immunoglobulin A; IgG, immunoglobulin G; ISS, International Staging System; LDH, lactate dehydrogenase; NA, not available.
β2 microglobulin available for ISS grading in 32 out of 36 NDMM patients and 8 out of 11 SMM patients.
High-risk genetics by FISH, including del17p, t(4;14), and t(14;16), were tested in 20 out of 36 NDMM patients.
Available in 28 out of 36 NDMM patients.
Fluorescence flow cytometry analyses
Fluorescence flow cytometry samples comprised fresh whole-blood samples, cryopreserved BM mononuclear cells (MNCs), and PB MNCs isolated by Ficoll-Hypaque density gradient. To analyze cytokine production, BM MNCs were rested overnight, sorted into CD3+CD69− and CD3+CD69+ cells (BDFACS Aria II, BD Biosciences), and stimulated with phorbol 12-myristate 13-acetate and ionomycin calcium salt (Sigma-Aldrich) for 4 hours with the addition of protein transport inhibitor cocktail (Thermo Fisher Scientific) for the last 3 hours of culture. Following stimulation, cells were labeled with monoclonal antibodies (mAbs) targeting surface antigens (supplemental Table 1), fixed and permeabilized (fixation/permeabilization buffer, BD Biosciences), and stained with mAbs specific to interferon-γ (IFN-γ) and tumor necrosis factor α (TNF-α) in Perm/Wash buffer (BD Biosciences).
TCR-Vβ usage was analyzed using the IOTest β Mark TCR-Vβ Repertoire Kit (Beckman Coulter Life Sciences). TCR-Vβ family–expressing populations within a patient’s TTE were determined to be oligoclonally expanded when the percentage of TTE cells expressing that TCR-Vβ family was more than 3 standard deviations (SDs) higher than the mean frequency within the naive CD8+T (TN) compartment of healthy blood donors. The dominant oligoclonal expansion in each patient was defined as the one representing the largest percentage of PB TTE cells.
To analyze the elimination of autologous CD38hiPCs (target) by CD69−TTE cells (effectors), 2 subsets of TTE cells were flow-sorted from PB MNCs: those expressing the dominant TCR-Vβ family and the remainder expressing all other TCR-Vβ families. Sorted cells were stimulated for 12 to 14 hours by CD2/CD3/CD28-loaded anti-biotin MACSiBead (Miltenyi Biotec) before the addition to the target cells; CD3-depleted autologous BM MNCs (effector/target ratio of 1:2). To assess degranulation and associated IFN-γ expression, αCD107a and protein transport inhibitor cocktail were added at the beginning of cell culture. Following 1 to 2 hours of culture, cells were labeled with mAbs targeting surface markers (supplemental Table 1), fixed and permeabilized, and stained depending on the experiment with mAbs specific to cleaved caspase-3 AF488, rabbit immunoglobulin G AF488, or IFN-γ in Perm/Wash buffer.
Mass cytometry staining and data acquisition
BM MNCs and PB MNCs from MGUS (n = 4) and NDMM (n = 8) patients were analyzed by mass cytometry. Cells were stained with 1.25 μL cisplatin (Fluidigm) followed by quenching and washing with fluorescence-activated cell sorter buffer. Cells were initially incubated with an AF647-labeled CD160 (BD Biosciences) mAb, followed by a cocktail of metal-conjugated mAbs targeting surface proteins (supplemental Table 2). Cells were subsequently fixed and permeabilized (Foxp3 fixation kit, Thermo Fisher Scientific) and stained with metal-conjugated mAbs targeting intracellular proteins (supplemental Table 2). Cell acquisition occurred at a rate of 200 to 400 cells per second using a CyTOF 2 Helios upgraded mass cytometer (Fluidigm, Charles Perkins Centre, University of Sydney, NSW, Australia).
Analysis of mass cytometry data
After exclusion of CD3−CD38hiPCs, CD56+NK cells, and CD19+B cells, BM-TTE and PB-TTE cells in MGUS and NDMM were manually gated based on CD57 expression within the CD8+T gate, imported into R studio (v1.2.1135), and analyzed using the CAPX (v0.3) script, which includes both the clustering algorithm FlowSOM19 and dimensionality reduction algorithm t-distributed stochastic neighbor embedding (tSNE)21 in a single script.22 tSNE plots were generated using the same 15 antigens selected for FlowSOM, with the addition of 3 TCR-Vβ antigens (supplemental Table 2) to allow visualization of oligoclonal expanded CD69−TTE cells expressing dominant TCR-Vβ family.
Statistical analysis
The nonparametric Mann-Whitney U test was used to compare 2 variables, and the Kruskal-Wallis and Friedman test with Dunn’s multiple comparisons was used to compare multiple unpaired and paired data sets, respectively. Relationships between 2 variables were analyzed by linear regression and between multiple variable by nonparametric Spearman correlation. Statistical significance was determined at Spearman R < −0.5 and > 0.5 and P < .05 for all statistical analyses using GraphPad Prism version 8.02 (GraphPad Software).
Results
TTE cells within BM and PB of controls and MGUS, SMM, and NDMM patients
We initially characterized CD8+CD57+TTE cells in the BM and PB of age-matched controls and patients with MGUS, SMM, and NDMM by fluorescence flow cytometry. In all subjects, BM-TTE cells contained both CD69− and CD69+ subsets, whereas PB TTE cells contained only the CD69− cell subset (Figure 1). Although CD69+TTE cells were “resident” within BM, only a minority expressed another marker of residency, CD10323 (supplemental Figure 1). CD69− and CD69+ cells accounted for comparable proportions of BM-TTE cells in controls, while CD69− cells dominated within BM-TTE cells of MGUS and SMM patients (Figure 1B). Neither of these trends was apparent in BM-TTE cells of NDMM patients; instead, CD69− cells in some patients and CD69+ cells in other patients represented the majority of BM-TTE compartment (Figure 1B). In addition, CD69− and CD69+ subsets, as well as total BM-TTE cells, accounted for highly variable proportions of BM-CD8+T cells in NDMM (Figure 1C; supplemental Figure 2A). Despite interpatient variability, CD69−TTE and total TTE cells were significantly increased in the BM of NDMM patients compared with controls (expressed as a percentage of CD8+T cells; Figure 1C; supplemental Figure 2A). In contrast to BM TTE cells, PB CD69−TTE and total PB TTE cells were present in similar proportions in all subjects (Figure 1B-C; supplemental Figure 2A). These data suggest that BM TTE cells possess inherent non–myeloma-related heterogeneity (based on CD69 expression) that is differently regulated during progression from MGUS and/or SMM to NDMM and that there is an accumulation of both CD69−TTE and total TTE cells (expressed as a percentage of CD8+T cells) in the BM of NDMM patients.
Figure 1.
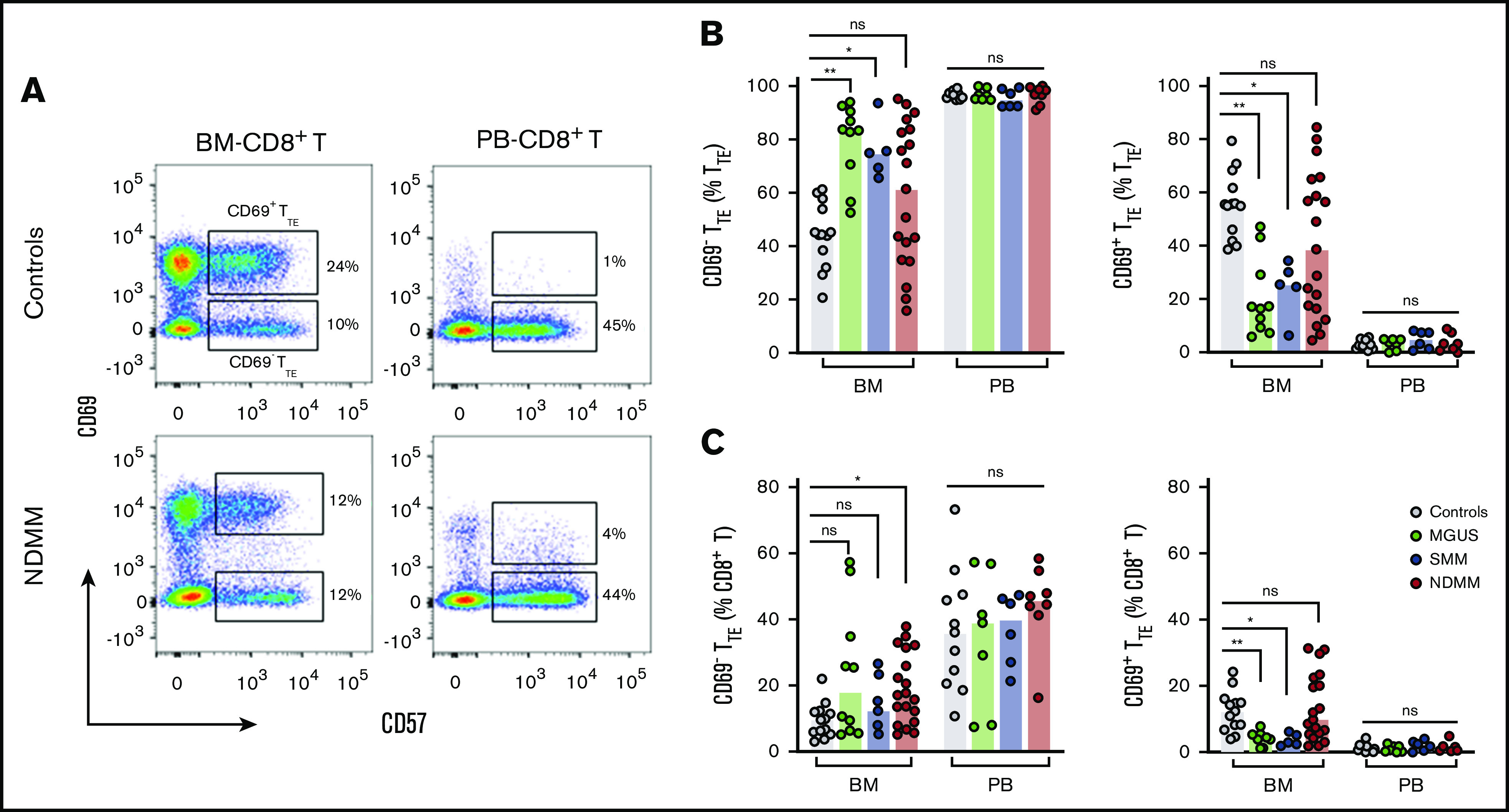
Tissue distribution of CD69−TTEand CD69+TTEcells in controls and MGUS, SMM, and NDMM patients. (A) Representative dot plots gated for CD8+T cells show distribution of CD69+TTE and CD69−TTE cells in the BM and PB of controls and NDMM patients. Regions occupied by CD69+TTE and CD69−TTE cells are indicated. Numbers indicate proportions of CD69+TTE and CD69−TTE cells within BM-CD8+T and PB-CD8+T cells. Bars (median with scatter plots) show proportions of CD69−TTE and CD69+TTE cells within TTE (B) and CD8+T (C) cells in the BM of controls (n = 13), MGUS patients (n = 10), SMM patients (n = 5), and NDMM patients (n = 19) and PB of controls (n = 13), MGUS patients (n = 7), SMM patients (n = 6), and NDMM patients (n = 8). *P < .05; **P < .01; Kruskal-Wallis test with Dunn’s multiple comparisons. ns, not significant.
Inverse relationship between CD69−TTE and CD69+TTE cells in BM of NDMM patients, but not in MGUS patients and controls
We next analyzed whether the different contribution of CD69− and CD69+ subsets to BM-TTE compartments of controls and MGUS and NDMM patients along with the increase in CD69−TTE and total TTE cells seen in BM of NDMM patients involve different relationships between various memory/effector subsets within CD8+T cells. We analyzed relationships between CD69−TTE cells, CD69+TTE cells, total TTE cells, conventional T memory cells (CD8+CD57−CD45RO+TM), and total CD8+T cells (Figure 2; supplemental Figure 3). SMM patients were excluded from this analysis due to limited numbers (SMM, n = 5, Figure 1B-C; supplemental Figure 2A). In controls, there was a positive correlation between CD69− and CD69+ subsets within the BM-TTE compartment. Further, there was a strong positive correlation between total BM-TTE cells and their CD69− and CD69+ subsets (Figure 2A,D-E). Compared with controls, MGUS patients maintained only a strong positive correlation between total BM-TTE cells and their CD69− subset (Figure 2B,D-E). None of the positive correlations observed in controls and MGUS patients were sustained in NDMM patients; instead, a negative correlation was established between CD69− and CD69+ subsets within the BM-TTE compartment (Figure 2C-E). These data suggest that inverse relationships established between CD69− and CD69+ subsets within the BM-TTE compartment discriminate NDMM from MGUS and controls.
Figure 2.
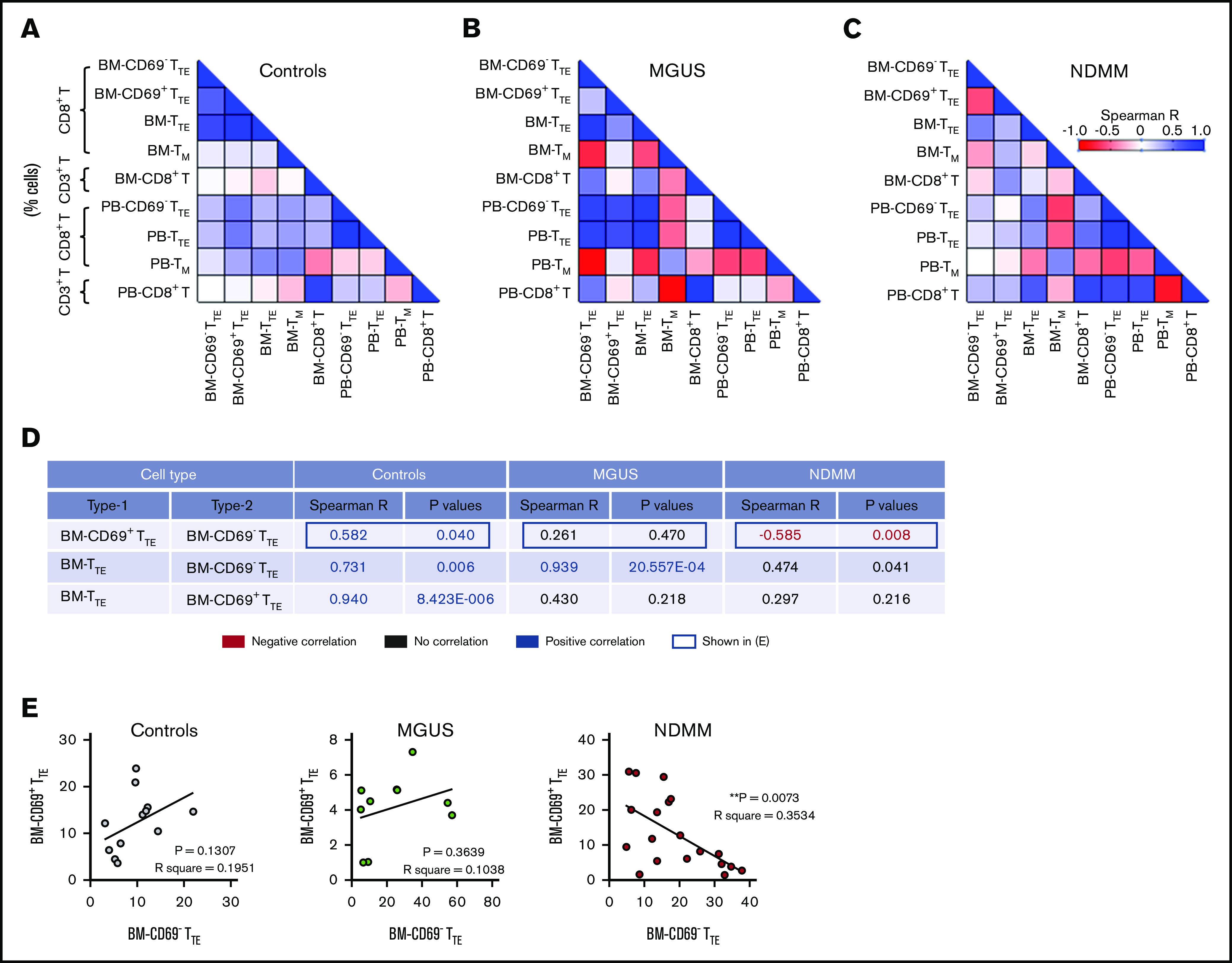
Relationships between proportions of CD69−TTE, CD69+TTE, TTE,TM, and CD8+T cells in controls and MGUS and NDMM patients. (A-B) The matrix shows correlations between proportions of CD69−TTE, CD69+TTE, TTE, TM (CD8+CD45RO+CD57−), and CD8+T cells in the BM and PB of controls (A), MGUS patients (B), and NDMM patients (C). Proportions of CD69−TTE, CD69+TTE, TTE and TM are presented as % CD8+T cells and proportion of CD8+T as % CD3+T cells. (D) The table shows selected significant negative (in red) and positive (in blue) correlations depicted from panels A-C with Spearman R < −0.5 and > 0.5 and P < .05. (E) Relationships between paired proportion of CD69+TTE and CD69−TTE cells in BM of controls (left), MGUS patients (middle), and NDMM patients (right) analyzed by linear regression model. **P < .01.
Oligoclonal expanded CD69−TTE cells contain myeloma-reactive cells capable of eliminating autologous CD38hiPCs
Our group has previously reported the presence of oligoclonal expansions expressing different TCR-Vβ families within PB-CD8+T cells in the majority of MM patients.7 To reveal oligoclonal expansions within CD8+TTE cells that may be overlooked at the level of CD8+T cells, we assessed TCR-Vβ usage within PB TTE cells in age-matched controls and patients with MGUS, SMM, and NDMM (Figure 3A). As expected based on the known oligoclonality of TTE cells,24 TCR-Vβ expansions were evident in 98.8% (84 out of 85) of analyzed samples, with an average of 3 expanded TCR-Vβ families detected per sample. The average size of the oligoclonal expansions expressed as a percentage of PB-TTE cells was higher in NDMM (12.2%) than in controls (10.3%), MGUS (8.7%), and SMM (8.6%) (Figure 3A). Although the data may be skewed due to differences in the numbers of subjects in each group, results suggest that expanded TCR-Vβ families account for higher proportions of PB TTE in NDMM than in MGUS (Figure 3A). Since the significance of oligoclonal expansions comprising <5% of PB-TTE cells maybe not conclusive, we excluded them from further analysis. The percentage of oligoclonal expansions of size >5% of PB TTE cells was higher in NDMM (83.7%) than in controls (74.7%), MGUS (76.9%), or SMM (76.3%) (supplemental Figure 4). Occasionally, very highly expanded TCR-Vβ families representing >50% of PB TTE cells were detected across cohorts (Figure 3A; supplemental Figure 4). In addition, the majority of expanded TCR-Vβ families were shared between BM-TTE and PB-TTE cells of NDMM patients (Figure 3B).
Figure 3.
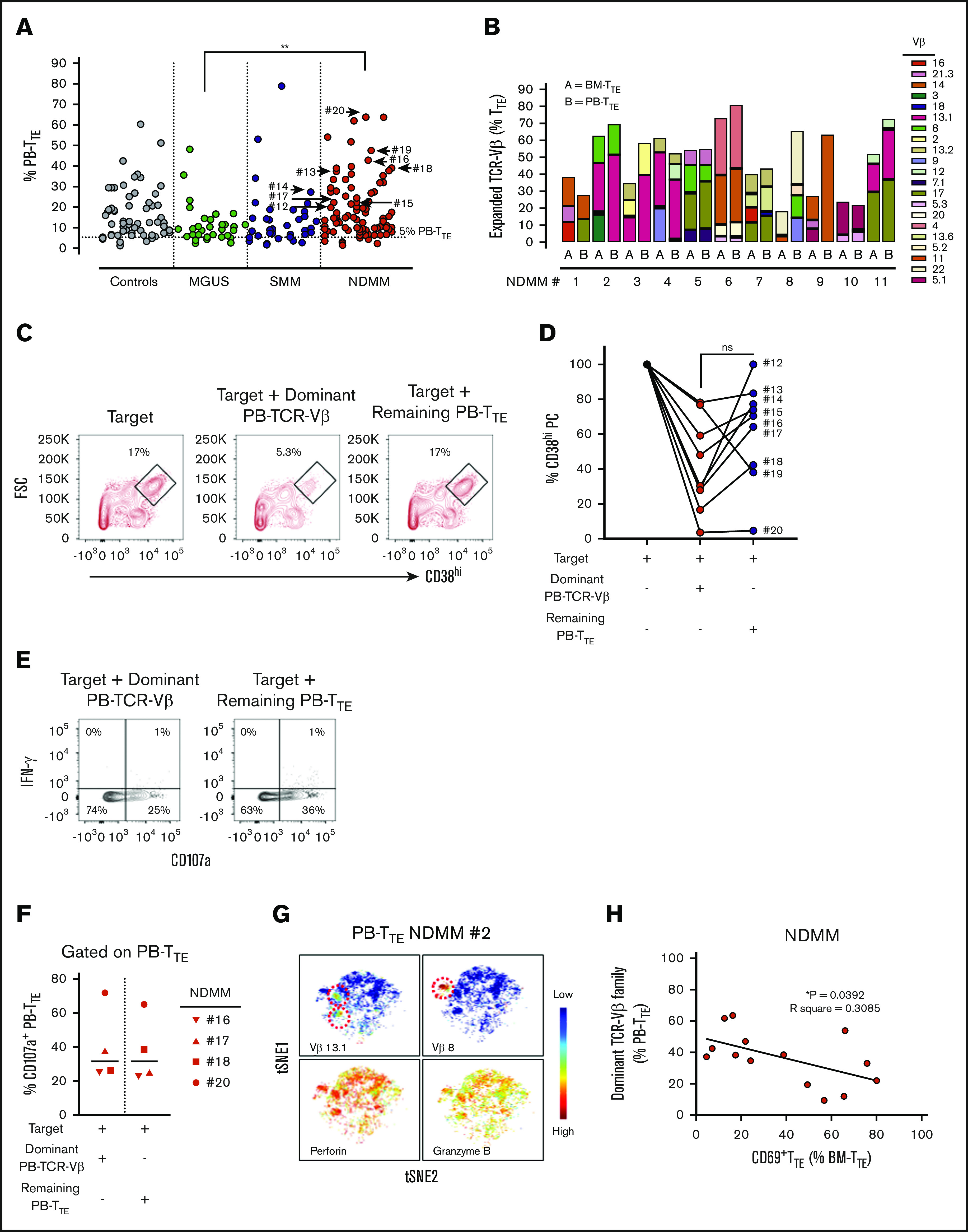
Distribution and function of oligoclonal expanded PB TTEcells. (A) Scatter plots of oligoclonal expansion of TCR-Vβ family–expressing populations within PB-TTE cells of controls (n = 26), MGUS patients (n = 13), SMM patients (n = 10), and NDMM patients (n = 36). Expanded TCR-Vβ family–expressing populations were defined when the percentage of TTE cells expressing that TCR-Vβ family was >3 SD higher than the mean frequency within CD8+TN compartment of healthy blood donors (see “Materials and methods”). Each dot represents the percentage of PB-TTE cells with an individual TCR-Vβ family expansion, with multiple expansions shown for each subject (average 3 expanded TCR-Vβ families per subject). The arrows indicate selected dominant TCR-Vβ family expansions that were tested for capacity to kill autologous CD38hiPCs (n = 9; #12 to #20; see below, panel D). (B) Oligoclonal expansion of TCR-Vβ family–expressing populations in paired BM-TTE and PB-TTE of NDMM patients (n = 11, #1 to #11). Each colored segment in a stacked vertical bar indicates the proportion of an individual oligoclonal expansion within total BM-TTE and PB-TTE compartment. (C) Dot plots gated for CD3−CD14− cells show CD38hiPCs after a 2-hour culture assay with the target (CD3+T-cell-depleted BM MNCs) alone, the target with flow-sorted autologous oligoclonal expanded PB-TTE cells expressing the subject’s dominant TCR-Vβ family, or the target with flow-sorted autologous PB-TTE cells not expressing the dominant TCR-Vβ family (remaining PB-TTE cells). Boxes and numbers indicate the percentage of CD38hiPCs recovered in each culture condition. FSC, forward scatter. (D) Graph shows percentage of CD38hiPCs recovered in culture with PB-TTE cells expressing dominant TCR-Vβ family or with remaining PB-TTE cells, normalized to the percentage of CD38hiPCs recovered in culture with target only. The dominant TCR-Vβ family expansion from each of 9 tested patients (NDMM, #12-13,#15-20; SMM, #14) is indicated by an arrow in Figure 3A. (E) Dot plots show cell surface CD107a and intracellular IFN-γ expression. (F) Graph shows proportions of CD107a+TTE cells (n = 4) in PB-TTE cells expressing dominant TCR-Vβ family and remaining PB-TTE cells after a 2-hour culture assay with target. (G) tSNE plots show distribution of Vβ13.1 and Vβ8 family–expressing PB-TTE cells (top, indicated by red dotted circles) with high perforin and granzyme B expression (bottom) within PB-TTE cells of NDMM #2. (H) Relationship between paired proportion of PB-TTE cells expressing dominant TCR-Vβ family (presented as percentage of PB-TTE cells) and CD69+TTE cells (presented as percentage of BM-TTE cells) in NDMM patients (n = 14) analyzed by linear regression model. *P < .05; **P < .01.
The increase in the percentage of oligoclonal expanded TCR-Vβ families within PB-TTE of NDMM suggested intensified immune responses. To address the possibility that these responses may be mediated by PB-TTE cells against the patient’s myeloma, we purified oligoclonal expanded PB-TTE cells expressing the dominant TCR-Vβ family from the remaining PB-TTE cells within that individual and tested both populations for their capacity to kill autologous CD38hiPCs in a 2-hour culture assay (Figure 3C-D). Contamination by natural killer T (NKT) cells (CD3+CD56+CD16+ cells) and γδ T cells was negligible, as NKT cells represented <5% and γδ T cells were undetectable in PB-TTE compartment (data not shown). PB-TTE cells expressing the dominant TCR-Vβ families eliminated on average 70% of autologous CD38hiPCs within the last hour of a 2-hour culture assay (Figure 3D). Elimination of CD38hiPCs involved caspase-3 activation; however, we were not able to demonstrate an increase in caspase-3 activity in CD38hiPCs cultured with oligoclonal expanded PB-TTE (either at 1- or 2-hour time points), likely due to the rapid cell death following caspase-3 activation (data not shown). In 8 out of 9 patients, oligoclonal expanded PB-TTE cells expressing the dominant TCR-Vβ family were superior to the remaining PB-TTE cells in eliminating autologous CD38hiPCs (Figure 3D). However, PB-TTE cells that did not contain the dominant oligoclonal expansion also exhibited some cytotoxic activity against autologous CD38hiPCs (eliminating on average 30% of autologous CD38hiPCs), suggesting that smaller oligoclonal expansions also contribute to antimyeloma activity (Figure 3D). At the 2-hour time point of coculture, both PB-TTE cells expressing the dominant TCR-Vβ family and the remaining PB-TTE cells exhibited comparable levels of degranulation, as measured by CD107a expression, without concordant IFN-γ production (Figure 3E-F). These results indicate that the enhanced cytotoxic capacity of PB-TTE cells expressing the dominant TCR-Vβ family against autologous CD38hiPCs likely relies on increased perforin and granzyme B content (Figure 3G) rather than on increased activity of degranulation. This is the first direct evidence supporting autologous antimyeloma responses mediated by oligoclonal expanded PB-TTE cells. It is worth noting that myeloma-reactive PB-TTE cells expressing the dominant TCR-Vβ family inversely correlate with CD69+TTE (Figure 3H), implying that antimyeloma responses may be limited in NDMM patients with increased proportions of CD69+TTE cells in their BM.
Phenotypic and functional differences between CD69−TTE and CD69+TTE cells revealed by mass cytometry
We used mass cytometry18 and FlowSOM clustering analyses19 to obtain a more comprehensive signature of CD69−TTE and CD69+TTE cells in NDMM. In agreement with flow cytometry data, we confirmed the presence and tissue distribution of CD69−TTE and CD69+TTE cells in NDMM (Figure 4A). By including mAbs to detect dominant oligoclonal expansions in matched PB and BM samples, we demonstrated that oligoclonal expanded TTE cells were restricted to the CD69−TTE subset in BM and PB of NDMM patients (Figure 4B-C). This suggests that BM-CD69−TTE cells could be the direct counterpart of the PB-CD69−TTE cells capable of killing autologous CD38hiPCs in vitro.
Figure 4.
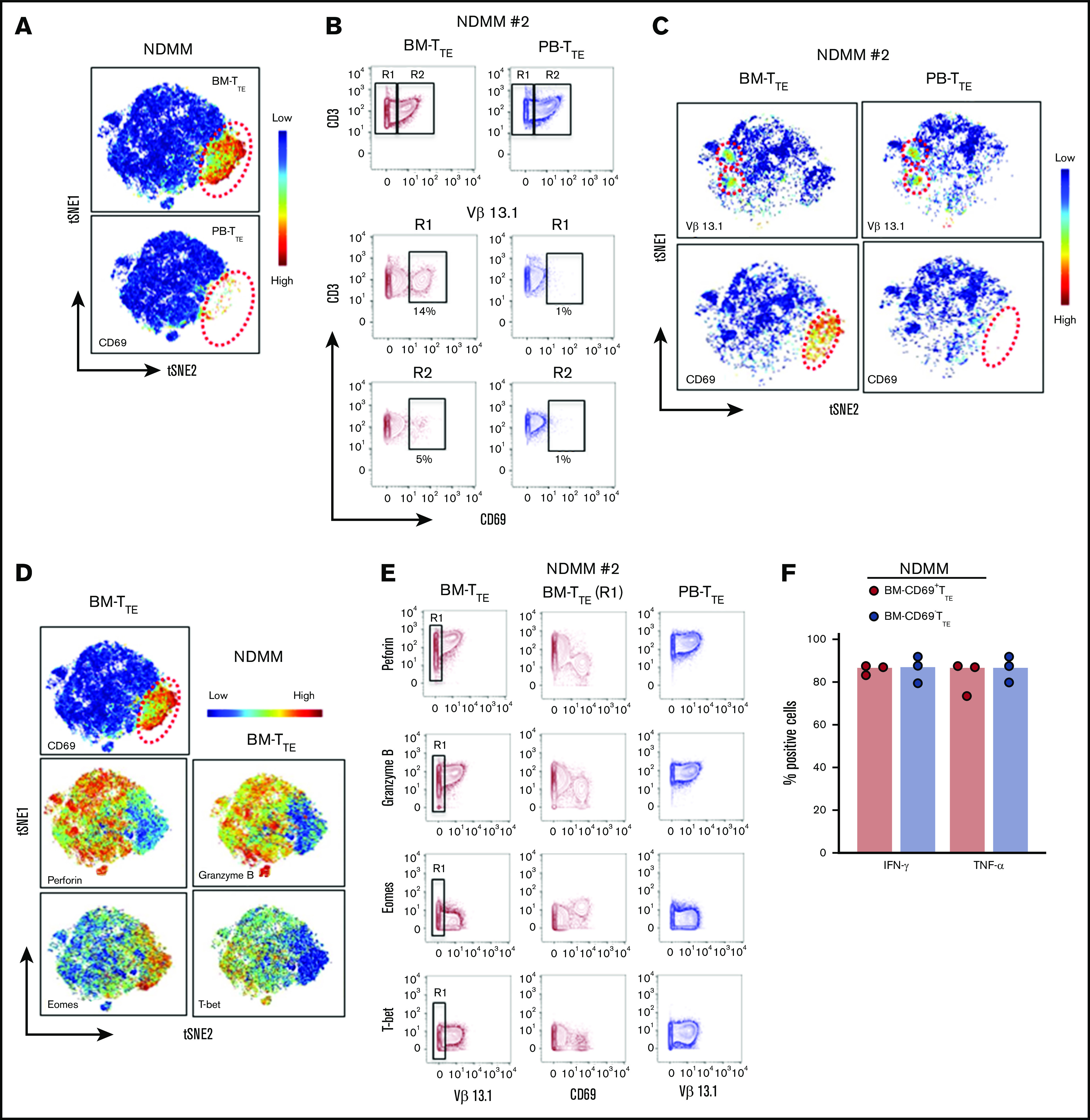
Detailed phenotype of CD69−TTEand CD69+TTEcells in NDMM patients. (A-E) Mass cytometry data. (F) Fluorescence flow cytometry data. (B-C,E) Data from 1 representative NDMM patient (NDMM #2; Figure 3B,G). (A) tSNE plots show distribution of CD69−TTE and CD69+TTE cells within pooled BM-TTE and PB-TTE cells of NDMM patients (n = 5). The area occupied by CD69+TTE cells is indicated by the red dotted circle. (B) Biaxial contour plots show BM-TTE and PB-TTE cells containing oligoclonal expanded Vβ13.1 family–expressing cells of NDMM patient #2. Regions indicate Vβ13.1−TTE cells (R1) and Vβ13.1+TTE cells (R2, top panel). Biaxial contour plots gated for Vβ13.1−TTE cells (R1, middle panels) and Vβ13.1+TTE cells (R2, bottom panels) show presence of CD69+TTE cells indicated by boxes and numbers. (C) tSNE plots show distribution of Vβ13.1 family–expressing TTE cells (top) and CD69+TTE cells (bottom) within BM-TTE and PB-TTE cells of NDMM #2 indicated by red dotted circles. (D) tSNE plots show distribution of CD69, perforin, granzyme B, Eomes, and T-bet within pooled BM-TTE cells of NDMM patients (n = 5). (E) Biaxial contour plots showing expression of perforin, granzyme B, Eomes, and T-bet in oligoclonal expanded Vβ13.1 family–expressing cells and remaining TTE cells in BM and PB of NDMM patient #2. Gated BM-TTE cells that do not express Vβ13.1 family (R1) are shown in the middle panels. (F) Bars (median with scatter plots) show proportion of BM-CD69+TTE and BM-CD69−TTE cells producing IFN-γ and TNF-α (NDMM, n = 3).
To further define the phenotype of CD69−TTE and CD69+TTE cells, we examined the expression of additional molecules within the CD69+TTE and oligoclonal expanded CD69−TTE cells expressing the dominant TCR-Vβ family in PB and BM of NDMM patients. BM-CD69+TTE cells expressed a TbetloEomeshi transcriptional signature and low perforin and granzyme B (Figure 4D-E). In contrast, the phenotype of BM-CD69−TTE cells strongly indicated cytotoxic function, with high expression of perforin and granzyme B and the reciprocal TbethiEomeslo/neg signature (Figure 4D-E). The dominant TCR-Vβ13.1+ oligoclonal expansion within BM-CD69−TTE and PB-CD69−TTE cells shared the cytotoxic phenotype and transcriptional signature of the CD69−TTE population as a whole (Figure 4E). Both BM-CD69−TTE and BM-CD69+TTE cells produced the cytokines IFN-γ and TNF-α at equivalent levels (Figure 4F). BM-CD69+TTE were further distinguished from oligoclonal expanded CD69−TTE expressing the dominant TCR-Vβ family in PB and BM of NDMM by decreased expression of CD45RA, retention of CD27 and CD28, and higher expression of several inhibitory checkpoints, CD279 (PD-1), TIGIT, CD223 (Lag3), and CD160 (supplemental Figure 5). Overall, based on the expression of CD69, BM residency, cytokine production, and inhibitory checkpoint expression, CD69+TTE cells appear closely related to TRM cells,23 while the phenotype of the oligoclonal expanded CD69−TTE cells was that of highly cytotoxic CD8+TTE cells.
CD69+TTE cells account for a small proportion of TTE cells in BM of MGUS patients
Using mass cytometry, we next compared CD69−TTE, CD69+TTE, and total TTE cells in small cohorts of premalignant MGUS and NDMM patients, aiming to understand CD69−TTE-cell and CD69+TTE-cell development during disease progression. We used 2 approaches: manual gating and FlowSOM clustering of mass cytometry data. Although data are preliminary (due to limited availability of BM samples from MGUS patients), both CD69−TTE and CD69+TTE cells were clearly identified within total BM-TTE cells in MGUS and maintain the same tissue distribution as their counterparts in NDMM (Figure 5A-C). CD69+TTE and total TTE cells accounted for a significantly lower proportion of BM-CD8+T cells in MGUS than in NDMM (Figure 5D; supplemental Figure 2B). In particular, CD69+TTE cells were very sparse, representing on average 13.8% of TTE cells and 2.1% of CD8+T cells in BM of MGUS patients (Figure 5C-D). Expression of CD69 on TM cells, and the extent of its downregulation during the transition from the TM to the TTE stage of differentiation, appeared to be related to the proportions of CD69+TTE cells in BM of NDMM and MGUS patients (Figure 5E). Our data suggest that CD69 expression may be amplified on TM and TTE cells in BM of NDMM compared with their counterparts in BM of MGUS patients.
Figure 5.
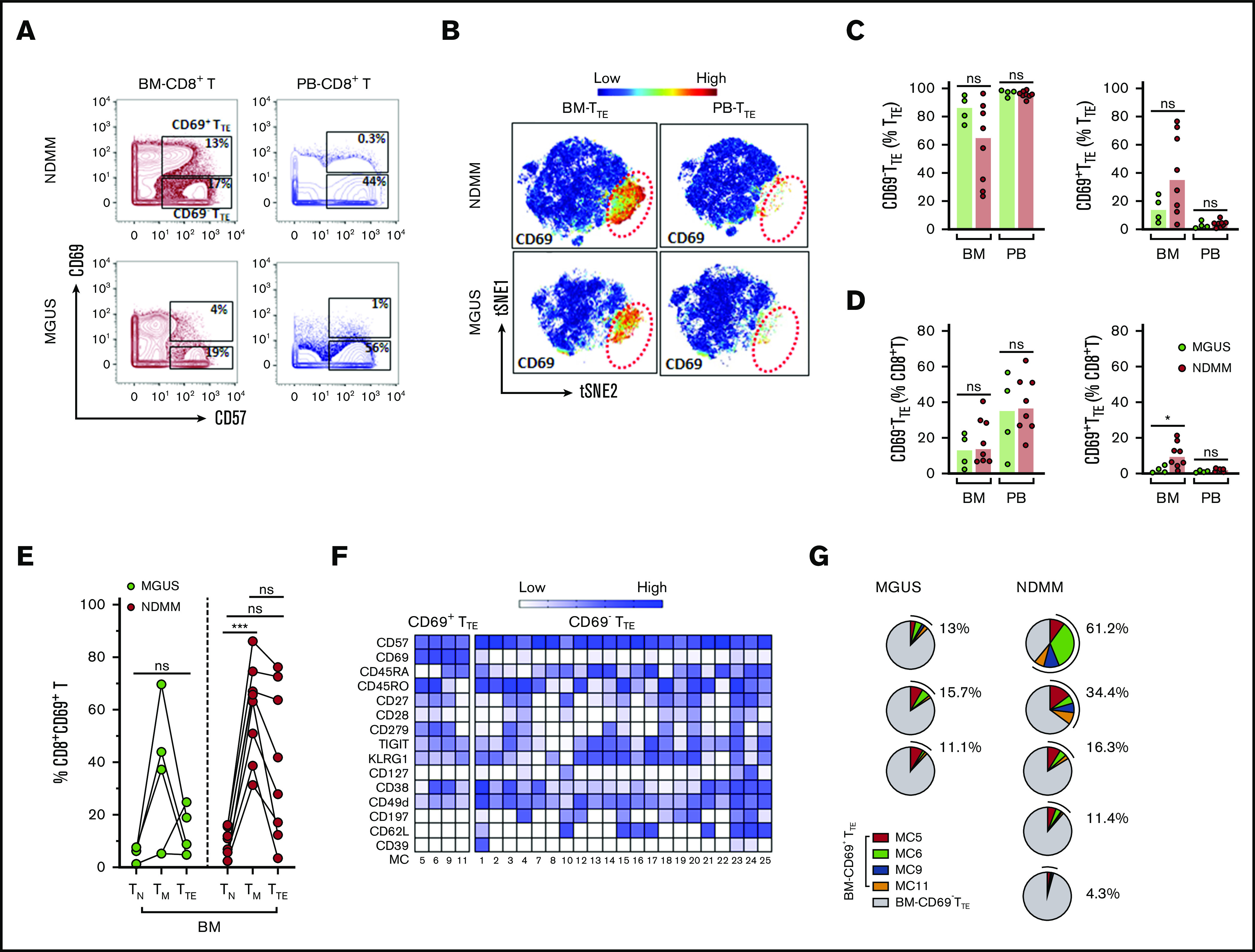
Mass cytometric analysis of TTEcells in MGUS and NDMM patients. (A) Biaxial contour plots gated for CD8+T cells show distribution of CD69+TTE and CD69−TTE cells in NDMM and MGUS patients. Regions occupied by CD69+TTE and CD69−TTE cells are indicated. Numbers indicate proportions of CD69+TTE and CD69−TTE cells within BM-CD8+T and PB-CD8+T cells of NDMM and MGUS patients. (B) tSNE plots show distribution of CD69−TTE and CD69+TTE cells within pooled BM-TTE and PB-TTE cells of NDMM patients (n = 5, shown in Figure 4A) and MGUS patients (n = 3). The area occupied by CD69+TTE cells is indicated by the red dotted circle. Bars (median with scatter plots) show proportions of CD69−TTE and CD69+TTE cells within TTE (C) and CD8+T (D) cells in the BM and PB of NDMM (n = 8) and MGUS (n = 4) patients. *P < .05. (E) Graph shows proportion of CD69+ cells within the TN, TM, and TTE compartments in the BM of MGUS (n = 4) and NDMM (n = 8) patients. TN, TM, and TTE cells from the same patient are connected by lines. ***P < .001; Friedman test with Dunn’s multiple comparators. (F) Heat map showing the phenotype of the 25 MCs defined by FlowSOM analysis of TTE cells. MC5, MC6, MC9, and MC11 contain CD69+TTE cells, and CD69−TTE cells are distributed within the remaining 21 MCs. The intensity of the color in each cell indicates the median signal intensity for an individual marker (row) in an individual MC (column). (G) Pie charts show the contribution of CD69+TTE cells assigned to MC5, MC6, MC9, and MC11 to the BM-TTE cells in NDMM (n = 5) and MGUS (n = 3) patients. Numbers indicate total CD69+TTE cells within MC5, MC6, MC9, and MC11 in each patient, expressed as a percentage of total BM-TTE cells.
Finally, to compare the degree of heterogeneity of BM-TTE cells in both MGUS and NDMM, we analyzed the phenotypes of the 25 metaclusters (MCs) generated by FlowSOM clustering (Figure 5F-G). This revealed that BM-CD69+TTE cells, like their CD69−TTE counterparts, are still phenotypically heterogeneous in both MGUS and NDMM. CD69+TTE can be subdivided into 4 MCs based on differences in expression of CD45RA, CD45RO, CD27, CD28, CD279, and CD38 while uniformly expressing TIGIT, KLRG1, and CD49d and lacking expression of CD127, CD197 (CCR7), CD62L, and CD39 (Figure 5F). These 4 MC are variable in size but persist in all MGUS and NDMM patients (Figure 5G).
Discussion
This study provides the first definitive evidence that circulating oligoclonal expanded cytotoxic CD69−TTE cells are myeloma-reactive cells capable of eliminating autologous CD38hiPCs in vitro. It also suggests the novel concept that balance between cytotoxic oligoclonal expanded CD69−TTE cells and noncytotoxic CD69+TTE cells, which resemble TRM cells with BM residency, may regulate immune responses in NDMM patients.
The most clinically important outcome of this study is the demonstration that antimyeloma responses occur in NDMM patients and are executed by oligoclonal expanded PB-CD69−TTE cells. Thus, oligoclonal expanded PB-CD69−TTE cells, which we previously reported as “T-cell clones,”7 are indeed myeloma reactive. Elimination of autologous CD38hiPCs occurs rapidly within a 2-hour culture, likely through degranulation and release of preformed perforin and granzyme from lytic granules of PB-CD69−TTE cells. As expected, no IFN-γ production was detected after 2 hours of culture, as it requires a longer time to be produced de novo following T-cell activation.25 The potent cytotoxic functions of myeloma-reactive PB-CD69−TTE cells are further reinforced by their lower expression of inhibitory receptors, CD279, TIGIT, Lag 3, and CD160, consistent with our previous studies.11,26 Mass cytometric analysis indicated a high degree of similarity between CD69−TTE cells in PB and BM, and this similarity is further extended to the dominant oligoclonal expansions present in both tissues (BM and PB) of NDMM patients. Direct testing of oligoclonal expanded CD69−TTE cells in the BM for their myeloma reactivity remains challenging due to their limited numbers and small volume of diagnostic BM samples available for research. However, our data are consistent with the concept that myeloma-reactive cells likely arise within MIL in the MM microenvironment and undergo oligoclonal expansion and terminal differentiation into CD69−TTE cells, which then circulate between BM and PB.
We found that myeloma reactivity is not restricted to oligoclonal expanded PB-CD69−TTE cells expressing dominant TCR-Vβ families, since remaining PB-CD69−TTE cells expressing less expanded TCR-Vβ families also retain some capacity to eliminate autologous CD38hiPCs. Also, highly expanded TCR-Vβ families representing >50% of PB-TTE cells are not restricted to patients, as they are occasionally seen in age-matched controls, perhaps induced by aging-associated autoimmune and infective processes.27 We ruled out the possibility that contaminating NKT and γδ T cells play a significant role in the elimination of autologous CD38hiPCs. This is consistent with the observation that NKT and γδ T cells are not required for antimyeloma immunity in mice bearing Vk*MYC myeloma and that CD8+T-cell clones of rare, small, and medium size are protective.1 Nonetheless, antigen specificity of readily accessible TCR-Vβ restricted myeloma-reactive PB-CD69−TTE cells could be further explored, in particular by testing for restriction to major histocompatibility complex class I–related molecule (MR1), which is expressed on myeloma cells.28 The possibility that PB-CD69−TTE cells include MR1-reactive T cells capable of killing a variety of cancers expressing MR129,30 is an interesting future research topic.
Our finding that oligoclonal expanded PB-CD69−TTE cells in NDMM patients are highly functional conflicts with a number of reports that T cells in MM patients are dysfunctional, senescent, and/or exhausted.13,14,16 Our data strongly support the concept that potent antimyeloma immunity mediated by oligoclonal expanded CD69−TTE cells persists in NDMM. However, how these naturally induced circulating myeloma-reactive CD69−TTE cells can be retained within MIL and protected from the harmful effects of myeloma therapeutics is not clear. In particular, it will be important to determine the sensitivity of myeloma-reactive CD69−TTE cells to myeloma therapies and to understand their role in the immunological aspects of autologous stem cell transplantation, a front-line therapy for transplant-eligible MM patients.20
In addition to defining myeloma-reactive cytotoxic CD69−TTE cells, this study provides a detailed analysis of noncytotoxic, proinflammatory BM-CD69+TTE cells that are present in controls and all patient groups. Correlative analysis performed in this study revealed that noncytotoxic BM CD69+TTE cells maintained inverse relationships with their CD69−TTE counterparts within BM-TTE cells of NDMM patients, but not MGUS patients and controls. They also maintain negative relationships with myeloma-reactive oligoclonal expanded PB-CD69−TTE cells expressing dominant TCR-Vβ families indicating altered immune homeostasis over and above the direct effect of oligoclonal expansion within the CD69−TTE subset. How the development of CD69+TTE and CD69−TTE cells within the BM-TTE compartment is regulated remains unclear. However, our mass cytometry data suggest that the development of BM-CD69+TTE cells may be more closely related to the transition from TM cells expressing CD69 rather than from CD69−TTE cells and that regulation of this transition may differ between NDMM and MGUS patients.
We demonstrated that CD69+TTE cells have markedly different properties from cytotoxic CD69−TTE cells, including low expression of the cytotoxic molecules perforin and granzyme B, an EomeshiTbetlow/neg transcriptional signature, and high expression of multiple inhibitory checkpoints, such as CD279, TIGIT, Lag 3, and CD160, suggesting CD69+TTE cells may be a suitable target for checkpoint inhibition immunotherapy.17 CD69+TTE cells reside in the BM and appear closely related to TRM cells.23 Small proportions of CD8+CD69+CD57+TRM cells have been reported in human lung and spleen23 and may be equivalent to the CD69+TTE cells described in this study. To the best of our knowledge, this is the first documentation of BM-resident CD69+TTE cells, which likely belong to the resident CD8+CD69+ cells seen in human BM.31
We found that CD69+TTE cells account for highly variable proportions of the BM-TTE compartment in NDMM patients (5% to 84%), a finding that may relate to clinical heterogeneity. CD69+TTE cells appear to be less frequent within BM-TTE cells of MGUS and SMM patients (6% to 47% MGUS; 6% to 34% SMM), suggesting that progression to clinical MM, at least in some patients, may be associated with an accumulation of noncytotoxic CD69+TTE cells within MILs. Accumulation of CD69+TTE within MILs may contribute to local inflammation through the production of the proinflammatory cytokines IFN-γ and TNF-α, impair the development of cytotoxic CD69−TTE cells, and thus promote myeloma growth. It has already been demonstrated by a study in CD69-knockout mice that CD69 expression on T cells impaired the antitumor immune response, suggesting CD69 is an attractive target for cancer immunotherapy.32 Understanding the role of CD69+TTE cells within MILs throughout disease progression has the potential to lead to the development of novel immune-based approaches for the management of MM.
Our study suggests that changes in TTE cells contributing to MILs, without apparent changes in the PB-TTE compartment, are associated with myeloma progression from premalignant MGUS or asymptomatic SMM. Data also suggests that the development of cytotoxic CD69−TTE cells, which mediate antimyeloma responses in NDMM patients, can be affected by the accumulation of noncytotoxic CD69+TTE cells within MILs. Tracing CD69+TTE cells within MILs and correlating their numbers with clinical outcome in MM patients receiving MILs as adoptive T-cell therapy33,34 could provide essential insights into role of BM-CD69+TTE cells in antimyeloma immunity.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
The authors thank Alberto Catalano for excellent research management support, RPAH Medical Registrars for collection of patient samples and informed consent, and patients and their families for donating samples for research. They would also like to thank all the support staff at Sydney Cytometry and the Ramaciotti Facility for Human Systems Biology for their assistance with the mass cytometry studies.
This work is funded by a Brian D. Novis research grant from the International Myeloma Foundation (C.E.B.).
Footnotes
Data sharing statement: e-mails to the corresponding authors, Slavica Vuckovic (slavica.vuckovic@health.nsw.gov.au) and Christian E. Bryant (christian.bryant@health.nsw.gov.au).
Authorship
Contribution: S.V. and C.E.B. designed and performed the research, analyzed the data, and wrote the paper; K.H.A.L. and S.Y. performed the research and analyzed the data; J.F. performed the research, wrote the human ethics, and assisted in writing the paper; H.M.M. designed and performed mass cytometry assays; G.C. assisted research design and collection of the control samples; B.F.d.S.G. assisted in research design, mass cytometry data analysis, and writing the paper; F.M.-W. assisted with the flow/CAPX approach and analyzed FlowSOM data; N.N. assisted in research design and data analysis; C.E.B., E.A., V.V., D.M., C.B., S.L., S.D., L.K., J.G., and P.J.H. assisted in research design, reviewed patients, and assisted with the collection of patient samples and clinical information; R.B. reviewed patients undergoing hip arthroplasty and designed research; and D.J. and P.J.H. assisted in research design and writing the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Slavica Vuckovic, Royal Prince Alfred Hospital, Level 5, Building 77, Missenden Rd, Camperdown, NSW 2050, Australia; e-mail: slavica.vuckovic@health.nsw.gov.au; and Christian E. Bryant, Royal Prince Alfred Hospital, Level 5, Building 77, Missenden Rd, Camperdown, NSW 2050, Australia; e-mail: christian.bryant@health.nsw.gov.au.
References
- 1.Vuckovic S, Minnie SA, Smith D, et al. Bone marrow transplantation generates T cell-dependent control of myeloma in mice. J Clin Invest. 2019;129(1):106-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dong S, Ghobrial IM. Autologous graft versus myeloma: it’s not a myth. J Clin Invest. 2019;129(1):48-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Byrne JL, Carter GI, Ellis I, Haynes AP, Russell NH. Autologous GVHD following PBSCT, with evidence for a graft-versus-myeloma effect. Bone Marrow Transplant. 1997;20(6):517-520. [DOI] [PubMed] [Google Scholar]
- 4.Tricot G, Vesole DH, Jagannath S, Hilton J, Munshi N, Barlogie B. Graft-versus-myeloma effect: proof of principle. Blood. 1996;87(3):1196-1198. [PubMed] [Google Scholar]
- 5.Joshua D, Suen H, Brown R, et al. The T cell in myeloma. Clin Lymphoma Myeloma Leuk. 2016;16(10):537-542. [DOI] [PubMed] [Google Scholar]
- 6.Perumal D, Imai N, Laganà A, et al. Mutation-derived neoantigen-specific T-cell responses in multiple myeloma. Clin Cancer Res. 2020;26(2):450-464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sze DM, Giesajtis G, Brown RD, et al. Clonal cytotoxic T cells are expanded in myeloma and reside in the CD8(+)CD57(+)CD28(-) compartment. Blood. 2001;98(9):2817-2827. [DOI] [PubMed] [Google Scholar]
- 8.Mahnke YD, Brodie TM, Sallusto F, Roederer M, Lugli E. The who’s who of T-cell differentiation: human memory T-cell subsets. Eur J Immunol. 2013;43(11):2797-2809. [DOI] [PubMed] [Google Scholar]
- 9.Chattopadhyay PK, Betts MR, Price DA, et al. The cytolytic enzymes granyzme A, granzyme B, and perforin: expression patterns, cell distribution, and their relationship to cell maturity and bright CD57 expression. J Leukoc Biol. 2009;85(1):88-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown RD, Spencer A, Ho PJ, et al. Prognostically significant cytotoxic T cell clones are stimulated after thalidomide therapy in patients with multiple myeloma. Leuk Lymphoma. 2009;50(11):1860-1864. [DOI] [PubMed] [Google Scholar]
- 11.Suen H, Brown R, Yang S, Ho PJ, Gibson J, Joshua D. The failure of immune checkpoint blockade in multiple myeloma with PD-1 inhibitors in a phase 1 study. Leukemia. 2015;29(7):1621-1622. [DOI] [PubMed] [Google Scholar]
- 12.Badros A, Hyjek E, Ma N, et al. Pembrolizumab, pomalidomide, and low-dose dexamethasone for relapsed/refractory multiple myeloma. Blood. 2017;130(10):1189-1197. [DOI] [PubMed] [Google Scholar]
- 13.Zelle-Rieser C, Thangavadivel S, Biedermann R, et al. T cells in multiple myeloma display features of exhaustion and senescence at the tumor site. J Hematol Oncol. 2016;9(1):116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sponaas AM, Yang R, Rustad EH, et al. PD1 is expressed on exhausted T cells as well as virus specific memory CD8+ T cells in the bone marrow of myeloma patients. Oncotarget. 2018;9(62):32024-32035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bailur JK, McCachren SS, Doxie DB, et al. Early alterations in stem-like/resident T cells, innate and myeloid cells in the bone marrow in preneoplastic gammopathy. JCI Insight. 2019;5:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kourelis TV, Villasboas JC, Jessen E, et al. Mass cytometry dissects T cell heterogeneity in the immune tumor microenvironment of common dysproteinemias at diagnosis and after first line therapies. Blood Cancer J. 2019;9(9):72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Franssen LE, Mutis T, Lokhorst HM, van de Donk NWCJ. Immunotherapy in myeloma: how far have we come? Ther Adv Hematol. 2019;10:2040620718822660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bendall SC, Nolan GP, Roederer M, Chattopadhyay PK. A deep profiler’s guide to cytometry. Trends Immunol. 2012;33(7):323-332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Gassen S, Callebaut B, Van Helden MJ, et al. FlowSOM: using self-organizing maps for visualization and interpretation of cytometry data. Cytometry A. 2015;87(7):636-645. [DOI] [PubMed] [Google Scholar]
- 20.Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15(12):e538-e548. [DOI] [PubMed] [Google Scholar]
- 21.Laurens van der Matten HG. Visualizing data using t-SNE. J Mach Learn Res. 2008;9:2579-2605. [Google Scholar]
- 22.Ashhurst TM, Cox DA, Smith AL, King NJC. Analysis of the murine bone marrow hematopoietic system using mass and flow cytometry. Methods Mol Biol. 2019;1989:159-192. [DOI] [PubMed] [Google Scholar]
- 23.Kumar BV, Ma W, Miron M, et al. Human tissue-resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep. 2017;20(12):2921-2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dolstra H, Preijers F, Van de Wiel-van Kemenade E, Schattenberg A, Galama J, de Witte T. Expansion of CD8+CD57+ T cells after allogeneic BMT is related with a low incidence of relapse and with cytomegalovirus infection. Br J Haematol. 1995;90(2):300-307. [DOI] [PubMed] [Google Scholar]
- 25.Betts MR, Brenchley JM, Price DA, et al. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods. 2003;281(1-2):65-78. [DOI] [PubMed] [Google Scholar]
- 26.Suen H, Brown R, Yang S, et al. Multiple myeloma causes clonal T-cell immunosenescence: identification of potential novel targets for promoting tumour immunity and implications for checkpoint blockade. Leukemia. 2016;30(8):1716-1724. [DOI] [PubMed] [Google Scholar]
- 27.Strioga M, Pasukoniene V, Characiejus D. CD8+ CD28- and CD8+ CD57+ T cells and their role in health and disease. Immunology. 2011;134(1):17-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gherardin NA, Loh L, Admojo L, et al. Enumeration, functional responses and cytotoxic capacity of MAIT cells in newly diagnosed and relapsed multiple myeloma. Sci Rep. 2018;8(1):4159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lepore M, Kalinichenko A, Calogero S, et al. Functionally diverse human T cells recognize non-microbial antigens presented by MR1. eLife. 2017;6:e24476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crowther MD, Dolton G, Legut M, et al. Genome-wide CRISPR-Cas9 screening reveals ubiquitous T cell cancer targeting via the monomorphic MHC class I-related protein MR1 [published correction appears in Nat Immunol. 21:695]. Nat Immunol. 2020;21(2):178-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pascutti MF, Geerman S, Collins N, et al. Peripheral and systemic antigens elicit an expandable pool of resident memory CD8+ T cells in the bone marrow. Eur J Immunol. 2019;49(6):853-872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mita Y, Kimura MY, Hayashizaki K, et al. Crucial role of CD69 in anti-tumor immunity through regulating the exhaustion of tumor-infiltrating T cells. Int Immunol. 2018;30(12):559-567. [DOI] [PubMed] [Google Scholar]
- 33.Noonan K, Matsui W, Serafini P, et al. Activated marrow-infiltrating lymphocytes effectively target plasma cells and their clonogenic precursors. Cancer Res. 2005;65(5):2026-2034. [DOI] [PubMed] [Google Scholar]
- 34.Noonan KA, Huff CA, Davis J, et al. Adoptive transfer of activated marrow-infiltrating lymphocytes induces measurable antitumor immunity in the bone marrow in multiple myeloma. Sci Transl Med. 2015;7(288):288ra78. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.