

Abstract
The pochoximes, based on the radicicol pharmacophore, are potent inhibitors of heat shock protein 90 (HSP90) that retain their activity in vivo. Herein we report an extended library that broadly explores the structure–activity relationship (SAR) of the pochoximes with four points of diversity. Several modifications were identified that afford improved cellular efficacy, new opportunities for conjugation, and further diversifications. Cocrystal structures of pochoximes A and B with HSP90 show that pochoximes bind to a different conformation of HSP90 than radicicol and provide a rationale for the enhanced affinity of the pochoximes relative to radicicol and the pochonins.
Keywords: Hsp90, inhibitors, pochoxime, radicicol, structure–activity relationships
Graphical Abstract
Introduction
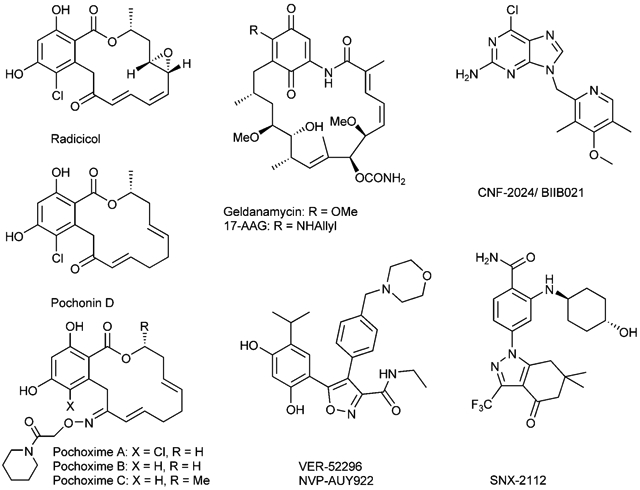
The heat shock protein 90 (HSP90) has emerged as an extremely promising therapeutic target in recent years.[1-3] Despite the seemingly ubiquitous functions of this highly expressed chaperone, its role in stabilizing conformationally labile proteins has implications in diverse pathologies. Inhibitors of HSP90 have been shown to be broadly effective for a number of cancer indications,[4,5] neurodegenerative diseases,[6-10] infectious diseases,[11] and inflammation-related disorders.[12] Two natural products, radicicol and geldanamycin, both of which disrupt the ATPase activity of Hsp90, have been instrumental in understanding the role of HSP90 in oncogenic processes and the therapeutic potential of its inhibition.[13-15] However, neither natural product has acceptable pharmacological properties for clinical application. Medicinal chemistry efforts have led to the discovery of novel scaffolds such as purines[16,17] (CNF-2024), resorcinol-isoxazoles[18-20] (VER-52296), and 2-aminobenzamides[21,22] (SNX-2112), which are currently in clinical or preclinical development.[5] However, improving the pharmacological properties and potency of the natural pharmacophores remains important. Indeed, the most advanced clinical candidate is a semisynthetic derivative of geldanamycin, 17AAG.[23,24] Another semisynthetic derivative with a hydroquinone functionality has recently been reported to have better pharmacological properties than 17AAG; although both compounds are in equilibrium with each other in vivo.[25]
We had previously demonstrated[26] that pochonin D represents a simplified pharmacophore of radicicol that recapitulates its activity. Furthermore, significant improvements in cellular efficacy could be achieved through the formation of oximes.[27] In fact, pochoximes A–C are amongst the most potent HSP90 inhibitors reported to date; inducing client protein degradation in SKBR3 cell lines at low-nm concentration. Pochoxime A treatment leads to tumor regression in xenografts bearing BT474 breast tumor cells. Herein we report a library that extends the diversity of the pochoximes, binding affinity to HSP90, and cellular efficacy. Crystal structures obtained by cocrystalization of pochoxime A and B with human HSP90α provide a rational for the significant improvement in efficacy.
Results and Discussion
The synthetic planning of the library was based on previously developed chemistry[27] and leveraged on the use of solid-phase synthesis and polymer-bound reagents. To broaden the structure–activity relationship (SAR) data on the pochoximes, a library with four points of diversity was envisioned (1, Scheme 1); the library would stem from a divergent coupling of fragment A to B followed by oxime formation and introduction of fragment C and then D. Our choice of fragments A–D was based on our preliminary SAR data[27-29] and the objective of biasing the conformational profile of the macrocycle through different ring sizes and additional small substituents. Preliminary experiments with the aryl moiety (fragment A) had shown that modifications of either phenol was detrimental; however, the presence or absence of a chlorine at R1 did have a subtle impact on the activity and both alternatives were included in the library. We[27] and others[30] had previously found that in the lower part of the macrocycle (derived from fragment B) the saturated fragment B2 was tolerated. However, additional substitutions had not been investigated and three new combinations (B3–5) were included. In the top part of the macrocycle (fragment C), eight modifications were considered that led to lactones (C1–2, C4–7) or a lactam (C3), different macrocycle size (C5–6) and an alkyne (C8), which would afford a diene as the product of the metathesis rather than the usual endocyclic alkene. The last group (D) included the largest diversity to probe the optimal oxime substituent.[27,31,32]
Scheme 1.
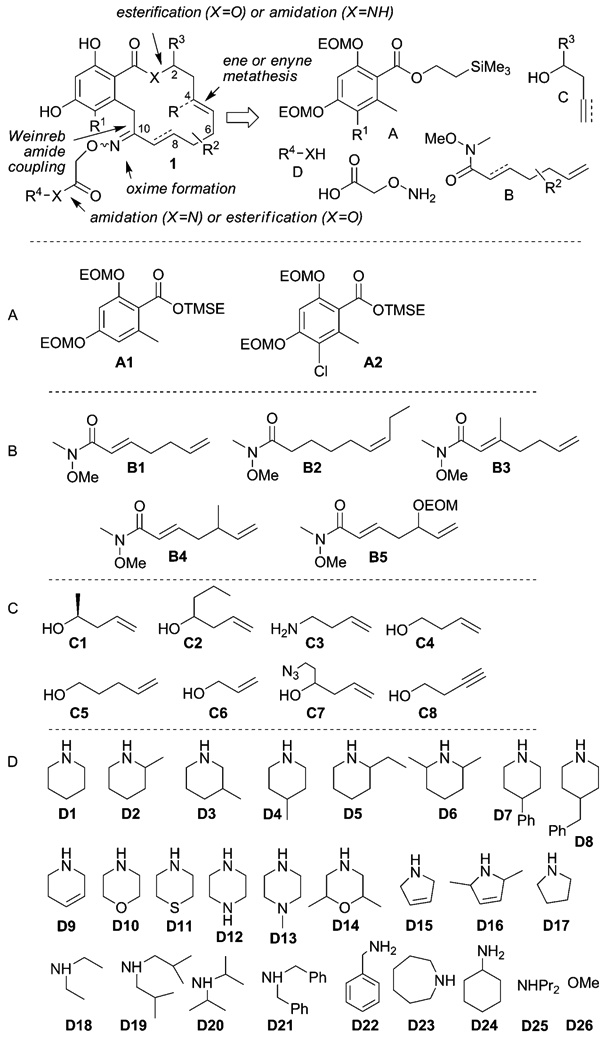
Synthetic planning of the library and the structures of library fragments.
As shown in Scheme 2, deprotonation of the toluate fragment A with LDA followed by the addition of fragment B afforded a total of ten different combinations of intermediate 2 in moderate to good yield (50–85 %), except for the coupling with fragment B2, which was afforded unacceptable yield (<10%); this is presumably due to competing enolization of the Weinreb amide. Alternatively, the same product could be obtained by conjugate reduction of the coupling product obtained with fragment B1 by using NaCNBH3 (25–50 %). Each product was treated with aminooxyacetic acid and, after filtration through a pad of silica to remove excess aminooxyacetic acid, the resulting oximes (2) were loaded on a chlorotrityl resin to afford polymer-bound intermediates (3). To ensure complete consumption of the starting material, a high loading resin (1.1 mmolg−1) was used in excess, and once all starting material had been consumed (24 h), the resin was capped with the addition of AcOH to afford ten resins of 3. The 2-(trimethyl-silyl)ethyl ester was then cleaved under the action of TBAF to afford 4, and each batch of resin was further divided into eight batches for the esterification or amide formation with fragments C. It was essential to thoroughly wash the resin with a 1% AcOH solution in CH2Cl2 after the TBAF deprotection to protonate the polymer-bound carboxylate and remove tetra-butyl ammonium salts. Each resin was then subjected to a metathesis reaction with the second generation Grubbs catalyst[33] under microwave irradiation at 120°C for 45 min to afford the desired polymer-bound macrocycles (5). To ensure completion of the reaction and equilibration to the thermodynamically more favorable E alkene, the reactions were repeated three times with a catalyst loading of 6% in each treatment. For the ene–yne metathesis (resins including fragment C8), an additional alkene was added in solution to facilitate catalyst turnover. The library of macrocycles (5) was then cleaved from the resin by using hexafluoroisopropanol (HFIP), which was found to preserve the integrity of the EOM groups to afford, after purification, the products in 20–30% yield over five steps. The macrocycles bearing a free carboxylic acid were then further divided in separate pools for coupling to fragments D by using polymer-bound carbodiimide and 4-DMAP with an excess of amine (>2.0 equiv); this afforded the products with excellent conversion (>90 %). The excess of amine was removed during the evaporation or sequestered along with DMAP during the subsequent treatment with a large excess (10 equiv) of sulfonic acid resin in MeOH to perform the EOM deprotection (>75% isolated yield for two steps). While not all permutations were pursued, a library containing at least one example of each fragment was prepared. All compounds were purified by preparative thin-layer chromatography (PTLC). However, no effort was made to separate the E and Z isomers of the oxime, which typically have very similar if not identical Rf values. The purity of all products was assessed by LC/MS and a sample of the library was analyzed by NMR spectroscopy. Generally, compounds were obtained as a 1:1 E/Z mixture of oxime geometry.
Scheme 2.
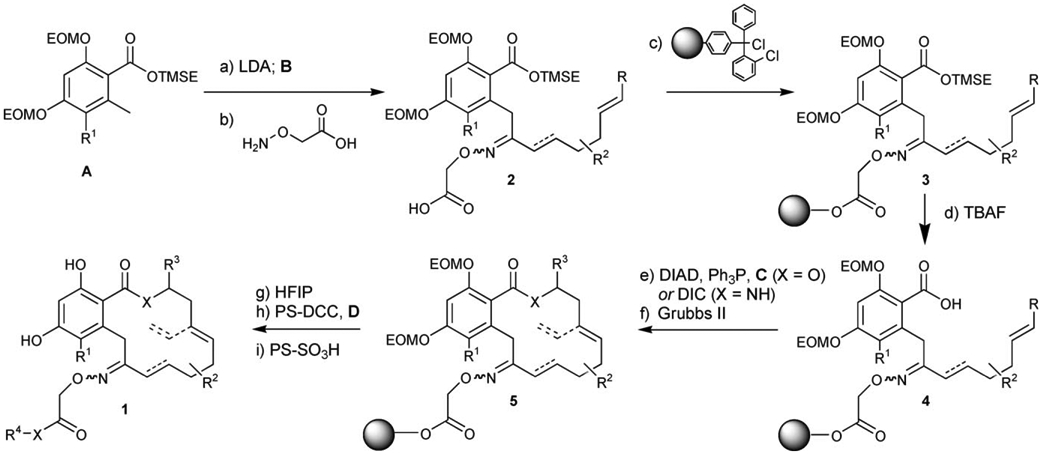
Reagents and conditions: a) LDA (2.0 equiv), B (0.9 equiv), THF, −78°C, 20 min, 50–85%; b) H2NOCH2CO2H (5.0 equiv), 40°C, py, 24–48 h, 85–95%; c) PS-ClTr-Cl (3.0 equiv), EtiPr2N (6.0 equiv), CH2Cl2, 23°C, 24 h; then AcOH (20 equiv), 23°C, 24 h; d) TBAF (4.0 equiv), 23°C 4 h; e) C (5.0 equiv), Ph3P (2.0 equiv), DIAD (2.0 equiv), toluene, 23°C, 12 h (for X=O) or C (2.0 equiv), DIC (5.0 equiv), 4-DMAP (5 mol%), CH2Cl2, 23°C, 12 h (for X=NH); f) Grubbs’ II (0.06 equiv), CH2Cl2, 120°C microwave, 3×45 min; g) HFIP/CH2Cl2 (1/4), 23°C, 3 h, 20–30% over five steps; h) PS-DCC (3.0 equiv), DMAP (cat), D (2.0 equiv), 23°C 72 h; i) PS-SO3H (10 equiv), MeOH, 23°C 4 h, >75% for two steps (>90% conversion).
A subset of this library was screened for its affinity to human HSP90α by using a competition assay with a fluorescein-labeled analogue of 17AAG[34] and in a cellular assay by using Her-2 as a pharmacodynamic marker of cellular efficacy.[35] We had previously observed that the Z isomers were less potent than the E isomers (particularly in cellular assays); however, as the E/Z ratio is consistent within the library, the results obtained should be qualitatively significant. The results are summarized in Table 1 and revealed interesting opportunities for modifications that are well tolerated as a single point modification or in conjunction with other modifications. As previously noted, there are only moderate differences in activity for compounds with the unsubstituted aryl ring (A1) and chlorinated aryl ring (A2; entry 1 vs. entry 40). However, this can become significant in conjunction with other modifications. For example, the presence of the chlorine atom is beneficial in conjugation with fragment C2 in which the combination A2C2 is roughly fivefold more potent in its affinity for HSP90 than A1C2 (entries 3 vs. 41, 29 vs. 46, and 33 vs. 48). On the other hand, it is detrimental in conjunction with B4 (entries 35 vs. 49 and 37 vs. 51). In the lower part of the macrocycles, an α,β-conjugated oxime is more active than the saturated one (B1 vs. B2). An additional methylene at the β position (B3) or γ position (B4) as well as a hydroxyl group at the γ position (B5) are generally well tolerated: The combinations A1B3C4D1 (entry 34), A1B4C4D1 (entry 37), and A1B5C1D1 (entry 39) are among the fittest ligands from the library. For the upper part of the macrocycle, there was generally little difference between the activity of compounds with the chiral methyl group (C1) or the simple primary ester (C4). Longer alkyl chains at carbon 2 (C2 and C7) were tolerated in some permutations (entries 41 and 44), but not with fragment B4 (entry 50). Replacement of the lactone with a lactam (C3) led to a significant decrease in affinity (entry 42 vs. 40), and different macrocycle size (13-member ring with C5 and 15-member ring with C6) also led to reduced affinity. Fragment C8, which affords a product substituted at carbon 4 with a vinyl group, also led to a significant decrease in affinity (entry 46). We had already noted that the aliphatic amide provided the best activity for fragment D. However, this larger panel of amides refined the SAR. Substituents on the piperidine ring are well tolerated at the β (D3) and δ (D4) positions, but not at the α position (D2, D5, D6) or if they are too large (D7 and D8). Five- (D15–17) or seven-member rings (D23) as well as acyclic secondary amides (D18–21, D25) lead to reduced affinity. One modification did provide a slight improvement in affinity: dehydropiperidine D9 (entry 11; Table 1).
Table 1.
HSP90 affinity (EC50 [μm] with statistical analysis (r2)) and pharmacodynamic (PD) evaluation of pochoximes (measurement of Her-2 depletion in SkBr3 cells treated with the inhibitor overnight (EC50, [μm]). See the Supporting Information for a table with structures.
| Entry | Compound | Affinity [μm] | r2 | PD [μm] | |||
|---|---|---|---|---|---|---|---|
| 1 | A1 | B1 | C1 | D1 | 0.034 | 0.996 | ≤5.0 |
| 2 | D18 | 0.540 | 0.994 | ||||
| 3 | C2 | D1 | 0.588 | 0.991 | > 10 | ||
| 4 | C4 | D2 | 0.886 | 0.997 | |||
| 5 | D3 | 0.060 | 0.991 | ≤ 0.1 | |||
| 6 | D4 | 0.070 | 0.983 | ≤ 0.5 | |||
| 7 | D5 | 0.243 | 0.994 | < 1.0 | |||
| 8 | D6 | 0.238 | 0.990 | < 5.0 | |||
| 9 | D7 | 2.240 | 0.990 | > 10 | |||
| 10 | D8 | 5.757 | 0.995 | > 10 | |||
| 11 | D9 | 0.022[a] | 0.988 | ≤ 0.5 | |||
| 12 | D10 | 0.124 | 0.995 | ≤ 0.5 | |||
| 13 | D11 | 0.070 | 0.998 | ≤ 0.5 | |||
| 14 | D12 | 0.640 | 0.997 | ≤ 5.0 | |||
| 15 | D13 | > 10 | 0.991 | ||||
| 16 | D14 | 0.337 | 0.995 | ≤ 5.0 | |||
| 17 | D15 | 0.128 | 0.997 | ≤ 1.0 | |||
| 18 | D16 | 0.119 | 0.982 | ≤ 1.0 | |||
| 19 | D17 | 0.219 | 0.998 | ≤ 0.5 | |||
| 20 | D18 | 0.196 | 0.991 | ≤ 1.0 | |||
| 21 | D19 | 0.094 | 0.982 | ≤ 5.0 | |||
| 22 | D20 | 0.162 | 0.993 | ≤ 1.0 | |||
| 23 | D21 | 2.992 | 0.993 | > 10 | |||
| 24 | D22 | 0.057 | 0.984 | ≤ 1.0 | |||
| 25 | D23 | 0.140 | 0.991 | ||||
| 26 | D24 | 0.155 | 0.987 | ≤ 5.0 | |||
| 27 | D25 | 0.116 | 0.980 | ≤ 1.0 | |||
| 28 | D26 | 0.372 | 0.994 | ≤ 10 | |||
| 29 | B2 | C2 | D1 | 1.936 | 0.997 | > 10 | |
| 30 | C6 | 3.027 | 0.989 | > 10 | |||
| 31 | B3 | C1 | D18 | 0.200 | 0.989 | ≤ 1.0 | |
| 32 | C2 | D1 | 0.196 | 0.994 | ≤ 1.0 | ||
| 33 | D18 | 4.663 | 0.993 | > 10 | |||
| 34 | C4 | D1 | 0.046 | 0.976 | ≤ 0.5 | ||
| 35 | B4 | C1 | 0.032 | 0.910 | |||
| 36 | C2 | D1 | 0.811 | 0.988 | |||
| 37 | C4 | 0.021[a] | 0.988 | ≤ 0.5 | |||
| 38 | C5 | 0.587 | 0.987 | ≤ 5.0 | |||
| 39 | B5 | C1 | D1 | 0.020[a] | 0.965 | ||
| 40 | A2 | B1 | C1 | 0.039 | 0.996 | ||
| 41 | C2 | 0.096 | 0.994 | ≤ 5.0 | |||
| 42 | C3 | 0.198 | 0.980 | ≤ 1.0 | |||
| 43 | C5 | D1 | 0.182 | 0.984 | ≤ 5.0 | ||
| 44 | C7 | 0.079 | 0.931 | ||||
| 45 | C8 | 0.432 | 0.985 | ≤ 1.0 | |||
| 46 | B2 | C2 | D1 | 0.543 | 0.981 | ≤ 5.0 | |
| 47 | C4 | 0.510 | 0.986 | ≤ 10 | |||
| 48 | B3 | C2 | D18 | 0.900 | 0.976 | > 10 | |
| 49 | B4 | C1 | D1 | 0.154 | 0.995 | ≤ 1.0 | |
| 50 | C2 | 1.751 | 0.998 | ≤ 10 | |||
| 51 | C4 | 0.170 | 0.976 | ≤ 5.0 | |||
| 52 | C5 | 1.722 | 0.996 | > 10 | |||
| 53 | C6 | 0.599 | 0.995 | > 10 | |||
| 54 | B5 | C1 | 0.010[a] | 0.980 | |||
| 55 | C4 | 0.018[a] | 0.980 | ||||
| 56 | radicicol | 0.156 | 0.997 | ||||
| 57 | 17-AAG | 0.033 | 0.978 | ||||
Based on the fact that the assay is performed by using 20 nm of HSP90, an IC50 below 20 nm can not be reliably measured.
The cocrystal structure of the N-terminal part HSP90 bound to radicicol (1bgq, Figure 1, panels A)[36] is very similar to the structure of apo-HSP90 (1yer).[37] Crystal structures have also been reported for the functionally related ER chaperone GRP94 (1u0z).[38] Again, these structures show a similar conformation of the ATP-binding pocket of HSP90. Likewise, the cocrystal structure of several resorcylide analogues of radicicol with HSP90 have also been reported to bind to a similar conformation of HSP90.[30] Our own studies on the binding mode of pochoximes by using docking, binding free energy calculations, and NMR 15N chemical shifts with yeast HSP90 suggested a similar binding mode to radicicol.[39] However, they did not provide a rationale for the significant enhancement of activity from pochonin D to the pochoximes. While the ketone of pochonin D does point towards a small, unoccupied pocket, docking experiments suggested that its conversion to an oxime with large substituents (such as the piperidine amide present in pochoximes A–C) would make it too large to fit and it would be placed outside of the binding pocket. Cocrystalyzation of pochoxime A and B by using a twofold molar excess of ligand with the N-terminal domain of human HSP90α afforded crystals that diffracted at 1.95 Å (PDB ID: 3inw) and 1.75 Å (PDB ID: 3inx), respectively (see the Supporting Information for statistical analysis). In both cases, the inhibitor could be placed unambiguously in the calculated electron densities. Analysis of both structures revealed a significant rearrangement in the “ATP-lid,” with the peptide segment from Ile104 through Ala124 assuming a contiguous α-helix (Figure 1, panels B and C). This rearrangement creates a large lyophilic pocket at the interface of the side chains of Met98, Leu103, Phe138, and Trp162 with the piperidine moiety sandwiched between the aryl moiety of Thp162 and the side chains of Met98. The rearrangement thus creates the opportunity for favorable interactions with the oxime substituent and hence, a rationale for the enhancements in activity of the pochoximes as well as the preference of lipophilic groups (such a piperidine amides) on the hydoxylamine. Based on the similarity between the binding mode of pochonin and radicicol, extrapolation of these results to radicicol can also account for the benefits of oximes.[31,32] A similar change in HSP90 conformation has been noted for the purine-based inhibitor PU3 (PDB ID: 1uym);[37] in the later case, however, the pocket created by Met98, Leu103, and Thp162 is less open and only accommodates a methoxy group rather than the whole piperidine ring, while the aryl ring of Ph138 is involved in π-stacking interactions to the benzyl moiety of PU3. Likewise, the cocrystal structure of a representative 2-aminobenzamide (PDB ID: 3d0b)[40] was shown to have a contiguous α-helix in the “ATP-lid;” however, it does not exploit lipophilic interactions between Met98 and Trp162.
Figure 1.
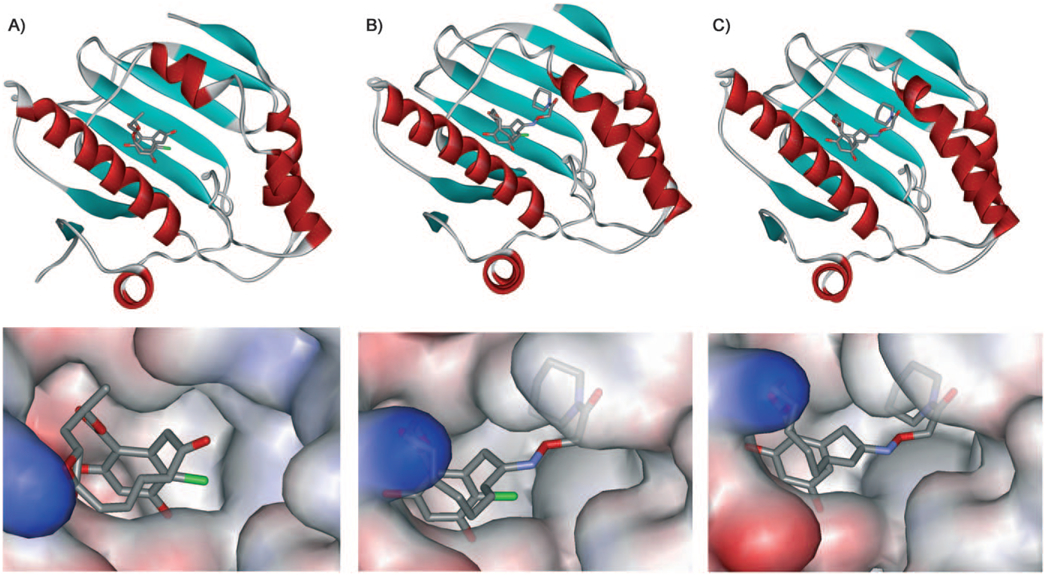
Cocrystal structures of HSP90 with A) radicicol (PDB: 1bgq), B) pochoxime A (PDB: 3inw), and c) pochoxime B (PDB: 3inx).
Based on this structure, substitution at the allylic position as in compounds that include fragment B4 and B5 should point towards the solvent. Compound 1A1B5C1D1, which bears a hydroxyl substitution at the allylic position (carbon 6), was deemed most interesting as the hydroxyl group should improve aqueous solubility relative to pochoximes A–C and provide a handle to label the inhibitor with a marker or affinity tag. This compound was prepared as a mixture of four diastereoisomers (two oxime geometries with either stereochemistry at carbon 6) that proved to be separable by HPLC. Interestingly, while the E and Z oximes were readily identifiable by NMR analysis, the diastereoisomers stemming from the different chirality of the allylic hydroxyl group had very similar NMR spectra and their stereochemistry could not be inferred. The four isomers were tested for their cellular efficacy (Figure 2). As previously noted, the E isomers of the oximes were considerably more potent than the Z oximes (60- to 100-fold). There was also a notable difference in activity (about tenfold) between the two different diastereoisomers. The most potent compound was active at 5 nm; this makes it the most potent pochoxime identified to date. (It has tentatively been assigned as the R isomer based on docking studies).
Figure 2.
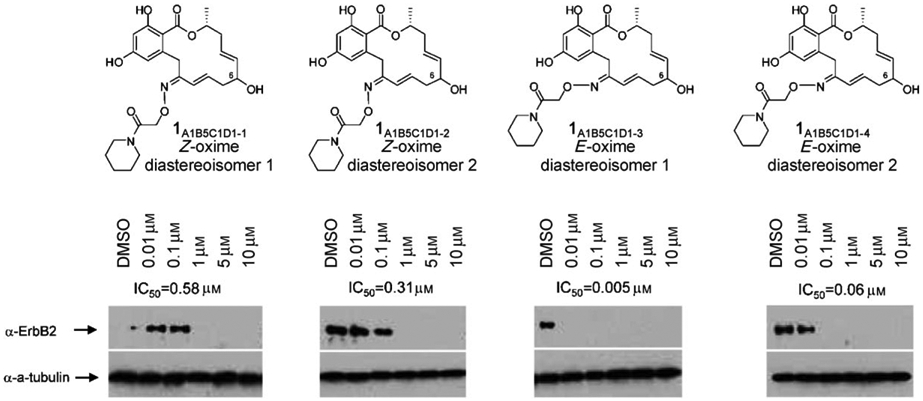
Cellular efficacy of pochoximes A1B5C1D1-1 to A1B5C1D1-4 (IC50). Depletion of Her-2 in SkBr3 cells that were treated with the inhibitor for 18 h.
In order to prepare derivatives of this newly identified analogue, compound 6, which bears a silyl protecting group on the allylic hydroxyl group, was prepared according to a previously developed protocol (see the Supporting Information for detailed procedures).[29] As shown in Scheme 3, selective silyl deprotection of 6 by using TBAF afforded 7. A short linker was introduced to 7 through alkylation, azide displacement, and reduction to afford amine 9. Labeling of 9 with Cy3 followed by EOM deprotection afforded the pochoxime–Cy3 conjugate 10. Alternatively, compound 7 was labeled with biotin by using a four step sequence that involved coupling to a short PEG linker, Fmoc deprotection, coupling to biotin, and global deprotection with TFA. It should be noted that in this case, global deprotection with sulfonic acid resin led to transesterification of the PEG-ester. The affinity of these derivatives for human HSP90α was found to be below 30 nm; this suggests that the linker to a fluorophore or biotin in the form of an ether or ester does not perturb the binding.
Scheme 3.
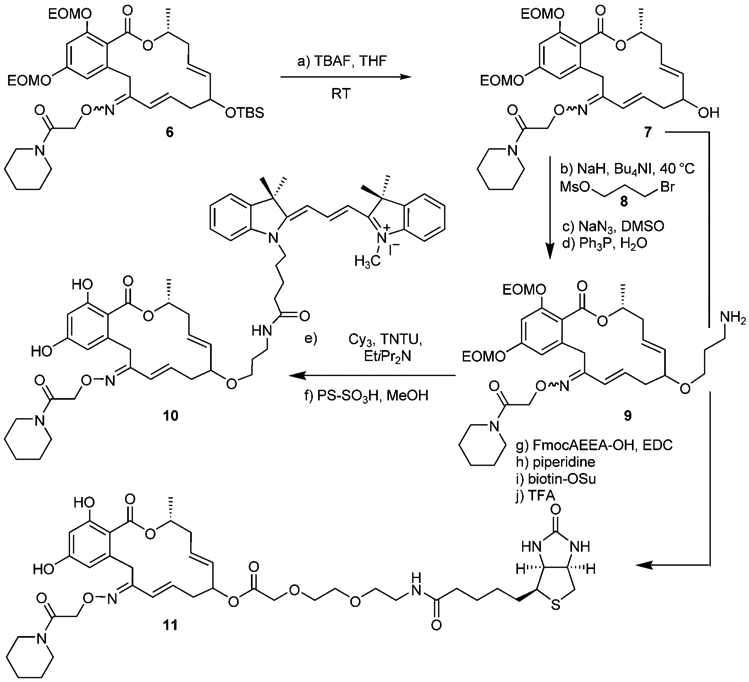
Reagents and conditions: a) TBAF (1.5 equiv), THF, 0–23°C, 3 h, 88%; b) NaH (7.2 equiv), 0°C, then Bu4NI (1.1 equiv), MsO(CH2)3Br (4.7 equiv), 0–40°C, 3 h; c) NaN3 (11 equiv), DMSO, 60°C, 2 h, 50% over two steps; d) Ph3P (2.0 equiv), THF/H2O (9:1) 40°C, 24 h, 54%; e) Cy-3 (1.5 equiv), TNTU (1.35 equiv), EtiPr2N (3.0 equiv) 0–23°C, 1 h, 95%; f) PS-SO3H (10 equiv), MeOH, 40°C, 2 h, >90%; g) FmocAEEA-OH (2.0 equiv), EDC (2.0 equiv), 4-DMAP (0.1 equiv), CH2Cl2, 23°C, 22 h, 50%; h) 20% piperidine in DMF, 10 min, 20°C,>95%; i) biotin-OSu (1.2 equiv), Et3N (4.6 equiv), DMF, 23°C, 12 h, 60%; TFA/cresol (4:1), 10 min, 23°C, >95%. FmocAEEA-OH=Fmoc-8-amino-3,6-dioxaoctanoic acid.
Conclusions
This extended library of pochoximes led to the identification of several analogues with enhanced activity. Introduction of a hydroxyl group at carbon 6 on the macrocycle afforded the most potent pochoxime to date; it exhibits a cellular efficacy below 10 nm. The cocrystal structures of pochoximes A and B with human HSP90α show that the pochoximes bind to a different conformation of HSP90 than the closely related radicicol. Whether this alternative binding mode translates to different biological activities is currently under investigation. The identification of derivatives with an affinity tag or marker should assist in this endeavor and could prove useful in diagnostics and imaging. Finally, the flexibility in the “ATP-lid” region[41] of human HSP90 as exemplified by the structures reported herein should be an important consideration in the design of inhibitors. Furthermore, differences in the flexibility in this region of the protein amongst the different HSP paralogues or HSPs from different species may be exploited to achieve selective inhibition.
Supplementary Material
Acknowledgements
This work was funded in part by a grant from the Agence National de la Recherche (ANR) and Conectus. A BDI fellowship (J.-G.F.) is also gratefully acknowledged.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cbic.200900494.
References
- [1].Workman P, Burrows F, Neckers L, Rosen N, Ann. N. Y. Acad. Sci 2007, 1113, 202–216. [DOI] [PubMed] [Google Scholar]
- [2].Neckers L, Neckers K, Expert Opin. Emerging Drugs 2005, 10, 137–149. [DOI] [PubMed] [Google Scholar]
- [3].Taldone T, Sun W, Chiosis G, Bioorg. Med. Chem 2009, 17, 2225–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Whitesell L, Lindquist SL, Nat. Rev Cancer 2005, 5, 761–772. [DOI] [PubMed] [Google Scholar]
- [5].Taldone T, Gozman A, Maharaj R, Chiosis G, Curr. Opin. Pharmacol 2008, 8, 370–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sittler A, Lurz R, Lueder G, Priller J, Lehrach H, Hayer-Hartl MK, Hartl FU, Wanker EE, Hum. Mol. Genet 2001, 10, 1307–1315. [DOI] [PubMed] [Google Scholar]
- [7].Waza M, Adachi H, Katsuno M, Minamiyama M, Sang C, Tanaka F, Inukai A, Doyu M, Sobue G, Nat. Med 2005, 11, 1088–1095. [DOI] [PubMed] [Google Scholar]
- [8].Luo W, Dou F, Rodina A, Chip S, Kim J, Zhao Q, Moulick K, Aguirre J, Wu N, Greengard P, Chiosis G, Proc. Natl. Acad. Sci. USA 2007, 104, 9511–9516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Auluck PK, Bonini NM, Nat. Med 2002, 8, 1185–1186. [DOI] [PubMed] [Google Scholar]
- [10].Luo W, Rodina A, Chiosis G, BMC Neurosci. 2008, 9, S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Geller R, Vignuzzi M, Andino R, Frydman J, Genes Dev. 2007, 21, 195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rice JW, Veal JM, Fadden RP, Barabasz AF, Partridge JM, Barta TE, Dubois LG, Huang KH, Mabbett SR, Silinski MA, Steed PM, Hall SE, Arthritis Rheum. 2008, 58, 3765–3775. [DOI] [PubMed] [Google Scholar]
- [13].Sharma SV, Agatsuma T, Nakano H, Oncogene 1998, 16, 2639–2645. [DOI] [PubMed] [Google Scholar]
- [14].Schulte TW, Akinaga S, Soga S, Sullivan W, Stensgard B, Toft D, Neckers LM, Cell Stress Chaperones 1998, 3, 100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Neckers L, Schulte TW, Mimnaugh E, Invest. New Drugs 1999, 17, 361–373. [DOI] [PubMed] [Google Scholar]
- [16].Chiosis G, Kang Y, Sun W, Expert Opin. Drug Discovery 2008, 3, 99–114. [DOI] [PubMed] [Google Scholar]
- [17].Lundgren K, Zhang H, Brekken J, Huser N, Powell RE, Timple N, Busch DJ, Neely L, Sensintaffar JL, Yang YC, McKenzie A, Friedman J, Scannevin R, Kamal A, Hong K, Kasibhatla SR, Boehm MF, Burrows FJ, Mol. Cancer Ther 2009, 8, 921–929. [DOI] [PubMed] [Google Scholar]
- [18].Sharp SY, Prodromou C, Boxall K, Powers MV, Holmes JL, Box G, Matthews TP, Cheung K-MJ, Kalusa A, James K, Hayes A, Hard-castle A, Dymock B, Brough PA, Barril X, Cansfield JE, Wright L, Surgenor A, Foloppe N, Hubbard RE, Aherne W, Pearl L, Jones K, McDonald E, Raynaud F, Eccles S, Drysdale M, Workman P, Mol. Cancer Ther 2007, 6, 1198–1211. [DOI] [PubMed] [Google Scholar]
- [19].McDonald E, Jones K, Brough PA, Drysdale MJ, Workman P, Curr. Top. Med. Chem 2006, 6, 1193–1203. [DOI] [PubMed] [Google Scholar]
- [20].Eccles SA, Massey A, Raynaud FI, Sharp SY, Box G, Valenti M, Patterson L, de Haven Brandon A, Gowan S, Boxall F, Aherne W, Rowlands M, Hayes A, Martins V, Urban F, Boxall K, Prodromou C, Pearl L, James K, Matthews TP, Cheung K-M, Kalusa A, Jones K, McDonald E, Barril X, Brough PA, Cansfield JE, Dymock B, Drysdale MJ, Finch H, Howes R, Hubbard RE, Surgenor A, Webb P, Wood M, Wright L, Workman P, Cancer Res. 2008, 68, 2850–2860. [DOI] [PubMed] [Google Scholar]
- [21].Chandarlapaty S, Sawai A, Ye Q, Scott A, Silinski M, Huang K, Fadden P, Partdrige J, Hall S, Steed P, Norton L, Rosen N, Solit DB, Clin. Cancer Res 2008, 14, 240–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Huang KH, Veal JM, Fadden RP, Rice JW, Eaves J, Strachan JP, Barabasz AF, Foley BE, Barta TE, Ma W, Silinski MA, Hu M, Partridge JM, Scott A, DuBois LG, Freed T, Steed PM, Ommen AJ, Smith ED, Hughes PF, Woodward AR, Hanson GJ, McCall WS, Markworth CJ, Hinkley L, Jenks M, Geng L, Lewis M, Otto J, Pronk B, Verleysen K, Hall SE, J. Med. Chem 2009, 52, 4288–4305. [DOI] [PubMed] [Google Scholar]
- [23].Chiosis G, Caldas Lopes E, Solit D, Curr. Opin. Invest. Drugs 2006, 7, 534–541. [PubMed] [Google Scholar]
- [24].Modi S, Stopeck AT, Gordon MS, Mendelson D, Solit DB, Baga-tell R, Ma W, Wheler J, Rosen N, Norton L, Cropp GF, Johnson RG, Hannah AL, Hudis CA, J. Clin. Oncol 2007, 25, 5410–5417. [DOI] [PubMed] [Google Scholar]
- [25].Sydor JR, Normant E, Pien CS, Porter JR, Ge J, Grenier L, Pak RH, Ali JA, Dembski MS, Hudak J, Patterson J, Penders C, Pink M, Read MA, Sang J, Woodward C, Zhang Y, Grayzel DS, Wright J, Barrett JA, Palombella VJ, Adams J, Tong JK, Proc. Natl. Acad. Sci. USA 2006, 103, 17408–17413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Moulin E, Zoete V, Barluenga S, Karplus M, Winssinger N, J. Am. Chem. Soc 2005, 127, 6999–7004. [DOI] [PubMed] [Google Scholar]
- [27].Barluenga S, Wang C, Fontaine JG, Aouadi K, Beebe K, Tsutsumi S, Neckers L, Winssinger N, Angew. Chem 2008, 120, 4504–4507 [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed 2008, 47, 4432–4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dakas PY, Barluenga S, Totzke F, Zirrgiebel U, Winssinger N, Angew. Chem 2007, 119, 7023–7026 [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed 2007, 46, 6899–6902. [DOI] [PubMed] [Google Scholar]
- [29].Moulin E, Barluenga S, Totzke F, Winssinger N, Chem. Eur. J 2006, 12, 8819–8834 [DOI] [PubMed] [Google Scholar]; Wang C, Barluenga S, Koripelly GK, Fontaine JG, Chen R, Yu JC, Shen X, Chabala JC, Heck JV, Rubenstein A, Winssinger N, Bioorg. Med. Chem. Lett 2009, 19, 3836–3840. [DOI] [PubMed] [Google Scholar]
- [30].Proisy N, Sharp SY, Boxall K, Connelly S, Roe SM, Prodromou C, Slawin AM, Pearl LH, Workman P, Moody CJ, Chem. Biol 2006, 13, 1203–1215. [DOI] [PubMed] [Google Scholar]
- [31].Ikuina Y, Amishiro N, Miyata M, Narumi H, Ogawa H, Akiyama T, Shiotsu Y, Akinaga S, Murakata C, J. Med. Chem 2003, 46, 2534–2541. [DOI] [PubMed] [Google Scholar]
- [32].Soga S, Neckers LM, Schulte TW, Shiotsu Y, Akasaka K, Narumi H, Agatsuma T, Ikuina Y, Murakata C, Tamaoki T, Akinaga S, Cancer Res. 1999, 59, 2931–2938. [PubMed] [Google Scholar]
- [33].Scholl M, Ding S, Lee CW, Grubbs RH, Org. Lett 1999, 1, 953–956. [DOI] [PubMed] [Google Scholar]
- [34].Kim J, Felts S, Llauger L, He H, Huezo H, Rosen N, Chiosis G, J. Biomol. Screening 2004, 9, 375–381. [DOI] [PubMed] [Google Scholar]
- [35].Xu W, Mimnaugh E, Rosser MF, Nicchitta C, Marcu M, Yarden Y, Neckers L, J. Biol. Chem 2001,276, 3702–3708. [DOI] [PubMed] [Google Scholar]
- [36].Roe SM, Prodromou C, O′Brien R, Ladbury JE, Piper PW, Pearl LH, J. Med. Chem 1999, 42, 260–266. [DOI] [PubMed] [Google Scholar]
- [37].Wright L, Barril X, Dymock B, Sheridan L, Surgenor A, Beswick M, Drysdale M, Collier A, Massey A, Davies N, Fink A, Fromont C, Aherne W, Boxall K, Sharp S, Workman P, Hubbard RE, Chem. Biol 2004, 11, 775–785. [DOI] [PubMed] [Google Scholar]
- [38].Soldano KL, Jivan A, Nicchitta CV, Gewirth DT, J. Biol. Chem 2003, 278, 48330–48338. [DOI] [PubMed] [Google Scholar]
- [39].Spichty M, Taly A, Hagn F, Kessler H, Barluenga S, Winssinger N, Karplus M, Biophys. Chem 2009, 143, 111–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Barta TE, Veal JM, Rice JW, Partridge JM, Fadden RP, Ma W, Jenks M, Geng L, Hanson GJ, Huang KH, Barabasz AF, Foley BE, Otto J, Hall SE, Bioorg. Med. Chem. Lett 2008, 18, 3517–3521. [DOI] [PubMed] [Google Scholar]
- [41].Richter K, Moser S, Hagn F, Friedrich R, Hainzl O, Heller M, Schlee S, Kessler H, Reinstein J, Buchner J, J. Biol. Chem 2006, 281, 11301–11311. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.