

Abstract
Unlike health, parenchymal macrophages in retinal degeneration are not solely comprised of microglia but may include monocyte-derived recruits. Recent studies applied transgenics, lineage-tracing and transcriptomics to decipher their distinct roles in the diseases of inherited retinal degeneration and age-related macular degeneration. Literature discussed here focuses on the disease ectopic presence of both macrophage types in the subretinal space, an extracellular site surrounding outer photoreceptor cells, which is crucially involved in the pathobiology. From these studies we propose a working model in which perturbed photoreceptor states cause a microglial dominant migration to the subretinal space for protection, whereas abundant presence of subretinal monocyte-derived cells instead drives pathogenesis and accelerated disease. The latter, we propose, is underpinned by specific genetic and non-genetic determinants that lead to a maladaptive macrophage state.
Keywords: macrophages, inherited retinal dystrophy, retinitis pigmentosa, age-related macular degeneration, AMD
Introduction
As part of the central nervous system (CNS), the retina is endowed with mononuclear phagocytes (MNPs) that continually surveil its neuronal parenchyma and border tissues. These consist of microglia, a specialized type of yolk-sac derived macrophage that reside in the parenchyma [1–3] and border-associated resident macrophages [4] (i.e. long-lived retinal perivascular macrophages and short-lived choroidal macrophages adjacent to the retina [3, 5]). In certain disease states, blood derived monocytes can transiently invade the retina and differentiate into macrophages [2, 6, 7]. These monocyte-derived cells (MdCs) and retinal microglia, have received considerable research attention in retinal degenerative disease, including inherited retinal degenerations (IRDs) and age-related macular degeneration (AMD) (Box 1). In these conditions, MNPs infiltrate the affected photoreceptor layers and other outer retinal structures, and manipulation of MNPs in mouse models can alter the disease course. Given the new appreciation that microglia and monocytes represent distinct MNP lineages [1, 8–10], the field recognizes their non-redundant functions in disease and the importance of studying these cells as separate entities. As such, the current review focuses on the distinctive roles of MdCs and microglia in retinal degenerative diseases. Here, we discuss recent studies that have elucidated differential contributions of these cells in the context of the subretinal space (SRS), delineated by the outer segments of the photoreceptors and the retinal pigment epithelium (RPE), which is crucially involved in pathobiology. From these findings in mice, we propose a unified model that classifies MNP responses as adaptive versus maladaptive, and further extrapolate this idea to retinal degenerative diseases in humans.
Box 1. IRDs and AMD.
Inherited retinal degenerations (IRDs) and age-related macular disease (AMD) are both characterized by the degeneration of rods, cones, the RPE, and the choroid, while the overlying inner retina remains morphologically intact to a certain extent. However, from an etiological and symptomatic standpoint, IRDs and AMD are very different. IRDs are rare (1/4000) monogenetic diseases [112], most often due to mutations of rod genes (rod-cone dystrophy). The lack of the essential gene (recessive e.g. phosphodiesterase 6B, rd10 mutation) or the accumulation of a dysfunctional variant (dominant e.g. rhodopsin P23H mutation) leads to rod cell death and night blindness (age of onset 10–20 years), followed by a centripetal degeneration of the RPE and cones that lead to tunnel vision and eventually blindness. AMD is a late onset (above 55 years), prevalent, complex and multifactorial disease caused by the interplay of age, environmental, and genetic risk factors. The common AMD risk variants are not specific for the RPE or photoreceptors but instead concern factors of the complement cascade and lipoproteins, among others [107, 114]. Early AMD is characterized by lipoproteinaceous debris in the subretinal space (termed pseudodrusen, reviewed in [115]) or below the RPE (soft drusen). Advanced or ‘late’ debilitating AMD is characterized by central choroidal neovascularization (wet or ‘exudative’ AMD, late form) and ultimately a disciform scar (wet AMD end stage), or by a central, centrifugally extending lesion of the photoreceptors, RPE, and choroid that often starts parafoveally (geographic atrophy, GA, late form [33]). Loss of rods and to a lesser extent of cones, occur prior to RPE loss above pseudodrusen [116] and in a transitional zone adjacent to GA lesions [34] and disciform scars [117, 118]. Finally, the degeneration of the RPE and choroid leads to the typical atrophic lesions in the fundus appearance, which extends centrifugally. AMD only affects the central retina that contains the macula and the cone-rich fovea, necessary for high acuity vision that does not exist in mice.
Roles of MNPs in Retinal Health and Photoreceptor Degenerative Disease
Type and pattern of retinal MNPs are largely homologous to brain and spinal cord parenchyma. Microglia are the principal MNPs in the normal retina parenchyma [11]. The perivascular spaces also have resident macrophages that are long-lived [12], albeit radio-sensitive [11]. Monocytes (Box 2) and dendritic cells are not thought to gain access to the retinal parenchyma in non-diseased states. Like elsewhere in the CNS, retinal microglia are highly ramified, tiled, and express C-X3-C Motif Chemokine Receptor-1 (Cx3cr1), a homeostatic microglial immune checkpoint factor that inhibits expression of inflammatory cytokines such as Interleukin-1β (IL-1β) [13, 14] and C-C Motif Chemokine-2 (Ccl2) [6]. However, also like in brain and spinal cord, retinal degeneration can result in large-scale monocyte recruitment, along with microglial reactivity. Yet, due to overlapping phenotypic markers of these two MNP lineages, tools to study their isolated contributions have only recently been available, which we summarize in Table 1.
Box 2. Monocyte Derived Cells in Health and Disease.
Monocytes are bone marrow-derived myeloid cells that circulate in the blood. They are precursors of MdCs that participate in the resident MNP pool of certain organs (such as lung and gut) and are recruited during tissue damage or infection of any tissues [62]. They comprise three major subsets (classical, intermediate and non-classical) and are likely sequentially transitioning from one into another [119]. The short-lived classical monocytes constitute 90% of all circulating monocytes in humans and 60–70% in mice. They express high levels of CCR2, CD14, and Ly6C and low levels of CX3CR1 and CD16 and circulate the blood for 24h before 99% of them disappear. One percent of classical monocytes evolve into longer-lived intermediate (CCR2/CD14high CX3CR1/CD16high) monocytes, which finally become CCR2/CD14low CX3CR1/CD16high ‘patrolling’ monocytes [119]. During tissue injury, damage- and pathogen-associated molecular pattern (DAMPs and PAMPs) activate resident mast cells and resident macrophages that produce chemokines, such as CCL2 (the main ligand of CCR2), which recruit classical monocytes that differentiate into anti-microbial and pro-inflammatory macrophages. These cells release reactive oxygen species and complement components to kill, opsonize and phagocytose pathogens and secrete inflammatory cytokines (such as IL-1β, TNF-α, IL-6, and CCL2) that alter the tissue to facilitate the immune response [120]. Although these mediators are crucial to combat infection and ensure the survival of the organism, they can also cause considerable collateral damage, in particular in the CNS (including the retina) with poor regenerative potential [33]. Once the lesion is disinfected, the infiltrating macrophages produce cytokines such as VEGF that facilitate angiogenesis and tissue repair, scar formation, and inflammation resolution [121, 122]. Finally, pro-inflammatory macrophages disappear from the site and the tissue is left with the tissue-specific resident macrophages, as before the injury [123]. If the inflammatory response is not quickly controlled, it can become pathogenic and contribute to degeneration, pathological neovascularization and fibrosis, as seen in many diseases such as metabolic diseases (obesity, atherosclerosis), neurodegenerative diseases and cancers [120, 124–126].
Table 1.
Technical Challenges for Discerning MNP Lineages in Retinal Degeneration.
| Tools | Pros | Cons | Refs |
|---|---|---|---|
| Marker staining | |||
| Conventional markers, such as Iba-1, F4/80, Cd11b |
|
|
[2] |
| Microglial specific markers, such as P2ry12, Tmem119 |
|
|
[3,92,95] |
| Chimaeras | |||
| Bone marrow chimera |
|
|
[2] |
| Bone marrow chimera with lead head shielding |
|
|
[11] |
| Circulating monocyte-labeling | |||
| Systemic 5- ethynyl-2′- deoxyuridine (EdU) |
|
|
[6] |
| Intravenously injected fluorescent tracers |
|
|
[72] |
| Reporter mice | |||
| Cx3cr1GFP or Cx3cr1YFP |
|
|
[11,14,105] |
| Ccr2RFP |
|
|
[6,7,34] |
| Cx3cr1CreER R26fl-STOP-fl-reporter |
|
|
[9,11,34,84,85] |
| Depletion / recruitment inhibition studies | |||
| Csf1r antagonists |
|
|
[97,98,127] |
| Intravenous clodronate liposome |
|
|
[6,98] |
| Cx3cr1CreER R26fl-STOP-fl-DTA |
|
|
[37,113] |
| Cx3cr1CreER R26fl-STOP-fl-DTR |
|
|
[3,84] |
| Ccl2- and Ccr2- deletion / inhibition |
|
|
[6–7, 73–80] |
Normal Physiological Conditions
The retina is highly organized into distinct laminae. Inner layers are endowed with blood vessels that have vascular endothelial cells with tight junctions making up the inner blood retina barrier (BRB). Within the inner most layers, some microglia reside around somata of retinal ganglion cell (RGC) and their axon projections within the nerve fiber layer, which connect the eye to the brain lateral geniculate nucleus and the superior colliculus via the optic nerve. However, most microglia reside within the two distinct synaptic layers, inner and outer plexiform layers (OPL and IPL, respectively). These are where photoreceptors synapse with bipolar cells and bipolar cells synapse with RGC, respectively. Recent work has revealed key differences amongst IPL versus OPL microglial pools in the steady state [3]. The maintenance of IPL microglia is mostly dependent on RGC-produced IL-34 (alternate ligand of colony stimulating factor-1 receptor, Csf1r) [15–17], whereas OPL microglia are IL-34 independent (presumably maintained by glial-derived Csf1). Importantly, these respective pools have distinct electrophysiological contributions to visual processing (Box 3).
Box 3. Microglial Roles in Visual Function.
There is a growing appreciation for the role of microglia in adult retinal neurophysiology. Work by Saban and colleagues dissected microglial contributions to visual processing by their distinct anatomical location in the retina [3]. Mice that lack of Il34, which is normally expressed by RGC, have a specific microglial loss in the IPL but not OPL. This IPL-specific defect is associated with a diminished electrophysiologic responses to light stimuli by cone bipolar cells, but normal photoreceptor responses [3]. Il34 knockout mice have no obvious quantitative loss of synapses, thus pointing to a possible qualitative role for IL-34-dependent microglia in synaptic function within the IPL. Hence, this study established the existence of an IL-34 dependent pool of microglia restricted to the IPL that plays a key role in cone bipolar cell responses during visual processing. Prior to this study, Wong and colleagues demonstrated that transient microglial depletion leads to reduced electrophysiological responses to light stimuli in both photoreceptors and bipolar cells [113]. Interestingly, transient microglial depletion does not result in major retinal morphological changes or neuronal loss [113]. Their subsequent study showed that microglial repopulation after depletion restored the impaired visual functions [127]. Collectively, in addition to RPE [128] and Muller glia [129], these studies identified microglia as another non-neuronal cell that is involved in neuro-retinal electrophysiology.
Outer most retinal layers of adult human and pigmented rodents are physiologically devoid of MNPs and blood vessels [6, 18, 19]. This is the case throughout the photoreceptor length, from their soma (outer nuclear layer) to their ciliated segments, all the way through to the SRS and the RPE, a specialized monolayer of neural crest-derived border cells [20] (Fig.1). Absence of microglia in the SRS is of critical importance, as this site is essential for photoreceptor homeostasis and function. It contains the interphotoreceptor matrix and surrounds apical microvilli of the RPE. There, the RPE delivers neurotrophic factors, nutrients, ions, and visual cycle chromophores to photoreceptors. The RPE also phagocytoses spent photoreceptor outer segment discs in the SRS, a process carried out via conserved phagocytic machinery, such as with certain integrins [21, 22] and Mer tyrosine kinase (Mertk) [23] in a circadian-dependent manner. Lastly, RPE cells are laterally connected via tight junctions forming the outer BRB. The RPE is highly immunosuppressive [24–26], and MNPs reaching the SRS that ligate thrombospondin-1 (Tsp-1) via their CD47 receptor are eliminated by the RPE expressing Fas ligand (FasL) [26, 27] (Figure 1).
Figure 1: Physiological distribution of retinal MNPs and main functions of the subretinal space:
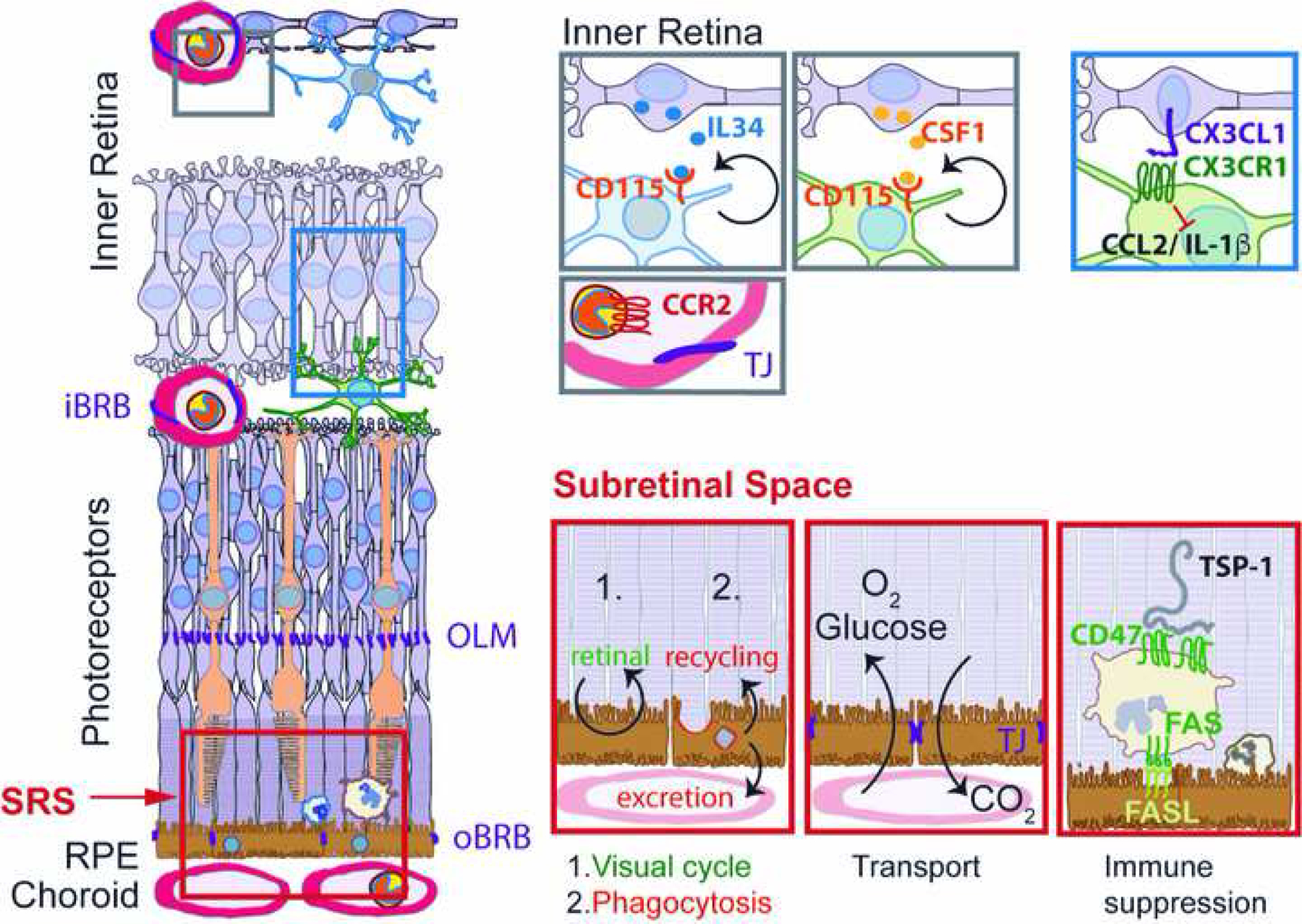
Physiologically the retina is only populated by IL34-dependent and CSF-1-dependent microglial cells. CCR2-positive Mos are restricted to the circulation separated from the retina by the inner and outer blood retinal barrier. CX3CL1 is constitutively expressed as a transmembrane protein in inner retinal neurons and provides a tonic inhibitory signal to the microglial checkpoint factor CX3CR1, expressed by retinal MCs. The subretinal space, demarcated by the tight junctions of the outer limiting membrane and of the RPE is the site of 1.) the visual cycle; 2.) phagocytosis of used photoreceptor outer segments and excretion or recycling the material; 3.) Oxygen and glucose uptake and CO2 excretion; 4.) immune suppression, in which FasL induces elimination of infiltrating leukocytes after activation of the integrin associated protein CD47 by thrombospondin 1 (TSP-1). iBRB: inner blood brain barrier; oBRB: outer blood brain barrier; OLM: outer limiting membrane; SRS: subretinal space; RPE: retinal pigment epithelium; IL: interleukin; CSF: colony stimulating factor; TJ; tight junction.
Subretinal Space in Retinal Degenerative States
Invasion by MNPs of the photoreceptor layers and SRS is a disease phenotype of IRDs and AMD [6, 14, 18, 19, 28]. It is accompanied by the loss of ramified microglia (to ameboid shaped cells) [29] and a general increase in total MNPs due to microglia replication and monocyte infiltration in certain settings [5, 30], while a Cd11c+ subpopulation was also described [31, 32]. Histological studies of human postmortem retinas suggest that subretinal MNPs are invariably present in intermediate and advanced (or ‘late’) forms of AMD [6, 19, 33–35] and pronounced in mouse mutants with accelerated age-associated photoreceptor degeneration [14], RPE injury model [36] and laser-induced choroidal neovascularization (CNV), an experimental model for wet AMD [37]. Subretinal MNPs have also been described in human postmortem IRD subjects [18], and in adult rodents with IRD mutations [3, 7, 38, 39], as well as other injury models [7, 11]. In fact, the presence of subretinal MNPs in IRDs was appreciated as early as the 1970’s using Royal College of Surgeons rats that have dystrophic retinas [40] - a degenerative condition later determined to be caused by a Mertk mutation that renders RPE incapable of phagocytosing spent photoreceptor discs [41], and Mertk mutation cause IRDs in humans [42]. However, it took several more decades before the distinction between endogenous microglia and recruited MdCs was definitively made [1, 9].
An important advancement to the field came in 2007 by Sennlaub et al. [14]. Authors demonstrated the association of a variant of CX3CR1 with AMD, later supported in a meta-analysis [43], and that Cx3cr1-deficient mice develop age-related MNP infiltration of the SRS accompanied by photoreceptor degeneration and exaggerated CNV. Given that Cx3cr1 is a homeostatic microglial checkpoint gene of MNPs [44], this phenotype revealed a link between intrinsic MNP dysregulation and disease pathogenesis. In 2013, the same group showed that AMD patients are characterized by increased intraocular CCL2 concentrations and that MdCs are present in the SRS of intermediate and late AMD patients, as well as in Cx3cr1-deficient mice, where they were shown to mediate photoreceptor toxicity [6]. Importantly, authors demonstrated that two key human AMD genetic risk gene variants, the Apolipoprotein E2 isoform (APOE) and Complement Factor H variant Y402H (CFH) directly promote pathogenic inflammation in the SRS in mouse models [26, 27]. These findings emphasized the role of AMD-risk variants in inflammation and the pathogenic role of inflammation in AMD (Fig.2a).
Figure 2: Molecular mechanisms of the role of MNPs in retinal degenerations:
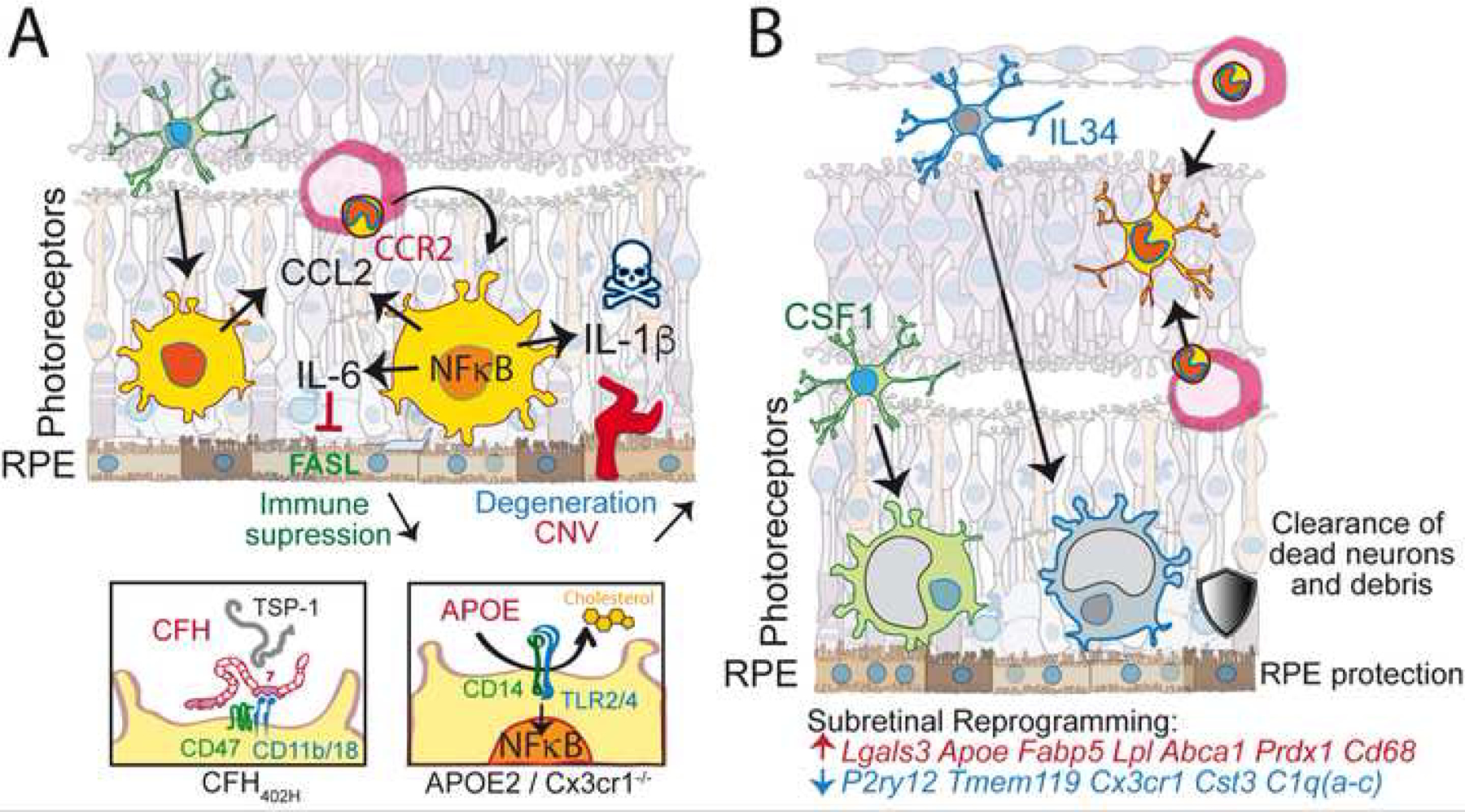
(A) participation of mononuclear-derived cells in the infiltration of the SRS: loss of microglial checkpoint genes (such as in Cx3cr1 deficiency) lowers the threshold of microglial activation and induces NF-κB signaling and inflammatory cytokine secretion in conditions of normally sub-threshold stress. Increased levels of CCL2, IL-6 and IL-1β lead to monocyte recruitment, down-regulation of FasL and immune-suppression and photoreceptor degeneration and neovascularization, respectively. (A’) In AMD this NF-κB activation can be due to the AMD-associated APOE2 allele that, similar to Cx3cr1-deficiency in mice, leads to increased APOE concentrations, lipid-raft destabilization of the plasma membrane and the activation of the innate immunity receptor cluster (CD14 and TLR2/4). (A”) Furthermore, the AMD-associated Complement Factor H 402H variant inhibits MNP elimination as it blocks TSP-1 mediated CD47 activation particularly efficiently. Both AMD-risk factors thereby promote pathogenic subretinal inflammation. (B) Dominance of resident microglia in the SRS in Cx3cr1 sufficient settings: both IL34-dependent and independent microglia migrate to the SRS, which is poorly accessible to MdCs. These subretinal microglia are transcriptionally reprogrammed by downregulating homeostatic marker genes and upregulating genes involved in lipid metabolism, anti-oxidant activity and others as indicated. Functionally, these cells clear dead neurons and cellular debris and protect the RPE and photoreceptors.
In 2019, another advancement in understanding the cellular make-up and contribution of subretinal MNPs in degeneration was made by O’Koren and Yu et al [3]. Building on their study in 2016 [11], authors applied Cx3cr1CreER/+ microglial lineage tracing mice and single cell RNA-seq (scRNA-seq) to decipher the ontogeny and function of subretinal MNPs in the Cx3cr1-sufficient setting. In a humanized IRD model [45] and in the toxic light-induced photoreceptor degeneration model (LD) [46], authors demonstrated that microglia are the dominant MNP lineage which occupies the SRS, whereas recruited MdCs were largely confined to the parenchyma. This was similarly demonstrated by another lab in a RPE injury model [36]. Moreover, authors showed direct evidence that these subretinal microglia restrict, not accelerate, disease progression. Opening the door to future elucidation of these disease restricting activities, the authors also identified gene specific inductions of subretinal microglia in these settings (Fig.2b). Collectively, their study now offers an alternative view to the cell type and function of subretinal MNPs in the course of retinal degenerative diseases.
The next sections will discuss the results that led to the independent observations of pathogenic MdCs versus protective microglia in the SRS during retinal degenerative diseases.
Pathogenic Role of MdCs in the Subretinal Space
Infiltration of MNPs in wet AMD (and laser-induced experimental CNV) characterized by a breach of the BRB (edema and subretinal hemorrhages), has long been recognized as an important factor of the pathogenesis (reviewed in [33]). In contrast, in slowly evolving geographic atrophy (GA) and IRDs, MNPs, in particular MdCs, were not commonly assumed to infiltrate the photoreceptor cell layer or inner retina, as no clinical signs of BRB rupture are visible. Over recent years, it has become increasingly clear that MNPs, including MdCs, infiltrate the outer retina in intermediate AMD and GA [33]. However, it is still passionately debated whether and how the MNP infiltration influences CNV and degeneration.
Evidence for pathogenic subretinal MNP accumulation
One of the first pieces of evidence that MNPs could be more than bystanders in retinal degeneration came from the study of Cx3cr1-deficient mice. Cx3cr1 receptor is strongly expressed by MNPs, where it mediates a tonic inhibitory signal, but not on neurons or the RPE in the adult retina. Cx3cr1-deficient mice develop accelerated and exaggerated subretinal MNP accumulation in conditions that do not trigger inflammation in wildtype mice (such as 12–18m old mice under normal light conditions, and a non-toxic light-challenge that does not induce degeneration in pigmented wildtype mice) [6, 14, 26, 27]. In aged Cx3cr1-deficient mice, the prolonged presence of MNPs in the SRS and their continued ingestion of lipid-, retinol-rich photoreceptor outer segments [14] leads to sizeable, ‘foam cell’ macrophages that are visible in vivo by fundus examination [47] and share a similar appearance with pseudodrusen in AMD (Box 1). Interestingly, pseudodrusen are also associated with subretinal MNPs in patients [28]. Importantly, this age-dependent accumulation of subretinal MNPs in Cx3cr1-deficient mice is associated with a significant degeneration of rods and cones [6, 14, 26, 27, 34, 48], but not with RPE atrophy (Fig.2A). They therefore quite accurately model a subtype of GA called incomplete outer retinal atrophy [49].
Mechanistically, Cx3cr1-deficient MNPs are characterized by an over-expression of APOE. The APOE excess activates toll-like receptor (TLR) signaling on MNPs in the absence of their natural ligands (damage- and pathogen-associated molecular patterns) [35]. This is likely due to APOE-induced cholesterol extraction from the lipid rafts of the plasma membrane of MNPs [33, 50]. Physiologically, TLR2 and TLR4 are separated from their co-receptor CD14 as they locate to non-raft plasma membrane. The cholesterol extraction from the rafts levitates this separation, and trigger intracellular NF-κB activation and cytokine expression similar to ligand induced dimerization (Fig.2A’) [33]. A similar mechanism has been previously described for APOA1 [51]. The resulting increased secretion of IL-6 and CCL2 observed in subretinal Cx3cr1-deficient MNPs reduces their RPE-induced elimination and increases monocyte recruitment, respectively (see below) [14, 35]. Additionally, Cx3cr1-deficient MNPs overexpress P2rx7, which constitutively activates their inflammasome and facilitates IL-1β maturation and secretion [48, 52]. The increased IL-1β secretion from this dysregulation mediates rod and cone degeneration, and significantly increases CNV [34, 48, 53] (Fig.2A). Collectively, Cx3cr1 deficiency is sufficient to trigger a pathogenic non-resolving subretinal inflammation, and deletion of Apoe in Cx3cr1-deficient mice prevented the subretinal MNP accumulation, and attenuated photoreceptor degeneration and CNV after laser-injury [35].
The Cx3cr1-deficient mice used in these studies did not carry the rd8, a spontaneous frameshift mutation in Crb1 found on C57BL6/N mice that causes late-onset photoreceptor degeneration and CNV [54]. Indeed, a number of publications describing degeneration and/or CNV formation in Ccr2−/−-, Ccl2−/−- and Cx3cr1−/−Ccl2−/− mice were in fact due to inadvertent rd8 contamination.
In summary, it can be argued that Cx3cr1-deficiency induces a model of ‘primary’ subretinal inflammation, where inflammation is not the consequence of a tissue injury but solely due to ‘over reacting’ MNPs, triggered by a non-toxic initiating stimulus (e.g. aging, a non-toxic light challenge [6, 14]). In ‘secondary’ inflammation models induced by a tissue injury, Cx3cr1-deletion often exaggerates MNP accumulation and associated degenerative changes: e.g. laser-induced CNV [14], paraquat-induced retinopathy [55], and rd10 mice [56, 57], a recessive IRD model.
AMD risk factors promote subretinal pathogenic inflammation
AMD is a highly heritable disease and several haplotypes encoding for protein variants have been established as risk-determinants in AMD. An isoform of APOE, along with a common variant of CFH accounts for an important part of the genetic risk [58, 59].
With respect to APOE, carriers of the APOE2-isoform, that is associated with higher APOE concentrations, are at increased risk for AMD and carriers of the APOE4-isoform, associated with lower APOE concentrations and impaired cholesterol transport, are protected against AMD compared to the common APOE3-allele [60, 61]. The MNPs of transgenic mice expressing human APOE2 (TRE2-mice) were shown to express high levels of APOE that activate TLR signaling and induce inflammatory cytokines, similar to aforementioned Cx3cr1-deficient MNPs. Analogous to Cx3cr1-knockout mice, TRE2-mice develop age- and stress-related pathogenic subretinal MNP accumulation [26, 35]. In Cx3cr1-deficient mice, in the context of APOE-dependent subretinal inflammation, the APOE4-allele led to diminished APOE and CCL2 levels and protected against harmful subretinal MNP accumulation observed in Cx3cr1-deficient APOE3-allele carrying mice [26] (Fig.2A’).
Concerning CFH, it’s binding to integrin CD11b/CD18 complex, strongly expressed on MNPs, curbs TSP-1 activation of its integrin-associated CD47 receptor that is necessary for MNP elimination. The AMD-associated CFH(H402) variant markedly increased this effect, promoting chronic subretinal inflammation [27] (Fig.2A”).
Taken together, these results showed that APOE2 and the CFH(H402) variant promote subretinal pathogenic inflammation. The fact that causative genetic AMD risk variants that provide an essential trigger for disease development, promote subretinal MNP accumulation and associated degeneration, provides strong evidence for a pathogenic role of subretinal inflammation in AMD.
Evidence for subretinal MdCs as a major driver for AMD pathology
Most studies do not allow a differentiation of MdCs and microglia, as non-specific markers were used for their identification (Table 1). The receptor CCR2, strongly expressed on inflammatory monocytes, constitutes one of the few specific markers that distinguish monocytes and early MdCs, as it is not expressed by microglia [6, 62, 63]. Studies of postmortem sections showed that CCR2+ MdCs invariably participated in the SRS in intermediate AMD and GA [6]. Additionally, increased intra-ocular levels of CCL2, a cytokine that recruits CCR2+ monocytes to the tissue [62], were found in neovascular [64–69] and atrophic-AMD [6, 68].
Experimentally, Cx3cr1-deficient MNPs overexpress CCL2 in aged and light-challenged mice [6]. After a light-challenge of Cx3cr1GFP/GFPCcr2RFP/+ mice that express RFP under the Ccr2 promoter, around 10% of subretinal MNPs were RFP+ and therefore monocyte-derived [6]. However, Ccr2 is quickly down-regulated when monocytes differentiate to macrophages [3, 6, 30, 70], which leads to an underestimation of MdCs. To overcome this, Sennlaub et al. permanently labeled monocytes and MdCs by repeated systemic injections of the traceable nucleotide 5-ethynyl-2′-deoxyuridine (EdU). These experiments revealed that ~50% of the subretinal MNPs are MdCs [6]. Monocyte depletion with systemic clodronate liposome completely prevented EdU+ MdC recruitment. Concordantly, deletion of Ccl2 or Ccr2, inhibition of Ccr2, diminished the subretinal MNPs by ~50–60% in acute and chronic models. Interestingly, the inhibition of CCR2+-monocyte recruitment in Cx3cr1-deficient mice nearly completely prevented their inflammation-associated photoreceptor degeneration, suggesting that MdCs are responsible for the inflammation induced degeneration. Similar results were observed in the carboxyethyl pyrrole immunization-induced AMD model [71].
In laser-induced CNV, MdCs participate significantly in subretinal MNP accumulation as determined in vivo imaging of mice injected with an intra-peritoneal or subcutaneous depot tracer that labels monocytes and MdCs [72], and in bone marrow transplantation experiments [37, 73]. Functionally, the depletion of circulating monocytes [73–75] and the inhibition of monocyte-recruitment by genetic deletion of Ccr2 and Ccl2 [76–78], very significantly inhibited subretinal MNP accumulation and CNV formation [75] in numerous studies.
In IRDs, the participation of MdCs seems to largely depend on the underlying genetic defect and possibly the intensity and speed of the degeneration. For example, Ccr2 deletion diminished MNP infiltration and had a minor protective effect in progressively degenerating rd10 mice [79]. The faster degeneration in Arrestin-1 (Arr1) knockout mice [7], could not be rescued when the monocyte recruitment was prevented by CCL2 inhibition [7]. However, in Mertk−/− mice, which lack this important phagocytosis receptor on the RPE but also on MNPs, MdCs were recruited to the SRS [38] and Ccl2 deletion very significantly protected Mertk−/− mice from degeneration [80].
Taken together, MdCs participate in the subretinal MNP accumulation under certain circumstances. In fast-evolving monogenetic IRDs such as in Arr1 knockout mice, they do not aggravate the degeneration. Following laser-induced CNV, MdCs are strongly involved in CNV formation, similar to other wound-healing models. In ‘primary’ inflammation, where the genetic deficit primarily activates MNPs, such as Cx3cr1−/− mice and in TRE2-mice, they are the main culprits of photoreceptor degeneration. Mertk−/− mice represent a mixed situation, where the genetic deficit affects retinal homeostasis and MNP function directly. The fact that well-established causative AMD-risk variants directly affect MNP function and promote inflammation, emphasizes the role of the AMD-risk variants in inflammation and the role of MNPs, in particular MdCs, in AMD. In IRDs, this might not necessarily be the case.
Protective Role of Microglia in the Subretinal Space
With the new appreciation in the late 2000’s that microglia and MdCs represent distinct lineages [1] and early studies pointing to their possible differential functions in experimental autoimmune encephalomyelitis (EAE) and other models of CNS conditions [81–83], the need for new tools to distinguish these two cell types had become imminent. A breakthrough came in 2013 with development of Cx3cr1CreER transgenic mice [9, 84, 85], now enabling conditional genetic manipulation of microglia by taking advantage of their long half-lives and self-renewability [84]. Importantly, these mice could be used to decipher the ontogeny of microglia versus MdCs in disease, conditionally knockout genes, or deplete microglia in an isolated fashion when paired with inducible diphtheria toxin receptor (DTR) gene (Table 1). In this section, we review recent findings showing that isolated microglia dominantly take up the SRS in Cx3cr1-sufficent retinal degenerative disease settings and have cytoprotective functions.
Microglial Predominance of the Subretinal Space
O’Koren et al. [11] was one of the first studies that applied Cx3cr1CreER/+ mice to decipher in situ spatial distributions of microglia versus MdCs during disease. They used confocal microscopy of retinal tissues from mice subjected to the LD model with Cx3cr1eYFP-CreER/+R26flox-STOP-flox-tdTomato mice. This approach allowed microglia to be traced as YFP+ tdTomato+ and MdCs as YFP+ tdTomato−. The findings were striking as microglia and MdCs were found distributed in distinct areas of the retina: microglia migrated to the SRS, whereas monocytes poured into the neuroparenchyma (presumably via retinal vasculature [7]). This finding was subsequently recapitulated by Wong and colleagues using the model of sodium iodate induced RPE injury, showing microglial dominance of the SRS through 30 days post injury [36]. Collectively, both studies demonstrated the spatial distinction between microglia and MdCs and now potentially uncovered a key role for microglia within the SRS in disease.
More comprehensive studies dissecting the isolated migration patterns of microglia were later carried out in a series of Cx3cr1CreER/+R26fl-STOP-fl-DTR mouse experiments by O’Koren and Yu et al [3]. Authors used human rhodopsin P23H knock-in mice (rhodopsin mutant model of a major autosomal dominant IRD form [45, 86]), demonstrating again that microglia predominantly occupied the SRS and were adherent to the apical aspect of the RPE [3]. These subretinal microglia were absent when administrating diphtheria toxin, indicating that they are indeed DTR-expressing microglia (Fig.2B). In stark contrast, MdCs were largely incapable of migrating to the SRS in both the LD and P23H mice, even when microglia were depleted. Hence, microglia do not outcompete MdCs for the SRS, but have restricted access to it in these disease settings. The same case is also likely in the NaIO3-induced RPE injury model [36].
A similar observation was described in the context of Alzheimer’s disease (AD), where microglia encapsulate amyloid-β plaques in the cortex and hippocampus. Two groups independently demonstrated that peripheral-derived myeloid cells do not similarly cluster around these plaques and fail to modify amyloid load when microglia were depleted in the APP23 and APP/PS1 transgenic mouse models of AD [87, 88]. Another example can be gleaned in the normal mouse brain, where a distinctive microglial population, but not the choroid plexus macrophage population, was found to reside on the apical epithelium of the choroid plexus [89]. Together, these reports reveal restricted microglial access to specific parenchymal or non-parenchymal niches throughout the CNS.
Reprogramming of Subretinal Microglia
With this novel characterization of the SRS as a microglial-dominant immune niche in retinal degeneration conditions, the door now opened to addressing whether the SRS microenvironment shape the role of these microglia in disease pathobiology. Through analysis of Cx3cr1YFP+ sorted cells using scRNA-seq from retinas, authors were able to demonstrate that microglia underwent transcriptional reprogramming in the SRS [3], indicating these cells are indeed altered in this niche. Changes included upregulated antioxidant and lipid activity genes (e.g. Srxn1, Apoe, Lpl, Abca1) and downregulation of some immune response genes (e.g. Ifngr1, C1qa, Aif1, Cx3cr1, Jun). This profile suggests an ability for subretinal microglia to respond to increased extracellular lipids and oxidative stress levels consistent with a neurodegenerative environment, and to do so in a manner that involves reduced pro-inflammatory responses (Fig.2B). Interestingly, downregulation of Abca1 with senescence impairs cholesterol efflux and promotes AMD [90]. Also, supporting a protective reprogramming is the upregulation of Ifnar1 by subretinal microglia, the loss of which increases lesion size and vascular leakage in CNV model [91]. Collectively these findings suggested against a pathogenic role for subretinal microglia.
Further countering a pathogenic role, subretinal microglia had a similar transcriptional profile with disease-associated microglial (DAM) cells. Amit and colleagues discovered this microglial subtype in the 5xFAD model of AD surrounding brain amyloid-β plaques in a Trem2-dependent fashion and concluded that DAM cells play a role in restricting disease progression [92]. Subretinal microglia have similar signature gene expressions, such as increased expression of Lgals3, Lpl, Spp1, Apoe, Trem2, and Cstb, and downregulated of Tmem119, P2ry12, Cx3cr1 and Siglech. Furthermore, Cx3cr1CreER/+ mice utilized by O’Koren and Yu et al. in retinal degeneration experiments allowed to conclude that microglia, as opposed to MdCs, indeed carry this DAM signature [3]. Interestingly, a similar microglia gene signature was observed during normal postnatal brain development [93], and further thought to be crucial in normal RGC development [94]. It is tempting to hypothesize that DAM cells represent a stereotypic response that supports physiological processes under stressful environments of large-scale cell death shared in postnatal development and neurodegenerative disease. However, because a similar microglial gene profile was also reported in the EAE model which was characterized as a pathological response rather than a disease restricting one [95], more work is still needed to fully resolve the functional significance of all aspects of the DAM signature.
RPE and Photoreceptor Protection by Microglia
The discovery of microglial reprogramming in the SRS and their adherent nature to the apical RPE now begged the question as to what is the isolated contribution of microglia in degeneration. Cx3cr1eYFP-CreER/+R26fl-STOP-fl-DTR mice were used for conditional depletion of microglia in LD and P23H models to address this question. The absence of microglia resulted in massive structural damage of the RPE, thereby directly linking their subretinal localization and transcriptional reprogramming with RPE protection (Fig.2B). Moreover, the damage of RPE in this depletion setting included both lateral and/or apical defects to F-actin, resembling key features of RPE dysmorphogenesis observed in human AMD and certain forms of IRDs [96]. A better preserved RPE function is generally believed to help maintain the function and survival of photoreceptors. Hence, these findings may mean that subretinal microglia could potentially be involved in restricting degenerative disease progression.
Still, the question remained as to exactly how do microglia confer protection to the RPE. A clue was also revealed by O’Koren and Yu et al [3]. They showed that the absence of microglia led to the accumulation of dead photoreceptors and debris in the SRS in both LD and P23H models. Interestingly, as MdCs are spared in this system (as are astrocytes and Müller glia ought to be as well), these findings implicated a microglial-dominant role in this debris clearance. The same may be gleaned from a study that depleted microglia in a model of retinal detachment [97], and another study using a NMDA model of RGC excitotoxicity [98], as well as an IRD model of progressive rod-cone degeneration [99]. Also, a recent study on complement C3 and its CR3 dependent phagocytosis supports this conclusion in an IRD model of rd10 mice [100], although there also reports suggesting that C3 (and C1q) are pathological in LD [101, 102]. Likewise, Kipnis and colleagues described microglial-dominant clearance of neuronal debris in the optic nerve crush model [103], and a similar conclusion was made by Ransohoff and colleagues in the spinal cord of EAE mice [81]. Hence, the failure to clear this debris shown by O’Koren and Yu et al. could have cytotoxic effects, potentially explaining the observed RPE defect. In contrast to these findings, Zhao et al. used continual tamoxifen administration in Cx3cr1eYFP-CreERR26DTA to deplete microglia in the rd10 mice [39]; however, it is more likely that this effect was mostly due to tamoxifen alone [104]. Together, these findings expand our knowledge about roles of microglia beyond neuroinflammation and illuminate the differential roles between microglia and MdCs in neurodegeneration.
In summary, these studies demonstrate that the SRS is a microglial-dominant macrophage niche in Cx3cr1-sufficient mice in LD, P23H mice, and sodium iodate induced RPE injury, where few recruited MdCs were observed in the SRS. The net-effect of Cx3cr1-sufficient microglia during LD and P23H degeneration is the restriction of disease progression. Evidence suggests that this protective response is mediated at the SRS via swift clearance of otherwise toxic debris, but whether other activities that are involved in protection remains to be explored.
Unified MNP Model in Retinal Degenerative Diseases
From these studies we propose a new conceptual framework to help understand the roles of MNPs in retinal degeneration. Though the model we put forth here is based on conclusions made in different experimental settings, our thinking is guided by the understanding of microglial checkpoint genes, such as Cx3cr1 [44], whose physiologic expression exerts a tonic inhibitory stimulus. In Cx3cr1-deficient mice, a normally sub-threshold stimulus (such as normal aging or a light challenge) is sufficient to drive pathogenesis [6, 14]. By contrast, in Cx3cr1-sufficient mice, (due to acute or progressive photoreceptor death) microglia predominantly accumulate in the SRS [3, 11], where they restrict disease progression (Fig.3). The constitutive expression of Cx3cr1 in microglia results in responses in degeneration that have a beneficial net-effect, such as controlled clearance of otherwise toxic neuronal debris [105]. By contrast, the absence of Cx3cr1 primes microglia to drive responses that have a harmful net-effect, such as uncontrolled proinflammatory cytokine production and further recruitment of inflammatory MdCs [6, 13]. We can therefore categorize MNP responses as beneficial/’adaptive’ versus harmful/’maladaptive’ and that a loss of certain factors, such as Cx3cr1, can ‘tip the scale’ from the former to the latter.
Figure 3: A unified hypothesis for MNP responses in IRD and AMD:
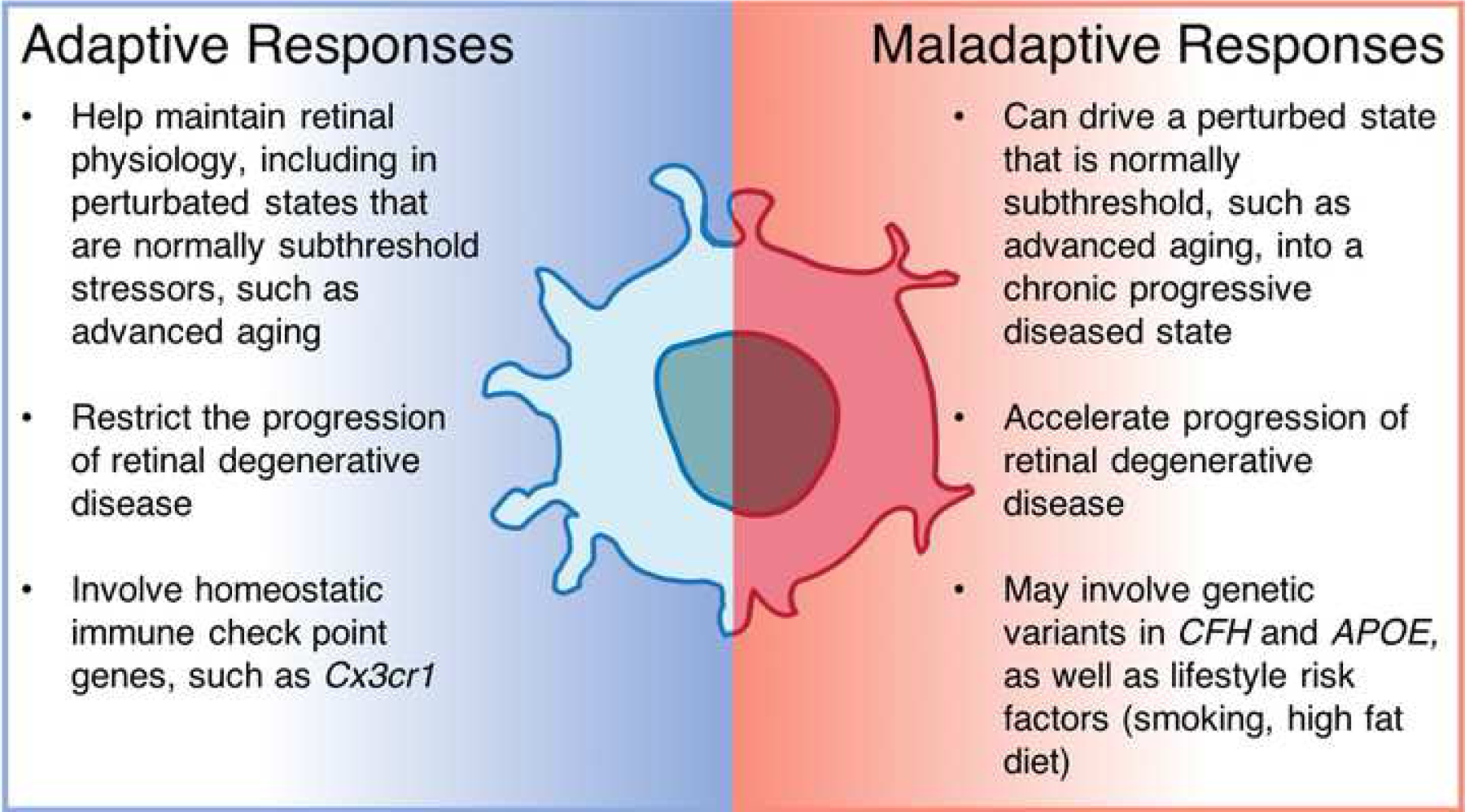
Adaptive responses by resident microglia express low levels of pathogenic cytokines, help eliminate toxic waste and slow down degeneration. When confounded by risk factors, such as genetic variants and environmental factors, microglia more readily express pathogenic cytokines and recruit monocytes. Such maladaptive responses by MNPs exacerbate retinal degenerative diseases.
To extrapolate this model for human disease, factors that help drive maladaptive responses could include genetic variants that confer risk in developing AMD. Indeed, CX3CR1 polymorphisms T280M or V249I in humans have been shown to be a risk factor in AMD, which may be independent or dependent on other factors [43, 106]. Other major risk factors, APOE2 isoform and CFH402H variant, may also be involved in driving harmful MNP responses [26, 27]. Likewise, TGFBR1 polymorphisms found in GWAS studies that confer AMD risk [107] may act in a similar manner. This receptor is phosphorylated by TGFBR2 in order to propagate TGF-β1 signaling [108], which in turn maintains microglia identity and function [109, 110]. Indeed, induced microglia conditional Tgfbr2 deletion in mice was shown to cause a degenerative disease phenotype [111]. These respective genetic factors might not be sufficient alone to drive harmful MNP responses, but possibly can with the addition of non-genetic determinants that confer risk in AMD (advanced age, western diet, and/or smoking). While AMD pathogenesis is multifactorial, MNPs are usually not driven into maladaptive in IRDs that have a monogenic Mendelian etiology involving genes of the visual system [112]. One exception may be IRDs caused by MERTK mutations, a gene that regulates phagocytosis of the RPE and MNPs such as microglia. The isolated role of these mutations in MNPs and their influence on retinal degeneration is not understood, but could conceivably contribute to infiltration of subretinal MdCs that may be involved in the disease phenotype.
There are limitations to our proposed model. Certain situations exist in which a robust inflammatory response could be categorized as adaptive, rather than maladaptive. For example, the control of ocular infections and the inhibition of some malignancy types likely benefit from prominent immune responses for host survival. Another limitation is that not all activities in either Cx3cr1-sufficient or -deficient settings are binary, i.e. solely beneficial or harmful. Microglial migration out of plexiform layers in order to take up the SRS is likely to cause synaptic deficits at the inner retina [3, 113]. Likewise, these negative changes may be curtailed by transient MdC recruitment to these evacuated plexiform niches.
Concluding Remarks
Not only is it important to study MNP responses in retinal degeneration, but it is also crucial to probe monocyte and microglial lineages as distinct entities (Outstanding Questions). We discussed how these two cell types even display functional disparities within the same microenvironment of the SRS, further highlighting the importance of ontogeny. Lastly, we proposed a unified model that classifies MNP responses in the retina as beneficial versus harmful. With this framework we posit that there are factors acting in concert which can ‘tip the scale’ from adaptive to maladaptive responses that help cause disease. Continued research is critical to better understand microglia and monocytes in health and disease, particularly given the immense disease burden that retinal degenerative diseases have on our society.
Outstanding Questions.
Are there markers that can distinguish subretinal monocyte-derived cells versus microglia in patients with degeneration?
Does an association exist between the macrophage type in the subretinal space and progression of degeneration in AMD or IRDs?
What is the specific ‘tipping point’ that makes a maladaptive response pathogenic in individuals with AMD genetic and non-genetic risk determinants?
Highlights.
The subretinal space — an extracellular site that hosts essential activities for photoreceptors — is invaded by mononuclear phagocyte (MNPs) in retinal degeneration, but the role of these cells in patients is controversial
In mice, “adaptive” MNP responses in retinal degeneration are those in which reactive microglia migrate into the subretinal space to help restrict disease progression, whereas recruited monocyte-derived cells have limited subretinal access
“Maladaptive” MNP responses are triggered in mice with certain genetic deficiencies by a normally sub-threshold insult that results in dysregulated microglia and an abundance of pathogenic monocyte-derived cells in the subretinal space
Applying this concept of adaptive and maladaptive responses may help unravel the complex roles of subretinal MNPs in inherited retinal degenerations and age-related macular degeneration
Acknowledgement
This work was supported by grants from NIH R01EY021798, NIH P30EY005722, Bright Focus MDR, Research to Prevent Blindness (Unrestricted) for Saban. For Sennlaub, INSERM, ANR MACLEAR (ANR-15-CE14-0015-01), LABEX LIFESENSES [ANR-10-LABX-65] supported by the ANR (Programme d’Investissements d’Avenir [ANR-11-IDEX-0004-02]), BrightFocus Foundation grant (M2018096), AVIESAN-UNADEV Maladies de la Vision 2018–2019 (N°UU159-00-C18/1616, N°UU113-00-C17/2086), Carnot, and a generous donation by Doris and Michael Bunte.
Reference
- 1.Ginhoux F et al. (2010) Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330 (6005), 841–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reyes NJ et al. (2017) New insights into mononuclear phagocyte biology from the visual system. Nat Rev Immunol 17 (5), 322–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Koren EG et al. (2019) Microglial Function Is Distinct in Different Anatomical Locations during Retinal Homeostasis and Degeneration. Immunity 50 (3), 723–737 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goldmann T et al. (2016) Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol 17 (7), 797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McMenamin PG et al. (2019) Immune cells in the retina and choroid: Two different tissue environments that require different defenses and surveillance. Prog Retin Eye Res 70, 85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sennlaub F et al. (2013) CCR2(+) monocytes infiltrate atrophic lesions in age-related macular disease and mediate photoreceptor degeneration in experimental subretinal inflammation in Cx3cr1 deficient mice. EMBO Mol Med 5 (11), 1775–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karlen SJ et al. (2018) Monocyte infiltration rather than microglia proliferation dominates the early immune response to rapid photoreceptor degeneration. J Neuroinflammation 15 (1), 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Z et al. (2019) Fate Mapping via Ms4a3-Expression History Traces Monocyte-Derived Cells. Cell 178 (6), 1509–1525 e19. [DOI] [PubMed] [Google Scholar]
- 9.Yona S et al. (2013) Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38 (1), 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gomez Perdiguero E et al. (2015) Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 518 (7540), 547–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Koren EG et al. (2016) Fate mapping reveals that microglia and recruited monocyte-derived macrophages are definitively distinguishable by phenotype in the retina. Sci Rep 6, 20636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lapenna A et al. (2018) Perivascular macrophages in health and disease. Nat Rev Immunol 18 (11), 689–702. [DOI] [PubMed] [Google Scholar]
- 13.Cardona AE et al. (2006) Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci 9 (7), 917–24. [DOI] [PubMed] [Google Scholar]
- 14.Combadiere C et al. (2007) CX3CR1-dependent subretinal microglia cell accumulation is associated with cardinal features of age-related macular degeneration. J Clin Invest 117 (10), 2920–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin H et al. (2008) Discovery of a cytokine and its receptor by functional screening of the extracellular proteome. Science 320 (5877), 807–11. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y et al. (2012) IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat Immunol 13 (8), 753–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greter M et al. (2012) Stroma-derived interleukin-34 controls the development and maintenance of langerhans cells and the maintenance of microglia. Immunity 37 (6), 1050–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gupta N et al. (2003) Activated microglia in human retinitis pigmentosa, late-onset retinal degeneration, and age-related macular degeneration. Exp Eye Res 76 (4), 463–71. [DOI] [PubMed] [Google Scholar]
- 19.Lad EM et al. (2015) Abundance of infiltrating CD163+ cells in the retina of postmortem eyes with dry and neovascular age-related macular degeneration. Graefes Arch Clin Exp Ophthalmol 253 (11), 1941–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Strauss O (2005) The retinal pigment epithelium in visual function. Physiol Rev 85 (3), 845–81. [DOI] [PubMed] [Google Scholar]
- 21.Finnemann SC et al. (1997) Phagocytosis of rod outer segments by retinal pigment epithelial cells requires alpha(v)beta5 integrin for binding but not for internalization. Proc Natl Acad Sci U S A 94 (24), 12932–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu C et al. (2019) Annexin A5 regulates surface alphavbeta5 integrin for retinal clearance phagocytosis. J Cell Sci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dransfield I et al. (2015) Mer receptor tyrosine kinase mediates both tethering and phagocytosis of apoptotic cells. Cell Death Dis 6, e1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Griffith TS et al. (1995) Fas ligand-induced apoptosis as a mechanism of immune privilege. Science 270 (5239), 1189–92. [DOI] [PubMed] [Google Scholar]
- 25.Wenkel H and Streilein JW (2000) Evidence that retinal pigment epithelium functions as an immune-privileged tissue. Invest Ophthalmol Vis Sci 41 (11), 3467–73. [PubMed] [Google Scholar]
- 26.Levy O et al. (2015) APOE Isoforms Control Pathogenic Subretinal Inflammation in Age-Related Macular Degeneration. J Neurosci 35 (40), 13568–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Calippe B et al. (2017) Complement Factor H Inhibits CD47-Mediated Resolution of Inflammation. Immunity 46 (2), 261–272. [DOI] [PubMed] [Google Scholar]
- 28.Greferath U et al. (2016) Correlation of Histologic Features with In Vivo Imaging of Reticular Pseudodrusen. Ophthalmology 123 (6), 1320–31. [DOI] [PubMed] [Google Scholar]
- 29.Silverman SM and Wong WT (2018) Microglia in the Retina: Roles in Development, Maturity, and Disease. Annu Rev Vis Sci 4, 45–77. [DOI] [PubMed] [Google Scholar]
- 30.Ronning KE et al. (2019) Molecular profiling of resident and infiltrating mononuclear phagocytes during rapid adult retinal degeneration using single-cell RNA sequencing. Sci Rep 9 (1), 4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McPherson SW et al. (2019) The retinal environment induces microglia-like properties in recruited myeloid cells. J Neuroinflammation 16 (1), 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dando SJ et al. (2019) Regional and functional heterogeneity of antigen presenting cells in the mouse brain and meninges. Glia 67 (5), 935–949. [DOI] [PubMed] [Google Scholar]
- 33.Guillonneau X et al. (2017) On phagocytes and macular degeneration. Prog Retin Eye Res 61, 98–128. [DOI] [PubMed] [Google Scholar]
- 34.Eandi CM et al. (2016) Subretinal mononuclear phagocytes induce cone segment loss via IL-1beta. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Levy O et al. (2015) Apolipoprotein E promotes subretinal mononuclear phagocyte survival and chronic inflammation in age-related macular degeneration. EMBO Mol Med 7 (2), 211–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma W et al. (2017) Monocyte infiltration and proliferation reestablish myeloid cell homeostasis in the mouse retina following retinal pigment epithelial cell injury. Sci Rep 7 (1), 8433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caicedo A et al. (2005) Blood-derived macrophages infiltrate the retina and activate Muller glial cells under experimental choroidal neovascularization. Exp Eye Res 81 (1), 38–47. [DOI] [PubMed] [Google Scholar]
- 38.Kohno H et al. (2015) Expression pattern of Ccr2 and Cx3cr1 in inherited retinal degeneration. J Neuroinflammation 12, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao L et al. (2015) Microglial phagocytosis of living photoreceptors contributes to inherited retinal degeneration. EMBO Mol Med 7 (9), 1179–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Essner E and Gorrin G (1979) An electron microscopic study of macrophages in rats with inherited retinal dystrophy. Invest Ophthalmol Vis Sci 18 (1), 11–25. [PubMed] [Google Scholar]
- 41.D’Cruz PM et al. (2000) Mutation of the receptor tyrosine kinase gene Mertk in the retinal dystrophic RCS rat. Hum Mol Genet 9 (4), 645–51. [DOI] [PubMed] [Google Scholar]
- 42.Gal A et al. (2000) Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nat Genet 26 (3), 270–1. [DOI] [PubMed] [Google Scholar]
- 43.Zhang R et al. (2015) Associations Between the T280M and V249I SNPs in CX3CR1 and the Risk of Age-Related Macular Degeneration. Invest Ophthalmol Vis Sci 56 (9), 5590–8. [DOI] [PubMed] [Google Scholar]
- 44.Deczkowska A et al. (2018) Microglial immune checkpoint mechanisms. Nat Neurosci 21 (6), 779–786. [DOI] [PubMed] [Google Scholar]
- 45.Sakami S et al. (2011) Probing mechanisms of photoreceptor degeneration in a new mouse model of the common form of autosomal dominant retinitis pigmentosa due to P23H opsin mutations. J Biol Chem 286 (12), 10551–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Noell WK et al. (1966) Retinal damage by light in rats. Invest Ophthalmol 5 (5), 450–73. [PubMed] [Google Scholar]
- 47.Raoul W et al. (2008) Lipid-bloated subretinal microglial cells are at the origin of drusen appearance in CX3CR1-deficient mice. Ophthalmic Res 40 (3–4), 115–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu SJ et al. (2015) Upregulation of P2RX7 in Cx3cr1-Deficient Mononuclear Phagocytes Leads to Increased Interleukin-1beta Secretion and Photoreceptor Neurodegeneration. J Neurosci 35 (18), 6987–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sadda SR et al. (2018) Consensus Definition for Atrophy Associated with Age-Related Macular Degeneration on OCT: Classification of Atrophy Report 3. Ophthalmology 125 (4), 537–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smoak KA et al. (2010) Myeloid differentiation primary response protein 88 couples reverse cholesterol transport to inflammation. Cell Metab 11 (6), 493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sullivan PM et al. (2011) Reduced levels of human apoE4 protein in an animal model of cognitive impairment. Neurobiol Aging 32 (5), 791–801. [DOI] [PubMed] [Google Scholar]
- 52.Netea MG et al. (2009) Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood 113 (10), 2324–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lavalette S et al. (2011) Interleukin-1beta inhibition prevents choroidal neovascularization and does not exacerbate photoreceptor degeneration. Am J Pathol 178 (5), 2416–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mattapallil MJ et al. (2012) The rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest Ophthalmol Vis Sci 53, 2921–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen M et al. (2013) Paraquat-Induced Retinal Degeneration Is Exaggerated in CX3CR1-Deficient Mice and Is Associated with Increased Retinal Inflammation. Invest Ophthalmol Vis Sci 54 (1), 682–90. [DOI] [PubMed] [Google Scholar]
- 56.Zabel MK and Kirsch WM (2013) From development to dysfunction: microglia and the complement cascade in CNS homeostasis. Ageing Res Rev 12 (3), 749–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peng B et al. (2014) Suppression of microglial activation is neuroprotective in a mouse model of human retinitis pigmentosa. J Neurosci 34 (24), 8139–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Swaroop A et al. (2007) Genetic susceptibility to age-related macular degeneration: a paradigm for dissecting complex disease traits. Hum Mol Genet 16 Spec No. 2, R174–82. [DOI] [PubMed] [Google Scholar]
- 59.McKay GJ et al. (2011) Evidence of association of APOE with age-related macular degeneration: a pooled analysis of 15 studies. Hum Mutat 32 (12), 1407–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heeren J et al. (2004) Impaired recycling of apolipoprotein E4 is associated with intracellular cholesterol accumulation. J Biol Chem 279 (53), 55483–92. [DOI] [PubMed] [Google Scholar]
- 61.Mahley RW et al. (2009) Apolipoprotein E: structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. J Lipid Res 50 Suppl, S183–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Geissmann F et al. (2010) Development of monocytes, macrophages, and dendritic cells. Science 327 (5966), 656–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mizutani M et al. (2011) The fractalkine receptor but not CCR2 is present on microglia from embryonic development throughout adulthood. J Immunol 188 (1), 29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fauser S et al. (2015) Intraocular and systemic inflammation-related cytokines during one year of ranibizumab treatment for neovascular age-related macular degeneration. Acta Ophthalmol 93 (8), 734–8. [DOI] [PubMed] [Google Scholar]
- 65.Guymer RH et al. (2011) Identification of Urinary Biomarkers for Age-related Macular Degeneration. Invest Ophthalmol Vis Sci. [DOI] [PubMed] [Google Scholar]
- 66.Jonas JB et al. (2010) Monocyte chemoattractant protein 1, intercellular adhesion molecule 1, and vascular cell adhesion molecule 1 in exudative age-related macular degeneration. Arch Ophthalmol 128 (10), 1281–6. [DOI] [PubMed] [Google Scholar]
- 67.Kramer M et al. (2012) Monocyte chemoattractant protein-1 in the aqueous humour of patients with age-related macular degeneration. Clin Exp Ophthalmol 40 (6), 617–25. [DOI] [PubMed] [Google Scholar]
- 68.Newman AM et al. (2012) Systems-level analysis of age-related macular degeneration reveals global biomarkers and phenotype-specific functional networks. Genome Med 4 (2), 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rezar-Dreindl S et al. (2016) The Intraocular Cytokine Profile and Therapeutic Response in Persistent Neovascular Age-Related Macular Degeneration. Invest Ophthalmol Vis Sci 57 (10), 4144–50. [DOI] [PubMed] [Google Scholar]
- 70.Saederup N et al. (2010) Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS One 5 (10), e13693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cruz-Guilloty F et al. (2013) Infiltration of proinflammatory m1 macrophages into the outer retina precedes damage in a mouse model of age-related macular degeneration. Int J Inflam 2013, 503725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sim DA et al. (2015) A simple method for in vivo labelling of infiltrating leukocytes in the mouse retina using indocyanine green dye. Dis Model Mech 8 (11), 1479–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shi YY et al. (2011) Monocyte/macrophages promote vasculogenesis in choroidal neovascularization in mice by stimulating SDF-1 expression in RPE cells. Graefes Arch Clin Exp Ophthalmol 249 (11), 1667–79. [DOI] [PubMed] [Google Scholar]
- 74.Liu J et al. (2013) Myeloid cells expressing VEGF and arginase-1 following uptake of damaged retinal pigment epithelium suggests potential mechanism that drives the onset of choroidal angiogenesis in mice. PLoS One 8 (8), e72935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sakurai E et al. (2003) Macrophage depletion inhibits experimental choroidal neovascularization. Invest Ophthalmol Vis Sci 44 (8), 3578–85. [DOI] [PubMed] [Google Scholar]
- 76.Luhmann UF et al. (2009) The drusen-like phenotype in aging Ccl2 knockout mice is caused by an accelerated accumulation of swollen autofluorescent subretinal macrophages. Invest Ophthalmol Vis Sci 50 (12), 5934–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Robbie SJ et al. (2016) Enhanced Ccl2-Ccr2 signaling drives more severe choroidal neovascularization with aging. Neurobiol Aging 40, 110–9. [DOI] [PubMed] [Google Scholar]
- 78.Tsutsumi C et al. (2003) The critical role of ocular-infiltrating macrophages in the development of choroidal neovascularization. J Leukoc Biol 74 (1), 25–32. [DOI] [PubMed] [Google Scholar]
- 79.Guo C et al. (2012) Knockout of ccr2 alleviates photoreceptor cell death in a model of retinitis pigmentosa. Exp Eye Res 104, 39–47. [DOI] [PubMed] [Google Scholar]
- 80.Kohno H et al. (2014) CCL3 production by microglial cells modulates disease severity in murine models of retinal degeneration. J Immunol 192 (8), 3816–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yamasaki R et al. (2014) Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med 211 (8), 1533–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ajami B et al. (2007) Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci 10 (12), 1538–43. [DOI] [PubMed] [Google Scholar]
- 83.Ajami B et al. (2011) Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci 14 (9), 1142–9. [DOI] [PubMed] [Google Scholar]
- 84.Parkhurst CN et al. (2013) Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155 (7), 1596–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Goldmann T et al. (2013) A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat Neurosci 16 (11), 1618–26. [DOI] [PubMed] [Google Scholar]
- 86.Dryja TP et al. (1990) A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature 343 (6256), 364–6. [DOI] [PubMed] [Google Scholar]
- 87.Prokop S et al. (2015) Impact of peripheral myeloid cells on amyloid-beta pathology in Alzheimer’s disease-like mice. J Exp Med 212 (11), 1811–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Varvel NH et al. (2015) Replacement of brain-resident myeloid cells does not alter cerebral amyloid-beta deposition in mouse models of Alzheimer’s disease. J Exp Med 212 (11), 1803–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Van Hove H et al. (2019) A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci 22 (6), 1021–1035. [DOI] [PubMed] [Google Scholar]
- 90.Sene A et al. (2013) Impaired cholesterol efflux in senescent macrophages promotes age-related macular degeneration. Cell Metab 17 (4), 549–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Luckoff A et al. (2016) Interferon-beta signaling in retinal mononuclear phagocytes attenuates pathological neovascularization. EMBO Mol Med 8 (6), 670–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Keren-Shaul H et al. (2017) A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169 (7), 1276–1290 e17. [DOI] [PubMed] [Google Scholar]
- 93.Hammond TR et al. (2019) Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 50 (1), 253–271 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Anderson SR et al. (2019) Developmental Apoptosis Promotes a Disease-Related Gene Signature and Independence from CSF1R Signaling in Retinal Microglia. Cell Rep 27 (7), 2002–2013 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Krasemann S et al. (2017) The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 47 (3), 566–581 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Handa JT et al. (2019) A systems biology approach towards understanding and treating non-neovascular age-related macular degeneration. Nat Commun 10 (1), 3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Okunuki Y et al. (2018) Microglia inhibit photoreceptor cell death and regulate immune cell infiltration in response to retinal detachment. Proc Natl Acad Sci U S A 115 (27), E6264–E6273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Todd L et al. (2019) Reactive microglia and IL1beta/IL-1R1-signaling mediate neuroprotection in excitotoxin-damaged mouse retina. J Neuroinflammation 16 (1), 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Spencer WJ et al. (2019) PRCD is essential for high-fidelity photoreceptor disc formation. Proc Natl Acad Sci U S A 116 (26), 13087–13096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Silverman SM et al. (2019) C3- and CR3-dependent microglial clearance protects photoreceptors in retinitis pigmentosa. J Exp Med 216 (8), 1925–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Natoli R et al. (2017) Retinal Macrophages Synthesize C3 and Activate Complement in AMD and in Models of Focal Retinal Degeneration. Invest Ophthalmol Vis Sci 58 (7), 2977–2990. [DOI] [PubMed] [Google Scholar]
- 102.Jiao H et al. (2018) Subretinal macrophages produce classical complement activator C1q leading to the progression of focal retinal degeneration. Mol Neurodegener 13 (1), 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Norris GT et al. (2018) Neuronal integrity and complement control synaptic material clearance by microglia after CNS injury. J Exp Med 215 (7), 1789–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang X et al. (2017) Tamoxifen Provides Structural and Functional Rescue in Murine Models of Photoreceptor Degeneration. J Neurosci 37 (12), 3294–3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zabel MK et al. (2016) Microglial phagocytosis and activation underlying photoreceptor degeneration is regulated by CX3CL1-CX3CR1 signaling in a mouse model of retinitis pigmentosa. Glia 64 (9), 1479–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Schaumberg DA et al. (2014) Prospective study of common variants in CX3CR1 and risk of macular degeneration: pooled analysis from 5 long-term studies. JAMA Ophthalmol 132 (1), 84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fritsche LG et al. (2013) Seven new loci associated with age-related macular degeneration. Nat Genet 45 (4), 433–9, 439e1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wrana JL et al. (1994) Mechanism of activation of the TGF-beta receptor. Nature 370 (6488), 341–7. [DOI] [PubMed] [Google Scholar]
- 109.Butovsky O et al. (2014) Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci 17 (1), 131–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zoller T et al. (2018) Silencing of TGFbeta signalling in microglia results in impaired homeostasis. Nat Commun 9 (1), 4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ma W et al. (2019) Absence of TGFbeta signaling in retinal microglia induces retinal degeneration and exacerbates choroidal neovascularization. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Duncan JL et al. (2018) Inherited Retinal Degenerations: Current Landscape and Knowledge Gaps. Transl Vis Sci Technol 7 (4), 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wang X et al. (2016) Requirement for Microglia for the Maintenance of Synaptic Function and Integrity in the Mature Retina. J Neurosci 36 (9), 2827–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ratnapriya R et al. (2019) Retinal transcriptome and eQTL analyses identify genes associated with age-related macular degeneration. Nat Genet 51 (4), 606–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Spaide RF et al. (2018) Subretinal drusenoid deposits AKA pseudodrusen. Surv Ophthalmol 63 (6), 782–815. [DOI] [PubMed] [Google Scholar]
- 116.Spaide RF (2013) Outer retinal atrophy after regression of subretinal drusenoid deposits as a newly recognized form of late age-related macular degeneration. Retina 33 (9), 1800–8. [DOI] [PubMed] [Google Scholar]
- 117.Bird AC et al. (2014) Geographic atrophy: a histopathological assessment. JAMA Ophthalmol 132 (3), 338–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Curcio CA (2001) Photoreceptor topography in ageing and age-related maculopathy. Eye (Lond) 15 (Pt 3), 376–83. [DOI] [PubMed] [Google Scholar]
- 119.Patel AA et al. (2017) The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J Exp Med 214 (7), 1913–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wynn TA et al. (2013) Macrophage biology in development, homeostasis and disease. Nature 496 (7446), 445–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fadok VA et al. (2001) Phagocyte receptors for apoptotic cells: recognition, uptake, and consequences. J Clin Invest 108 (7), 957–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Huynh ML et al. (2002) Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest 109 (1), 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gautier EL et al. (2013) Local apoptosis mediates clearance of macrophages from resolving inflammation in mice. Blood 122 (15), 2714–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Glass CK et al. (2010) Mechanisms underlying inflammation in neurodegeneration. Cell 140 (6), 918–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Grivennikov SI et al. (2010) Immunity, inflammation, and cancer. Cell 140 (6), 883–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hotamisligil GS (2010) Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140 (6), 900–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhang Y et al. (2018) Repopulating retinal microglia restore endogenous organization and function under CX3CL1-CX3CR1 regulation. Sci Adv 4 (3), eaap8492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jin M et al. (2005) Rpe65 is the retinoid isomerase in bovine retinal pigment epithelium. Cell 122 (3), 449–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Morshedian A et al. (2019) Light-Driven Regeneration of Cone Visual Pigments through a Mechanism Involving RGR Opsin in Muller Glial Cells. Neuron 102 (6), 1172–1183 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]