

Abstract
Diffuse large B-cell lymphoma (DLBCL) is a molecularly heterogeneous group of malignancies with frequent genetic abnormalities. G-quadruplex (G4) DNA structures may facilitate this genomic instability through association with activation-induced cytidine deaminase (AID), an antibody diversification enzyme implicated in mutation of oncogenes in B-cell lymphomas. Chromatin immunoprecipitation sequencing analyses in this study revealed that AID hotspots in both activated B cells and lymphoma cells in vitro were highly enriched for G4 elements. A representative set of these targeted sequences was validated for characteristic, stable G4 structure formation including previously unknown G4s in lymphoma-associated genes, CBFA2T3, SPIB, BCL6, HLA-DRB5 and MEF2C, along with the established BCL2 and MYC structures. Frequent genome-wide G4 formation was also detected for the first time in DLBCL patient-derived tissues using BG4, a structure-specific G4 antibody. Tumors with greater staining were more likely to have concurrent BCL2 and MYC oncogene amplification and BCL2 mutations. Ninety-seven percent of the BCL2 mutations occurred within G4 sites that overlapped with AID binding. G4 localization at sites of mutation, and within aggressive DLBCL tumors harboring amplified BCL2 and MYC, supports a role for G4 structures in events that lead to a loss of genomic integrity, a critical step in B-cell lymphomagenesis.
INTRODUCTION
Diffuse large B-cell lymphoma (DLBCL) is an aggressive non-Hodgkin’s lymphoma (NHL) with a higher incidence relative to other NHL subtypes in both the United States and Europe (1,2). While the standard immunochemotherapy regimen involving rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone (RCHOP) improved outcome for patients with DLBCL, roughly 40% of patients experience a rapid clinical progression and drug-resistant disease (3). The failure to achieve long-term disease-free survival is likely due to the complex genetic heterogeneity of DLBCL, which is highlighted by multiple molecular subtypes with varying patient outcomes. For example, the germinal center B-cell (GCB)-derived cell-of-origin (COO) subtype is characterized by frequent mutations in genes involved in epigenetic and germinal center (GC) reaction regulation, while the activated B-cell (ABC)-like subtype is associated with mutations leading to an altered B-cell receptor signaling and an inferior patient outcome (4–8). In addition, a particularly refractory subset of DLBCL tumors consists of BCL2 and MYC translocations and/or amplifications resulting in double positive BCL2/MYC protein expression with most patients surviving <5 years due to the lethal combination of highly proliferating and chemotherapy-resistant tumors (3,9–12). The molecular mechanism as to why the DLBCL genome is prone to genetic deregulation is unclear, though an enzyme, activation-induced cytidine deaminase (AID), is thought to be a key factor (13–18).
Normally, AID-directed mutations in immunoglobulin (Ig) genes generate the diverse antibody repertoire via somatic hypermutation and class switch recombination (19,20). AID induces point mutations via deaminating cytosine to uracil predominantly at AGCT motifs, which most commonly leads to dC>dT transition when left unrepaired, but depending on how the lesion is recognized, processed and repaired, dA>dG and dA>dC mutations can also occur (21,22). Mismatch repair often facilitates what is referred to as the second phase of somatic hypermutation where a patch DNA synthesis process causes accumulation of A:T substitutions (22). When the altered nucleotide occurs within Ig variable regions, somatic hypermutation results and enhances antibody affinity. Lesions within Ig constant switch regions produce double-strand breaks, subsequent recombinational deletion of the IgM constant exons and Ig class switching. Together, these processes yield high-affinity, specific antibodies following antigen presentation (21,22). Aberrant AID activity outside Ig genes, however, is associated with mutation and rearrangements linked to lymphoma. Previous sequencing studies in DLBCL revealed recurrent non-Ig genes that contain mutation hotspots with AID hypermutation signatures (7,8,16). The classic features of AID hotspots seen in oncogenes are similar to those in Ig genes and are characterized by single-stranded DNA regions near promoters, sites undergoing active transcription and super-enhancer (SE) regions particularly those within convergent transcription (23–27). A recent study demonstrated that AID recognizes an alternative DNA conformation for class switch recombination (28), called the G-quadruplex (G4), which supports previous findings of AID interaction with guanine (G)-looped stretches of single-stranded DNA (29,30).
The influence of DNA topology on its dynamic function and maintaining genomic processes is becoming increasingly recognized. The rapidly progressing G4 field demonstrates their discovery in nuclear activities such as replication and transcription, and links G4 DNA with regions of gene amplification (31–33). Our research efforts previously showed that DNA secondary structures within the BCL2 and MYC promoter regions serve as therapeutic targets for modulating oncogene expression (34–36). G4 secondary DNA structures arise from at least four contiguous runs of Gs separated by intervening loop sequences of variable length and are frequently found in the promoters of oncogenes (32,33,37). A novel antibody probe for detecting G4 structures in DNA indicated that in some cancer tissues G4 structures occur more frequently in malignant cell nuclei compared to their benign counterparts (38). Together with AID recognition of G4s, these studies strongly implicate a role for such alternative DNA structures in the loss of genomic integrity that might lead to oncogenesis in B cells. However, the prevalence and molecular associations of G4 formation within AID hotspots and other genetic abnormalities, particularly in lymphoma, are unknown.
Here, we sought to address whether G4s are involved in the recruitment of AID to targeted regions within B-cell and DLBCL genomes and can identify tumors with genetic events such as mutations and amplifications. We evaluated the presence of G4 structures in DLBCL tissues, the frequency of G-based structure-forming sequences within AID-targeted genomic loci and the impact these patterns have on the presence of genetic alterations in BCL2 and MYC as well as patient outcome. This study provides insight into the involvement of these highly G-rich DNA sequences in AID mutational activity and genomic instability.
MATERIALS AND METHODS
Patient samples
A total of 277 pre-treatment DLBCL biopsy formalin-fixed, paraffin-embedded tissues (FFPETs) from previous Leukemia/Lymphoma Molecular Profiling Project (LLMPP) studies (5,12,39) were used in this study for different analyses. Overall, there were 162 male and 115 female patients with an average age of 60 years at diagnosis (range: 14–92 years). In the AID mRNA analyses, 216 cases were used with the COO assigned by the gene expression profiling (GEP) algorithm (5) as 91 ABC, 100 GCB and 25 unclassified (UNC). A subset of 90 cases with available blocks for section and scroll preparation was utilized for immunohistochemical and sequencing analyses. The COO for the subset cohort was assigned by GEP or the Hans algorithm (40) for 80 cases as 38 ABC, 35 GCB, 6 UNC and 1 non-GCB. There were 10 in-house cases without COO determination. The unknown and non-GCB COO cases were only used in the overall DLBCL analyses and grouped together as unknown. These 11 cases were excluded from COO-specific analyses. BCL2 and MYC amplification and translocation status was obtained in our previous studies (12,39). Sections of benign tonsil served as control tissue. The use of human tissues and clinical data for this study was approved from the University of Arizona Institutional Review Boards in accordance with the Declaration of Helsinki.
Gene expression analyses
Raw cel files were downloaded from the Gene Expression Omnibus (GEO) website (accession number GSE10846). The cel files for the additional nine in-house cases along with the normal B-cell and DLBCL cell lines were obtained from the NIH LLMPP consortium. The cel files were imported into Partek Genomics Suite software and subjected to a robust multichip averaging, GC and quantile normalization followed by a log2 transformation as previously described (41).
Immunohistochemistry
FFPET slides were prepared at 4 μm and stained using the BenchMark® XT instrument (Ventana Medical Systems, Tucson, AZ) for AID with an anti-AID antibody (ab59361, Abcam; 1:2000 dilution). Cytoplasmic staining was considered positive and no nuclear staining was observed. Similar staining patterns and protein identification were previously reported using this same antibody (42,43). For G4 detection, we adapted a manual cell-based assay with a FLAG-tagged single-chain Fv antibody (BG4) to develop an automated colorimetric protocol on the BenchMark® XT instrument. Each slide was deparaffinized with a 56-min CC1 cell-conditioning regimen, incubated for 32 min at 37°C and detected with the OptiView DAB immunohistochemistry (IHC) detection kit. Slides were scored for the presence of AID or BG4 staining within a 150 cell count. Out of the 90 cases available, 74 were successfully stained for AID and 86 for BG4 scoring due to loss of tissue integrity during staining protocol or poor staining (i.e. expected positive control cells did not stain).
Immunofluorescence
U2932 DLBCL cells were treated with 10 μM pyridostatin (PDS) for 2 min and then washed with phosphate-buffered saline (PBS) before seeding onto poly-l-lysine-coated coverslips. The coverslipped cells were fixed with 3.7% paraformaldehyde for 30 min at room temperature followed by a PBS wash and then permeabilized with cold methanol at −20°C for 5 min. Cells were treated with 50 μg/ml RNase A for 1 h at room temperature before subjecting to immunofluorescence with the same antibodies used for IHC, BG4 and AID. Secondary antibodies conjugated to Alexa Fluor 488 (goat anti-rabbit) and Alexa Fluor 594 (goat anti-mouse) from Invitrogen at a dilution of 1:500 were applied with a counterstain of DAPI (Thermo Fisher Scientific). Coverslips were mounted onto cover glass with an anti-fade solution [0.02% p-phenylenediamine (Sigma, P6001) in 90% glycerol in PBS]. Samples were visualized and captured using a Ti-2 inverted C2+ confocal microscope and AID and BG4 foci were quantified in 100–150 nuclei with ImageJ software. The resulting number of foci in each nucleus was plotted and assumed to follow a Poisson distribution rather than a normal distribution. Accordingly, to evaluate whether PDS had an effect on the number of foci in each cell, we performed an exact test based on the Poisson distribution.
BCL2 sequencing and mutational analyses
Eleven primer pairs were designed for amplification of exon 1, exon 2 and exon 3 of BCL2 using the Primer3 software (Supplementary Table S1). Samples were prepared at 10 ng of template DNA per PCR reaction following the Illumina two-step PCR protocol. Multiple indexed libraries were pooled and sequenced on the Illumina MiSeq using 150-bp paired-end reads. The BWA-mem aligner v0.7.5a (44) was used to align the amplicon sequencing reads against the human reference genome (hg19). Variants were detected using VarScan version 2.3.6 (45) and filtered for non-silent coding and non-coding BCL2 mutations. Germline mutations were removed based on the presence in dbSNP version 137 (46) or having an allele frequency >75%. Potentially artifactual mutations were manually inspected using the Integrated Genome Viewer version 2.3.25 (47), flagging the removal of a mutation at chr18:60985870 occurring at an allele frequency of ∼10% and in 93% of patients.
In silico analyses and evaluation of potential G4-forming sequence stability
We constructed a fasta file containing the genomic regions of previously identified AID targeting sequences (25,26) and performed an in silico search for potential G4-forming sequence (PQS). The same genome versions for mouse (mm9) and Homo sapiens (hg19) as previously used (25,26) were downloaded from the University of California, Santa Cruz genome browser and the mouse B-cell SE BED file (GSE62063_aB_H3K27Ac_ SuperEnhancers.bed) from the NCBI GEO. The fastaFromBed tool, a part of the BEDTools package, created a fasta file from the mouse mm9 genome using the genomic locations found in the GSE62063 BED file. A custom perl script extracted the G4 sequences, number of sequences (counts) and locations of the G4 sequence from the fasta file. The perl script used the regular expression (regex) function to search the fasta file for the G4 sequence pattern GGGN(1–7)GGGN(1–7)GGGN(1–7)GGG, using the established PQS algorithm where N can be any nucleotide (37). Likewise, to evaluate the non-SE AID target regions in mouse ABCs and the human Burkitt’s lymphoma cell line AID targets, separate BED files specific for the AID target mm9 and the hg19 loci, respectively, were created and subjected to the same custom perl script. Sequences were selected using a positive lookahead assertion (i.e. /? = pattern/gi). All possible candidates were selected that matched the regex pattern query, enabling overlap of matched patterns.
To assess the quadruplex propensity score for the identified PQSs, we performed G4 motif identification in hg19 using the G4Hunter algorithm (48) with a score >1 and window size of 25. The G4Hscore of each PQS was calculated, and the G4Hscore represents the propensity of a sequence to form a G4. The maximum score of G4Hscore was assigned to each SE. A Wilcoxon rank-sum test was used to determine a significant difference between the G4Hscore of PQS within SE loci targeted by AID and those loci not targeted by AID.
Chromatin immunoprecipitation sequencing
U2932 DLBCL cells obtained from the American Type Culture Collection were grown in RPMI 1640 medium (Corning) supplemented with 10% fetal bovine serum (Atlanta Biologicals) and 1% Pen/Strep (Gibco). Chromatin immunoprecipitation (ChIP) was carried out using Peirce Magnetic ChIP kit according to the manufacturer’s instructions (Thermo Scientific). Exponentially growing cells were cross-linked with 1% formaldehyde and then quenched with glycine for 5 min at room temperature followed by lysis and MNase digestion. Samples were sonicated for 60 min using the Bioruptor (Diagenode). For each immunoprecipitation reaction, 4 × 106 chromatin cell equivalents were incubated overnight with either an AID antibody (Abcam, ab59361) or a rabbit IgG negative control antibody, subjected to protein A/G magnetic bead binding, IP elution and DNA recovery. The ChIP DNA was used to generate ChIP sequencing (ChIP-seq) libraries with the ChIP-seq sample preparation kit (Illumina) for sequencing at the Arkansas Children’s Research Institute Genomics Core. The human genome hg19 version was used for mapping AID ChIP-seq reads and PQS prediction.
AID ChIP data analysis
Raw sequence reads were preprocessed to ensure that only the high-quality reads will be used for further bioinformatics analysis; adapter trimming and quality filtering were performed using fastp software v0.19.5 with default parameters (49). The resulting high-quality reads were then aligned to the human genome version hg19, using BWA-MEM v0.7.17 with ‘-M’ as parameter (50). The PCR duplicate reads were then removed using MarkDuplicates from Picards package v2.9.2 (http://broadinstitute.github.io/picard) with default parameter. The uniquely mapped reads were filtered (mapping quality score MAPQ ≥30), sorted and converted to BAM files using SAMtools (http://samtools.sourceforge.net/). The BAM files were subjected to subsequent processing with model-based analysis for ChIP-seq (MACS2 v2.1.1, https://pypi.python.org/pypi/MACS2). Enriched peak regions of the genome were identified by comparing the AID antibody and a rabbit IgG (a mock pull-down) sample to whole cell extract samples using callpeak from MACS2 with modified parameters ‘-f BAMPE --broad --broad-cutoff 0.1’. The AID enriched peaks were filtered out if they overlap with the mock pull-down peaks and the blacklist (ENCODE blacklisted regions), which is combined from a consensus list empirically defined by the Encyclopedia of DNA Elements (ENCODE) consortium, available at http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeMapability/wgEncodeDacMapabilityConsensusExcludable.bed.gz. Data are available from the GEO website (accession number GSE146695).
Random permutation tests for assessing the association of AID peaks with PQS and activated mouse B-cell SEs, and BG4 peaks with AID-targeted loci
The activated mouse B-cell SE BED files were converted to the human genome (hg19) coordinates using CrossMap (51) with the mm9 to hg19 liftover chain file from UCSC. The permutation tests were used to assess the association of AID peaks with PQS and activated mouse B-cell SEs. We generated 60 random genomic location sets (control sets), preserving the number per chromosome and size of the original AID peaks. The shuffleBed in the BEDTools v2.25 (52) was used to randomly permute the genomic locations of AID peaks with parameters (-noOverlapping -chrom). Number of each control set containing PQS and activated mouse B-cell SEs was calculated and used as a null distribution. The number of the observed AID peaks containing PQS and activated mouse B-cell SEs was then compared to that from control sets and one-sample Student’s t-test was performed. The BEDTools Fisher’s test function was used to determine the number of observed AID peaks that overlapped with BG4 peaks from our previous BG4 ChIP (GSE99205).
Circular dichroism
Circular dichroism (CD) analyses were conducted on a Jasco-1100 spectropolarimeter (Jasco, Easton, MD) as previously described (35). The G4-forming oligonucleotides were prepared at a 5 μM strand concentration in a 10 mM Tris–HCl buffer (pH 7.4) with various KCl concentrations (0–100 mM). Melting curves were recorded at the wavelength of the maximum spectral molar ellipticity over 4–95°C and then plotted against temperatures for Tm determination (35). Tm values were calculated within ±1°C error using the log(inhibitor) versus response nonlinear regression fit on Prism software.
Survival analyses
Cut points for high and low expression of AID mRNA and protein, and BG4 staining were determined by the Youden index via a receiver operating characteristic (ROC) curve analysis using MedCalc software (53). Overall survival (OS) and progression-free survival (PFS) were defined as the time from date of diagnosis to the date of death or last follow-up and as the time from date of diagnosis to the date of DLBCL progression or death due to any cause, respectively. OS and PFS curves were estimated using the Kaplan–Meier method and plotted with GraphPad Prism software. Significant differences (P < 0.05) were assessed using the log-rank test and the Cox proportional hazards model was applied to determine independence from other covariates.
Statistical analysis
For mRNA expression, differences were assessed by a multiple-test corrected one-way ANOVA using the Benjamini and Hochberg false discovery rate with the Partek Genomics Suite software v7. All other statistical analyses were performed with GraphPad Prism software v6 and v7. Significance (P < 0.05) was evaluated for two-group comparisons using a two-tailed Student’s t-test unless multiple testing occurred and then the Šídák–Bonferroni correction was used to adjust the P-value. Fisher’s exact test was applied to determine significant differences in the genomic loci with PQS present or absent. The Mann–Whitney test compared PQS per locus within AID targets according to overlap with SE regions and BCL2 mutation counts between ABC-DLBCL and GCB-DLBCL samples. Data are represented as either mean ± standard error of the mean (SEM) or median ± minimum and maximum values unless otherwise stated.
RESULTS
G4 formation is detectable in DLBCL nuclei, occurs more frequently in ABC-DLBCL and tends to correlate with a lower helicase expression
Given the extensive occurrence of G4 sequences within the genome (32,33) and detection of G4 structure formation in cancer tissues (38), we sought to determine the frequency of G4 formation in DLBCL tissues. To visualize formed G4 complexes within DLBCL FFPET, we adapted the single-chain Fv BG4 antibody (38,54) to develop a semi-automated colorimetric IHC protocol. A non-malignant tonsil biopsy was used for optimization and we observed BG4-positive staining of B cells within the GC, mantle zone (MZ) and interfollicular regions (Figure 1A). The majority of DLBCL tumors displayed positive malignant cell nuclei indicating the frequent presence of formed G4 structures with intense staining observed in mitotic nuclei (Figure 1A, inset). G4 formation was detected in 85% (73/86) of the DLBCL FFPET (Supplementary Figure S1). The median percent BG4-positive malignant cells within a given tumor was 50%, and using ≥50% as a cutoff, there was a strong trend for a higher incidence of G4 formation in ABC-DLBCL in comparison to GCB-DLBCL (21/37, 57% versus 11/34, 32%; P = 0.06). ABC-DLBCL cases also tended to exhibit more BG4-positive malignant cells on average, although the difference was not significant (Figure 1B).
Figure 1.
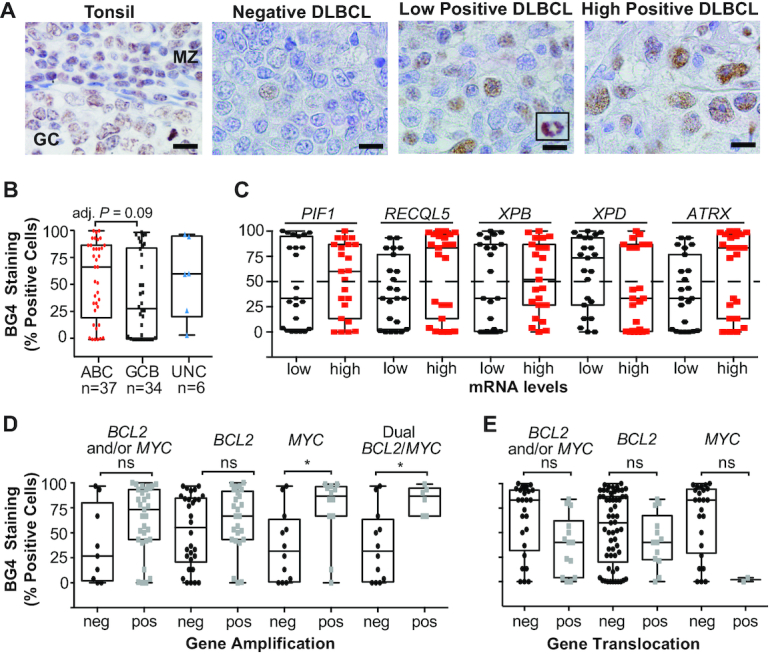
BG4 staining indicates G4 formation is frequent in DLBCL tissues, particularly in non-GCB subtypes, and correlates with BCL2 and MYC amplification. (A) Representative BG4 staining in a non-malignant tonsil biopsy with positive staining within a normal GC and the MZ, and DLBCL tissues that stained negative, low or highly positive with intense staining in mitotic nuclei (inset). Original magnification is ×1000. Scale bar represents 10 μm. (B) Percent BG4-positive DLBCL cells in DLBCL tissues (n = 77) according to COO. (C) Percent BG4-positive DLBCL cells according to low or high mRNA of the given DNA helicase. Percent DLBCL cells positive for BG4 staining in tumors with BCL2 and/or MYC amplification (D) or translocation (E). Box and whisker plot: line is the median; error bars are the minimum and maximum. *Adjusted P < 0.05 as determined by a two-tailed t-test with Šídák’s multiple test correction; ns, not significant.
We next investigated whether the mRNA expression of helicases known to unwind G4 structures, including PIF1 (55), RECQL5 (56), XPB/D (57) and ATRX (58), was lower in ABC-DLBCL and potentially accounted for the difference in G4 formation between the two DLBCL COO subtypes. Of the 86 cases with BG4 staining, 46 had available gene expression data; however, there were no significant differences in expression of the five helicases between the ABC-DLBCL and GCB-DLBCL (Supplementary Table S1). Despite an overall tendency for ABC-DLBCL to display more G4 formation, over half of the GCB-DLBCL and all of the UNC-DLBCL tissues consisted of detectable G4 staining. When the tissues were examined for helicase expression according to high G4 positivity (≥50%) irrespective of DLBCL subtype, tumors with low XPD mRNA levels were more likely to have G4 formation compared to cases with high XPD (15/23, 65% versus 8/23, 35%; P = 0.08; Figure 1C).
DLBCL with concurrent BCL2/MYC gene amplification has increased G4 formation
G4 formation, if left unresolved by helicases, may lead to DNA replication errors resulting in increased genome instability and gene amplification. Therefore, it is notable that elevated G4 formation was observed in DLBCL cases with dual BCL2/MYC amplification and with MYC amplification only (Figure 1D). BG4 staining was not significantly different in DLBCL tumors that consisted of either a BCL2 or MYC translocation compared to tissues negative for these translocations (Figure 1E). Amplification and translocation status was determined in previous publications (12,39). These findings suggest that G4 formation in DLBCL correlates with aberrant and dramatic amplification of these two critical oncogenes.
Genomic loci targeted by AID are highly enriched for G4 formation whether within or outside SE regions
Due to the potential for G4 structure involvement in mechanisms of genomic instability important in lymphoma biology such as somatic hypermutations and class switch recombination (28–30), we investigated whether sequences encoding G4s uniquely define AID targets relative to non-AID-targeted areas of the genome. First, we interrogated 1003 SEs previously identified in activated mouse embryo B cells (25), of which 167 distinct SE regions were targeted by AID. We investigated co-incidence between PQSs, SEs and AID-targeted regions. Considering all 1003 SEs, the 167 AID-targeted SEs consisted of a significantly higher average of PQSs per locus (28 versus 22; P < 0.004) and AID-targeted SEs are two times more likely to harbor at least 28 PQSs compared to the non-AID-targeted SEs (Figure 2A and Supplementary Table S2A). Of note, G4 sequences were detected in non-AID SE regions at a range of 0–121 PQSs per locus indicating that G4-forming sequences may be a common feature to SE regions. In the same mouse B cells, an additional 69 AID targets were identified, 6 SEs contained 2–3 targeted loci and 62 AID targets fell outside SE regions completely. We then assessed all 236 AID-targeted loci according to SE overlap and found that targets within SEs had a significantly higher number of PQSs per locus than targets outside an SE region (Figure 2B and Supplementary Table S2B). We next evaluated the 54 AID targets found in a parallel study using the Ramos Burkitt’s lymphoma cell line. Twenty-eight of these AID targets were associated with SEs, 42 with areas of convergent transcription and 10 were not associated with either SE or convergent transcription genomic regions (23). Similarly, we observed PQSs present regardless of genomic overlap of SE and/or convergent transcription with the highest frequency of PQSs in SE-associated AID targets (Supplementary Table S2C).
Figure 2.

AID targeting loci are enriched for G4-forming sequences. (A) Comparison of the percentage of SE regions previously detected in activated mouse B cells (25) that contain <28 PQSs (white bars) or ≥28 (black bars) according to whether the SE region contained an AID target. ***P < 0.001 as determined by Fisher’s exact test. (B) The mean PQSs per AID-targeted locus according to overlap with an SE region. ***P < 0.001 as determined by a two-tailed Mann–Whitney test. (C) UpSet chart generated using UpSetR package shows the intersection of AID peaks detected in this study with PQS and the activated mouse B-cell SEs, which were lifted over from mm9 to hg19. The observed number of AID peaks shared between PQSs and/or SEs is indicated in the top bar chart (black bars) corresponding to the solid points below the bar chart and each column represents shared AID peaks between the PQS and SEs (linked dots). The gray bars represent the mean of intersection of random shuffling AID peaks (n = 60) with PQSs and SEs. The vertical bar plot reports the intersection of either PQSs or SEs. Data represent mean ± standard deviation. ****P < 1e−57 as determined by a one-sample t-test. (D–H) UCSC genome annotations of stacked tracks beneath genome coordinate positions for each corresponding gene. PQS feature is overlaid on gene track. For gene tracks, solid black blocks connected by horizontal lines representing introns represent coding exons. The 5′ and 3′ UTRs are displayed as thinner blocks on the leading and trailing ends of the aligning regions. Arrowheads on the connecting intron lines indicate the direction of transcription. AID ChIP-seq peak detected in this study (red blocks) overlaps with previously known AID binding site (30) in MYC (D) and PQS located around the exon 1 of BCL2 (E). CBFA2T3 contained enriched AID ChIP binding sites and PQS compared to control reads (F). AID ChIP-seq peak overlapped to multiple PQSs in SPIB (G). AID ChIP-seq peak, PQS and SE were located upstream of IL21R (H). (I) Box plot representing the G4Hscore distribution in SE loci without and with AID-targeted regions. ***P = 4.88e−10 as determined by the Wilcoxon rank-sum test.
We then performed AID ChIP-seq to determine whether the genomic regions targeted by AID in DLBCL are also enriched for PQSs using the well-characterized U2932 ABC-DLBCL cell line. AID ChIP-seq peaks mapped to 4573 loci and overlapped with 638 PQSs. Since PQSs are found throughout the genome and in close proximity to transcription start sites similar to AID-targeted elements (25), we aimed to control for this abundance and further confirm the identified PQSs are enriched within AID targets. We therefore integrated the activated mouse B-cell SEs, from mm9 to hg19, with our identified DLBCL AID-targeted regions (4573 AID peaks) and performed the permutation tests of the association of the AID peaks with the two sets of PQSs and activated mouse B-cell SEs (see the ‘Materials and Methods’ section). We observed that the intersection of AID peaks with PQSs and SEs is significantly higher than expected by chance in all cases of comparisons and supports that the number of AID peaks sharing PQSs and/or SEs is not a random event (Figure 2C). Consistent with this strong association, analysis of genome-wide G4 sequences identified from a previous BG4 ChIP (59) demonstrated these PQSs were highly enriched for AID-targeted loci. We found that 252 peaks of AID-targeted loci overlapped with BG4 ChIP-seq peaks more often than expected by chance (odds ratio = 11.4; P = 1.16e−174; Supplementary Table S2D).
Next, we first validated ChIP-seq-based AID peaks with known AID targets, MYC and the IgH locus (25,26,30), and found overlapping of our signal to the previously reported AID peaks and PQSs (Figure 2D and Supplementary Figure S2). Interestingly, BCL2 is a known target of AID (16,17), but was not identified in the earlier studies as an AID-targeted locus (25,26). Here, for the first time, we detected AID peaks primarily in exon 1 of BCL2 (Figure 2E), most of which aligned with a PQS known to form stable G4s (60). In addition to these genes, we showed the overlapping of AID peaks and PQSs in two other genes, CBFA2T3 and SPIB (Figure 2F and G). One of the top significant AID ChIP-seq signal clusters mapped to CBFA2T3 (MTG16), a gene translocated in myeloid malignancies, and associated with a high density of PQSs. We found multiple PQSs overlapping with AID peaks in SPIB, an oncogene essential for ABC-DLBCL cell survival (6) that was also identified as an AID target in mouse ABCs (25). In agreement with SEs as preferred AID peaks, we also pulled down gene loci that overlapped with known SE regions, as observed with IL21R (Figure 2H), a gene that undergoes DNA double-strand breaks in the presence of AID (25). To assess the stability of the PQS on a more global scale, we applied the quadruplex propensity score or G4Hscore (48). The G4Hscore accounts for G-richness, G-skewness and the presence of G-blocks in a given sequence to indicate propensity to form. A higher G4Hscore favors highly stable G4 motifs. The PQSs in SE regions targeted by AID show significantly higher G4Hscores than those not targeted by AID (Figure 2I).
To confirm that PQSs within the previously identified AID targets and the newly detected sequences in the AID ChIP-seq can form G4s, we selected five G4 sequences from the Burkitt’s human lymphoma cell line and one from each of the SPIB and CBFA2T3 loci for biophysical analysis (Supplementary Table S3). The selected genes have relevance to lymphoma biology (BCL6, HLA-DRB5, MYC, MEF2C) or a canonical AID target (IgG Sμ) and represent different genomic overlap (i.e. SE, ConvT or none). The MYC sequence is an extended version of the well-characterized and inherently stable G4 structure (61) and provides a control to compare the structure formation and stability of the newly identified PQSs. All of the PQSs demonstrated the ability to form intramolecular G4 structures with the characteristic positive maxima around 262–266 nm and a negative maximum at 240 nm using CD spectral analysis (Figure 3A) that exhibited K+-dependent structure formation (Supplementary Figure S3A) and high thermal stability with melting temperatures ranging from 66 to 95°C (Figure 3B, Supplementary Figure S3B and Supplementary Table S3). With AID ChIP-seq identification of BCL2, we also included the well-characterized G4 sequence in the CD analyses as an additional control for G4 formation, particularly because the BCL2 G4 forms a mixed parallel–anti-parallel structure and provides a different CD spectral signature from the MYC parallel G4 conformation (60,61). The secondary peak at 290 nm observed for the IgG Sμ is similar to BCL2 and indicates a mixed anti-parallel–parallel strand orientation (Figure 3A). As further support for G4 formation in PQSs that overlap with AID-targeted regions, the selected SPIB and CBFA2T3 PQSs exhibit K+-dependent structure formation and thermal stability (Figure 3C and D, and Supplementary Figure S4).
Figure 3.
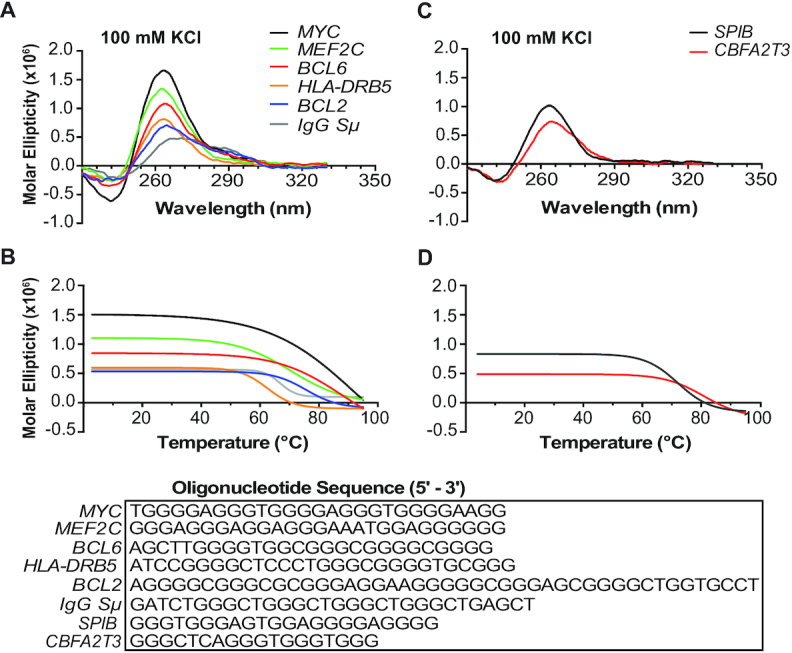
PQSs in AID-targeted and SE regions form stable G4 structures. (A) CD spectra of PQSs found within AID-targeted regions previously identified (25). The BCL2 sequence (blue) was included as a positive control for G4 formation. (B) Corresponding melt curves from the PQSs in (A). (C) CD spectra of selected PQSs found within AID ChIP binding sites in SPIB and CBFA2T3 and the corresponding melt curves (D). All oligomers were prepared in 100 mM KCl–Tris buffer. CD spectra and melt curves are representative of three independent experiments. Sequences are provided for each oligonucleotide.
AID co-localizes with G4 structure formation within DLBCL cell nuclei
In demonstrating a significant enrichment of both G4-forming sequences within AID-targeted loci and conversely AID-targeted loci within BG4 ChIP-sequenced genomic sites, we then assessed co-localization of AID with BG4 in DLBCL cell nuclei. First, we performed co-staining immunofluorescence to detect AID and BG4 in untreated U2932 DLBCL cells (Figure 4A, top panel). As expected based on the IHC and previous literature (42,43), the bulk of AID resides in the cytoplasm with some AID foci observed in the nucleus. Out of 100–150 individual nuclei, 1.2 AID foci were detected on average per nucleus (range: 0–8; Figure 4B). While a few discrete BG4 foci appeared in the cytoplasm, the majority of BG4 was located within the nucleus at an average of 7.7 foci per nucleus (range: 0–28; Figure 4A and C). When the images were merged, AID and BG4 foci were frequently found in close proximity within the nucleus and sometimes within the cytoplasm (Figure 4A, top panel). To address whether an increased G4 formation would recruit additional AID into the nucleus with co-localization to BG4 foci, we then treated the U2932 cells with PDS, a known G4 stabilizer (54). Treatment with PDS led to a significant increase in nuclear AID foci (mean: 1.9; range: 0–17; Figure 4B) and, to a greater extent, an increase in BG4 foci (mean: 9.9; range: 0–38; Figure 4C). The enhanced AID and BG4 nuclear foci resulted in notable co-localization that was evident not only by a close proximity of the AID and BG4 foci, but also as yellow, overlapping signals in the merged images (Figure 4C, bottom panel). The AID–BG4 co-localization, particularly after G4 stabilization, supports G4 formation within DLBCL cells and provides a nuclear environment for AID targeting.
Figure 4.

AID localizes to G4s in DLBCL cell nuclei. (A) Immunofluorescence for AID and BG4 in DLBCL cells without (top panel) and with (bottom panel) PDS treatment (10 μM for 2 min). Discrete AID (green) and BG4 (red) were observed in the nucleus. Nuclei were counterstained with DAPI (blue). Yellow arrowheads indicate co-localization. Scale bars represent 20 μm. (B) Quantification of AID foci number per nucleus in cells without and with PDS treatment. (C) Quantification of BG4 foci number per nucleus without and with PDS treatment. One hundred to one hundred fifty nuclei were counted and the SEM was calculated from three independent experiments. ***P < 0.001 as determined by a Poisson exact test.
BCL2 mutations overlap with both AID binding sites and areas of G4 sequences
A subset of the DLBCL tissues (n = 77) was sequenced for BCL2 mutations to explore a potential relationship between AID targets, G4s and mutations. Consistent with the previous literature (7), this analysis revealed a high number of recurrent BCL2 mutations (Figure 5A and Supplementary Table S4) in 58% of tumors (45/77) with a higher frequency in GCB-DLBCL relative to ABC-DLBCL (Figure 5B). The BCL2 mutations found in all 45 tumors mapped to two regions, exon 1 and exon 3, and aligned with the AID ChIP signals (Figure 5C). A high prevalence of mutations (229 mutations) was located in exon 1 and overlapped with AID-targeted regions and PQSs, including with the well-known G4 structure (60), while only 7 mutations were seen in exon 3 and did not overlap with an AID binding site or a PQS suggesting G4 involvement with AID-induced mutagenesis (Figure 5C).
Figure 5.

BCL2 mutations in DLBCL patient samples overlap with both AID binding sites and areas of G4 sequences. (A) BCL2 mutational profile in DLBCL. Frequency of SNPs found in the BCL2 oncogene from 45/77 DLBCL cases using a >75% frequency filter. (B) Violin plot showing the number of mutations in BCL2 for all ABC (n = 34) and GCB cases (n = 35). Median shown as a line with the upper 95% confidence interval (CI) and the black circles above the CI are outliers. *P < 0.05 as determined by a two-tailed Mann–Whitney test. (C) The 45 DLBCL tumors with detectable BCL2 mutations mapped to exon 1 and exon 3. The majority of mutations (229) are located within exon 1 and overlapped with AID peaks from the ChIP-seq (red bars) and PQS (pink bars). There were seven mutations detected in exon 3 that did not overlap with AID signals. Individual mutations are shown for each sample in the expanded panels as blue hash marks.
High AID mRNA and BG4 staining are associated with worse PFS
The inferior survival of patients with the ABC COO of DLBCL is well established (5). With our observation that G4 formation is more frequent in ABC-DLBCL together with the previous correlation with higher AID mRNA expression (5) in this DLBCL COO, we investigated whether G4 formation and/or AID mRNA expression can stratify patients according to poor outcome following RCHOP treatment. The investigated cases belong to the larger cohort from the Lenz study (5) and we confirmed that the level of AID mRNA was significantly elevated by 3.5-fold in the ABC-DLBCL relative to the GCB-DLBCL cases in the RCHOP cohort (Supplementary Figure S5A). We also determined whether the increased AID expression in ABC-DLBCL was seen at the protein level using IHC in a subset of these DLBCL cases (n = 36) along with the additional cases used in this study (n = 38). A benign tonsil sample was used as a control (Supplementary Figure S5B). Representative negative, low and high AID staining in DLBCL tissues is provided in Supplementary Figure S5C. Of note, AID staining was primarily cytoplasmic, which is in agreement with previous and recent studies, along with frequent interstitial staining (14,18).
When AID protein expression was evaluated according to COO, there was no significant difference in AID protein expression with ABC-DLBCL tumors exhibiting an average 64% AID-positive cells compared to 58% in GCB-DLBCL and 71% in UNC-DLBCL (Supplementary Figure S5D). We then plotted AID mRNA levels against protein expression from the 36 matched samples and did not see a correlation (Supplementary Figure S5E). Samples with discordant AID mRNA–protein expression, where high mRNA levels were associated with low protein expression, were predominantly ABC-DLBCL cases suggestive that AID may be differentially regulated post-transcriptionally in some ABC-DLBCL tumors (Supplementary Figure S5E, pink circles).
We then analyzed each of the variables, AID mRNA, AID IHC and BG4 IHC, independently and observed only high AID mRNA levels identified DLBCL patients with worse OS and PFS, which maintained a trend toward significance with DLBCL subtype as a covariate for PFS only (Figure 6A). The lack of association of AID protein levels with patient outcome is possibly due to post-transcriptional/translational modifications that result in no correlation between mRNA and protein levels (Supplementary Figure S5E). Although the correlation did not achieve statistical significance, there is a clear separation in the survival curves that lends a slight tendency for patients with higher BG4 staining DLBCL tumors to have a poor OS (Figure 6A). In 17 cases, 10 ABC-DLBCL, 6 GCB-DLBCL and 1 UNC-DLBCL, all three parameters were available for analysis. The Cox proportional hazards regression model demonstrated that both AID protein and G4 formation significantly correlated with inferior patient survival (Figure 6B) indicating elevated AID expression and G4 formation may confer poor response to current therapy.
Figure 6.

DLBCL OS and PFS according to AID expression and BG4 staining. (A) Kaplan–Meier curves of OS (top) and PFS (bottom) of DLBCL patients who received RCHOP treatment according to low or high AID mRNA, AID protein or BG4 staining. Cut points were determined using the Youden index derived from ROC curves: AID mRNA, OS > 9.95 and PFS > 10.94 log2 transformed; AID protein, OS > 5% and PFS > 30% positive staining cells; BG4 staining, OS > 61% and PFS > 0%. (B) Plot of the Cox proportional hazards model for 17 matched cases with data for all three variables where mean expression of the covariates is used in relation to survival probability. Corresponding coefficients are shown in the table. P-values were obtained from the log-rank test and the Cox proportional hazards multivariate analysis where DLBCL COO was a covariate.
DISCUSSION
Several lines of evidence demonstrate a role for G4 formation in facilitating genomic instability, as G4 sequences are often located at DNA breakpoints and sites of genomic amplification in cancer. Additionally, defects in G4-resolving helicases, as observed in diseases such as Bloom’s and Werner’s syndromes, result in susceptibility for cancer development (32,62–64). For the first time, we demonstrate a high frequency of G4 formation within DLBCL and that this phenomenon associates with concurrent gene amplification of two highly genetically altered and prognostically important oncogenes in DLBCL, BCL2 and MYC, as well as MYC amplification alone. This correlation supports previous work showing prominent G4 formation at sites of genomic amplification (33) and a recent study mapping G4s specifically to amplified regions that characterize certain breast cancer subtypes (65). The loss of genetic integrity through translocations, amplifications and mutations in these important oncogenes contributes to the poor therapeutic response of a significant portion of DLBCL patients (3). This association with patient outcome is becoming increasingly evident with the recent comprehensive studies identifying distinct molecular subtypes of DLBCL according to genetic lesions (66,67). The mechanism behind these genetic alterations is not well understood, although the enzyme AID is recognized as a key factor (18). We confirm that AID is frequently expressed within DLBCL tissues with higher mRNA levels in ABC-DLBCL. Patients with high-expressing AID DLBCL tumors experience poor survival and disease progression suggesting elevated AID confers an aggressive phenotype.
We then investigated the co-incidence of G4-forming sequences at genomic sites of AID targeting, and consistent with both AID and G4 involvement in genomic aberrations, we found that G4 motifs with potential to form secondary DNA structures are enriched within these regions. PQSs were a common feature of AID targets regardless of their association with other known AID preferential genomic environments of convergent transcription and SEs. A striking correlation occurred with BCL2, the most heavily mutated oncogene in DLBCL (68), where the alignment of AID peaks coincided with the majority of mutations identified in patient samples and PQSs, whereas loci with no observed AID–PQS overlap showed a substantially lower mutation rate. While the propensity of PQSs in all of the previously identified mouse B-cell AID-targeted SEs and many of the non-AID-targeted SEs indicates G4 structures may also be involved in SE biology, the significantly higher density of these motifs within AID targets suggests an involvement in AID recruitment to non-IgG loci. The incidence of G4-forming sequences in SEs is not altogether unexpected considering their association with enhanced chromatin accessibility, topological complexity and transcription activity (25), which are features conducive for G4 formation (33). We also observed this trend in the DLBCL cells further validating the previous studies; however, we were also able to detect additional AID-targeted genes, including BCL2, which had previously been inferred as targets for AID recruitment but never demonstrated. The dense G4 landscape within AID-targeted regions resembles that of its canonical target Ig sequences (28,69). Although the majority of AID-targeted regions were associated with PQSs, a small percentage did not appear to contain these G4 motifs. The PQS algorithm used to survey the AID targets did not detect non-canonical G4 DNA, which was recently recognized to function in minisatellite genomic instability of Saccharomyces cerevisiae (70). Thus, our analysis may underestimate the incidence of G4 DNA in AID targets. Furthermore, we did not consider upstream or downstream G4 motifs to the AID-targeted region or the density of the AGCT consensus AID targeting sequence. Another important feature for AID targeting is genome accessibility, and given that G4 formation most likely alters chromatin architecture, evaluating changes in chromatin accessibility in cells with high BG4 staining and AID co-localization will provide additional insight into the mechanisms of AID targeting. With these limitations to the current study, we cannot exclude that atypical G4 structures, the density of AGCT sites in AID-targeted regions or chromatin architecture plays a role in AID recruitment.
In direct support of the overlapping AID ChIP-seq peaks and identified PQSs, we show co-localization of AID with the G4-detecting probe BG4 within DBCL cell nuclei that was subsequently more pronounced after increasing G4 formation. While the precise mechanism for AID interaction with G4 structures requires further study, this finding further supports a model for G4 structure recruitment of AID to targeted loci. Since G4 structures most likely produce a looped-out single strand on the complementary cytosine-rich sequence (71), it is feasible that G4 formation creates a nuclear environment conductive for AID binding to its cytosine substrate on the opposite strand. Likewise, when the AID-targeted cytosine is contained within or in close proximity to the G-rich sequence itself (28,69), formation of the G4 structure may serve as a scaffold to enable accessibility of this cytosine within the G4 loop or capping structure. With the importance of R loops in AID biology (72) and the known interplay of G4s and these RNA structures during nuclear events, G4–R loop interactions may also contribute to the mechanism behind AID targeting (27,73).
As one of the few studies to date linking G4 formation across patient samples to genomic abnormalities and clinical outcome, our work greatly advances this field and demonstrates BG4 as a novel tool to directly measure G4 DNA within lymphoma tissue with potential for G4 staining to serve as a biomarker, especially given the recent findings that a particularly aggressive, lethal subset of DLBCL (MCD subtype) has frequent amplification of SPIB (66), which we show here consists of a substantial number of PQSs with at least one confirmed to form a stable G4. Additionally, coupling the detection of G4 formation with the use of emerging small molecules that recognize and regulate G4s in several different cancer types (29) has the potential to inform on translational efforts. Using this new methodology, we can address the biological and clinical significance of these structures in contributing to the genomic instability and aggressiveness in DLBCL with application to investigate other malignancies.
DATA AVAILABILITY
The data that support the findings of this study are available from the corresponding author upon reasonable request. The ChIP-seq data reported in this paper are available at the NCBI GEO repository under accession number GSE146695. The previous GEP and sequencing data queried in this paper are available under accession numbers GSE10846 and GSE62063.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank other members of the LLMPP for helpful discussions and providing the cel files for the additional nine in-house cases.
Author contributions: P.J., T.W. and S.K. designed the study. Y.-Z.X., V.S., E.A.C. and S.S.H. performed the experiments, and C.S. and L.M.R. acquired patient data. P.J., T.W., S.D.B., D.T., C.S., S.B., L.M.R. and S.K contributed to the analysis and interpretation of the data. D.T. and S.K wrote and revised the manuscript and all authors reviewed the manuscript.
Contributor Information
Ying-Zhi Xu, Department of Biochemistry and Molecular Biology, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA.
Piroon Jenjaroenpun, Department of Bioinformatics, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA; Division of Bioinformatics and Data Management for Research, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok 10700, Thailand.
Thidathip Wongsurawat, Department of Bioinformatics, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA; Division of Bioinformatics and Data Management for Research, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok 10700, Thailand.
Stephanie D Byrum, Department of Biochemistry and Molecular Biology, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA.
Volodymyr Shponka, Department of Pathology, University of Arizona, Tucson, AZ 85721, USA.
David Tannahill, Cancer Research UK Cambridge Institute, University of Cambridge, Cambridge CB2 0RE, UK.
Elizabeth A Chavez, British Columbia Cancer Agency, Vancouver, BC V5Z 1L3, Canada.
Stacy S Hung, British Columbia Cancer Agency, Vancouver, BC V5Z 1L3, Canada.
Christian Steidl, British Columbia Cancer Agency, Vancouver, BC V5Z 1L3, Canada.
Shankar Balasubramanian, Cancer Research UK Cambridge Institute, University of Cambridge, Cambridge CB2 0RE, UK; Department of Chemistry, University of Cambridge, Cambridge CB2 1EW, UK.
Lisa M Rimsza, Laboratory Medicine and Pathology, Mayo Clinic, Scottsdale, AZ 85259, USA.
Samantha Kendrick, Department of Biochemistry and Molecular Biology, University of Arkansas for Medical Sciences, Little Rock, AR 72205, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Cancer Online.
FUNDING
National Institutes of Health [P20GM121293 to S.K.]; UAMS Winthrop P. Rockefeller Cancer Institute Seeds of Science Award [to S.K.]; Cancer Research UK [C14303/A17197, C9681/A18618 and C9681/A29214 to S.B.]. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1. Tilly H., Gomes da Silva M., Vitolo U., Jack A., Melganan M., Lopez-Guillermo A., Walewski J., Andre M., Johnson P.W., Pfreundschuch M.et al. Diffuse large B-cell lymphoma (DLBCL): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2012; 23:78–82. [DOI] [PubMed] [Google Scholar]
- 2. Teras L.R., DeSantis C.E., Cerhan J.R., Morton L.M., Jemal A., Flowers C.R. 2016 US lymphoid malignancy statistics by World Health Organization subtypes. CA Cancer J. Clin. 2016; 66:443–459. [DOI] [PubMed] [Google Scholar]
- 3. Johnson N.A., Slack G.W., Savage K.J., Connors J.M., Ben-Neriah S., Rogic S., Scott D.W., Tan K.L., Steidl C., Sehn L.H.et al. Concurrent expression of MYC and BCL2 in diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J. Clin. Oncol. 2012; 30:3452–3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alizadeh A.A., Eisen M.B., Davis R.E., Ma C., Lossos I.S., Rosenwald A., Boldrick J.C., Sabet H., Tran T., Yu X.et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000; 403:503–511. [DOI] [PubMed] [Google Scholar]
- 5. Lenz G., Wright G., Dave S.S., Xiao W., Powell J., Zhao H., Xu W., Tan B., Goldschmidt N., Iqbal J.et al. Stromal gene signatures in large-B-cell lymphomas. N. Engl. J. Med. 2008; 359:2313–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lenz G., Wright G., Emre N.C., Kohlhammer H., Dave S.S., Davis R.E., Carty S., Lam L.T., Xiao W., Powell J.et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc. Natl Acad. Sci. U.S.A. 2008; 105:13520–13525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Morin R.D., Mungall K., Pleasance E., Mungall A.J., Goya R., Huff R.D., Scott D.W., Ding J., Roth A., Chiu R.et al. Mutational and structural analysis of diffuse large B-cell lymphoma using whole genome sequencing. Blood. 2013; 122:2156–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lohr J.G., Stojanov P., Lawrence M.S., Auclair D., Chapuy B., Sougnez C., Cruz-Gordillo P., Knoechel B., Asmann Y.W., Slager S.L.et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc. Natl Acad. Sci. U.S.A. 2012; 109:3879–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Green T.M., Young K.H., Visco C., Xu-Monette Z.Y., Orazi A., Go R.S., Nielsen O., Gadeberg O.V., Mourits-Andrsen T., Frederiksen M.et al. Immunohistochemical double-hit score is a strong predictor of outcome in patients with diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J. Clin. Oncol. 2012; 30:3460–3467. [DOI] [PubMed] [Google Scholar]
- 10. Hu S., Xu-Monette Z.Y., Tzankov A., Green T., Wu L., Balasubramanyam A., Liu W.M., Visco C., Li Y., Miranda R.N.et al. MYC/BCL2 protein coexpression contributes to the inferior survival of activated B-cell subtype of diffuse large B-cell lymphoma and demonstrates high-risk gene expression signatures: a report from the International DLBCL Rituximab-CHOP Consortium Program. Blood. 2013; 121:4021–4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Perry A.M., Alvarado-Bernal Y., Laurini J.A., Smith L.M., Slack G.W., Tan K.L., Sehn L.H., Fu K., Aoun P., Greiner T.C.et al. MYC and BCL2 protein expression predicts survival in patients with diffuse large B-cell lymphoma treated with rituximab. Br. J. Haematol. 2014; 165:382–391. [DOI] [PubMed] [Google Scholar]
- 12. Kendrick S.L., Redd L., Muranyi A., Henricksen L.A., Stanislaw S., Smith L.M., Perry A.M., Fu K., Weisenburger D.D., Rosenwald A.et al. BCL2 antibodies targeted at different epitopes detect varying levels of protein expression and correlate with frequent gene amplification in diffuse large B-cell lymphoma. Human Pathol. 2014; 45:2144–2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ramiro A.R., Jankovic M., Eisenreich T., Difillppantonio S., Chen-Klang S., Muramatsu M., Honjo T., Nussenzweig A., Nussenzweig M.C. AID is required for c-myc/IgH chromosome translocations in vivo. Cell. 2014; 118:431–438. [DOI] [PubMed] [Google Scholar]
- 14. Pasqualucci L., Guglielmino R., Houldsworth J., Mohr J., Aoufouchi S., Polakiewicz R., Chaganti R.S., Dalla-Favera R. Expression of the AID protein in normal and neoplastic B cells. Blood. 2004; 104:3318–3325. [DOI] [PubMed] [Google Scholar]
- 15. Pasqualucci L., Bhagat G., Jankovic M., Compagno M., Smith P., Muramatsu M., Honjo T., Morse H.C., Nussenzweig M.C., Dalla-Favera R. AID is required for germinal center-derived lymphomagenesis. Nat. Genet. 2008; 40:108–112. [DOI] [PubMed] [Google Scholar]
- 16. Khodabakhshi A.H., Morin R.D., Fejes A.P., Mungall A.J., Mungall K.L., Bolger-Munro M., Johnson N.A., Connors J.M., Gascoyne R.D., Marra M.A.et al. Recurrent targets of aberrant somatic hypermutation in lymphoma. Oncotarget. 2012; 3:1308–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pasqualucci L., Neumeister P., Goossens T., Nanjangud G., Chaganti R.S., Kuppers R., Dalla-Favera R. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature. 2001; 412:341–346. [DOI] [PubMed] [Google Scholar]
- 18. Teater M., Dominguez P.M., Redmond D., Chen Z., Ennishi D., Scott D.W., Cimmino L., Ghione P., Chaudhuri J., Gascoyne R.D.et al. AICDA drives epigenetic heterogeneity and accelerates germinal center-derived lymphomagenesis. Nat. Commun. 2018; 9:222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Muramatsu M., Kinoshita K., Fagarasan S., Yamada S., Shinkai Y., Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000; 102:553–563. [DOI] [PubMed] [Google Scholar]
- 20. Revy P., Muto T., Levy Y., Geissmann F., Plebani A., Sanal O., Catalan N., Forveille M., Dufourcq-Lagelouse R., Gennery A.et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the hyper-IgM syndrome (HIGM2). Cell. 2000; 102:565–575. [DOI] [PubMed] [Google Scholar]
- 21. Rebhandl S., Huemer M., Greil R., Geisberger R. AID/APOBEC deaminases and cancer. Oncoscience. 2015; 2:320–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Di Nola J.M., Neuberger M.S. Molecular mechanisms of antibody somatic hypermutation. Annu. Rev. Biochem. 2007; 76:1–22. [DOI] [PubMed] [Google Scholar]
- 23. Hackney J.A., Misaghi S., Senger K., Garris C., Sun Y., Lorenzo M.N., Zarrin A.A. DNA targets of AID: evolutionary link between antibody somatic hypermutation and class switch recombination. Adv. Immunol. 2009; 101:163–189. [DOI] [PubMed] [Google Scholar]
- 24. Neuberger M.S., Ehrenstein M.R., Klix N., Jolly C.J., Yelamos J., Rada C., Milstein C. Monitoring and interpreting the intrinsic features of somatic hypermutation. Immunol. Rev. 1998; 162:107–116. [DOI] [PubMed] [Google Scholar]
- 25. Qian J., Wang Q., Dose M., Pruett N., Kieffer-Kwon K.R., Resch W., Liang G., Tang Z., Mathe E., Benner C.et al. B cell super-enhancers and regulatory clusters recruit AID tumorigenic activity. Cell. 2014; 159:1524–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Meng F.L., Du Z., Federation A., Hu J., Wang Q., Kieffer-Kwon K.R., Meyers R.M., Amor C., Wasseman C.R., Neuberg D.et al. Convergent transcription at intragenic super-enhancers targets AID-initiated genomic instability. Cell. 2014; 159:1538–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Alinikula J., Schatz D.G. Super-enhancer transcription converges on AID. Cell. 2014; 159:1490–1492. [DOI] [PubMed] [Google Scholar]
- 28. Qiao Q., Wang L., Meng F.L., Hwang J.K., Alt F., Wu H. AID recognizes structured DNA for class switch recombination. Mol. Cell. 2017; 67:361–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Duquette M.L., Huber M.D., Maizels N. G-rich proto-oncogenes are targeted for genomic stability in B-cell lymphomas. Cancer Res. 2007; 67:2586–2594. [DOI] [PubMed] [Google Scholar]
- 30. Duquette M.L., Pham P., Goodman M.F., Maizels N. AID binds to transcription-induced structures in c-MYC that map to regions associated with translocation and hypermutation. Oncogene. 2005; 24:5791–5798. [DOI] [PubMed] [Google Scholar]
- 31. Bochman M.L., Paeschke K., Zakian V.A. DNA secondary structures: stability and function of G-quadruplex structures. Nat. Rev. Genet. 2012; 13:770–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hansel-Hertsch R., Di Antonio M., Balasubramanian S. DNA G-quadruplexes in the human genome: detection, functions, and therapeutic potential. Nat. Rev. Mol. Cell. Biol. 2017; 18:279–284. [DOI] [PubMed] [Google Scholar]
- 33. Hansel-Hertsch R., Beraldi D., Lensing S.V., Marsico G., Zyner K., Parry A., Di Antonio M., Pike J., Kimura H., Narita M.et al. G-quadruplex structures mark human regulatory chromatin. Nat. Genet. 2016; 48:1267–1272. [DOI] [PubMed] [Google Scholar]
- 34. Kendrick S., Muranyi A., Gokhale V., Hurley L.H., Rimsza L.M. Simultaneous drug targeting of the promoter MYC G-quadruplex and BCL2 i-motif in diffuse large B-cell lymphoma slows tumor growth. J. Med. Chem. 2017; 60:6587–6597. [DOI] [PubMed] [Google Scholar]
- 35. Kendrick S., Kang H.J., Alam M.P., Madathil M.M., Agrawal P., Gokhale V., Yang D., Hecht S.M., Hurley L.H. The dynamic character of the BCL2 promoter i-motif provides a mechanism for modulation of gene expression by compounds that bind selectively to the alternative DNA hairpin structure. J. Am. Chem. Soc. 2014; 136:4161–4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kang H.J., Kendrick S., Hecht S.M., Hurley L.H. The transcriptional complex between the BCL2 i-motif and hnRNP LL is a molecular switch for control of gene expression that can be modulated by small molecules. J. Am. Chem. Soc. 2014; 136:4172–4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huppert J.L., Balasubramanian S. G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 2007; 35:406–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Biffi G., Tannahill D., Miller J., Howat W.J., Balasubramanian S. Elevated levels of G-quadruplex formation in human stomach and liver cancer tissues. PLoS One. 2014; 9:e102711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Valentino C., Kendrick S., Johnson N., Gascoyne R., Chan W.C., Weisenburger D., Braziel R., Cook J.R., Tubbs R., Campo E.et al. Colorimetric in-situ hybridization identifies MYC gene signal clusters correlating with increased copy number, mRNA, and protein in diffuse large B-cell lymphoma. Am. J. Clin. Pathol. 2012; 139:242–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Meyer P.N., Fu K., Greiner T.C., Smith L.M., Delabie J., Gascoyne R.D., Ott G., Rosenwald A., Braziel R.M., Campo E.et al. Immunohistochemical methods for predicting cell of origin and survival in patients with diffuse large B-cell lymphoma treated with rituximab. J. Clin. Oncol. 2011; 29:200–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Irizarry R.A., Bolstad B.M., Collin F., Cope L.M., Hobbs B., Speed T.P. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003; 31:e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gion Y., Takeuchi M., Shibata R., Takata K., Miyata-Takata T., Orita Y., Tachibana T., Yoshino T., Sato Y. Up-regulation of activation-induced cytidine deaminase and its strong expression in extra-germinal centres in IgG4-related disease. Sci. Rep. 2019; 9:761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hasler J., Rada C., Neuberger M.S. Cytoplasmic activation-induced cytidine deaminase (AID) exists in stoichiometric complex with translation elongation factor 1α (eEF1A). Proc. Natl Acad. Sci. U.S.A. 2011; 108:18366–18371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 2013; arXiv doi:26 May 2013, preprint: not peer reviewedhttps://arxiv.org/abs/1303.3997.
- 45. Koboldt D.C, Zhang Q., Larson D.E., Shen D., McLellan M.D., Lin L., Miller C.A., Mardis E.R., Ding L., Wilson R.K. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012; 22:e576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sherry S.T., Ward M.H., Kholodov M., Baker J., Phan L., Smigielski E.M., Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001; 29:308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Robinson J.T, Thorvaldsdottir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011; 29:e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bedrat A., Lacroix L., Mergny J.-L. Re-evaluation of G-quadruplex propensity with G4Hunter. Nucleic Acids Res. 2016; 44:1746–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen S., Zhou Y., Chen Y., Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018; 34:i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li H., Durbin R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics. 2010; 26:589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhao H., Sun Z., Wang J., Huang H., Kocher J.P., Wang L. CrossMap: a versatile tool for coordinate conversion between genome assemblies. Bioinformatics. 2014; 30:1006–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim S.J., Myong J.P., Suh H., Lee K.E., Youn Y.K. Optimal cutoff age for predicting mortality associated with differentiated thyroid cancer. PLoS One. 2015; 10:e0130848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Biffi G., Tannahill D., McCafferty J., Balasubramanian S. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 2013; 5:182–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Byrd A.K., Bell M.R., Raney K.D. Pif1 helicase unfolding of G-quadruplex DNA is highly dependent on sequence and reaction conditions. J. Biol. Chem. 2018; 293:17792–17802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Voter A.F., Qiu Y., Tippana R., Myong S., Keck J.L. A guanine-flipping and sequestration mechanism for G-quadruplex unwinding by RecQ helicases. Nat. Commun. 2018; 9:4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gray L.Y., Vallur A.C., Eddy J., Maizels N. G quadruplexes are genome wide targets of transcriptional helicases XPB and XPD. Nat. Chem. Biol. 2014; 10:313–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wang Y., Yang J., Wild A.T., Wu W.H., Shah R., Danussi C., Riggins G.J., Kannan K., Sulman E.P., Chan T.A., Huse J.T. G-quadruplex DNA drives genomic instability and represents a targetable molecular abnormality in ATRX-deficient malignant glioma. Nat. Commun. 2019; 10:943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hansel-Hertsch R., Spiegel J., Marsico G., Tannahill D., Balasubramanian S. Genome-wide mapping of endogenous G-quadruplex DNA structures by chromatin immunoprecipitation and high-throughput sequencing. Nat. Protoc. 2018; 13:551–564. [DOI] [PubMed] [Google Scholar]
- 60. Dexheimer T.S., Sun D., Hurley L.H. Deconvoluting the structural and drug-recognition complexity of the G-quadruplex-forming region upstream of the bcl-2 P1 promoter. J. Am. Chem. Soc. 2006; 128:5404–5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Siddiqui-Jain A., Grand C.L., Bearss D.J., Hurley L.H. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl Acad. Sci. U.S.A. 2002; 99:11593–11598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. De S., Michor F. DNA secondary structures and epigenetic determinants of cancer genome evolution. Nat. Struct. Mol. Biol. 2011; 18:950–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Katapadi V.K., Nambiar M., Raghavan S.C. Potential G-quadruplex formation at breakpoint regions of chromosomal translocations in cancer may explain their fragility. Genomics. 2012; 100:72–80. [DOI] [PubMed] [Google Scholar]
- 64. Wu Y., Brosh R.M. Jr. G-quadruplex nucleic acids and human disease. FEBS J. 2010; 277:3470–3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hansel-Hersch R., Simeone A., Shea A., Hui W.W.I., Zyner K.G., Marsico G., Rueda O.M., Bruna A., Martin A., Zhang X.et al. Landscape of G-quadruplex DNA structural regions in breast cancer. Nat. Genet. 2020; 52:878–883. [DOI] [PubMed] [Google Scholar]
- 66. Chapuy B., Stewart C., Dunford A.J., Kim J., Kamburov A., Redd R.A., Lawrence M.S., Roemer M.G.M., Li A.J., Ziepert M.et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 2018; 24:679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Schmitz R., Wright G.W., Huang D.W., Johnson C.A., Phelan J.D., Wang J.Q., Roulland S., Kasbekar M., Young R.M., Shaffer A.L.et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. N. Engl. J. Med. 2018; 378:1396–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Schuetz J.M., Johnson N.A., Morin R.D., Scott D.W., Tan K., Ben-Nierah S., Boyle M., Slack G.W., Marra M.A., Connors J.M.et al. BCL2 mutations in diffuse large B-cell lymphoma. Leukemia. 2012; 26:1383–1390. [DOI] [PubMed] [Google Scholar]
- 69. Sen D., Gilbert W. Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature. 1988; 344:364–366. [DOI] [PubMed] [Google Scholar]
- 70. Piazza A., Cui X., Adrian M., Samazan F., Heddi B., Phan A.-T., Nicolas A.G. Non-canonical G-quadruplexes cause the hCEB1 minisatellite instability in Saccharomycescerevisiae. eLife. 2017; 6:e26884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cui Y., Kong D., Ghimire C., Xu C., Mao H. Mutually exclusive formation of G-quadruplex and i-motif is a general phenomenon governed by steric hindrance in duplex DNA. Biochemistry. 2016; 55:2291–2299. [DOI] [PubMed] [Google Scholar]
- 72. Zheng S., Vuong B.Q., Valdyanathan B., Lin J.-Y., Huang F.-T., Chaudhuri J. Non-coding RNA generated following lariat-debranching mediates targeting of AID to DNA. Cell. 2015; 161:762–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Maffia A., Ranise C., Sabbioneda S. From R-loops to G-quadruplexes: emerging new threats for the replication fork. Int. J. Mol. Sci. 2020; 21:1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request. The ChIP-seq data reported in this paper are available at the NCBI GEO repository under accession number GSE146695. The previous GEP and sequencing data queried in this paper are available under accession numbers GSE10846 and GSE62063.