

Key Points
Chemotherapy has lingering effects on surviving T cells, which are evident in damaged mitochondrial energy reserve.
This damage impairs proliferation required by adoptive cell therapies.
Abstract
Engineered T-cell therapies have demonstrated impressive clinical responses in patients with hematologic malignancies. Despite this efficacy, many patients have a transient persistence of T cells, which can be correlated with transient clinical response. Translational data on T cells from pediatric cancer patients shows a progressive decline in chimeric antigen receptor (CAR) suitability with cumulative chemotherapy regardless of regimen. We investigated the effects of chemotherapy on surviving T cells in vitro, describing residual deficits unique to each agent including mitochondrial damage and metabolic alterations. In the case of cyclophosphamide but not doxorubicin or cytarabine, these effects could be reversed with N-acetylcysteine. Specifically, we observed that surviving T cells could be stimulated, expanded, and transduced with CARs with preserved short-term cytolytic function but at far lower numbers and with residual metabolic deficits. These data have implications for understanding the effects of chemotherapy on mature T cells later collected for adoptive cell therapy, as chemotherapy-exposed T cells may have lingering dysfunction that affects ex vivo adoptive cell therapy manufacturing techniques and, ultimately, clinical efficacy.
Visual Abstract

Introduction
Immune-based therapies are emerging as a promising alternative or supplement to conventional chemotherapy.1,2 Most of these, such as chimeric antigen receptor (CAR) T cells or negative checkpoint receptor (NCR) inhibitors, are being used in the relapsed or refractory setting after intense multimodal chemotherapy.3-6 Little is known about the direct or lasting effects of chemotherapy on surviving T cells, and how this may impact the use of immune-engaging therapies, whether in the relapsed or upfront setting or even when integrated into chemotherapy backbones. Translational data from 2 landmark trials indicate that the presence of early memory or naive T cells, or conversely the lack of exhausted T cells, correlates with better expansion and persistence of the CAR T-cell products.7,8 These surface phenotypes, however, do not provide complete sensitivity as a standalone biomarker of CAR T-cell performance. We wanted to explore non–surface marker phenotypes of T cells that survive chemotherapy to investigate other signatures of potential dysfunction.
Prior reports in pediatric patients documented lower absolute T-cell counts in children with leukemia both at diagnosis and throughout therapy, and that chemotherapy can deplete T cells over time.9,10 Importantly, T cells recovered from children after chemotherapy cycles demonstrated an increased incidence of activation-induced cell death (AICD) in response to mitogens though the mechanism underlying this is not clear.10-14 The internal energy potential of T cells measured by oxygen consumption analysis and mitochondrial membrane potential (Δψ) have also been shown to be critical to the potency of T-cell therapies.15-17 This makes intuitive sense, as the mitochondrial mass and energy reserve of T cells changes with the transition from naive to memory to effector states.18 We used in vitro assays of chemotherapy exposure to characterize the effects on mature T cells and each subset to help elucidate the potential impact on T-cell energy reserve as a possible mechanism of poor T-cell function postchemotherapy. We found that 3 classes of chemotherapy acted in different ways to cripple T-cell energy reserve at the mitochondrial level as well as via the reliance on glycolysis, and that this varied between T-cell subsets as well. The ability of T cells and certain subsets to recover and expand varied depending on the nature and timing of this exposure. We assessed the use of N-acetylcysteine (NAC), a known antidote to reactive oxygen species damage caused by cyclophosphamide, to reverse the effects on proliferation. Using this model, we made CAR T cells from cyclophosphamide-exposed T cells with and without NAC, showing that NAC significantly increased the recovery of CAR+ cells and restored metabolic reserve. Adaptive cell manufacture, altering the media or reagents to grow T cells based on biomarkers for each product, is a subject of intense investigation.19,20 These data reveal lingering deficits in chemotherapy-exposed T cells with a metabolic signature that may potentially be targeted for reversal with adaptive manufacturing to improve cell therapies.
Materials and methods
Chemotherapy
Doxorubicin (CAS number 25316-40-9) and Cytarabine (CAS number 147-94-4) were purchased from Sigma-Aldrich; 4-hydroperoxycyclophosphamide (4-HPCP; CAS number 39800-16-3) was purchased from Santa Cruz Biotechnology.
Ex vivo T-cell expansion and culture
Lymphocytes were harvested from peripheral blood and expanded as previously described.21 T cells from normal donors were obtained under an institutional review board–approved protocol from the Human Immunology Core at the University of Pennsylvania and deidentified prior to receipt. For all chemotherapy experiments using normal donor T cells, a minimum of 3 different donors were tested in duplicate to ensure generalized results.
Flow cytometry
T cells were stained for cell-surface markers to differentiate T-cell lineage. Previous work22 has demonstrated that CCR7, CD62L, CD45RO, and CD95 can be used to differentiate the various T-cell phenotypes by using the following expression patterns: naive (TN): CCR7+, CD62L+, CD45RO−, CD95−; stem central memory (TSCM): CCR7+, CD62L+, CD45RO−, CD95+; central memory (TCM): CCR7+, CD62L+, CD45RO+, CD95+; effector memory (TEM): CCR7−, CD62L−, CD45RO+, CD95+; and terminal effector (TEff): CCR7−, CD62L−, CD45RO−, CD95+. The antibodies used for this analysis as well as quantitation of T-cell bulk described herein were CD8-fluorescein isothiocyanate (FITC) (BD Biosciences, Franklin Lakes, NJ), CD3-phycoerythrin (PE) (BD Biosciences), CD4-allophycocyanin (BD Biosciences), CCR7-FITC (BD Biosciences), CD95-PE (BD Biosciences), CD45RO (BD Biosciences) and CD62L-PE/Cy7 (BioLegend, San Diego, CA). Samples were then washed twice and flow cytometry acquisition was performed on a BD FACS Verse Flow Cytometer or BD Accuri C6 (BD Biosciences). Sorting was done using MoFlo Astrios (Beckman Coulter, Inc).
To determine the Δψ, JC1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolcarbocyanine iodide) (BD Biosciences) was used as per manufacturer’s instruction. MitoTracker Green (Molecular Probes) was used to determine the mitochondrial mass in different treatment groups using flow cytometry per manufacturer’s instructions.
For B-cell lymphoma 2 (BCL-2) staining (BD Bioscience) intracellular flow cytometry is performed after the stipulated time of treatment with chemotherapeutic agents using appropriate isotype control. Analysis was performed using FlowJo software (TreeStar, Inc, Ashland, OR).
Analysis of mitochondrial function
Seahorse XF Analyzers measure oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) of live cells in a multiwell plate, interrogating key cellular functions such as mitochondrial respiration and glycolysis. OCR and ECAR rates are key indicators of mitochondrial respiration and glycolysis and these measurements provide a systems-level view of cellular metabolic function in cultured cells and ex-vivo samples.
Seahorse XF Cell Mito Stress Test was performed to measure the effect of chemotherapeutic drugs on mitochondrial OCR and ECAR using an extracellular flux analyzer (Seahorse Bioscience). Each well of the XF96 cell culture plates was coated with Corning Cell-Tak cell and tissue adhesive in accordance with the manufacturer’s instructions. T cells were treated in 6-well tissue culture plate with different drugs and cells were transferred to 15ml tubes, centrifuged at 1200g for 5 minutes. Cell pellets were then resuspended in XF assay medium (nonbuffered RPMI 1640) containing 5.5 mM glucose, 2 mM l-glutamine, and 1 mM sodium pyruvate (pH 7.4), counted and seeded in an XF 96-well plate leaving the 4 corner wells as blank. The microplate was then centrifuged at 1000g for 5 minutes and incubated in standard culture conditions for 45 minutes for the proper attachment of the cells. The plate was then transferred into a 37°C non-CO2 incubator for at least 60 minutes before running the assay. Prior to the day of the assay the Agilent seahorse XFe96 sensor cartridge was hydrated in accordance with the manufacturer’s instructions. On the day of the assay, the hydrated cartridge was calibrated (∼30 minutes). After the calibration was done, the cell plate was loaded in the flux analyzer for running the assay. Mitochondrial functions were measured under basal conditions and in response to mitochondrial inhibitors oligomycin (inhibitor of ATP synthase), carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone (FCCP) (mitochondrial oxidative phosphorylation uncoupler), and rotenone with Antimycin-A (inhibitor of complex I and III, respectively) (Sigma-Aldrich, St. Louis, MO). All activities were normalized to the number of cells present in the wells for each treatment groups.17
Citrate synthase activity measurement
Citrate synthase (CS) is the initial enzyme of the tricarboxylic acid (TCA) cycle and an exclusive marker of the mitochondrial matrix. CS activity was determined in cell lysates using a Citrate Synthase Assay Kit (Sigma-Aldrich, St. Louis, MO). Total protein was determined by the method of Bradford, and the protein concentration of all samples was equalized. Citrate synthase activity was determined based on the formation of yellow color 5-thio-2-nitrobenzoic acid (TNB) and measured at a wavelength of 412 nm at 25°C on a spectrophotometer (Benchmark Plus Microplate Spectrophotometer; BioRad) according to the manufacturer’s protocol.
Glucose and lactate analysis
T cells after treatment with chemotherapeutic drugs were starved in PBS at room temperature for 30 to 45 minutes followed by incubation at 37°C in regular RPMI 1640 culture media supplemented with 11 mM glucose, 10% dialyzed FBS, 100 U/mL penicillin, 100 mg/mL streptomycin sulfate, and 2 mM glutamax. Five hundred microliter aliquots of cell culture were collected at indicated time points and spun down, and the supernatants were analyzed for glucose and lactate concentrations with the YSI 2950 Biochemistry Analyzer (YSI Life Sciences).
Glucose uptake assay
The fluorescently-labeled glucose analog (2-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-2-deoxyglucose [2-NBDG]; 30 mM stock) (Glucose Uptake Cell-Based Assay Kit; Cayman Chemical) was used to measure the relative glucose uptake according to the manufacturer’s protocol. Briefly, in each condition, T cells were incubated with 2-NBD-glucose (1/100 dilution) in glucose-free media for 30 minutes at 37°C, subsequently washed twice in cell-based assay buffer provided with the kit, and analyzed by flow cytometry in fluorescence channel FL1. Apigenin, a flavonoid that has been reported to be an inhibitor of glucose transport mediated by GLUT1, is included as a control.23
Transmission electron microscopy
For transmission electron microscopy (TEM), T cells were prepared with the Penn Electron Microscopy Resource Laboratory. Briefly, T cells with different treatments were fixed with 2.5% glutaraldehyde, 2.0% paraformaldehyde in 0.1 M sodium cacodylate buffer (pH 7.4) overnight at 4°C. After subsequent buffer washes, the samples were postfixed in 2.0% osmium tetroxide for 1 hour at room temperature, and then washed again in buffer followed by distilled water. After the dehydration through a graded ethanol series, the sample was then infiltrated and embedded in EMbed-812 (Electron Microscopy Sciences, Fort Washington, PA). Thin sections were stained with uranyl acetate and lead citrate and examined with a JEOL 1010 electron microscope fitted with a Hamamatsu digital camera and AMT (Advanced Microscopy Techniques Corp) Advantage image capture software.
CD107a and IFNγ
Assays were performed as previously described.24,25 Please see supplemental Methods for detailed descriptions of the CAR manufacturing and assay conditions.
Statistical analysis
All statistical analysis was performed using Prism 6 (version 6.02; Graphpad Software, La Jolla, CA) using analysis of variance testing for group comparisons. Regression analysis was used for analysis of trends over time. The Fisher's exact test was used for comparison of percentages. All comparisons that are reported as significant reached P < .05, or as calculated after Bonferroni corrections for multiple comparisons.
Results
Examination of mitochondrial integrity after chemotherapy exposure
Mitochondrial damage after chemotherapy is a common pathway and likely largely responsible for cell death via apoptosis. We wished to characterize the effects on mitochondrial integrity and function in the cells that survive chemotherapy exposure, as these are the T cells that would be collected for use in adoptive cell therapy. Cyclophosphamide (4HPCP was used for in vitro studies) had a devastating effect on the Δψ, and this effect was true across naive, CM, or EM) T cells that survived 24 hours of chemotherapy exposure and remained viable for 72 hours (Figure 1). Cytarabine had no effect on membrane potential, and doxorubicin strangely seemed to increase (polarize) membrane potential in CM T cells only. Using a mitochondrial matrix dye (Mitotracker Green), we saw an apparent increase in mitochondrial biomass with cyclophosphamide exposure, though follow-up examination with TEM revealed that this is not accurate. The mitochondria of T cells exposed to cyclophosphamide are small, round, and have short and widened cristae which results in more Mitotracker uptake but no increase in number or size of mitochondria. We attempted to correlate this with a patient sample that had enough T cells both prior to and after cyclophosphamide chemotherapy for primary mediastinal B-cell lymphoma. In this patient, mitochondria in T cells pretherapy appear to have normal long and narrow cristae whereas T cells collected 2 weeks after cyclophosphamide resembled those exposed to cyclophosphamide in vitro with small, round, and dense mitochondria with short, wide cristae (Figure 1). We were only able to assess a single patient, as getting the required number of T cells from a single peripheral blood draw in children frequently does not yield a sufficient number to perform TEM. Lastly, we examined the presence of antiapoptotic factor BCL-2 in T-cell subsets before and after chemotherapy. As expected from prior reports, naive T cells had higher levels of BCL-2 than CM and EM T cells.26 These levels remain high in naive T cells that survive chemotherapy, and CM and EM T cells that survive cyclophosphamide show increased levels of BCL-2 compared with prechemotherapy. BCL-2 may therefore be an important component of T cells that survive the mitochondrial damage from cyclophosphamide chemotherapy (Figure 2).
Figure 1.

Mitochondrial integrity after chemotherapy exposure. (A) T cells either in bulk or sorted into 3 subsets (naive, CM, or EM) were exposed to chemotherapy for 24 hours, recovered, and then the Δψ measured by JC1 dye. FCCP is used as a positive control to show ablation of mitochondrial potential in untreated T cells; unlabeled columns are no-treatment controls. Cyclophosphamide (4HPCP) ablates the membrane potential in all T-cell subsets. 4HPCP, cyclophosphamide at 2.5 μM; Cytarabine, cytarabine at 3 mM; Dox, doxorubicin at 400 nM. (B) There is no apparent change in mitochondrial biomass after chemotherapy exposure with the exception of more dye uptake in cyclophosphamide-exposed T cells. Doxo, doxorubicin. (C) Transmission electron microscopy (×75 000) of T cells reveals ultrastructural impact of cyclophosphamide. 1, T cells from a normal donor; 2, the same donor T cells exposed to cyclophosphamide for 24 hours; 3, T cells from a non-Hodgkin lymphoma patient prior to any chemotherapy; and 4, the same patient 2 weeks after cyclophosphamide-containing chemotherapy. The short and widened cristae are characteristic of cyclophosphamide exposure in vitro or in vivo. *P < .05. MFI, mean fluorescence intensity.
Figure 2.

BCL-2 expression in T cells exposed to chemotherapy. T cells from a normal donor either in bulk or sorted into subsets were exposed to chemotherapy and allowed to recover. Live cells were examined by intracellular flow cytometry for BCL-2 expression. Naive cells had higher baseline BCL-2 than memory cells, and memory T cells that survived cyclophosphamide had high BCL-2 levels compared with controls. *P < .01.
Chemotherapy alters T-cell mitochondrial respiration both before and after stimulation
We tested the OCR, spare respiratory capacity (SRC), and ECAR in T cells exposed to doxorubicin, cyclophosphamide, and cytarabine for 24 hours and then allowed to recover (see supplemental Figure 1 for dose finding). Cytarabine impaired all aspects in unstimulated T cells, whereas doxorubicin had no significant impact and cyclophosphamide decreased all 3 at only the higher dose (Figure 3). We assessed the same parameters in T cells exposed to chemotherapy for 24 hours and then stimulated for 24 hours with CD3/28 beads. Cytarabine again had significant reductions in all 3 parameters compared with controls. Any dose of chemotherapy impaired the maximum OCR, and had a modest reduction in ECAR. Effects on SRC were varied, with an increase noted for higher doses of cyclophosphamide. We also characterized the consumption of glucose and conversion to lactate under the same conditions. No lactate is generated in unstimulated cells whether exposed to chemotherapy or not. Lactate production is reduced by doxorubicin and nearly completely eliminated by cyclophosphamide and cytarabine in T cells that are stimulated with beads for 48 hours (Figure 3). Stimulated T cells would be expected to use glycolysis and increase lactate production. We wished to correlate these effects to the longer stimulation periods used in CAR T-cell manufacturing. We exposed T cells to chemotherapy for 24 hours, followed by 7 days of CD3/28 stimulation, and then rest until the cells reach 350 fL of volume. In control T cells exposed to chemotherapy and not stimulated, chemotherapy was devastating to all aspects of mitochondrial respiration in surviving T cells over this time frame (∼13 days; Figure 4). T cells that were stimulated as in CAR manufacture had altered mitochondrial respiration profiles, with doxorubicin- and cytarabine-exposed cells showing higher basal glycolysis rates and doxorubicin showing higher compensatory glycolysis rates and ECAR as well. Cyclophosphamide-exposed T cells had a significantly lower SRC compared with all others at this time point (Figure 4). These data indicate not only differences in the short-term and long-term consequences of chemotherapy exposure on T-cell mitochondrial reserve and metabolism, but also that each chemotherapy class has different effects.
Figure 3.

Mitochondrial respiration analysis of T cells exposed to chemotherapy (short-term analysis). (A) OCR analysis of T cells exposed to chemotherapy and either left unstimulated or stimulated with CD3/28 beads for 48 hours. Deficits in OCR are revealed in this short-term stimulation assay. (B) ECAR analysis from the same experiments. Only cytarabine (Cyta) lowers ECAR in stimulated cells. (C) SRC in the same experiments. This measure of mitochondrial reserve is lowered by higher doses of cyclophosphamide and cytarabine in unstimulated cells but these effects are largely gone after short-term stimulation. (D) Conversion of glucose to lactate after chemotherapy exposure and short-term stimulation. Chemotherapy reduces the capacity of T cells to produce lactate. Note: unstimulated cells made no lactate under any conditions. *P < .01.
Figure 4.

Mitochondrial respiration analysis of T cells exposed to chemotherapy (long-term stimulation). In this set of assays, T cells are exposed to chemotherapy for 24 hours, and then allowed to recover and left unstimulated or exposed to CD3/28 beads for 7 days followed by a rest down period of ∼6 days mimicking CAR manufacture. Parameters assessed by row are OCR (A), ECAR (B), SRC (C), basal glycolytic rate (D), and compensatory glycolytic rate (E). Under unstimulated conditions, any amount of chemotherapy is devastating to mitochondrial respiration and glycolytic energy production. With stimulation, surviving T cells have variable alterations characterized most notably by an increase in ECAR and glycolysis in doxorubicin- and cytarabine-exposed T cells and a loss of SRC in cyclophosphamide-exposed T cells. *P < .05.
T-cell subsets have different responses to chemotherapy and this affects their expansion potential
Given the potential importance of naive and early memory cells on the creation of effective CAR T cells, and the clinical data indicating depletion of these cells by chemotherapy, we sought to classify the effects of chemotherapy on each T-cell subset. Looking first at expansion potential, we confirmed our previous report that naive, SCM, and CM cells expand well with CD3/28 bead exposure. EM cells do not proliferate much or blast (reflected by maximal cell volume) when stimulated with CD3/28 beads. As before, we exposed T cells to chemotherapy for 24 hours, and then stimulated with CD3/28 beads for 7 days followed by bead removal and recovery over 6 days. Doxorubicin and cytarabine completely blocked the ability of any T-cell subset to expand (Table 1). Notably, chemotherapy can also affect a T cell’s ability to blast in response to stimulation, with SCM T cells retaining this ability except when exposed to cytarabine. Cyclophosphamide had no significant effect on SCM T cells, which resulted in preserved expansion in these donors. Due to the rarity of SCM cells, we could not perform additional experiments on this subset as we did not recover enough cells for the mitochondrial respiration studies. Nevertheless, we analyzed the OCR, ECAR, and SRC from each subset after chemotherapy exposure and with and without stimulation (Figure 5). In unstimulated cells, all chemotherapy reduced OCR/ECAR/SRC in naive cells far more than in CM or EM cells. Cytarabine was devastating to all T-cell subsets. Cyclophosphamide had little effect on memory T cells. Stimulation for 24 hours, however, largely mitigated the effects of doxorubicin on each parameter. Cyclophosphamide’s effects on naive T cells were largely mitigated, but memory T cells had significant reductions in OCR/ECAR/SRC after stimulation. Cytarabine continued to depress these functions even after stimulation. Additionally, we assessed glucose uptake under these conditions, finding that chemotherapy increases glucose uptake in unstimulated naive cells and reduces it in EM cells. Again, these effects are mitigated by stimulation in cells exposed to doxorubicin, and conversely glucose uptake is reduced in naive and CM exposed to cyclophosphamide and cytarabine. EM cells are largely unaffected (Figure 5). Correlating these findings with another measure of mitochondrial function, we assessed citrate synthase activity in the same conditions. Here, we observe a significant reduction in activity in naive cells exposed to cyclophosphamide and cytarabine but minimal effects on memory T cells (supplemental Figure 2).
Table 1.
T-cell subset expansion and cell size after chemotherapy exposure (long-term)
| Control | Dox | 4HPCP | Cytarabine | |
|---|---|---|---|---|
| Expansion (SD), fold | ||||
| Unstimulated | ||||
| Bulk | 0.71 (.22) | 0.52 (.26) | 0.42 (.10) | 0.50 (.10) |
| Naive | 0.99 (.13) | 0.82 (.20) | 0.41 (.11) | 0.60 (.10) |
| SCM | 1.29 (.11) | 1.10 (.24) | 0.53 (.10) | 0.73 (.11) |
| CM | 0.95 (.08) | 0.69 (.26) | 0.49 (.10) | 0.65 (.12) |
| EM | 0.76 (.24) | 0.67 (.19) | 0.82 (.15) | 0.66 (.10) |
| Stimulated | ||||
| Bulk | 4.10 (2.1) | 0.48 (.23) | 3.80 (.36) | 0.23 (.09) |
| Naive | 4.70 (1.4) | 0.75 (.24) | 0.89 (.12) | 0.33 (.10) |
| SCM | 4.00 (1.8) | 1.30 (.33) | 3.10 (.25) | 0.34 (.10) |
| CM | 5.70 (2.1) | 0.37 (.10) | 0.95 (.11) | 0.32 (.08) |
| EM | 2.10 (1.3) | 0.33 (.11) | 0.55 (.09) | 0.40 (.10) |
| Cell size (SD), fL | ||||
| Unstimulated | ||||
| Bulk | 186 (19) | 186 (20) | 182 (18) | 192 (18) |
| Naive | 186 (19) | 186 (21) | 189 (21) | 186 (18) |
| SCM | 186 (19) | 186 (20) | 190 (20) | 189 (18) |
| CM | 202 (21) | 204 (22) | 204 (20) | 203 (19) |
| EM | 203 (19) | 191 (20) | 208 (20) | 203 (19) |
| Stimulated | ||||
| Bulk | 405 (54) | 285 (26)* | 375 (36) | 208 (18)* |
| Naive | 460 (69) | 308 (42)* | 297 (37)* | 226 (19)* |
| SCM | 505 (59) | 365 (58) | 461 (59) | 189 (18)* |
| CM | 391 (49) | 263 (41)* | 230 (29)* | 203 (19)* |
| EM | 294 (31) | 245 (24) | 229 (24) | 229 (20) |
T cells from normal donors were sorted by flow cytometry and exposed to chemotherapy for 24 hours (doxorubicin at 400 nM, 4HPCP at 2.5 μM, and cytarabine at 3 mM), allowed to recover and then exposed to CD3/28 beads as in CAR manufacture (∼13 days total). Chemotherapy almost uniformly blocked expansion of all T-cell subsets with the exception of 4HPCP (cyclophosphamide)-exposed SCM cells. When looking at the ability of the cells to blast, doxorubicin reduced blasting in most subsets, 4HPCP blocked naive and CM blasting, and cytarabine blocked essentially all cells. This suggests that the action of chemotherapy on each subset is different. Bold values represent statistically significant changes to control (P < .05).
Dox, doxorubicin; SD, standard deviation.
P < .05.
Figure 5.
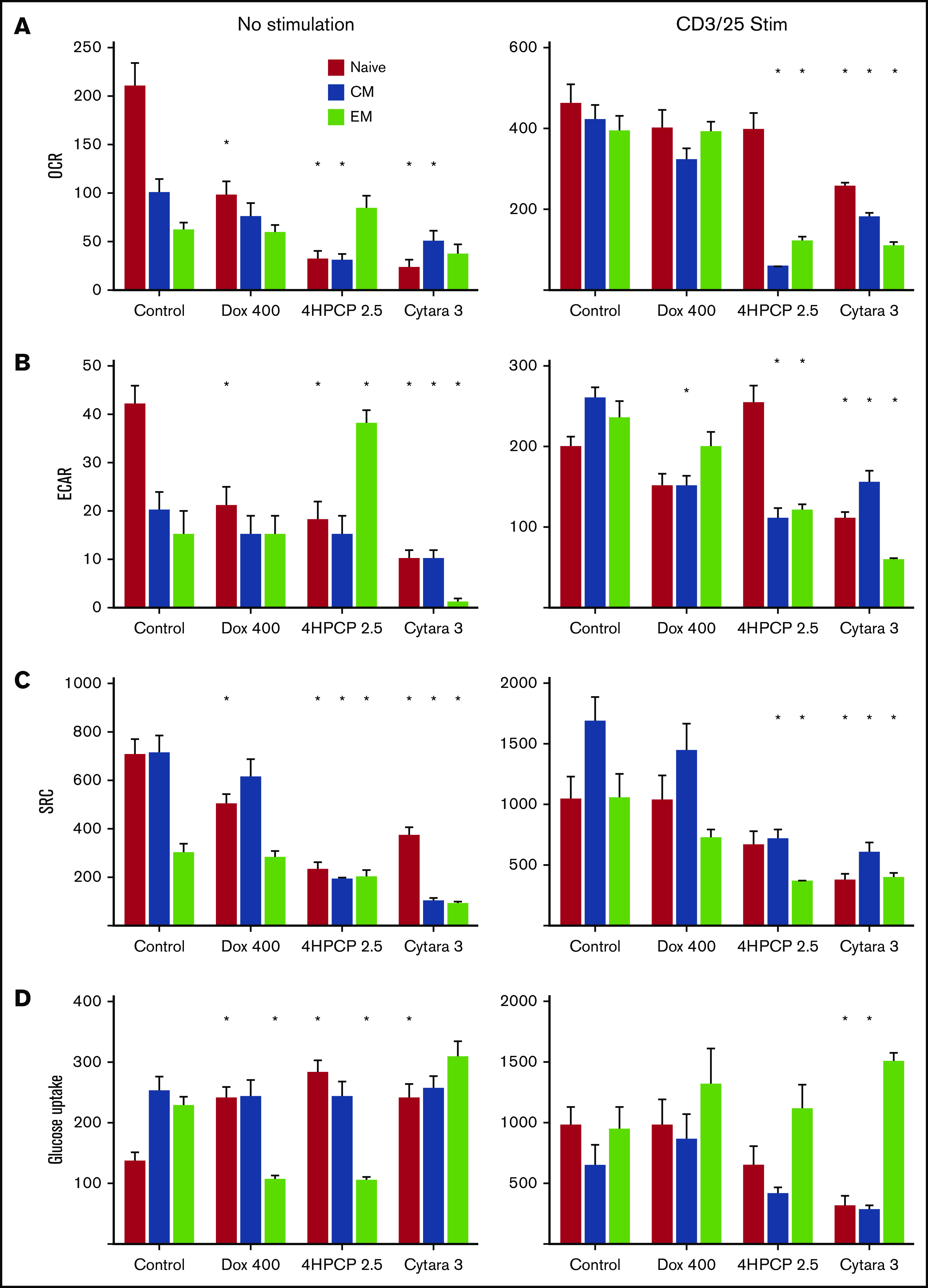
Mitochondrial respiration analysis in T-cell subsets. T cells from normal donors were sorted into 3 subsets and exposed to chemotherapy for 24 hours, allowed to recover, and then either left unstimulated or exposed to CD3/28 beads for 48 hours. Parameters assessed by row are OCR (A), ECAR (B), SRC (C), and glucose uptake (D). Doxorubicin at 400 nM reduces all aspects of respiration in unstimulated naive T cells with a corresponding increase in glucose uptake. Cyclophosphamide and cytarabine (Cytara) are detrimental to nearly all subsets. Stimulation largely rescues the doxorubicin effects but reveals deficits in cyclophosphamide- and cytarabine-exposed T cells. Glucose uptake after stimulation is only affected by cytarabine in naive and CM T cells. *P < .01.
NAC restores proliferation and SRC to cyclophosphamide-exposed T cells
Cyclophosphamide is known to induce reactive oxygen species as a method of damage, and so we applied the ROS scavenger NAC to cyclophosphamide-exposed T cells. The purpose was to explore whether chemotherapy-exposed T cells were defective functionally once made into CAR T cells or if the deficits were less related to cytotoxicity and more to energy reserve and proliferation. Preliminary testing of doxorubicin, cyclophosphamide, and cytarabine with NAC revealed that only cyclophosphamide reversed cell death or mitochondrial function and so only cyclophosphamide was pursued for CAR experiments (supplemental Figure 3). T cells from normal donors (n = 3) were exposed to cyclophosphamide, rescued with NAC, and then manufactured into CAR T cells targeting CD19 as previously described.19 No difference in CAR transduction or mean fluorescence intensity was noted (supplemental Figure 4). We noted a nearly 10-fold reduction in proliferation of cyclophosphamide-exposed T cells, but no reduction in degranulation or cytokine production in those cells that did survive the manufacturing process (Figure 6). The deficit in proliferation and in mitochondrial respiration (OCR and ECAR) were reversed by NAC rescue.
Figure 6.

NAC restores proliferation and metabolic reserve in cyclophosphamide-treated T cells. T cells from normal donors (n = 3) were exposed to cyclophosphamide, rescued with NAC, and then manufactured into CAR T cells targeting CD19 as previously described. We assessed CD107a degranulation against CD19 targets (Nalm-6) (A) as well as IFNγ production (B) in a 24-hour assay. There were no significant differences in short-term killing potential or cytokine release in the T cells that survived manufacture. Significant decreases with cyclophosphamide were seen in proliferation (C) during manufacture, however, with far fewer cell recovered. Peak blasting response measured by cell size (D) was also not affected. (E-F) NAC restores ECAR and OCR to near normal levels. Treatment with NAC restored proliferation and metabolic reserve. *P < .01. PMA+Ion, phorbol 12-myristate 13-acetate plus ionomycin.
Discussion
Although previous studies have assessed the effect of chemotherapy on T cells,9-12 these studies were focused on the depletion aspect in patients as opposed to the effect on surviving cells. We reported for most chemotherapy regimens the potential of T cells to respond to ex vivo stimulation declined over time, representing lingering deficits from chemotherapy exposure in surviving T cells.7 The mechanism behind this has not been explored, nor if the reason behind dysfunction is common or unique to each chemotherapy agent. This may have potential impact on T-cell engaging therapies or those that require harvesting of patient T cells for ex vivo manipulation. Because the deficit we previously described is related to strong stimulation, there is relevance to the field of immune-stimulating therapy. Most if not all CAR T-cell manufacturing strategies incorporate T-cell stimulation, whether using CD3/28 beads or surrogate antigen presentation to the CAR, as stimulation is often required to facilitate integration of retroviral vectors.1,27 Different centers and clinical products are made with different supportive media and reagents, highlighting that multiple support strategies are possible.13,20,28-31
A robust energy response is associated with T-cell stimulation, and we explore here the effects on mitochondrial energy reserve of 3 classes of chemotherapy; doxorubicin, cytarabine and cyclophosphamide. We found profound impact on mitochondrial function and energy reserve across all 3 classes, though the effects varied between agents and on individual T-cell subsets. This held true for both short and long-term stimulation, though the metabolic phenotype of poststimulation chemotherapy-exposed T cells was different than the prestimulation profile in most cases. Stimulation may result in the induction of AICD or the damaged T cells may activate other pathways to survive stimulation stresses, impacting long-term proliferative capacity.
The presence of a high percentage of naive and SCM cells still does not guarantee expansion or clinical efficacy meaning there may be qualitative defects not captured by surface phenotype. With this in mind, we attempted to characterize the internal metabolic reserve that may be associated with better expansion potential. This prompted us to investigate the mitochondrial and metabolic reserve of T cells, including the critical naive T-cell subset. We focused on the effects of chemotherapy, as this was able to be modeled in vitro. Chemotherapy has class specific effects on T cells, including naive T cells, and these effects vary in unstimulated and stimulated T cells. Cyclophosphamide has a devastating effect on all T cells and this is mediated by mitochondrial damage. T cells treated with cyclophosphamide show small, damaged mitochondria with low (depolarized) membrane potential and loss of many measures of mitochondrial reserve including SRC. Remarkably, early stimulation after chemotherapy can alleviate many of these findings over time, though our data suggests this may be due to the SCM subset specifically surviving and proliferating. This subset is rare, and difficult to study without artificial culture conditions so we will focus on further mechanistic studies in this subset in the future having established much of the baseline data here. The mechanism behind doxorubicin remains unclear, as the cells blast in response to stimulation, increasing their volume but then die in a process that appears similar to activation induced cell death. Doxorubicin has little to no effect on mitochondrial integrity or function, and T cells exposed to doxorubicin show short-term impaired lactate production but long-term increased used of glycolysis after stimulation. Although an obvious final pathway is AICD, the relatively intact mitochondrial membrane studies, normal OCR and ECAR rates argue that this process may be more complex and possibly suited to metabolic interventions targeting glucose use to reverse damage and preserve T-cell capacity.32 Cytarabine similarly lacks an easy explanation for how it cripples T-cell proliferation. Here, the pattern is 1 of impaired mitochondria with lower citrate synthase activity and impaired measurements of mitochondrial respiration but no impact on membrane potential or ultrastructural changes. Purine analogs have been reported to interfere with mitochondrial (mt) DNA, but we did not detect any mtDNA copy number changes in cytarabine-treated T cells (data not shown).33,34
All 3 chemotherapy agents have been previously characterized for their ability to deplete lymphocytes.35-39 Unique in our focus is the assessment of the lymphocytes that survive these agents in the short-term for their ability to respond to ex vivo stimulation relevant to adoptive cell therapy manufacturing. The importance of the “starting material,” that is, the T cells collected from patients, is becoming more clear as more clinical data are reported. Our previously reported clinical data very clearly suggest that cumulative chemotherapy depletes T-cell potential, as well as differences between diagnoses and chemotherapy regimens. This has been explored somewhat in adult chronic lymphocytic leukemia, where starting material from patients with a poor clinical response on a CD19 CAR trial were found to have higher glycolytic profiles. Blocking glucose use with 2-deoxyglucose during a research based remanufacture produced products with better in vitro profiles of expansion.32 Our data here suggest there are chemotherapy-induced deficits that can linger, impacting the ultimate quality of a cell-therapy product even when successfully manufactured.
In summary, we provide here the first examination of the effects of chemotherapy classes on mature T cells and their subsets, with a focus on the relevance to immune therapy for pediatric cancer. We demonstrate differences between 3 classes of chemotherapy on T-cell mitochondrial energy reserve and glycolytic function both with and without stimulation as well as short- and long-term. Targeted rescue of specific deficits as with cyclophosphamide and NAC underscore that we be able to overcome many deficits with adaptive cell manufacture techniques, and that short-term cytolytic function may not capture the effects of chemotherapy damage. A major limitation of our model is the short time of recovery after chemotherapy exposure, which is required in this in vitro model as T cells do not survive long in culture without additional stimulation. Ideally, studies would involve tracking recovery over weeks, which is not currently possible in vitro. We are eager to further our studies with in vivo models once additional rescue strategies for cytarabine and doxorubicin can be identified, as though our model is limited it has identified important avenues for further study in a more physiologic and clinically relevant model. In addition, we plan to examine T cells from patients on future trials to verify these signatures. Time from chemotherapy exposure is likely to be extremely important, as new thymic emigrants (recovering T cells not present during chemotherapy) may increase with time and be important to ultimate cell-therapy quality vs chemotherapy-exposed T cells with lingering mitochondrial deficits. These studies provide a foundation upon which to potentially rationally adapt T-cell collection or manufacturing strategies to ensure the highest possible activity for future cellular therapies.
Supplementary Material
The full-text version of this article contains a data supplement.
Acknowledgments
Jessica Perazzelli provided technical assistance, as did the Flow Cytometry Core at the Children’s Hospital of Philadelphia and the Electron Microscopy Core at the University of Pennsylvania.
This work was supported in part by The Doris Duke Charitable Foundation Clinical Scientist Development Award, the Jeffrey Pride Foundation, and an American Association for Cancer Research Stand Up to Cancer (AACR-SU2C) Innovative Research Grant (all to D.M.B.), as well as National Institutes of Health, National Cancer Institute grants U01CA232361-01A1 (D.M.B.) and RO1CA226983-03 (R.S.O.).
Stand Up to Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research.
Footnotes
Data-sharing requests may be e-mailed to the corresponding author, David M. Barrett, at barrettd@email.chop.edu.
Authorship
Contribution: D.M.B. and R.K.D. designed the research; D.M.B., R.S.O., and R.K.D. performed the research; D.M.B., R.K.D., and S.A.G. wrote the paper; and all authors reviewed the paper.
Conflict-of-interest disclosure: S.A.G. discloses research and/or clinical trial support from Novartis, Servier and Kite; consulting, study steering committees, or scientific advisory boards: Novartis, Adaptimmune, Eureka, TCR2, Juno, GlaxoSmithKline, Cellectis, Janssen, Roche; and a toxicity management patent managed by University of Pennsylvania policies. D.M.B. discloses participation in a scientific advisory board for Eureka. The remaining authors declare no competing financial interests.
Correspondence: David M. Barrett, Department of Pediatrics, Children’s Hospital of Philadelphia, 3032 CTRB, 3501 Civic Center Blvd, Philadelphia, PA 19104; e-mail: barrettd@email.chop.edu.
References
- 1.Barrett DM, Singh N, Porter DL, Grupp SA, June CH. Chimeric antigen receptor therapy for cancer. Annu Rev Med. 2014;65:333-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Badar T, Shah NN. Chimeric antigen receptor T cell therapy for acute lymphoblastic leukemia [opinion]. Curr Treat Options Oncol. 2020;21(2):16. [DOI] [PubMed] [Google Scholar]
- 3.Geoerger B, Kang HJ, Yalon-Oren M, et al. . Pembrolizumab in paediatric patients with advanced melanoma or a PD-L1-positive, advanced, relapsed, or refractory solid tumour or lymphoma (KEYNOTE-051): interim analysis of an open-label, single-arm, phase 1-2 trial. Lancet Oncol. 2020;21(1):121-133. [DOI] [PubMed] [Google Scholar]
- 4.Fucà G, Reppel L, Landoni E, Savoldo B, Dotti G. Enhancing chimeric antigen receptor T-cell efficacy in solid tumors. Clin Cancer Res. 2020;26(11):2444-2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McGowan E, Lin Q, Ma G, Yin H, Chen S, Lin Y. PD-1 disrupted CAR-T cells in the treatment of solid tumors: promises and challenges. Biomed Pharmacother. 2020;121:109625. [DOI] [PubMed] [Google Scholar]
- 6.Signorelli D, Giannatempo P, Grazia G, et al. ; Immune-Oncology YOUNG Group . Patients selection for immunotherapy in solid tumors: overcome the naive vision of a single biomarker. BioMed Res Int. 2019;2019:9056417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Das RK, Vernau L, Grupp SA, Barrett DM. Naive T-cell deficits at diagnosis and after chemotherapy impair cell therapy potential in pediatric cancers. Cancer Discov. 2019;9(4):492-499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Finney OC, Brakke HM, Rawlings-Rhea S, et al. . CD19 CAR T cell product and disease attributes predict leukemia remission durability. J Clin Invest. 2019;129(5):2123-2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Komada Y, Zhang SL, Zhou YW, et al. . Cellular immunosuppression in children with acute lymphoblastic leukemia: effect of consolidation chemotherapy. Cancer Immunol Immunother. 1992;35(4):271-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mackall CL, Fleisher TA, Brown MR, et al. . Lymphocyte depletion during treatment with intensive chemotherapy for cancer. Blood. 1994;84(7):2221-2228. [PubMed] [Google Scholar]
- 11.Mackall CL, Fleisher TA, Brown MR, et al. . Distinctions between CD8+ and CD4+ T-cell regenerative pathways result in prolonged T-cell subset imbalance after intensive chemotherapy. Blood. 1997;89(10):3700-3707. [PubMed] [Google Scholar]
- 12.Haining WN, Neuberg DS, Keczkemethy HL, et al. . Antigen-specific T-cell memory is preserved in children treated for acute lymphoblastic leukemia. Blood. 2005;106(5):1749-1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fuks Z, Strober S, Bobrove AM, Sasazuki T, McMichael A, Kaplan HS. Long term effects of radiation of T and B lymphocytes in peripheral blood of patients with Hodgkin’s disease. J Clin Invest. 1976;58(4):803-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hakim FT, Cepeda R, Kaimei S, et al. . Constraints on CD4 recovery postchemotherapy in adults: thymic insufficiency and apoptotic decline of expanded peripheral CD4 cells. Blood. 1997;90(9):3789-3798. [PubMed] [Google Scholar]
- 15.Sukumar M, Liu J, Mehta GU, et al. . Mitochondrial membrane potential identifies cells with enhanced stemness for cellular therapy. Cell Metab. 2016;23(1):63-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scharping NE, Menk AV, Moreci RS, et al. . The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction [published correction appears in Immunity. 2016;45(3):701-703]. Immunity. 2016;45(2):374-388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawalekar OU, O’ Connor RS, Fraietta JA, et al. . Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells [published correction appears in Immunity. 2016;44(3):712]. Immunity. 2016;44(2):380-390. [DOI] [PubMed] [Google Scholar]
- 18.Buck MD, O’Sullivan D, Klein Geltink RI, et al. . Mitochondrial dynamics controls T cell fate through metabolic programming. Cell. 2016;166(1):63-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barrett DM, Singh N, Liu X, et al. . Relation of clinical culture method to T-cell memory status and efficacy in xenograft models of adoptive immunotherapy. Cytotherapy. 2014;16(5):619-630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gardner RA, Finney O, Annesley C, et al. . Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood. 2017;129(25):3322-3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh N, Perazzelli J, Grupp SA, Barrett DM. Early memory phenotypes drive T cell proliferation in patients with pediatric malignancies. Sci Transl Med. 2016;8(320):320ra3. [DOI] [PubMed] [Google Scholar]
- 22.Gattinoni L, Lugli E, Ji Y, et al. . A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17(10):1290-1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Melstrom LG, Salabat MR, Ding XZ, et al. . Apigenin inhibits the GLUT-1 glucose transporter and the phosphoinositide 3-kinase/Akt pathway in human pancreatic cancer cells. Pancreas. 2008;37(4):426-431. [DOI] [PubMed] [Google Scholar]
- 24.Singh N, Hofmann TJ, Gershenson Z, et al. . Monocyte lineage-derived IL-6 does not affect chimeric antigen receptor T-cell function. Cytotherapy. 2017;19(7):867-880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh N, Liu X, Hulitt J, et al. . Nature of tumor control by permanently and transiently modified GD2 chimeric antigen receptor T cells in xenograft models of neuroblastoma. Cancer Immunol Res. 2014;2(11):1059-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhan Y, Carrington EM, Zhang Y, Heinzel S, Lew AM. Life and death of activated T cells: how are they different from naive T cells? Front Immunol. 2017;8:1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vormittag P, Gunn R, Ghorashian S, Veraitch FS. A guide to manufacturing CAR T cell therapies. Curr Opin Biotechnol. 2018;53:164-181. [DOI] [PubMed] [Google Scholar]
- 28.Gill S, June CH. Going viral: chimeric antigen receptor T-cell therapy for hematological malignancies. Immunol Rev. 2015;263(1):68-89. [DOI] [PubMed] [Google Scholar]
- 29.Maude SL, Frey N, Shaw PA, et al. . Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507-1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neelapu SS, Locke FL, Bartlett NL, et al. . Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531-2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ruella M, June CH. Chimeric antigen receptor T cells for B cell neoplasms: choose the right CAR for you. Curr Hematol Malig Rep. 2016;11(5):368-384. [DOI] [PubMed] [Google Scholar]
- 32.Fraietta JA, Lacey SF, Orlando EJ, et al. . Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat Med. 2018;24(5):563-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang L. Mitochondrial purine and pyrimidine metabolism and beyond. Nucleosides Nucleotides Nucleic Acids. 2016;35(10-12):578-594. [DOI] [PubMed] [Google Scholar]
- 34.Jin Z, Kinkade A, Behera I, et al. . Structure-activity relationship analysis of mitochondrial toxicity caused by antiviral ribonucleoside analogs. Antiviral Res. 2017;143:151-161. [DOI] [PubMed] [Google Scholar]
- 35.Binotto G, Trentin L, Semenzato G. Ifosfamide and cyclophosphamide: effects on immunosurveillance. Oncology. 2003;65(suppl 2):17-20. [DOI] [PubMed] [Google Scholar]
- 36.El-Serafi I, Abedi-Valugerdi M, Potácová Z, et al. . Cyclophosphamide alters the gene expression profile in patients treated with high doses prior to stem cell transplantation [published correction appears in PLoS One. 2014;9(3):e93239]. PLoS One. 2014;9(1):e86619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Breithaupt H, Pralle H, Eckhardt T, von Hattingberg M, Schick J, Löffler H. Clinical results and pharmacokinetics of high-dose cytosine arabinoside (HD ARA-C). Cancer. 1982;50(7):1248-1257. [DOI] [PubMed] [Google Scholar]
- 38.Yabe M, Yabe H, Hamanoue S, et al. . In vitro effect of fludarabine, cyclophosphamide, and cytosine arabinoside on chromosome breakage in Fanconi anemia patients: relevance to stem cell transplantation. Int J Hematol. 2007;85(4):354-361. [DOI] [PubMed] [Google Scholar]
- 39.Ferreri AJ, Reni M, Foppoli M, et al. ; International Extranodal Lymphoma Study Group (IELSG) . High-dose cytarabine plus high-dose methotrexate versus high-dose methotrexate alone in patients with primary CNS lymphoma: a randomised phase 2 trial. Lancet. 2009;374(9700):1512-1520. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.