

SUMMARY
Glycan recognition is typically studied using free glycans, but glycopeptide presentations represent more physiological conditions for glycoproteins. To facilitate studies of glycopeptide recognition, we developed Glyco-SPOT synthesis, which enables the parallel production of diverse glycopeptide libraries at microgram scales. The method employs a closed system for prolonged reactions required for coupling Fmoc-protected glycoamino acids, including O-, N-and S-linked glycosides, and release conditions to prevent side reactions. To optimize reaction conditions and sample reaction progress, we devised a biopsy testing method. We demonstrate the efficient utilization of such micro-scale glycopeptide libraries to determine the specificity of glycan-recognizing antibodies (e.g. CTD110.6) using microarrays, enzyme specificity on-array and in solution (e.g. ST6GalNAc1, GCNT1 and T-Synthase), and binding kinetics using fluorescence polarization. We demonstrated that the glycosylation on these peptides can be expanded using glycosyltransferases both in-solution and on-array. This technology will promote the discovery of biological functions of peptide modifications by glycans.
Graphical Abstract
eTOC Blurb
Mehta et al. demonstrate a new method called Glyco-SPOT synthesis, to generate microgram quantities of glycopeptides with various O-, N- and S- linked glycoamino acids in parallel. The libraries produced are utilized for high-throughput microarray analysis, studying enzyme reactions, measuring binding affinities and as mass spectrometry standards.
INTRODUCTION
Glycosylation accounts for a majority of all post-translational modifications of mammalian proteins(Apweiler et al., 1999; Saraswathy and Ramalingam, 2011). Protein-bound glycans function as structure modifiers, binding modulators, and ligands, in intrinsic and extrinsic recognition events(Varki, 2017). Glycan presentation can also affect the nature of the interaction being studied(Grant et al., 2014). Recent studies have demonstrated that both the glycan and peptide portions of glycopeptides play important roles in biological recognition of a number of binding partners, including PSGL-1/P-selectin and podoplanin/CLEC2(Buskas et al., 2006; Leppanen et al., 2003; Pan et al., 2014; Qu et al., 2016). Synthetic glycopeptides offer a key approach to study the various roles of glycans attached to peptides found in nature, presenting glycans in their more natural environment.
The major limitation to exploring glycopeptide recognition, however, is the lack of large glycopeptide libraries, due to the severely limited technologies for high throughput glycopeptide synthesis at low scale (<1 μmole). While solid phase peptide synthesis (SPPS) has advanced the uses of peptides, Fmoc-protected glycoamino acids (FPGAs) are hundreds of times more expensive than Fmoc-protected amino acids on a per gram basis (Figure 1A) and derivatization and incorporation strategies are often challenging for different derivatives. Furthermore, <1% of the chemical diversity of FPGAs is commercially available (Figure 1B). As a result, using FPGAs in traditional resin-based peptide synthesis to produce libraries of glycopeptides is not feasible at the micromole scale typical of conventional resin-based SPPS.
Figure 1.

(A) Cost of Fmoc-protected glycoamino acids (FPGAs) is >1500 fold higher than their non-glycosylated counterparts on a dollar-per-gram basis. This makes glycopeptide synthesis at milligram scale expensive. (B) FPGAs available commercially account for <1% of the total chemical space of glycoamino acids. The data of total chemical space of glycoamino acids was collected from GlyTouCan database (https://glytoucan.org/). This accounts for 14,239 N-linked and O-linked glycan structures which have been reported in GlyTouCan database. The numbers for commercially available FPGAs was obtained by counting the available structures from vendors like Sigma Aldrich, Sussex Research, GlyTech, CarboSyn. This accounts for 128 FPGA structures. These vendors may have overlapping structures, which could bring this number down even more. (C) Flow chart of the Glyco-SPOT synthesis process. Blue: highlighted modifications to the method in order to incorporate FPGAs into the synthesis to produce glycopeptides.
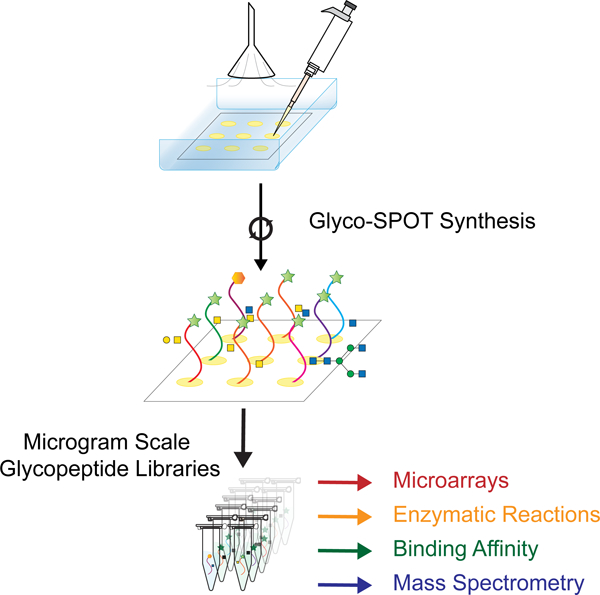
The availability of such glycopeptide libraries could be transformative. Glycopeptide libraries can provide structure-activity relationships for optimized ligand binding, which can lead to production of more potent molecules and possible therapeutics. Such libraries can also offer a means of high throughput screening of a number of glycan binding proteins (GBPs), as well as a resource to explore enzyme specificity in a glycopeptide context. To address this unmet need for such microscale glycopeptide libraries, here we present our development of Glyco-SPOT synthesis. This approach is ideal for generating glycopeptide libraries using parallel synthesis, and builds on the prior success of SPOT synthesis for production of nanomole amounts of non-glycosylated peptide libraries(Frank, 2002; Hilpert et al., 2007; Winkler and Campbell, 2008). Our development of Glyco-SPOT allows efficient, parallel glycopeptide production utilizing cellulose (filter paper) as the solid support for SPPS (Figure 1C and Figure 2A). The method allows the generation of multiple glycopeptides simultaneously using FPGAs. We also developed a novel biopsy method to check the efficiency of each synthesis cycle, thereby enabling modifications of the synthesis in real-time.
Figure 2.

(A) An overview of Glyco-SPOT synthesis strategy and its applications. Briefly, the spot synthesis is carried out on a piece of cellulose filter paper in an enclosed container under a flow of nitrogen using the setup shown above. During the several synthesis cycles, biopsy testing of the spots can be carried out as shown the on the left panel to check the efficacy and completeness of the reaction (See also Figure S1). After all the cycles are complete, the glycopeptide side chains are deprotected, the glycopeptides are released, purified and characterized using analytical techniques such as HPLC and MALDI-MS. For comparison of Glyco-SPOT synthesis strategy to traditional peptide library synthesis strategies see also Table S5. These glycopeptides can then be utilized for a number of applications: (a) The glycopeptides synthesized can further be treated with glycosyltransferase enzymes to either expand the structures in the library, or to explore the enzymatic specificity. (b) To print glycopeptide microarrays that can be utilized to probe glycan binding protein interactions. (c) Such glycopeptide microarrays can also be treated with enzymes, and be probed with lectins to probe the specificity of enzymes or to use such modified microarrays to probe binding of other proteins. (d) Using appropriate tags one can also utilize the glycopeptides to measure binding affinity. (e) The glycopeptides synthesized using this technique have been characterized using LC-MS and can potentially be used as standards in future experiments. (B) Example set of glycopeptides synthesized. The prototypical sequences of a series are shown, along with * indicating variations of amino acid sequences made for the peptides or translucent glycan symbols showing the different glycoforms synthesized.  indicates 5(6)-carboxyfluorescein on the N-terminus. For complete list of sequences see also Tables S1 and S2.
indicates 5(6)-carboxyfluorescein on the N-terminus. For complete list of sequences see also Tables S1 and S2.
With such glycopeptide libraries (example sequences shown in Figure 2B), one can print glycopeptide microarrays, perform fluorescence polarization assays for binding studies, and develop mass spectrometry (MS) standards. In addition, synthetic glycopeptide libraries offer core building blocks whose glycosylation can be further elaborated using glycosyltransferases, as well as helping to define glycosyltransferase specificity, both on-array and in-solution. These new tools should advance functional glycomics aimed at deciphering the structures, recognition, and functions of glycans on glycoproteins.
RESULTS
The complete list of sequences synthesized in the GP1 library and the GP2 library can be found in Supplementary Tables S1 and S2 respectively. The rationale for the choice of sequences and a note for synthesis of Cys-linked glycoamino acids is provided in the methods.
GLYCO-SPOT SYNTHESIS
This method exploits several aspects of classic SPOT synthesis protocols for peptides(Hilpert et al., 2007; Winkler and Campbell, 2008), but we have incorporated special modifications for FPGAs to enable efficient coupling and release of glycopeptides (Figure 1C). The classic SPOT synthesis protocol is performed in a relatively open environment without sealing and uses N,N-dimethylformamide (DMF) as the solvent of choice. These conditions work well for short reaction times (20 min), but are not useful for incorporation of FPGA, which generally required >3 times longer reaction times (depending on the glycan size, along with equivalents and concentration of reactants). This results in solvent evaporation along with side reactions with atmospheric moisture to produce dimethylamine, which can deprotect Fmoc groups on the FPGAs to cause self-polymerization(Buncel and Symons, 1970). In order to improve the yields during FPGA coupling steps, we devised a simple and effective strategy of using a sealable glass food container (snap lock air tight type) (Figure 2A) and replaced the solvents with N-methylpyrrolidone (NMP), which has a boiling point of 202°C (reducing evaporation and prolonging reagent contact) and promotes significantly less formation of amines. The membrane was placed at the bottom of the container and flushed with nitrogen prior to sealing. This makes the environment inert, preventing oxidative degradation of the solvent and reduces further evaporation. Together, these precautionary steps enable reaction times of >2 h, which are required for the glycopeptide synthesis.
Classic SPOT synthesis monitors the reaction using bromophenol blue stain. Although this stain method is very useful after each step to give the user qualitative monitoring of the reaction, it gives no information about the extent of completion of a reaction. In comparison, resin-based SPPS has a number of tests which can be performed to check the progress of the synthesis such as the Kaiser test, and even complete release of peptide to check by MS-based methods. However, visual observation of resin synthesis at the scale performed with SPOT synthesis is very difficult, as the high density of loading utilizes very little resin for small scale synthesis. Thus, we devised a biopsy method for testing the progress of the reaction in medio (Figure 2A left side panel and Figure S1) for the SPOT synthesis. In this method, a small punch biopsy (0.5 mm) of the sample is taken from the membrane and is deprotected, released and analyzed. This allows at least 20 samples to be taken from a single spot. A recommendation is to have 1 or 2 test spots with the same sequence just for testing with the biopsy method so as to not drop the overall yield of the synthesis for the main spots. This method allows fine-tuning of the synthesis, as it can be performed at any time during the synthesis after drying the membrane, and enabled screening of numerous conditions from a single synthetic run to identify ideal conditions during method optimization. Additionally, if the N-capping step of synthesis was skipped, a biopsy could identify incomplete reactions and we could reapply the reaction solution on those spots, to push the reactions to completion.
The final optimization involves the release of the peptide from the surface. Peptide cleavage from cellulose involves the base-mediated hydrolysis of the ester bond. Such basic solutions would also cause deacetylation of the protected glycan. Traditional SPOT synthesis protocols utilizes gaseous ammonia followed by aqueous washing in order to release the peptides(Hilpert et al., 2007). However, such conditions are difficult to control and can therefore cause unwanted β-elimination side products, especially in the case of O-GlcNAc, which is known to be more susceptible to this side reaction(Greis et al., 1996; Rademaker et al., 1998). To reduce such side effects, we investigated various conditions for their ability to cleave the peptide, as well as deprotect the acetylated glycan (Table S3), while noting any side effects. While all other methods showed drawbacks, a sodium bicarbonate-mediated ester hydrolysis exhibited no side effects.
The use of sodium bicarbonate for ester hydrolysis has been reported sparingly(Bora, 2011; Kaestle et al., 1991). In our hands, sodium bicarbonate in water yielded release of the peptide from the surface; however, the acetyl groups on the glycans remain intact (Supplementary Data S1 - MALDI spectra 1) and deprotection occurs very slowly requiring long reaction times to become fully deprotected glycopeptides for the GP1 library. This suggests that the peptide-cellulose ester bond is more labile as compared to the acetyl-ester bond. This could be useful in certain cases (for example, post-release modification of the peptide or cyclized peptides). However, we wanted to accelerate the reaction to yield completely deprotected glycopeptides. To do this, we found that addition of methanol to the reaction promoted the trans-esterification reactions and caused complete deacetylation of the glycan due to slightly lower pKa and stronger nucleophilicity (Figure 3A) (Phan and Mayr, 2005). The methyl peptide ester could potentially be attacked by cellulose alkoxide to reattach to the support, however, we tackled this issue by removing the cleaved peptide periodically and adding fresh sodium bicarbonate methanol:water mixture, which drives the equilibrium towards cleavage. Interestingly, when the process was carried out in the methanol:water mixture, the C-terminus of the peptide was a mix of –OMe and –OH as we observed an additional +14 Da peak in the MALDI spectra (Figure 3B). In toto, 3 washes of sodium bicarbonate (NaHCO3) in 1:2 methanol:water at 4 h, 8 h, and 18 h provided the most comprehensive deprotection-release.
Figure 3.

(A) Proposed mechanism of ester hydrolysis when utilizing sodium bicarbonate in methanol water. (B) Sample MALDI-MS spectra zoomed in to show the methyl ester with a difference of m/z +14 Da for GP2.11.
The released glycopeptides were purified and desalted using HPLC (for GP1 library) or SepPak C18 (for GP2 library) and characterized using MALDI-MS and HPLC (Supplementary Information and HPLC Reports). Finally, glycopeptides were quantified using either A205 method of the peptide bond, absorbance of the fluorescein group or BCA assay to give yields ranging from 50 – 300 μg/spot (median: 83 nmoles or 163 μg) (See Table S4 for all yields and specific quantification method used).
APPLICATIONS OF MICROGRAM-SCALE SYNTHESIS
We demonstrated the application of small-scale synthesis in biological assays and as standards in analytical chemistry.
Microarray Printing and Assays
Microarray technology has found wide utility due to the low amounts of compounds required. The GP1 and GP2 libraries were printed on NHS-coated slides as described in the methods.
Both arrays were initially probed with well-characterized plant lectins and showed binding (Figure S2). The GP1 array showed no binding with lectins GSLII, VVL as expected. WGA is known to bind terminal GlcNAc, however, peptides with single O-linked GlcNAc may not be recognized. However, control glycans on the array such as chitin and Blood Group A exhibited lectin binding with WGA. On the GP2 array, a cocktail of ConA, VVL, GSLII and WGA lectins was applied and showed binding to most glycans. Interestingly on this array, the lectins did not show any binding to the O-GlcNAc containing glycopeptides (GP2.27, GP2.30, GP2.33) (similar to GP1 array), however, they did bind at a low level to the S-GlcNAc containing glycopeptides (GP2.22, GP2.25, GP2.28, GP2.29). This binding was due to WGA, which bound to only some sequences with S-GlcNAc (Figure S3A).
Next, the arrays were probed with CTD110.6 antibody, which has been previously reported to bind O-linked GlcNAc(Comer et al., 2001). However, epitope specificity has not been explored against such a wide variety of glycopeptides. CTD110.6 bound to most O-GlcNAc containing glycopeptides on the GP1 preferentially as compared to non-glycosylated sequence (Figure 4A). The only sequence not recognized is one in which the acidic amino acid Glu is present before the glycosylation site (GP1.5). Interestingly, having an acidic residue after the O-GlcNAcylation site does not affect binding (GP1.8). The binding of the antibody to the array was completely abolished when the assay was performed in the presence of 20 mM GlcNAc, suggesting that binding is glycan mediated (Figure S3B, S3C).
Figure 4.

Binding of CTD110.6 antibody on (A) GP1 array at 10 μg/ml, and (B) and (C) GP2 array at 10 μg/ml and 1 μg/ml, respectively. The type of sugar on each peptide on the array is highlighted with color and symbol as shown in the legend. The antibody shows binding to most GlcNAc containing glycopeptides, but dislikes O-GlcNAc containing glycopeptides which have adjacent acidic residues in some sequences. The antibody is unable to differentiate between O-GlcNAc and S-GlcNAc, and in some cases may bind better to S-GlcNAc containing sequences than O-GlcNAc. See also Figures S2 and S3A (lectin binding on microarray), Figure S3B and S3C (hapten competition on GP1 array), Figure S4 (non-specific binding on GP2 array and hapten studies), Figure S5 (testing other antibodies on GP2 array). Error bars represent +/− 1 standard deviation.
When the CTD110.6 antibody was applied to the GP2 array printed at 100 μM print concentration, it exhibited significant non-specific binding regardless of concentration of antibody used (Figure S4). The results demonstrate that the antibody also binds GalNAc-containing glycopeptides when printed at higher concentrations. For ManNAc-containing glycopeptides, the antibody showed mixed specificity, binding to the IgA sequence with Cys-ManNAc, however, it bound less to the 14–3-3 peptide containing Cys-ManNAc. Interestingly, the specificity can be restored by addition of 20 mM GalNAc, leaving binding to only GlcNAc-containing glycopeptides, suggesting that the non-specific binding is also glycan mediated (Figure S4D).
When the GP2 array was printed at 25 μM, the CTD110.6 antibody shows specificity without the need for GalNAc-competition (Figure 4B,C), suggesting that the antibody binds to GlcNAc with higher affinity. However, if GalNAc-containing sequences of sufficient concentration are present they can also bind the antibody. The non-specific binding to GalNAc-containing sequences can be prevented by addition of free GalNAc. These results illustrate the utility of such small scale glycopeptides arrays, as such in-depth characterization of this antibody has not been performed previously using a library of glycopeptides. We also screened other GlcNAc-binding antibodies, specifically the 6D93 and RL2 antibodies on these arrays(Zachara et al., 2011). However, we did not see appreciable binding to either array except for a few glycopeptides (Figure S5), possibly due to their higher preference for specific sequence(Comer et al., 2001).
Additionally, we tested the recombinant BaGs6 (ReBaGs6) antibody produced by our laboratory(Matsumoto et al., 2019), which binds CD175, the Tn antigen. Indeed, this antibody binds specifically to O-linked α-GalNAc on our microarrays and does not recognize GlcNAc-containing or even Cys-β-GalNAc containing glycopeptides. Interestingly, reBaGs6 antibody showed higher binding to Ser-α-GalNAc containing peptides (GP2.02, GP2.04, GP2.05, GP2.36) as compared to corresponding Thr-α-GalNAc containing peptides (GP2.01, GP2.03, GP2.06, GP2.37), giving us previously unknown insights into the specificity of this antibody.
On-Array Glycosylation
We wanted to investigate the possibility elaborating structures on the microarrays using glycosyltransferases in order to (i) characterize enzyme specificity and (ii) expand the structures of glycans on the microarrays. We attempted glycosylation using ST6GalNAc1 to produce the Sialyl-Tn structure (Neu5Acα2–6GalNAcα1-S/T) and T-synthase glycosyltransferases to produce the T-antigen (Galβ1–3GalNAcα1-S/T) (Figure 5A).
Figure 5.

(A) On-array glycosylation using glycosyltransferase enzymes. The sequences of all the relevant IgA1 glycopeptides containing GalNAcα1- glycosylation are shown on the left where the * indicating the sites of glycosylation and the GP IDs are provided next to the sequence. The figure demonstrates how the binding pattern of lectins change when the array is treated with different glycosyltransferases either (i) ST6GalNAc1: The readout is using the binding of VVL lectin to the GalNAc on the peptides. As the reaction proceeds further, the binding of VVL lectin decreases as the product of the reaction is Neu5Acα2–6GalNAcα1- glycosylation, which is not recognized by the lectin. It was observed that the serine containing glycopeptides (highlighted in light blue or purple for the mixed glycosylation) are more resistant to sialylation by the enzyme. (ii) T-synthase: The readout is using PNA lectin which detects formation of the T-antigen (Galβ1–3GalNAcα1-S/T). Since PNA binds to the product of the reaction we see an increase in the glycosylation for all GalNAc containing peptides. As can be seen, T-synthase is not as selective about glycosylation. The data was normalized to the tallest peak in the dataset to highlight the differences clearly. Error bars represent +/− 1 standard deviation. Full array data is provided in Figure S6. (B) In-solution glycosylation using glycosyltransferase ST6GalNAc1. GP2.05 and GP2.06 were treated with ST6GalNAc1 in solution and the reaction was monitored over time using HPLC and MALDI-MS. As can be seen from the HPLC profiles, GP2.05 which contains GalNAcα1-Ser is resistant to conversion and shows little conversion (<25%) after 18 hours. In comparison GP2.05 contains GalNAcα1-Thr is converted (>80%) within 1.5 hours.
For the ST6GalNAc1 reaction, we monitored the reaction using VVL lectin, which recognizes the GalNAcα1-S/T selectively over the product Sialyl-Tn. Therefore, we observed a loss in signal when more reaction occurred. We observed that all of the GalNAcα1-Thr containing peptides were completely converted to Sialyl-Tn, while the GalNAcα1-Ser (GP2.02, GP2.04, GP2.05, GP2.36)-containing glycopeptides were resistant to conversion. Interestingly for the GalNAcα1-Thr containing peptides, virtually all signal was reduced to less than 5% at higher enzyme concentration, indicating near complete consumption of starting material, demonstrating the efficacy of the on-array glycosylation. For the T-synthase reaction, all peptides containing GalNAcα1 were converted to the Core-1 (T-antigen) structure as detected by the increase in PNA lectin binding. This shows that in contrast to the ST6GalNAc1, T-synthase is not as dependent on the amino acid residue to which the GalNAc is linked.
In-Solution Glycosylation
To test the relative recognition of these synthetic glycopeptides in solution versus on microarray, we performed glycosylation reactions using glycosyltransferase ST6GalNAc1 in solution. We treated the acceptors GP2.05 (containing GalNAcα1-Ser) and GP2.06 (containing GalNAcα1-Thr) with ST6GalNAc1 and the donor CMP-sialic acid. Based on the HPLC chromatograms (Figure 5B) at 3 time points (0h, 1.5h and 18h), we observed that GP2.05 showed very little product formation even after 18h, while GP2.06 showed that a majority of the acceptor was converted to product within 1.5h. We further confirmed that the product formed is sialylated by MALDI analysis on the reaction samples (Figure 6A, 6B). These results validate the substrate specificity observed in our on-array glycosylation.
Figure 6.
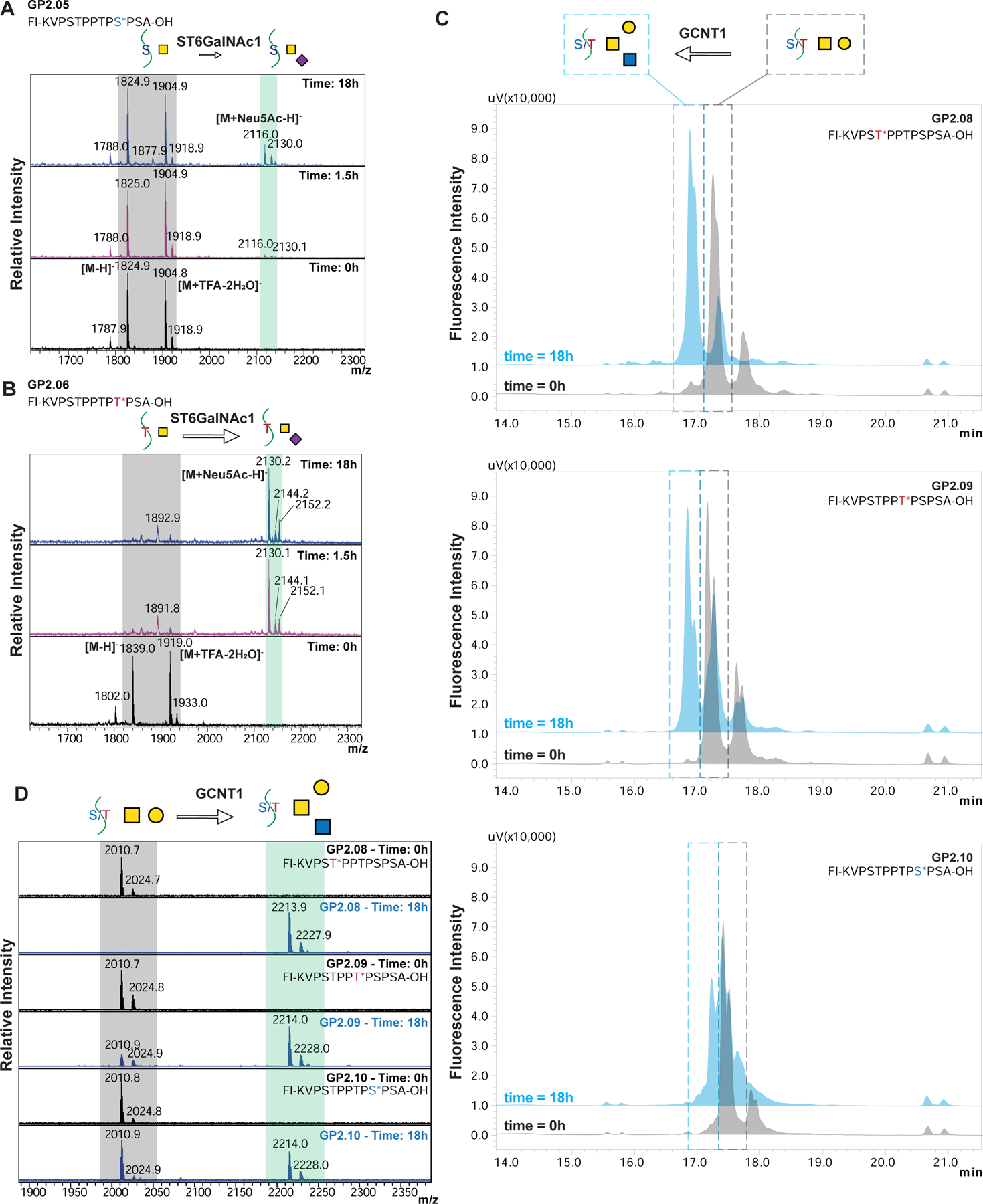
(A, B) MALDI-MS of GP2.05 (A) and GP2.06 (B) when reacted with ST6GalNAc1 at time 0, 1.5 and 18h. The peaks observed in the starting material are highlighted in grey, while the product mass is highlighted in blue. The spectra are acquired in negative mode and the starting material shows two major peaks for [M-H]- and [M+TFA-2H2O]- for both GP2.05 and GP2.06 as annotated in the Time: 0hr spectra. As time increases, GP2.05 shows a very small peak for +Neu5Ac peak at 18h suggesting a very sluggish reaction. In comparison, GP2.06 shows a strong signal for the +Neu5Ac peak at 1.5h indicative of a fast reaction with the threonine containing GP2.06. (C) HPLC chromatograms of GCNT1 reaction at time 0h (gray) and 18h (blue) for GP2.08, GP2.09 and GP2.10. The peak in the grey dashed box is the starting material core-1 structure, while the peak in the blue dashed box is the product peak for the core-2 structure. Peaks were confirmed by MALDI-MS. Order of reactivity appears to be GP2.8 > GP2.9 > GP2.10. (D) MALDI-MS of GCNT1 reactions at time 0h and 18h for GP2.08, GP2.09 and GP2.10. The peaks observed in the starting material are highlighted in grey, while the product mass is highlighted in blue. GP2.08 shows practically complete conversion to Core-2 structure in 18h, while GP2.9 and GP2.10 show partial conversion at 18h, with significant starting material peak still remaining. Order of reactivity seems to be GP2.8 > GP2.9 > GP2.10.
To further study glycosyltransferases, we incubated the T-antigen (Galβ1–3GalNAcα1)-containing peptides GP2.08, GP2.09 and GP2.10 with GCNT1 (or Core2-GlcNAc-transferase) to investigate how the different positions of acceptor substrates within a glycopeptide affect glycosylation. We monitored the reaction using HPLC and MALDI-MS (Figures 6C, 6D). We observed that the amount of Core2 (GlcNAcβ1–6(Galβ1–3)GalNAcα1) product formed is in the order GP2.08 > GP2.09 > GP2.10, and that GP2.08 shows almost complete conversion to the Core-2 structure. This suggests that the Thr5 in the sequence can undergo both sialylation or extension, while Ser10 is most likely not acted upon efficiently by either of these enzymes, and may be more limited to forming the Sialyl T- (Neu5Acα2–3Galβ1–3GalNAcα1) or di-Sialyl T-antigens (Neu5Acα2–3Galβ1–3(Neu5Acα2–6)GalNAcα1).
Fluorescence Polarization Binding Assays
We investigated the utility of fluorescent glycopeptides to measure the binding affinity of Vicia villosa lectin (VVL), which recognizes terminal a-linked GalNAc residues (Tollefsen and Kornfeld, 1983).
The previously reported affinity for VVL against glycopeptides was 0.1 – 0.4 μM, using Surface Plasmon Resonance (SPR), with simple 11-mer sequences mainly composed of Gly, Ser and Thr residues(Osinaga et al., 2000). We tested VVL binding to 4 fluorescein-modified glycopeptides containing Thr-GalNAc (GP2.3), Ser-GalNAc (GP2.4), Cys-GalNAc (GP2.21), and Cys-GlcNAc (GP2.22) (Figure 7A). We observed that the affinity was 3–4 fold higher than prior reports for our 13-mer GalNAcα1-Ser/-Thr sequences, possibly due to being a significantly different sequence. Moreover, VVL showed lower binding for GalNAcβ-Cys residues, as we observed a slight increase in the polarization but could not attain saturation (Figure 7A, GP2.21). With GlcNAcβ-Cys, no increase in polarization was observed, demonstrating the selectivity of VVL to recognize GalNAc over GlcNAc. While in our experiments we used a regular-volume 384-well plate, this protocol can easily be adapted to a 1536-well low volume plate with an appropriate plate reader, and is limited only by amount of protein required, along with the solubility of the glycopeptides and protein to be tested.
Figure 7.
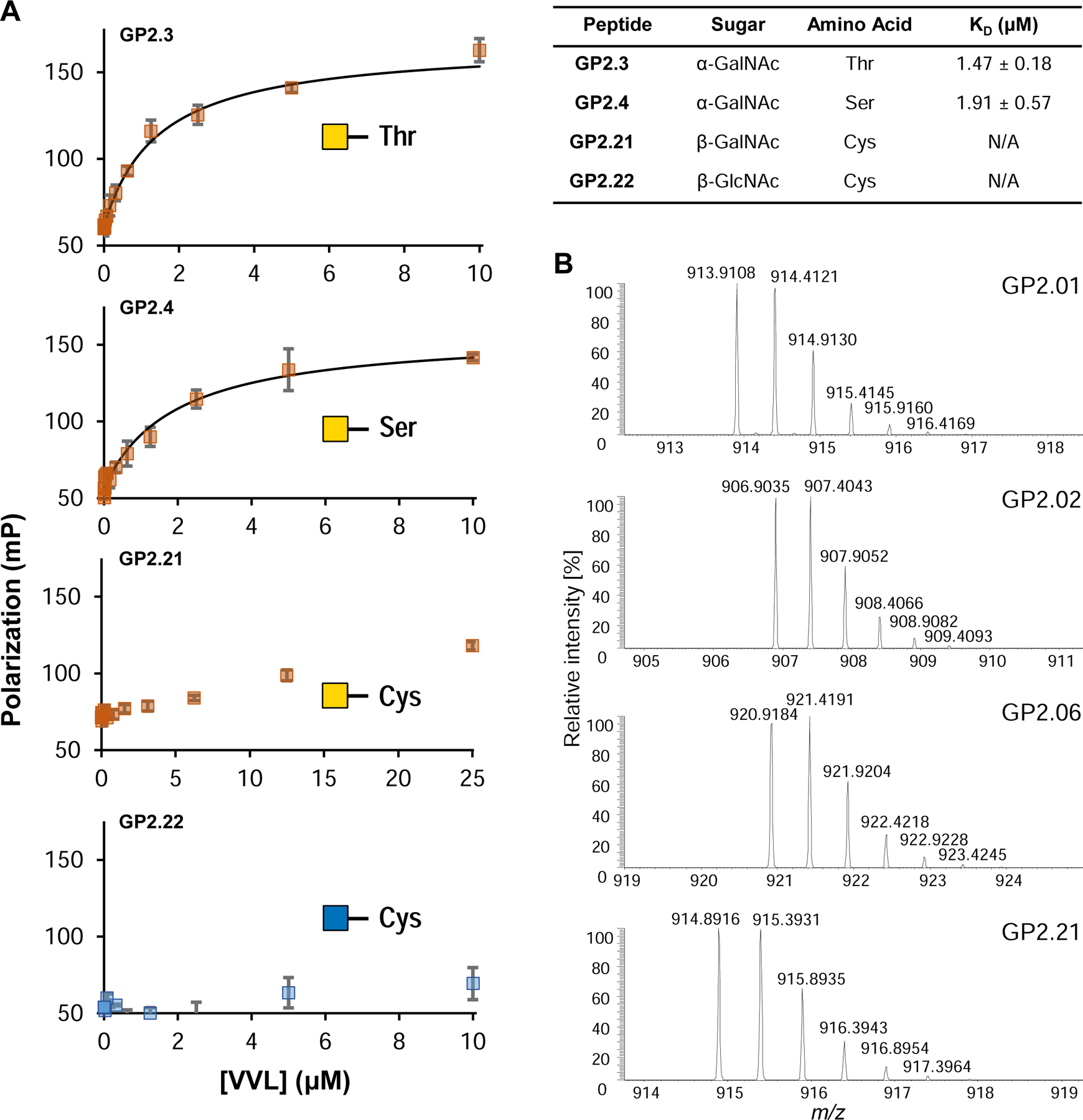
(A) Fluorescence polarization binding isotherms for 4 glycopeptides against VVL lectin. The lectin shows low micromolar affinity for GalNAcα -Ser/-Thr (GP2.3, GP2.4), while the GalNAcβ- Cys (GP2.21) shows an increase in polarization but is unable to reach saturation under these conditions and therefore the KD is not available. GlcNAcβ-Cys (GP2.22) shows no increase in the polarization at all. KD error represented as +/− SEM. (B) Example mass spectra obtained on Orbitrap LC-MS for 4 glycopeptides from the GP-2 library. The peaks show the parent [M+2H]2+ peak along with the isotope peaks. The spectra of all glycopeptides tested can be found in Supplementary Data S1. See also Figure S7.
Glycopeptides as Standards for Mass Spectrometry
We analyzed the 40 (glyco)peptides of the GP-2 library by high resolution Orbitrap LC-MS analyses. Figure 7B shows a sample of 4 spectra out of the entire dataset as a representative of the [M+2H]2+ ions observed. We estimate that for each Glyco-SPOT synthesis we can perform more than 2000 nano-LC-MS runs using these peptides. The fluorescein attached to the glycopeptides helps in following the peptides during manual sample preparation. These fluorescent peptides therefore can be used to spike complex samples and help in following glycopeptides during the sample preparation process. In addition, the glycopeptide HCD spectra library showed diagnostic oxonium ion pattern, which confirm known ratios of GlcNAc vs GalNAc(Halim et al., 2014) and will allow further evaluation of diagnostic ManNAc-specific ratios (Figure S7).
DISCUSSION
Glyco-SPOT synthesis represents a novel and robust method to create microgram-scale glycopeptide libraries without the need for expensive peptide synthesis instruments. We have demonstrated that the method permits generation of a variety of glycopeptide sequences which are either short (8–13mer) or long (22mer), with single or multiple glycosylation sites with different linkages (O-, N- and even S-). These synthetic libraries also incorporate non-natural amino acids (e.g. fluoro-prolines) and fluorophores (fluorescein) to expand their utility. To facilitate such diverse synthesis, we have developed a biopsy analysis method which enables sampling of SPOTs in the course of synthesis to ensure optimal synthesis. While we performed the synthesis manually, this could be adapted to existing SPOT synthesis robotics (capable of synthesizing up to 2,400 peptides simultaneously) in the future(Lopez-Perez et al., 2017). We further demonstrate the applicability of such small-scale synthesis in microarray assay development (~500 arrays/synthesis), use of fluorophore tags for binding studies using fluorescence polarization assays (~50 affinity measurements/spot per synthesis), and use as LC-MS standards (>2,000 runs/synthesis) to make unique discoveries in terms of antibody specificities, enzyme specificities, binding affinities and analytical characterization.
We found that these libraries can be expanded by enzymatic methods, as well as being useful for identifying glycosyltransferase preferences both on-array and in-solution. In particular the sialylation of IgA sequences has been known to contain Tn and Sialyl-Tn structures(Lehoux et al., 2014). Traditionally, ST6GalNAc1 was thought to act relatively similarly to both threonine and serine linked GalNAcα1 to synthesize the Sialyl-Tn(Kurosawa et al., 1994; Marcos et al., 2004). However recently, ST6GalNAc1 was reported to exhibit specificity for threonine over serine containing residues for MUC1 peptides(Yoshimura et al., 2019). We therefore questioned whether similar specificity would be observed for IgA1 hinge peptides. For the IgA1 sequences in our study, we observed a similar preference with respect to amino-acid type, i.e. the enzyme typically prefers threonine over serine. Interestingly, it has been reported previously that the methyl group of the threonine causes a dramatic change in the glycan orientation and even affects the hydration shell of the molecule(Corzana et al., 2007), which could explain the structural effects contributing to such specificity. ST6GalNAc1 is also known to play a role in various cancers(Ogawa et al., 2017; Wang et al., 2019). Such glycopeptide libraries could potentially help identify glycopeptides which can possibly be useful in developing biomarkers or therapeutics.
Advances in genetic engineering such as CRISPR/CAS9 have greatly impacted fields in biology. Yet site specific control of glycosylation at individual sites using such techniques is still not feasible. Synthetic glycopeptides enables dissection of biological function at single site and glycoform level. Historically, such glycopeptides have been costly and difficult to generate. Yet, they are an essential tool for the exploration of glycan recognition and glycopeptide function in glycobiology(Buskas et al., 2006), as glycosylation is known to play key roles in protein structure and function(Seitz, 2000). Over the years, several advances have been made in the fields of peptide chemistry and glycopeptide chemistry(Hojo and Nakahara, 2000, 2007). Yet most of these advances have focused on synthesis of limited and specific peptide structures, or specific FPGAs at scales which are relatively large, and not readily amenable for synthesis of highly diverse glycopeptide libraries. There has been relatively scant research in recent years on utilizing purely synthetic chemistry to synthesize highly diverse glycopeptide libraries (>20 glycopeptides) at microgram scale. To our knowledge, previous efforts have commonly used resin-based SPPS at scales >1 μmole, which usually requires >5 μmole FPGA per peptide synthesized(Blixt et al., 2010; Ohyabu et al., 2009; Risinger et al., 2017).
Such work was also limited in terms of glycosylation types. Additionally, most of these methods utilized automation in some form, requiring purchase of expensive machines. While in some cases, final characterization and purity/efficacy is not reported, making it impossible to judge the utility of techniques used. In comparison, we focused on better utilization of the precious FPGA resources and a method to synthesize libraries manually, with the efforts to democratize glycopeptide synthesis. A comparison of the Glyco-SPOT synthesis method to other resin-based methods as well as the 96-well synthesis is highlighted in Supplementary Table S5. The Glyco-SPOT method utilizes ~10-fold less FPGA/peptide to produce sub-micromole levels of synthesis, thereby reducing the cost of generating glycopeptide libraries. Other approaches recently have involved phage-display or chemical ligation strategies(Horiya et al., 2017; Izumi et al., 2017; Ng et al., 2012), both of which use glycans which are non-natural in structure in terms of their linkages. While researchers have envisaged such ideas of utilizing SPOT synthesis for glycopeptides(Günther and Ziegler, 2014; Jobron and Hummel, 2000; Norrlinger and Ziegler, 2014; Ziegler and Schips, 2006), there have been no reports of synthesis of diverse and extensive libraries having natural glycosidic linkages to amino acids utilizing the SPOT technique. We believe this has largely been due to lack of optimization of SPOT synthesis protocols for FPGA coupling and release/deacetylation conditions which can cause side effects such as β-elimination, especially in O-linked glycans.
While the current method demonstrates applicability to a wide range of peptides, the method may need modification for other peptides which may pose different intrinsic challenges due to their sequence or glycan structure, or perhaps solubility. Additionally, our method has only utilized alanine as the residue for the C-terminus as the first residue to coat the membrane for synthesis, but of course other residues could also be used. We have not investigated the efficiency of synthesis with other residues at the C-terminus, however, we believe that the efficiency should be comparable. Furthermore, we have only demonstrated glycosylation at 1 to 2 sites on a peptide, however certain proteins contain many glycosylation sites within short sequences. Peptides from such proteins may pose challenges intrinsic to glycopeptide synthesis. We strongly feel, however, that these obvious limitations can be easily addressed in future studies. Purification of such glycopeptides requires chromatographic techniques (such as HPLC) which can be a bottleneck. However, the small scale involved in Glyco-SPOT synthesis should facilitate automated fraction collection for multiple chromatographic runs in a batch provided such equipment is available. Furthermore, while in this study we elaborated structures using glycosyltransferases and expensive sugar nucleotides, we hope future developments in synthetic methods make reagents more readily available, and the Glyco-SPOT method would support efficient use of such reagents.
We can envision a wide range of applications of such glycopeptide libraries, which can be used in analytical chemistry, biochemistry or biological experiments. For example, isotope-labelled glycopeptides could be used as mass spectrometry standards, or for NMR studies, and the microscale nature of Glyco-SPOT synthesis could enable efficient use of costly isotope-labeled precursors. Another potential modification could be the attachment of a biotin tag to a glycopeptide, as biotinylated peptides have wide range of applications. Glycopeptides could also be used as “bait” to capture glycan- or glycopeptide-binding proteins from biological samples. Synthesis of multi-post-translationally modified (PTM) peptides would also be possible, i.e. glycopeptides containing O- and N-glycans and other PTMs such as phosphate or sulfate groups. Overall, the Glyco-SPOT technique miniaturizes the synthesis of libraries of glycopeptides and will greatly accelerate the field of glycoscience by making glycopeptide library synthesis a more accessible technology.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Richard D. Cummings (rcummin1@bidmc.harvard.edu).
Materials Availability
Glycopeptides generated in this study are available within reasonable limits of quantity, due to the small scale of synthesis upon request to the Lead Contact. The Cys-linked glycoamino acids are available from Dr. Nicola L. B. Pohl (npohl@indiana.edu).
Data and Code Availability
The glycopeptide microarray datasets supporting the current study are not deposited in a public repository as there is no repository currently built for glycopeptide microarrays, but are available from the Lead Contact on request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
This work did not involve any animals, human subjects, plants, microbe strains, cell lines, or primary cell culture models.
METHOD DETAILS
Synthesis of Cys-linked Glycoamino acids
Materials and instrumentation:
Chemicals and reagents were purchased from Sigma-Aldrich, Alfa Aesar, and Acros Chemicals and used without further purification. InBr3 was purchased from Sigma–Aldrich. Peracetyl-β-D-GlcNAc, peracetyl-β-D-GalNAc, peracetyl-β-D-ManNAc, and Fmoc-Cys-OH hydrated were purchased from Chem-Impex Inc. All solvents were kept over 4 Å molecular sieves for 12 h and used without purification. Reactions were monitored via thin layer chromatography (TLC) on silica gel glass slides (Sorbent Technologies, Norcross, GA). Detection was effected by UV light charring with 18:1:1 ethanol/p-anisaldehyde/sulfuric acid. Purification of compounds was achieved by performing medium pressure liquid chromatography using Teledyne ISCO instrument. Proton (1H) NMR and carbon (13C) NMR spectra were recorded on a 500 MHz or 600 MHz Varian instrument using the residual signals from CD3OD, δ3.31ppm, and 49.0 ppm as an internal reference for 1H and 13C chemical shifts, respectively (Bubb, 2003; Duus et al., 2000). Chemical shifts in 1H-NMR spectra are reported in ppm (δ) with reference to the signal of Me4Si, which was adjusted to δ 0.00 ppm unless otherwise noted. All NMR spectra were analysed and interpreted using MestReNova® software. ESI-HRMS mass spectrometry was obtained using Agilent 6540 QTOF instrument with Bruker AVANCE III 500 spectrometer. All reactions were carried out in a fume hood.
Nα-Fluoren-9-ylmethoxycarbonyl-S-(3,4,6-tetra-O-acetyl-2-acetamido-2-deoxy)-β-D-glucopyranosyl)-L-cysteine (3):
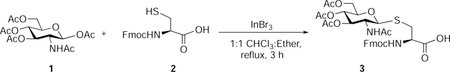
β-D-Glucosamine pentaacetate (850 mg, 2.18 mmol, 1.5 eq), InBr3 (232 mg, 0.65 mmol, 0.3 eq), and Nα-fluoren-9-ylmethoxycarbonyl-L-cysteine (500 mg, 1.47 mmol, 1 eq) were suspended in 1:1 CHCl3: diethyl ether (30 mL). The reaction mixture was stirred at reflux for 3 hours. Gradually precipitation was observed and the precipitate was filtered and slurried with diethyl ether for 1 h. Filtration and washed with ether to afford 3 as an off-white powder (744 mg, 76 %). Rf 0.3 (CH2Cl2/methanol, 9:1 v/v with 0.5 % AcOH)(Hsieh et al., 2012).
1H NMR (500 MHz, Methanol-d4) δ 7.82 (d, J = 7.5 Hz, 2H), 7.70 (t, J = 6.5 Hz, 2H), 7.41 (t, J = 7.4 Hz, 2H), 7.34 (ddd, J = 8.2, 6.8, 2.8 Hz, 2H), 5.22 (t, J = 9.7 Hz, 1H), 5.01 (t, J = 9.7 Hz, 1H), 4.80 (d, J = 10.5 Hz, 1H), 4.46 (dd, J = 8.9, 4.2 Hz, 1H), 4.40 – 4.36 (m, 2H), 4.27 (t, J = 7.0 Hz, 1H), 4.22 – 4.11 (m, 2H), 4.02 (d, J = 10.3 Hz, 1H), 3.84 – 3.73 (m, 1H), 3.38 (dd, J = 14.3, 4.2 Hz, 1H), 2.94 – 2.85 (m, 1H), 2.03 (s, 2H), 2.02 (s, 3H), 2.00 (s, 3H), 1.86 (s, 2H).
13C NMR (126 MHz, Methanol-d4) δ 171.3, 171.1, 171.0, 169.5, 143.5, 141.1, 127.6, 127.0, 124.8, 119.8, 83.4, 75.6, 73.7, 68.4, 67.0, 62.1, 52.6, 52.6, 46.9, 30.5, 22.3, 20.3, 20.3, 20.3.
HRMS-ESI-TOF (m/z):
Calcd. for [(C32H37N2O12S+] 673.2067 (M+H+), Found 673.2067 (100%).
Nα-Fluoren-9-ylmethoxycarbonyl-S-(3,4,6-tetra-O-acetyl-2-acetamido-2-deoxy)-β-D-galactopyranosyl)-L-cysteine (5):
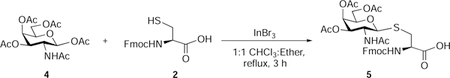
β-D-Galactosamine pentaacetate (850 mg, 2.18 mmol, 1.5 eq), InBr3 (232 mg, 0.65 mmol, 0.3 eq), and Nα-fluoren-9-ylmethoxycarbonyl-L-cysteine (500 mg, 1.47 mmol, 1 eq) were suspended in 1:1 CHCl3: ethyl ether (30 mL). The reaction mixture was stirred at reflux for 3 hours. The solvent was evaporated and ether was added to the crude solid, the mixture was stirred for 1 h at 0 °C, filtered and washed with cold ether. Again the crude product was slurred with dichloromethane for 30 min and filtered and washed with cold CH2Cl2 to afford 5 as an off-white powder (725 mg, 74 %). Rf 0.3 (CH2Cl2/methanol, 9:1 v/v with 0.5 % AcOH)
1H NMR (500 MHz, Methanol-d4) δ 7.81 (d, J = 7.5 Hz, 2H), 7.71 (dd, J = 18.5, 7.5 Hz, 2H), 7.45 – 7.39 (m, 2H), 7.38 – 7.31 (m, 2H), 5.37 (d, J = 3.3 Hz, 1H), 5.07 (dd, J = 10.8, 3.2 Hz, 1H), 4.72 (d, J = 10.4 Hz, 1H), 4.47 (dd, J = 9.4, 4.0 Hz, 1H), 4.43 – 4.32 (m, 2H), 4.32 – 4.24 (m, 2H), 4.17 – 4.07 (m, 2H), 3.98 (t, J = 6.5 Hz, 1H), 3.45 (dd, J = 14.4, 4.0 Hz, 1H), 2.88 (dd, J = 14.4, 9.4 Hz, 1H), 2.12 (s, 3H), 1.98 (s, 2H), 1.97 (s, 3H), 1.88 (s, 2H).
13C NMR (126 MHz, Methanol-d4) δ 172.19, 170.80, 170.66, 170.24, 157.03, 143.90, 143.75, 141.16, 127.42, 126.87, 126.83, 124.98, 124.85, 119.55, 119.53, 83.76, 74.34, 71.61, 66.99, 66.76, 61.74, 54.22, 48.48, 46.91, 31.04, 21.34, 19.16, 19.14, 19.13.
HRMS-ESI-TOF (m/z):
Calcd. for [(C32H37N2O12S+] 673.2067 (M+H+), Found 673.2060 (100%).
Nα-Fluoren-9-ylmethoxycarbonyl-S-(3,4,6-tetra-O-acetyl-2-acetamido-2-deoxy)-α-D-mannopyranosyl)-L-cysteine (7):
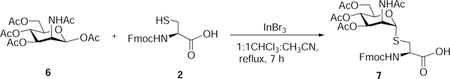
β-D-Mannosamine pentaacetate (850 mg, 2.18 mmol, 1.5 eq), InBr3 (232 mg, 0.65 mmol, 0.3 eq), and Nα-fluoren-9-ylmethoxycarbonyl-L-cysteine (500 mg, 1.47 mmol, 1 eq) were suspended in CH3CN:CHCl3 (15 mL). The reaction mixture was stirred at reflux for 7 hours. The reaction mixture was concentrated in vacuo, and the residue was purified by flash chromatography ISCO (SiO2, 0 – 1.0 % of methanol in CH2Cl2 with 0.5% Acetic acid) to afford 7 as an off-white powder (712 mg, 73%). Rf 0.3 (CH2Cl2/methanol, 9:1 v/v with 0.5 % AcOH).
1H NMR (500 MHz, Methanol-d4) δ 7.80 (d, J = 5.2 Hz, 2H), 7.69 (t, J = 7.1 Hz, 2H), 7.39 (t, J = 7.8 Hz, 2H), 7.32 (t, J = 7.6, 5.3 Hz, 2H), 5.33 (s, 1H), 5.20 (dt, J = 9.9, 2.6 Hz, 1H), 5.14 (dd, J = 10.1, 4.6 Hz, 1H), 4.64 (s, 1H), 4.50 – 4.37 (m, 3H), 4.32 – 4.23 (m, 3H), 4.13 (dt, J = 12.1, 2.6 Hz, 1H), 3.22 – 3.16 (m, 1H), 3.15 – 3.07 (m, 1H), 2.01 (s, 3H), 2.00 (s, 3H), 1.98 (s, 3H), 1.95 (s, 3H).
13C NMR (126 MHz, Methanol-d4) δ 172.09, 171.14, 170.14, 170.12, 156.96, 143.82, 141.17, 127.42, 126.83, 126.83, 124.96, 124.86, 119.52, 84.79, 69.80, 69.18, 66.70, 66.40, 62.64, 54.40, 51.12, 48.10, 47.00, 33.72, 20.93, 19.33, 19.21, 19.17.
HRMS-ESI-TOF (m/z):
Calcd. for [(C32H37N2O12S+] 673.2067 (M+H+), Found 673.2062 (100%).
Note on the synthesis of Cys-linked Glycoamino Acids
Synthetic methods used to access S-GlcNAcylated amino acids involve multiple steps that require the prior synthesis of sugar donors with proper selection of a C-2 protecting group for neighboring group participation (NGP) to get exclusively the β-isomer, followed by protecting group manipulation to obtain the final building block for SPPS(Hsieh et al., 2012). Protection of the Fmoc-Cys carboxylic acid, the use of toxic promotors, the need for extremely anhydrous conditions, and the subsequent deprotection steps make these routes too time-consuming and arduous for general use. Ideally, a straightforward, practical method for S-GlcNAcylated amino acid synthesis could be developed from commercially available materials. Recently, InBr3 was reported to activate anomeric acetates of glucose, resulting in formation of a variety of glycosides(Lefever et al., 2012; Szabo et al., 2016). Therefore, this promoter was tested for its ability to activate commercially-available 2-acetamido-2-deoxy-1,3,4,6-tetra-O-acetyl-β-D-glucopyranose (β-Ac4GlcNAc) to form β-glycosides through NGP. Our investigation started with Fmoc-Cys(SH)-COOH as acceptor and glycosylation with 2.0 eq donor and 0.2 eq InBr3 as previously reported. This reaction was carried out for 12 h, but only 30% conversion was seen as monitored by thin layer chromatography. Reaction optimization studies revealed solubility issues when using chloroform, therefore mixtures of solvents were tested with lower equivalents of donor and InBr3. Fortunately, several combinations of solvents significantly improved reaction conversions. A 1:1 ratio of chloroform:diethyl ether as solvent with only 0.3 mol% InBr3 in relation to the GlcNAc donor provided 100% conversion to the desired product. After only 2–3 hours of reaction time, the reaction product was precipitated, filtered, and washed with diethyl ether to provide the desired product in 75–80% yield with exclusive β–selectivity. The product proved to be of sufficient purity for successful use in SPPS. This new method was also tested with peracetylated galactosamine and mannosamine donors. With galactosamine, the reaction was also completed in only 2–3 hours, but the same purification sequence resulted in a product significantly contaminated with other sugar intermediates. A slurry with dichloromethane dissolved the remaining sugar-based byproducts quickly and thereby allowed filtration of a pure product in 70–75% yield with exclusive β-selectivity. However, the same protocol with the mannosamine donor required more than 24 hours to complete. An additional solvent screen revealed that the reaction could be completed in only 6–7 hour using a 1:1 ratio of chloroform and acetonitrile as reaction solvent; direct column purification afforded 70–75% of the desired product with exclusive α-selectivity.
Characterization Data
NMR spectra for Cys-Linked FPGAs provided in a separate chemical characterization Data S1 file.
Glycopeptide Synthesis
Prior works in SPOT synthesis offer a good guidance to the basic procedure(Frank, 2002; Hilpert et al., 2007; Winkler and Campbell, 2008). We recommend reading these articles in order to gain insight on the basic procedures and familiarity with the classical workflow.
Safety Statement:
No unexpected or unusually high safety hazards were encountered during these procedures, and the reagents used are routinely used by most chemical laboratories.
However, as most reagents used during synthesis are rated to be flammable, toxic and/or corrosive, it is advisable to perform all steps in a certified chemical fume-hood, while wearing appropriate personal protective equipment.
We used flame and chemical resistant lab coats (Workrite FR/CP), along with chemical resistant disposable gloves with extended cuffs (MICROFLEX® 93–260 from Ansell) and chemical safety goggles at all times. Additional use of chemical respirator was encouraged while pouring volatile solvents from large containers to smaller dispensing containers.
Rationale for Sequences:
The glycopeptides synthesized were diverse in terms of structure, glycosidic linkage, amino acid residue of attachment, sequence, tags, length of peptide and glycan. Furthermore, the FPGA’s used were mainly those available from commercial sources (except the Cys-linked FPGA’s) and these were strategic building blocks which can be expanded using glycosyltransferases.
The sequences for the first glycopeptide library designated GP1 to demonstrate proof-of-principle were modifications of the bait sequence used to pull down GlcNAc-binding proteins previously (Supplementary Table S1)(Toleman et al., 2018). This bait sequence was based on the consensus O-GlcNAcylated peptide sequences by aligning 802 mapped Ser-O-GlcNAc sites. The modifications were based on exchanging the flanking amino acid residues to modify the properties with different side chains. The reported chemistry was first optimized on this library of products.
The second glycopeptide library designated GP2 was used to further test the robustness of the method by testing a more diverse library, including peptides from IgA, 14–3-3 protein, PCBP1, antigen KI-67 and SGP sequences (Supplementary Table S2). The GP2 library also included O-, N- and S- linked glycans which consisted of GlcNAc, GalNAc, ManNAc, GalGalNAc and A2G0 (N-linked heptasaccharide) sequences. We also included fluoro-proline analogs in order to restrict the conformations in some IgA sequences, based on previous literature which shows that 4S-Fluoro-Pro relatively restricts peptides in the cis conformation, while the 4R-Fluoro-Pro relatively restricts peptides in the trans conformation as compared to regular Pro residues(Newberry and Raines, 2017). Furthermore, some sequences in the GP2 library were modified on the N-terminus with a fluorescein label to explore utility of such small scale synthesis in fluorescence-based assays and as standards for mass spectrometry (MS) sample preparation. These modifications demonstrate how adaptations to this method expand the scope of accessible glycopeptides to provide possible structural and biophysical insights when presented in a physiologically relevant manner.
The IgA sequences are of particular interest based on our previous work on IgA nephropathy where in there is observed Tn (GalNAcα1-) and Sialyl-Tn (Neu5Acα2–6GalNAcα1-) epitopes in this sequence in both healthy and patient IgA1(Lehoux et al., 2014). The different positional variations of the GalNAcα1- glycosylation can be studied using these peptides to check which positions show sialylation most efficiently and to test which enzymes would create larger glycan structures. Such tests should provide predictions as to which types of glycosylation patterns would be observed in this sequence region.
Materials
Whatman® quantitative filter paper, hardened low-ash, Grade 50 (Cat #: WHA1450916) 1-Methyl-2-pyrrolidinone (N-Methyl-2-pyrrolidone) (NMP) biotech. grade, ≥99.7% (Cat #: 494496–1L), 1-Hydroxybenzotriazole hydrate (HOBt) wetted with not less than 14 wt. % water, 97% (Cat #: 157260–100G), Bromophenol blue (Cat #: 114391–5G), were all purchased from Sigma-Aldrich (St. Louis, MO). All regular fmoc-protected amino acids were purchased from Novabiochem a subsidiary of Millipore Sigma (Burlington, MA). The following regular Fmoc-protected amino acids with side chain protecting groups were used from Novabiochem: Fmoc-Ala-OH, Fmoc-Asp(OtBu)-OH, Fmoc-Phe-OH, Fmoc-Gly-OH, Fmoc-Ile-OH, Fmoc-Lys(Boc)-OH, Fmoc-Leu-OH, Fmoc-Pro-OH, Fmoc-Gln(Trt)-OH, Fmoc-Arg(Pbf)-OH, Fmoc-Ser(tBu)-OH, Fmoc-Thr(tBu)-OH, Fmoc-Val-OH, Fmoc-Trp(Boc)-OH, Fmoc-Tyr(tBu)-OH. Anhydrous amine-free N,N-Dimethylformamide (DMF) (Cat #: AA43465-K7) was purchased from VWR (Radnor, PA). N,N’-Diisopropylcarbodiimide (DIC) 99%, AcroSeal™, ACROS Organics™ (Cat #: AC446181000) was purchased from Fisher Scientific (Pittsburgh, PA). Fmoc - Ser(β - D - GlcNAc(Ac)3) - OH and Fmoc - Thr(β - D - GlcNAc(Ac)3) - OH was purchased from Anaspec (Fremont, CA). Fmoc-Ser(α-D-GalNAc(Ac)3)-OH, Fmoc-Thr(α-D-GalNAc(Ac)3)-OH, Fmoc-Ser(Galβ(1–3)GalNAc)-OH, peracetate, Fmoc-Thr(Galβ(1–3)GalNAc)-OH, peracetate, were purchased from Sussex Research (Ottawa, Ontario Canada). Fmoc-trans-4-fluoro-Pro-OH and Fmoc-cis-4-fluoro-Pro-OH was purchased from Bachem (Torrance, CA). Fmoc-Asn(A2G0)-OH was prepared as previously described (Gao et al., 2019). Box-shaped snap-lock type glass food container was purchased from the local retail store. Reusable biopsy punch (0.5mm tip) was purchased form World Precision Instruments (Sarasota, FL) (Cat #: 504639). Nexterion® Slide H 3D NHS (Schott) slides were purchased from Applied Microarrays (Tempe, AZ) (Cat #: 1070936). All other reagents and chemicals were purchased from either Sigma-Aldrich or Fisher Scientific and used without purification.
Preparation of the Membrane
A Whatman quantitative filter paper with the dimensions as per the synthetic needs was cut out. Each spot of 1–1.5 μL occupies less than 1.6 cm x 1.6 cm in dimensions. Knowing this, we cut out the paper so as to get sufficient number of spots and also ensuring that the dimensions fit in the box we use so as to perform the washes and coupling. Using a pencil, a grid pattern was marked such that each spot has a 1.6 cm x 1.6 cm square and a 0.5 cm margin on all four sides of the sheet, which we can use to tape the membrane to the surface during coupling.
The membrane was dried for 30–60 minutes in a lyophilizer/vacuum desiccator. We recommend leaving the membrane under vacuum while other reagents are prepared. This drying was performed before each coupling and at the end of the day prior to storage.
Testing NMP / DMF Solvent Quality Before Synthesis
It is important that the solvents used be absolutely free of amines to get maximum efficiency for the reactions. N,N-Dimethylformamide (DMF) and N-methyl-2-pyrrolidone (NMP) react with atmospheric moisture to produce free amines which can be detrimental to the reaction. For this reason, only amine-free and anhydrous grades of solvents should be used and aliquots of solvents taken during synthesis steps should be checked for amines using bromophenol blue test. To perform the test, we took 1 ml of solvent and added 1 μL of 1% w/v bromophenol blue (in methanol) solution. If the color is green or blue, it indicates there is significant amines present and the solvent is discarded or used for final washes only. Only yellow color test qualifies for coupling and blocking steps, where amine-free requirements are essential.
To maintain the amine free nature of the solvents, it is necessary to flush the stock bottle and aliquot bottles with anhydrous inert gas such as nitrogen or argon.
Coupling the first amino acid on to the paper
The method previously described in the protocol was used to perform the coupling with modification(Hilpert et al., 2007). Briefly, for a 12 cm x 10 cm Whatman quantitative filter paper 960 mg of Fmoc-Ala-OH was weighed and dissolved in 15 ml of NMP. To this was added 560 μL of DIC (N,N’-Diisopropylcarbodiimide) and 475 microliters of 1-methylimidazole. The solution was poured into the box-shaped air tight reaction container (box-shaped snap-lock type glass food container) and inert gas was flowed over it using setup as shown in Figure 2A. Using forceps, the dried membrane was gently placed into this solution making sure there were no air bubbles. The container was closed and placed on a shaker overnight.
Spot Synthesis
Planning:
We create a list of the number of spots per cycle of specific amino acids. Each spot receives 1–2 μL of volume of amino acids. For each cycle the number of spots was multiplied by the volume/spot to give us the total volume of the amino acid required for that cycle. We weighed out the required quantity of amino acids so as to get 450 mM solution of the desired volume. HOBt was weighed out in individual tubes so as to make a 0.6 M solution of the total volume to be used each day. The solids were stored in a desiccator or in an air tight container in freezer.
For each cycle, the HOBt was dissolved using the volume of NMP to make 0.6 M of HOBt bulk solution to be used for the day. A 1.8 M solution of DIC in NMP was also prepared in bulk for each day. The final volume required to make 450 mM of amino acid solution was retrieved or calculated. The HOBt solution and DIC solution was added in a ratio of 3:1 HOBt solution : DIC solution to make the final volume. This gives a final concentration of 450 mM for the amino acids, HOBt and DIC. For FPGAs, the concentrations used were 100 mM and appropriate calculations were done to have similar ratio of HOBt and DIC (100 mM each). Following this the amino acids were ready for spotting.
Spotting:
For cycles in which only regular amino acids were used, the membrane was taped to a clear glass surface on its borders using regular office tape. The tape was first put on a paper towel and peeled off to reduce its adhesive strength, so that when it is applied to the synthesis membrane it can be removed easily. Carefully, using a micropipette 1–2 μL of the amino acid mixture was deposited on the membrane. This was repeated for all the spots.
The membrane was covered with a box-shaped glass food container placed upside down after a short flush with anhydrous inert gas argon/nitrogen and allowed to incubate for 20 minutes. A second coupling can be performed by repeating these steps to increase efficiency.
Glyco-SPOT Spotting:
For spotting FPGAs, the membrane was taped to the bottom of a box-shaped snap-lock type glass food container, such that the membrane sat flush with the glass. Then with the setup shown in Figure 2A a continuous stream of nitrogen/argon gas was supplied over the container, while performing the spotting. Immediately after, the container was covered with the snap-lock seal lid and incubated for 2 hr at room temperature. A second coupling was performed by repeating this step, in order to increase efficiency.
Blocking:
Two capping solutions were prepared in sufficient volume to submerge the paper (30ml for 10×12 cm paper). Note-proportionately smaller/larger volumes can be used for smaller/larger sheets of paper, as long as the paper can be completely submerged in the box-shaped container being used.
Capping solution A: 1% Acetic anhydride in amine-free DMF
Capping solution B: 1% Acetic anhydride with 1% diisopropylethylamine (DIPEA) in amine-free DMF
Capping solution A was poured in another box-shaped flat-bottom glass container which can hold the synthesis sheet. Then with tweezers, the membrane was carefully placed upside down into the container so as to submerge the entire membrane in the liquid and avoid bubbles. Do not shake the container. After 2 minutes, the solution was discarded and capping solution B was added. Carefully, the membrane was lifted with tweezers, turned face up and placed back into the solution, taking care that there are no air bubbles. The membrane was incubated for an additional 5 minutes, after which the solution is discarded. The membrane was immediately washed using the DMF wash steps described below.
DMF wash:
30 ml of amine-free DMF was added to the membrane in a box-shaped glass container and was shaken on an orbital shaker at 300 rpm for 2 minutes. The solution was discarded and additional 30 ml of DMF was added, followed by shaking the membrane for 30–60s and discarding the solution. This was repeated 2 additional times. Thus, there are total of 4 washes with DMF.
Fmoc Deprotection:
30 ml of 20% 4-methylpiperidine in DMF was added to the membrane and shaken on an orbital shaker at 300 rpm for 5 minutes. This solution is discarded and an additional treatment of 30 ml of 20% 4-methylpiperidine in DMF was given to the membrane with shaking for 5 minutes. The solution was discarded following which the DMF wash (above) and the methanol wash (below) were performed.
Methanol wash:
The membrane was treated with 30 ml of methanol with shaking on an orbital shaker at 300 rpm for 2 minutes. The solution is discarded and the step is repeated 3 additional times. Thus, there are total of 4 washes with methanol every time.
Quality Checks During Synthesis
Staining:
To check if synthesis is successful, a bromophenol blue stain was performed on the membrane after every step. For this, a stock 1L solution of 10 mg/L bromophenol blue in methanol was prepared. 30 ml of this solution was added to the membrane and shaken for 30s or less, until the appearance of a blue color on the spots. If a blue color does not appear on any spot, incubate for a longer time, but no more than 1 minute (permanent staining can occur). A blue color indicates that the free amine is present, and therefore, the coupling and Fmoc-deprotection steps were successful. To wash off the stain, methanol is added and the membrane is shaken for 5 minutes, and repeat it twice if needed. Note, in some conditions if the stain still does not come off, we would wash for 5 minutes with DMF, and repeat it twice if needed. Following this, another Methanol Wash (as above) was performed and then the membrane was dried in a stream of air followed by a vacuum desiccator or lyophilizer. Notes- (i) Certain amino acids do not show a blue color with the bromophenol blue stain. For these spots, it is recommended to proceed with the next step and check after the next the step or use the biopsy method (below) to check the extent of the reaction. (ii) If the stain does not work for 2 consecutive steps it is possible that you may need to use higher concentration of a bromophenol blue stain (up to 20 mg/L).
Biopsy:
To check if the synthesis is proceeding well, in a more detailed manner, a biopsy of the membrane was taken for a small-scale sampling. For this, a 0.5 mm diameter biopsy punch was used (World Precision Instruments Cat #504639). The membrane was placed on a clean cutting mat and 2–3 biopsies were taken from the sample spot off-center at different locations. It is important to take more than 1 biopsy per sample in order to ensure that the synthesis is going well across the region of the membrane. The biopsied samples were then treated for global deprotection (as given below) using 50 microliter of cleavage solution A and cleavage solution B. Following which, the peptide release (as given below) was performed using 20 microliter of release solution. After the incubation time, the release solution was sampled directly, diluted 1:10 in 50% ACN:Water with 0.1% TFA and spotted for MALDI analysis. Note- due to high salt content of the release solution, a dilution of 1:20 may also be performed in order to get better crystallization for MALDI. Alternatively, a ZipTip C18 desalting of the release solution may be performed to facilitate the MALDI analysis.
Global Side Chain Deprotection
To deprotect the side chain protecting groups of the amino acids a two-step cleavage procedure is used as described previously, with modified reaction times (Hilpert et al., 2007). The membrane was washed 4 times with 30 ml of dichloromethane (DCM) before starting the deprotection. First, the membrane was treated with cleavage 50 ml of solution A (TFA:TIPS:Water:Phenol:DCM :: 90:3:2:1:4) for 20 mins, making sure there were no air bubbles and the membrane was completely covered. Care was taken not to shake the membrane as shaking the membrane at this stage could disintegrate the membrane. After this, the membrane was washed 3 times with 30 mL of DCM. In the second step, the membrane was treated with 50 ml of cleavage solution B (TFA:TIPS:Water:Phenol:DCM :: 50:3:2:1:44) for 2 h (longer time up to 3 h if sequence has more than one Arginine residue) again without shaking. We ensured there were no air bubbles when the solution was first added, however, bubbles may form later, but that did not affect the reaction. The solution is discarded carefully. The membrane was washed 4 times with 30 mL of DCM followed by Methanol wash procedure. After this, the membrane was dried in a desiccator or lyophilizer for at least 5 hours to remove any residual trapped TFA. This step is required to ensure the efficiency of the release step which follows.
Peptide Release
A wide variety of release solutions were tested as described in supplementary table S3. The GP1 library used the milder release condition of sodium bicarbonate with water alone (no methanol). This method required longer reaction times. We optimized this condition by adding methanol for the GP2 library which significantly increased the rate of reaction (See Notes About MALDI Spectra in chemical characterization Data S1 document). The optimal release conditions are as follows: A solution of 200 mM Sodium Bicarbonate was made in 1:2 methanol:water as the release solution. The spots were cut out and placed in individual 2.0 ml microcentrifuge tubes. 1 ml of release solution was added to the tubes, and mixed on a shaker for 5 minutes to neutralize any residual acids on the surface. This solution was discarded, and a fresh 1 ml of release solution was added to the tubes. The tubes were placed on an incubator shaker for 6 h at 30 °C. These 1 ml releases were collected in separate tubes and neutralized with acetic acid solution. The spots were further washed with 1 ml of release solution overnight. The following day, the two release solutions for each spot were combined and dried using a centrifugal evaporator. The dried powder can be reconstituted as an aqueous solution for desalting using Sep-Pak C18 (Waters) cartridge, and for HPLC purification and analysis. The reconstituted sample can also be directly used for mass spectrometry after appropriate dilution. Note- Spots released with methanol:water mixtures produced methyl ester at C-terminus as a mixture, see Figure 3.
HPLC and MALDI Characterization
HPLC method and results are shared together in a separate HPLC-reports supplement. GP1 library was purified with semi-preparative method 1, while GP2 library was purified using Sep-Pak C18 and characterized by analytical method 2. MALDI-MS method and characterization is provided as the last part of the supplementary information.
Quantification of Peptides
Peptides from the GP1 array were quantified using the absorption at 205nm (A205) on a Nanodrop using the Scopes method, which is known to yield more accurate results for proteins and peptides containing tryptophan residues. Briefly 2 μL of sample is placed on the nanodrop instrument as instructed in the user manual and the absorption is read automatically by the software to yield the peptide concentration in mg/ml. This value is then used to calculate the total amount of peptide.
For peptides in the GP2 array which contain the fluorescein, we utilized the absorbance at 453 nm for the fluorescein as the means to quantify it. Previously it has been shown that the absorption maximum for fluorescein at pH 6.5 is at 453 nm with a molar absorptivity of 29,000 M−1cm−1(Sjöback et al., 1995). As a result, we dissolved the compounds in pH 6.5 buffer and quantified it using the nanodrop using similar technique as above.
For the peptides in the GP2 array without fluorescein, we utilized a BCA assay using the Pierce™ BCA Protein Assay Kit (ThermoFisher Scientific) following standard protocol provided by the manufacturer. Full quantification results provided in Supplementary Table S4.
Microarray Printing
Arrays were printed using sciFLEXARRAYER S11 microarray printer (Scienion) onto Nexterion® Slide H 3D NHS (Schott) slides. Briefly, the peptides were diluted into PBS (phosphate buffered saline) buffer pH 7.4 at 100 μM or 25 μM concentrations for printing. Following this, the peptides were put into a 384-well polypropylene microplate which can be interfaced with the instrument. The printer was used with the PDC 70 type 3 nozzles (70 μm inner diameter) and the voltage and pulse parameters were adjusted to dispense 330 pl per spot. All samples were printed as 4 spots on individual arrays in either 8-array or 16-array per slide format (dimensions based on Grace Biolabs ProPlate® Multi-Well Chambers), depending on the number of spots. The GP1 library was printed at 50, 100 and 200 μM concentrations on the same array (each concentration 4 spots), while the GP2 library was printed at 100 μM and 25 μM concentrations on separate arrays (each concentration 4 spots). Following the printing, the slides were incubated overnight at room temperature at 70% humidity and then blocked with 50 mM ethanolamine in 100 mM borate buffer for 1 hr, dip washed 10 times in PBS and then 10 times in water. The slides were then centrifuged in a slide centrifuge to dry, prior to storage at −20°C.
Microarray Assay
The general protocol to perform the assay follows guidelines previously described in detail(Heimburg-Molinaro et al., 2011). Briefly, the slides were removed from the freezer and allowed to come to room temperature in a vacuum desiccator. The multi-well chamber (either 8-well or 16-well) (Grace Biolabs) was assembled on the slide. TSM Wash (TSMW) buffer (20 mM Tris-HCl, 150 mM NaCl, 2 mM CaCl2, 2 mM MgCl2, 0.05% Tween-20, pH 7.4) was added and incubated for 5 minutes to rehydrate the slide. The biotinylated lectins or primary antibodies (Glycan Binding Protein-GBP) were diluted to the corresponding concentrations in TSM Binding Buffer (TSMW buffer + 1% Bovine Serum Albumin (BSA) (protease free)). Wash solutions were aspirated from the wells and 50 or 100 μL (16-well format or 8-well format) of the GBP was added to the corresponding wells. The slide was incubated for 1 hr at room temperature with orbital agitation, after which the wells were washed 4 times with TSMW buffer. The secondary detector (i.e. either streptavidin or secondary antibody) was diluted to the appropriate concentration and 50 or 100 μL was added to the appropriate wells. The slide was incubated for 1 hour at room temperature after which the wells were washed 4 times with TSMW buffer, then 4 times with TSM buffer (TSMW buffer without 0.05% Tween-20), then 4 times with water. The multi-well chamber was disassembled and the slide was scanned using the GenePix 4300A Scanner (Molecular Devices) at the appropriate wavelength with laser power 70% and PMT 450–500 at 5 μm/pixel resolution. Spots were aligned and analyzed to the print layout using the GenePix Pro software and data was processed using Microsoft Excel. For the GP1 array, spots printed at 100 μM showed consistent results and hence only data for those spots were analyzed for comparison. The trend for spots printed at 25 and 200 μM remained similar to that of 100 μM.
Enzymatic Glycosylation Reactions
Materials:
Enzyme constructs for glycosyltransferases ST6GalNAc1, GCNT1 were obtained from Dr. Kelley Moremen (University of Georgia) and the Repository of Glyco-enzyme Expression Constructs (http://glycoenzymes.ccrc.uga.edu/), which also provides details for expression(Moremen et al., 2018). Expression, purification and extensive characterization of recombinant T-synthase and Cosmc was as described previously(Aryal et al., 2010; Hanes et al., 2017). Sugar donors used were obtained from commercial sources as follows: CMP-Sialic Acid and UDP-GlcNAc were obtained from Chemily Glycoscience (Georgia, USA), while UDP-galactose was obtained from Carbosynth (San Diego, USA). Stock reaction buffer used was a 5X Cacodylate buffer consisting of 250 mM Cacodylate, 50 mM MnCl2, 5 mM CaCl2, pH 7.2.
On-Array Glycosylation
1). With ST6GalNAc1:
The microarray slides printed for GP2 array were assembled with multi-well chamber (16 well). 100 μL of a solution of ST6GalNAc1 at 0, 5 or 100 μg/ml with 2 mM CMP-Sialic acid in 1x Cacodylate buffer (50 mM Cacodylate, 10 mM MnCl2, 1 mM CaCl2, pH 7.2.) was premixed and added to a microarray well each. The array was sealed using ProPlate® Slide Module Seal Strips (Grace Biolabs) to minimize evaporation. The sealed slide was placed in a container with a wet tissue to maintain humidity and the container was sealed and placed in an incubator at 37°C for 18h (overnight). The arrays were probed with lectins as described below.
2). With T-synthase and Cosmc:
The microarray slides printed for GP2 array were assembled with multi-well chamber (16 well). 100 μL of a solution of T-synthase at 0, or 50 μg/ml with Cosmc at 0 or 1 μg/ml, with 2.5 mM UDP-Galactose in 1x Cacodylate buffer (50 mM Cacodylate, 10 mM MnCl2, 1 mM CaCl2, pH 7.2.) was premixed and added to a microarray well each. The array was sealed using ProPlate® Slide Module Seal Strips (Grace Biolabs) to minimize evaporation. The sealed slide was placed in a container with a wet tissue to maintain humidity and the container was sealed and placed in an incubator for 18h (overnight). The arrays were probed with lectins as described below.
3). Probing with Lectins:
After incubation with the enzymes, the slides were taken out of the incubator and the solution was diluted with TSMW buffer (composition in Microarray Assay methods above). The solution was aspirated and the arrays were washed with slight agitation 4 additional times with TSMW buffer. The biotinylated lectins Vicia villosa Lectin (VVL) and Peanut Agglutinin (PNA) were diluted in TSM binding buffer (composition in Microarray Assay methods above) at 5 μg/ml. 100 μl of VVL was added to each array treated with ST6GalNAc1 while 100 μl of PNA was added to each array treated with T-synthase. The arrays were incubated at room temperature for 1 hour on an orbital shaker. The arrays were then washed 4 times with TSMW buffer and incubated with 1 μg/ml Streptavidin-Cy5 for 1 hour. Following the incubation, the arrays were washed 4 times with TSMW buffer, then 4 times with TSM buffer and finally 4 times with water and then dried. The slides were then scanned on a GenePix 4300A scanner at appropriate wavelengths (635nm) with settings for PMT 450 and laser power set to 70.
In-Solution Glycosylation
To perform in-solution glycosylation with ST6GalNAc1, glycopeptides GP2.5 (GalNAca1-S) and GP2.6 (GalNAca1-T) at 30 μM were incubated with ST6GalNAc1 (5 μg/mL) and CMP-Sialic acid (2 mM) in reaction buffer (50 mM Cacodylate, 10 mM MnCl2, 1 mM CaCl2, pH 7.2). Final reaction volume was 100 μL. A sample was taken before adding the donor sugar for time 0. The mixture was incubated at 37°C overnight. Samples of 5 μl were taken at time points 0, 1.5 hrs and 18 hrs and diluted with 45 μl of 50% acetonitrile:water with 0.1% trifluoroacetic acid, to quench/denature the enzyme. These samples were then analyzed by MALDI-MS or by analytical HPLC.
To perform in-solution glycosylation with GCNT1, glycopeptides GP2.08, GP2.09 and GP2.10 at 30 μM and was incubated with GCNT1 (40 μg/ml) with UDP-GlcNAc 20 mM in reaction buffer (50 mM Cacodylate, 10 mM MnCl2, 1 mM CaCl2, pH 7.2). Final reaction volume was 100 μL. A sample was taken before adding the donor sugar for time 0. The mixture was incubated at 37°C overnight. Samples of 5 μl were taken at time points 0, and 18 hrs and diluted with 45 μl of 50% acetonitrile:water with 0. 1% trifluoroacetic acid, to quench/denature the enzyme. These samples were then analyzed by MALDI-MS or by analytical HPLC.
Fluorescence Polarization Assay
To perform the fluorescence polarization assay, 500 μL of 1 μM glycopeptide solution in buffer (50 mM Tris-HCl, 150 mM NaCl, 5 mM CaCl2, 5 mM MgCl2, pH 7.4) containing 2 mg/mL BSA was prepared. Unconjugated Vicia villosa lectin (VVL) (Vector Labs, Burlingame, CA) was prepared in buffer, ranging from concentrations from 0–25 μM, and put in a 384 plate at 76 μL. 4 μL of above glycopeptide solution was added to the wells making the effective concentration of glycopeptide within each well 50 nM and that of BSA in each well 0.1 mg/ml. The plate was incubated at room temperature for 15 minutes, after which the polarization was measured using the plate-reader.
Orbitrap LC-MS Analysis
All forty compounds from the glycopeptide synthetic library were analyzed separately for their accurate molecular weight and to assess compound purity on a high-resolution accurate mass platform; Lumos (Thermo Fisher Scientific). Instrument parameters for the experiment was data dependent acquisition with Full MS spectrum followed by 20 fragment ion spectra. Mass range for the Full MS experiment was held at m/z 400 – 1600 to ensure lack of contaminants. We acquired Full MS spectrum at 60,000 resolution at m/z 200 with automated gain control (AGC) 4e5. For the MS2 data, we acquired HCD data on the compounds at 15K resolution at 27eV. The AGC for the MS2 experiments was 2e5 and fragment ions were collected in the Orbitrap. MS2 data was acquired with HCD fragmentation; spectra were acquired at 15,000 resolution in centroid mode and stepping energy mode with 25% (+/− 5%). The starting mass for the MS2 data was at m/z 110 to enable analysis of low molecular weight oxonium ions.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data Quantitation for the Array Analysis
The signal intensities for microarray experiments were quantified using GenePix Pro 7 software which is provided with the scanner. Data in terms of relative fluorescence units (RFUs) for 4 replicate spots of a glycopeptide were background-subtracted and averaged in Excel to show as bar graphs and the standard deviation was used to represent the error bars in the bar graphs.
Data Quantitation for Fluorescence Polarization
The polarization was measured as mP (milli-polarization units) and an average of triplicate parallel readings was taken for each concentration of protein VVL. These were plotted using Origin (OriginLab®) and fit to the standard equation to obtain the KD:
Where, Pmin and Pmax are the minimum and maximum polarization signals; X is the concentration of VVL and the Signal is the observed polarization signal, KD is the binding affinity.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CTD110.6 | Santa Cruz Biotechnology | Cat#sc-59623 |
| RL2 | Santa Cruz Biotechnology | Cat#sc-59624 |
| 6D93 | Santa Cruz Biotechnology | Cat#sc-71736 |
| ReBaGs6 | Richard Cummings (Matsumoto et al., 2019) | N/A |
| Goat anti-Mouse IgG (H+L), Alexa Fluor 633 | Thermo Fisher | Cat#A-21052 |
| Goat anti-Mouse IgM Heavy Chain Cross-Adsorbed, Alexa Fluor 633 | Thermo Fisher | Cat#A-21046 |
| Goat anti-Human IgG (H+L), Alexa Fluor 633 | Thermo Fisher | Cat#A-21091 |
| Rabbit polyclonal anti-Mouse IgG (H+L), Alexa Fluor 488 | Thermo Fisher | Cat#A-11059 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Indium(III) bromide | Sigma Aldrich | Cat#545082 |
| β-D-Glucosamine pentaacetate (peracetyl-β-D-GlcNAc) | Sigma Aldrich | Cat#G4533 |
| β-D-Galactosamine pentaacetate (peracetyl-β-D-GalNAc) | Sigma Aldrich | Cat#G5644 |
| β-D-Mannosamine pentaacetate (peracetyl-β-D-ManNAc) | Sigma Aldrich; Carbosynth | N/A; MA04038 |
| Fmoc-Cys-OH | Chem-Impex | Cat#14009 |
| Whatman® quantitative filter paper, hardened low-ash, Grade 50 | Sigma Aldrich | Cat# WHA1450916 |
| 1-Methyl-2-pyrrolidinone (NMP) biotech grade, >99.7% | Sigma Aldrich | Cat#494496-1L |
| 1-Hydroxybenzotriazole hydrate (HOBt) wetted with not less than 14 wt. % water, 97% | Sigma Aldrich | Cat#157260-100G |
| Bromophenol blue | Sigma Aldrich | Cat#114391-5G |
| Anhydrous amine-free N,N-Dimethylformamide (DMF) | VWR | Cat#AA43465-K7 |
| N,N’-Diisopropylcarbodiimide (DIC) 99%, AcroSeal™ | ACROS Organics | Cat#AC446181000 |
| Fmoc - Ser(β - D - GlcNAc(Ac)3) - OH | Anaspec | Cat#AS-65495–100 |
| Fmoc - Thr(β - D - GlcNAc(Ac)3) - OH | Anaspec | Cat#AS-65495–100 |
| Fmoc-Ser(α-D-GalNAc(Ac)3)-OH | Sussex Research | Cat#GA121000 |
| Fmoc-Thr(α-D-GalNAc(Ac)3)-OH | Sussex Research | Cat#GA131000 |
| Fmoc-Ser(Galβ(1–3)GalNAc)-OH, peracetate | Sussex Research | Cat#GA121010 |
| Fmoc-Thr(Galβ(1–3)GalNAc)-OH, peracetate | Sussex Research | Cat#GA131010 |
| Fmoc-trans-4-fluoro-Pro-OH | Bachem | Cat#F-4035.1000 |
| Fmoc-cis-4-fluoro-Pro-OH | Bachem | Cat#B-4050.1000 |
| Fmoc-A2G0 (N-glycan) | Richard Cummings (Gao et al., 2019) | N/A |
| 5(6)-Carboxyfluorescein | Sigma | Cat#8510820005 |
| UDP-GlcNAc | Chemily | Cat#SN02009 |
| UDP-Galactose | Carbosynth | Cat#MU06699 |
| CMP-sialic acid | Chemily | Cat#SN02001 |
| Biotinylated Vicia villosa Lectin (VVL) | Vector Labs | Cat#B-1235-2 |
| Biotinylated Concanavalin A (ConA) | Vector Labs | Cat#B-1005-5 |
| Biotinylated Griffonia simplicifolia Lectin II (GSLII) | Vector Labs | Cat#B-1215-2 |
| Biotinylated Wheat Germ Agglutinin (WGA) | Vector Labs | Cat#B-1025-5 |
| Biotinylated Peanut Agglutinin (PNA) | Vector Labs | Cat#B-1075 |
| Vicia villosa Lectin, Unconjugated (VVL) | Vector Labs | Cat#L-1230-5 |
| T-Synthase | Richard Cummings (Aryal et al., 2010; Hanes et al., 2017) | N/A |
| Cosmc | Richard Cummings (Aryal et al., 2010; Hanes et al., 2017) | N/A |
| Critical Commercial Assays | ||
| Pierce BCA Protein Assay Kit | Thermo Fisher | Cat#23225 |
| Recombinant DNA | ||
| Plasmid: ST6GalNAc1-pGEn2 | Kelley Moremen (Moremen et al., 2018) | N/A |
| Plasmid: GCNT1-pGEn2 | Kelley Moremen | HsCD00413067 |
| Software and Algorithms | ||
| Excel | Microsoft | N/A |
| Prism | Graphpad | N/A |
| Origin | OriginLab | N/A |
| Other | ||
| Snap-lock type glass food container | Target | N/A |
| Reusable biopsy punch (0.5mm tip) | World Precision Instruments | Cat#504639 |
| Nexterion® Slide H 3D NHS (Schott) slides | Applied Microarrays | Cat#1070936 |
SIGNIFICANCE.
Synthesis of glycopeptide libraries is challenged by multi-faceted issues including cost and availability of starting materials, efficiency of reactions and inability to scale-down traditional synthetic methods to < 1 μmole scale. Glyco-SPOT synthesis offers a method to generate libraries of glycopeptides in microgram (nmole) scale with small amounts of expensive and rare starting materials and without the need for expensive instrumentation. It therefore attempts to democratize glycopeptide synthesis. Such glycopeptide libraries offer a unique opportunity to study the role of glycans present on glycoproteins in conjunction with the polypeptide in its natural form rather than using artificial linkers. Synthetic glycopeptides offer site specific control of glycosylation enabling structure activity relationship studies. This method also enables production of labelled glycopeptides with O-, N- and S-linked glycosylation. Libraries produced by this method can be efficiently utilized (i) in high-throughput glycopeptide microarrays, (ii) to study enzyme reactivity and expansion of libraries using enzymes, (iii) in fluorescence polarization assays to determine binding affinity (iv) as mass spectrometry standards. This method is poised to yield more glycopeptide libraries in the future to study variety of glycopeptide-protein interactions which can yield important biological discoveries.
HIGHLIGHTS.
The Glyco-SPOT synthesis technique was developed to generate glycopeptide libraries
Libraries consisted of variable length glycopeptides with O-, N-, S- linked glycans
Used for microarray screening, enzyme specificity, affinity, mass spectrometry
Technique opens up ability to study biology of glycopeptides
ACKNOWLEDGEMENTS
This work was supported by National Institutes of Health Grant P41GM103694. N. P. also thanks the Joan and Marvin Carmack Fund at Indiana University for support of this work and the Radcliffe Institute for Advanced Study at Harvard for the Edward, Frances, and Shirley B. Daniels Fellow position (2017–2018) that aided this collaboration. The authors would like to thank Dr. Kelley Moremen (University of Georgia) and the Repository of Glyco-enzyme Expression Constructs (http://glycoenzymes.ccrc.uga.edu/) (the National Institutes of Health Grant P41GM103390 and P01GM107012) for the glycosyltransferase constructs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no conflicts of interest.
SUPPLEMENTAL INFORMATION
Supplemental information includes rationale for the selected sequences synthesized in this manuscript, 7 figures, 5 tables. In addition, chemical characterization data is presented in a separate file Data S1.
REFERENCES
- Apweiler R, Hermjakob H, and Sharon N (1999). On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim Biophys Acta 1473, 4–8. [DOI] [PubMed] [Google Scholar]
- Aryal RP, Ju T, and Cummings RD (2010). The endoplasmic reticulum chaperone Cosmc directly promotes in vitro folding of T-synthase. J Biol Chem 285, 2456–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blixt O, Clo E, Nudelman AS, Sorensen KK, Clausen T, Wandall HH, Livingston PO, Clausen H, and Jensen KJ (2010). A high-throughput O-glycopeptide discovery platform for seromic profiling. J Proteome Res 9, 5250–5261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bora U (2011). An Eco-friendly and Mild Process for Deacetylation Reactions in Water. Asian J Chem 23, 941–942. [Google Scholar]
- Bubb WA (2003). NMR spectroscopy in the study of carbohydrates: Characterizing the structural complexity. Concepts in Magnetic Resonance Part A 19A, 1–19. [Google Scholar]
- Buncel E, and Symons EA (1970). The inherent instability of dimethylformamide–water systems containing hydroxide ion. Journal of the Chemical Society D: Chemical Communications, 164–165.
- Buskas T, Ingale S, and Boons GJ (2006). Glycopeptides as versatile tools for glycobiology. Glycobiology 16, 113R–136R. [DOI] [PubMed] [Google Scholar]
- Comer FI, Vosseller K, Wells L, Accavitti MA, and Hart GW (2001). Characterization of a mouse monoclonal antibody specific for O-linked N-acetylglucosamine. Anal Biochem 293, 169–177. [DOI] [PubMed] [Google Scholar]
- Corzana F, Busto JH, Jimenez-Oses G, Garcia de Luis M, Asensio JL, Jimenez-Barbero J, Peregrina JM, and Avenoza A (2007). Serine versus threonine glycosylation: the methyl group causes a drastic alteration on the carbohydrate orientation and on the surrounding water shell. J Am Chem Soc 129, 9458–9467. [DOI] [PubMed] [Google Scholar]
- Duus J, Gotfredsen CH, and Bock K (2000). Carbohydrate structural determination by NMR spectroscopy: modern methods and limitations. Chem Rev 100, 4589–4614. [DOI] [PubMed] [Google Scholar]
- Frank R (2002). The SPOT-synthesis technique. Synthetic peptide arrays on membrane supports--principles and applications. J Immunol Methods 267, 13–26. [DOI] [PubMed] [Google Scholar]
- Gao C, Hanes MS, Byrd-Leotis LA, Wei M, Jia N, Kardish RJ, McKitrick TR, Steinhauer DA, and Cummings RD (2019). Unique Binding Specificities of Proteins toward Isomeric Asparagine-Linked Glycans. Cell Chem Biol. [DOI] [PMC free article] [PubMed]
- Grant OC, Smith HM, Firsova D, Fadda E, and Woods RJ (2014). Presentation, presentation, presentation! Molecular-level insight into linker effects on glycan array screening data. Glycobiology 24, 17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greis KD, Hayes BK, Comer FI, Kirk M, Barnes S, Lowary TL, and Hart GW (1996). Selective detection and site-analysis of O-GlcNAc-modified glycopeptides by beta-elimination and tandem electrospray mass spectrometry. Anal Biochem 234, 38–49. [DOI] [PubMed] [Google Scholar]
- Günther KU, and Ziegler T (2014). Synthesis of 1,2,3-Triazole-Linked Glycoconjugates of N-(2-Aminoethyl)glycine: Building Blocks for the Construction of Combinatorial Glycopeptide Libraries. Synthesis 46, 2362–2370. [Google Scholar]
- Halim A, Westerlind U, Pett C, Schorlemer M, Ruetschi U, Brinkmalm G, Sihlbom C, Lengqvist J, Larson G, and Nilsson J (2014). Assignment of saccharide identities through analysis of oxonium ion fragmentation profiles in LC-MS/MS of glycopeptides. J Proteome Res 13, 6024–6032. [DOI] [PubMed] [Google Scholar]
- Hanes MS, Moremen KW, and Cummings RD (2017). Biochemical characterization of functional domains of the chaperone Cosmc. PLoS One 12, e0180242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimburg-Molinaro J, Song X, Smith DF, and Cummings RD (2011). Preparation and analysis of glycan microarrays. Curr Protoc Protein Sci Chapter 12, 12.10.11–12.10.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilpert K, Winkler DF, and Hancock RE (2007). Peptide arrays on cellulose support: SPOT synthesis, a time and cost efficient method for synthesis of large numbers of peptides in a parallel and addressable fashion. Nat Protoc 2, 1333–1349. [DOI] [PubMed] [Google Scholar]
- Hojo H, and Nakahara Y (2000). Recent progress in the solid-phase synthesis of glycopeptide. Curr Protein Pept Sci 1, 23–48. [DOI] [PubMed] [Google Scholar]
- Hojo H, and Nakahara Y (2007). Recent progress in the field of glycopeptide synthesis. Biopolymers 88, 308–324. [DOI] [PubMed] [Google Scholar]
- Horiya S, Bailey JK, and Krauss IJ (2017). Directed Evolution of Glycopeptides Using mRNA Display. Methods Enzymol 597, 83–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh YS, Wilkinson BL, O’Connell MR, Mackay JP, Matthews JM, and Payne RJ (2012). Synthesis of the bacteriocin glycopeptide sublancin 168 and S-glycosylated variants. Org Lett 14, 1910–1913. [DOI] [PubMed] [Google Scholar]
- Izumi M, Kuruma R, Okamoto R, Seko A, Ito Y, and Kajihara Y (2017). Substrate Recognition of Glycoprotein Folding Sensor UGGT Analyzed by Site-Specifically (15)N-Labeled Glycopeptide and Small Glycopeptide Library Prepared by Parallel Native Chemical Ligation. J Am Chem Soc 139, 11421–11426. [DOI] [PubMed] [Google Scholar]
- Jobron L, and Hummel G (2000). Solid-Phase Synthesis of Unprotected N-Glycopeptide Building Blocks for SPOT Synthesis of N-linked Glycopeptides. Angew Chem Int Ed Engl 39, 1621–1624. [DOI] [PubMed] [Google Scholar]
- Kaestle KL, Anwer MK, Audhya TK, and Goldstein G (1991). Cleavage of esters using carbonates and bicarbonates of alkali metals: synthesis of thymopentin. Tetrahedron Lett 32, 327–330. [Google Scholar]
- Kurosawa N, Kojima N, Inoue M, Hamamoto T, and Tsuji S (1994). Cloning and expression of Gal beta 1,3GalNAc-specific GalNAc alpha 2,6-sialyltransferase. J Biol Chem 269, 19048–19053. [PubMed] [Google Scholar]
- Lefever MR, Szabo LZ, Anglin B, Ferracane M, Hogan J, Cooney L, and Polt R (2012). Glycosylation of alpha-amino acids by sugar acetate donors with InBr3. Minimally competent Lewis acids. Carbohydr Res 351, 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehoux S, Mi R, Aryal RP, Wang Y, Schjoldager KT, Clausen H, van Die I, Han Y, Chapman AB, Cummings RD, et al. (2014). Identification of distinct glycoforms of IgA1 in plasma from patients with immunoglobulin A (IgA) nephropathy and healthy individuals. Mol Cell Proteomics 13, 3097–3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppanen A, Yago T, Otto VI, McEver RP, and Cummings RD (2003). Model glycosulfopeptides from P-selectin glycoprotein ligand-1 require tyrosine sulfation and a core 2-branched O-glycan to bind to L-selectin. J Biol Chem 278, 26391–26400. [DOI] [PubMed] [Google Scholar]
- Lopez-Perez PM, Grimsey E, Bourne L, Mikut R, and Hilpert K (2017). Screening and Optimizing Antimicrobial Peptides by Using SPOT-Synthesis. Front Chem 5, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcos NT, Pinho S, Grandela C, Cruz A, Samyn-Petit B, Harduin-Lepers A, Almeida R, Silva F, Morais V, Costa J, et al. (2004). Role of the human ST6GalNAc-I and ST6GalNAc-II in the synthesis of the cancer-associated sialyl-Tn antigen. Cancer Res 64, 7050–7057. [DOI] [PubMed] [Google Scholar]
- Matsumoto Y, Kudelka MR, Hanes MS, Lehoux S, Dutta S, Jones MB, Stackhouse KA, Cervoni GE, Heimburg-Molinaro J, Smith DF, et al. (2019). Identification of Tn Antigen O-GalNAc-expressing glycoproteins in human carcinomas using novel anti-Tn recombinant antibodies. Glycobiology. [DOI] [PMC free article] [PubMed]
- Moremen KW, Ramiah A, Stuart M, Steel J, Meng L, Forouhar F, Moniz HA, Gahlay G, Gao Z, Chapla D, et al. (2018). Expression system for structural and functional studies of human glycosylation enzymes. Nat Chem Biol 14, 156–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newberry RW, and Raines RT (2017). 4-Fluoroprolines: Conformational Analysis and Effects on the Stability and Folding of Peptides and Proteins. Top Heterocycl Chem 48, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng S, Jafari MR, Matochko WL, and Derda R (2012). Quantitative synthesis of genetically encoded glycopeptide libraries displayed on M13 phage. ACS Chem Biol 7, 1482–1487. [DOI] [PubMed] [Google Scholar]
- Norrlinger M, and Ziegler T (2014). Synthesis of aromatic glycoconjugates. Building blocks for the construction of combinatorial glycopeptide libraries. Beilstein J Org Chem 10, 2453–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa T, Hirohashi Y, Murai A, Nishidate T, Okita K, Wang L, Ikehara Y, Satoyoshi T, Usui A, Kubo T, et al. (2017). ST6GALNAC1 plays important roles in enhancing cancer stem phenotypes of colorectal cancer via the Akt pathway. Oncotarget 8, 112550–112564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohyabu N, Hinou H, Matsushita T, Izumi R, Shimizu H, Kawamoto K, Numata Y, Togame H, Takemoto H, Kondo H, et al. (2009). An essential epitope of anti-MUC1 monoclonal antibody KL-6 revealed by focused glycopeptide library. J Am Chem Soc 131, 17102–17109. [DOI] [PubMed] [Google Scholar]
- Osinaga E, Bay S, Tello D, Babino A, Pritsch O, Assemat K, Cantacuzene D, Nakada H, and Alzari P (2000). Analysis of the fine specificity of Tn-binding proteins using synthetic glycopeptide epitopes and a biosensor based on surface plasmon resonance spectroscopy. FEBS Lett 469, 24–28. [DOI] [PubMed] [Google Scholar]
- Pan Y, Yago T, Fu J, Herzog B, McDaniel JM, Mehta-D’Souza P, Cai X, Ruan C, McEver RP, West C, et al. (2014). Podoplanin requires sialylated O-glycans for stable expression on lymphatic endothelial cells and for interaction with platelets. Blood 124, 3656–3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan TB, and Mayr H (2005). Comparison of the nucleophilicities of alcohols and alkoxides. Can J Chem 83, 1554–1560. [Google Scholar]
- Qu J, Yu H, Li F, Zhang C, Trad A, Brooks C, Zhang B, Gong T, Guo Z, Li Y, et al. (2016). Molecular basis of antibody binding to mucin glycopeptides in lung cancer. Int J Oncol 48, 587–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rademaker GJ, Pergantis SA, Blok-Tip L, Langridge JI, Kleen A, and Thomas-Oates JE (1998). Mass spectrometric determination of the sites of O-glycan attachment with low picomolar sensitivity. Anal Biochem 257, 149–160. [DOI] [PubMed] [Google Scholar]
- Risinger C, Sorensen KK, Jensen KJ, Olofsson S, Bergstrom T, and Blixt O (2017). Linear Multiepitope (Glyco)peptides for Type-Specific Serology of Herpes Simplex Virus (HSV) Infections. ACS Infect Dis 3, 360–367. [DOI] [PubMed] [Google Scholar]
- Saraswathy N, and Ramalingam P (2011). 16 - Glycoproteomics. In Concepts and Techniques in Genomics and Proteomics, Saraswathy N, and Ramalingam P, eds. (Woodhead Publishing; ), pp. 213–218. [Google Scholar]
- Seitz O (2000). Glycopeptide synthesis and the effects of glycosylation on protein structure and activity. ChemBioChem 1, 214–246. [DOI] [PubMed] [Google Scholar]
- Sjöback R, Nygren J, and Kubista M (1995). Absorption and fluorescence properties of fluorescein. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy 51, L7–L21. [Google Scholar]
- Szabo LZ, Hanrahan DJ, Jones EM, Martin E, Pemberton JE, and Polt R (2016). Preparation of S-glycoside surfactants and cysteine thioglycosides using minimally competent Lewis acid catalysis. Carbohydr Res 422, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toleman CA, Schumacher MA, Yu SH, Zeng W, Cox NJ, Smith TJ, Soderblom EJ, Wands AM, Kohler JJ, and Boyce M (2018). Structural basis of O-GlcNAc recognition by mammalian 14–3-3 proteins. Proc Natl Acad Sci U S A 115, 5956–5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollefsen SE, and Kornfeld R (1983). The B4 lectin from Vicia villosa seeds interacts with N-acetylgalactosamine residues alpha-linked to serine or threonine residues in cell surface glycoproteins. J Biol Chem 258, 5172–5176. [PubMed] [Google Scholar]
- Varki A (2017). Biological roles of glycans. Glycobiology 27, 3–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WY, Cao YX, Zhou X, Wei B, Zhan L, and Sun SY (2019). Stimulative role of ST6GALNAC1 in proliferation, migration and invasion of ovarian cancer stem cells via the Akt signaling pathway. Cancer Cell Int 19, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler DF, and Campbell WD (2008). The spot technique: synthesis and screening of peptide macroarrays on cellulose membranes. Methods Mol Biol 494, 47–70. [DOI] [PubMed] [Google Scholar]
- Yoshimura Y, Denda-Nagai K, Takahashi Y, Nagashima I, Shimizu H, Kishimoto T, Noji M, Shichino S, Chiba Y, and Irimura T (2019). Products of Chemoenzymatic Synthesis Representing MUC1 Tandem Repeat Unit with T-, ST- or STn-antigen Revealed Distinct Specificities of Anti-MUC1 Antibodies. Sci Rep 9, 16641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachara NE, Vosseller K, and Hart GW (2011). Detection and analysis of proteins modified by O-linked N-acetylglucosamine. Curr Protoc Mol Biol Chapter 17, Unit 17 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler T, and Schips C (2006). An efficient Mitsunobu protocol for the one-pot synthesis of S-glycosyl amino-acid building blocks and their use in combinatorial spot synthesis of glycopeptide libraries. Nat Protoc 1, 1987–1994. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The glycopeptide microarray datasets supporting the current study are not deposited in a public repository as there is no repository currently built for glycopeptide microarrays, but are available from the Lead Contact on request.