

Abstract
Chronic lymphocytic leukemia is a well-defined lymphoid neoplasm with very heterogeneous biological and clinical behavior. The last decade has been remarkably fruitful in novel findings, elucidating multiple aspects of the pathogenesis of the disease including mechanisms of genetic susceptibility, insights into the relevance of immunogenetic factors driving the disease, profiling of genomic alterations, epigenetic subtypes, global epigenomic tumor cell reprogramming, modulation of tumor cell and microenvironment interactions, and dynamics of clonal evolution from early steps in monoclonal B-cell lymphocytosis to progression and transformation into diffuse large B-cell lymphoma. All this knowledge has offered new perspectives that are being exploited therapeutically with novel, targeted agents and management strategies. In this review we provide an overview of these novel advances and highlight questions and perspectives that need further progress to translate this biological knowledge into the clinic and improve patients’ outcome.
History
Chronic lymphocytic leukemia (CLL) is a lymphoid malignancy characterized by the proliferation and accumulation of mature CD5+ B cells in the blood, bone marrow and lymphoid tissues. The diagnosis of CLL requires the presence of ≥5 x109/L mono - clonal B cells of typical phenotype in the blood. Patients with <5 x109/L circulating CLL-type cells may be diagnosed with small lymphocytic lymphoma if they also present with either lymphadenopathy, organomegaly or extramedullary disease; or with monoclonal B-cell lymphocytosis (MBL) if they do not.1 CLL is the most prevalent type of leukemia in adults in Western countries, with an age-adjusted incidence rate of 4.9 cases per 100,000 inhabitants per year. There is a stark difference between the incidence in men (6.8 cases per 100,000/year) and women (3.5 cases per 100,000/year) and also between Caucasians (7.3 and 3.8 cases per 100,000/year for men and women, respectively), African Americans (4.9 and 2.4 cases per 100,000/year for men and women, respectively) and Asian Americans (1.5 and 0.7 cases per 100,000/year for men and women, respectively).2 The disease may have a stable course but also become aggressive, with frequent relapses, or even transform into an aggressive lymphoma, typically diffuse large B-cell lymphoma (DLBCL) (Richter transformation).
In the last decade, genomic and epigenomic studies have expanded our knowledge of the pathogenesis of CLL remarkably, unraveling a large number of novel alterations that might drive the evolution of the disease.3–7 Moreover, understanding the crosstalk between tumor cells and their microenvironment has been fundamental in the development of new, targeted agents, which are transforming the way we manage the disease. In this review we provide an overview of these novel advances and how they relate to our understanding of the pathogenesis and current management of CLL.
Pathogenesis
Genetic predisposition
Family studies have consistently shown that first-degree relatives of patients with CLL have a 2- to 8-fold increased risk of developing the disease.8 Genomewide association studies have identified up to 45 susceptibility loci, mostly mapping to non-coding regions of the genome.8 The mechanisms linking these susceptibility variants and the development of the disease are being elucidated thanks to integrated genome-wide association/ transcriptome/epigenome studies. These analyses recently revealed that 93% of the susceptibility loci are located in active promoters or enhancers and modify the binding sites of a number of transcription factors (e.g., FOX, NFAT and TCF/LEF) that, in turn, alter the expression of more than 30 genes involved in immune response, cell survival, or Wnt signaling (Figure 1).9 Despite these advances, molecular analysis for predisposition to CLL remains investigational.
Cell of origin
Hematopoietic stem cells derived from patients with CLL seem epigenetically primed to clonal expansions of CLL-like cells when implanted in mice. Interestingly, these clonal expansions do not always carry the same genomic aberrations as the original disease.10 Moreover, hematopoietic stem cells derived from patients with CLL express higher levels of transcription factors, such as TCF3, IKZF1 or IRF8, than those from healthy donors, which is intriguing if we consider that some susceptibility loci increase TCF3 binding or IRF8 expression.9 Mutations in driver genes such as NOTCH1 or SF3B1 may be acquired by hematopoietic stem cells, but also at more advanced stages of B-cell differentiation, explaining why these genomic aberrations are frequently subclonal.11–13 These alterations observed in early steps of B-cell development are also consistent with the identification of shared mutations in CLL and myeloid cells and the detection of oligo- and multi-clonality in patients with MBL/CLL.14–16
The B-cell receptor (BCR) is crucial for CLL pathogenesis and is composed of immunoglobulin (IG) molecules plus CD79a/b subunits. From an immunogenetic point of view, two major molecular subgroups have been identified: those harboring unmutated IG heavy-chain variable region (IGHV) genes (U-CLL, ≥98% identity with the germline) and those with mutated IGHV genes (MCLL). 17,18 U-CLL originates from B cells that have not experienced the germinal center, whereas M-CLL originates from post-germinal center B cells.19 In addition, around 30% of patients have highly homologous amino acid sequences derived from almost identical IG rearrangements, known as stereotypes.20 Several hundred stereotypes have been identified, of which 19 are considered major due to their frequency. The prognostic importance of several stereotypes has been prospectively validated.21 The presence of stereotypes and the remarkable bias in the use of certain IGHV genes highlight the relevance of antigen selection in CLL clonal expansion. Interestingly, the IG portion of the BCR may also recognize homotypic epitopes that trigger downstream signaling.22,23 In this sense, the acquisition of the mutation at position 110 (G>C, glycine-arginine) of IGLV3-21*01 mediated by somatic hypermutation confers autonomous BCR signaling. 24 This change is present in 7-18% of CLL and seems responsible for the adverse outcome associated with the use of IGLV3-21 independently of the mutational status of the IGHV.24,25
Figure 1.

Genetic susceptibility mechanisms. Most susceptibility loci map to non-coding regions of the genome, are mainly located in active promoters or enhancers, and modify the binding sites of a number of transcription factors. As a consequence, the binding sites for SPI1 and NF-κB are disrupted, whereas there is an increased affinity for members of the FOX, NFAT and TCF/LEF families. This, in turn, alters the expression of genes involved in the immune response (SP140, IRF8), cell survival (BCL2, BMF, CASP8, BCL2L11) or Wnt signaling (UBR5, LEF1).9
Epigenetic studies have shown that, although both CLL subtypes are antigen-experienced, M-CLL keeps a methylation signature of germinal center-experienced cells (memory-like B cells), whereas U-CLL has a pre-germinal center, naïve-like methylation signature.5,26 Of note, these epigenetic studies also identified a third subtype with an intermediate profile made of cases with moderate IGHV mutation levels. All three epigenetic subsets have different usage of IGHV genes, stereotypes, genomic aberrations and clinical outcome (Table 1).27 Their prognostic relevance has been validated in retrospective cohorts and clinical trials.26–28 The intermediate epigenetic subtype may be more heterogeneous than initially thought since it includes most stereotype subset 2 cases with aggressive behavior whereas other cases may behave more indolently. The understanding of the biological significance of this subtype requires further analysis.
The microenvironment in chronic lymphocytic leukemia
CLL cells are highly dependent on signals coming from the microenvironment for proliferation and survival. Tumor cells proliferate primarily in lymph nodes, and to less extent in bone marrow,29 where they are in intimate contact with extracellular matrix, T cells, nurse-like cells, follicular dendritic cells and other stromal cells (Figure 2). The interactions between CLL cells and this complex microenvironment are mediated by a network of adhesion molecules, cell surface ligands, chemokines, cytokines, and their respective receptors. CLL cells organize their supportive inflammatory milieu and promote an immunosuppressive microenvironment through different mechanisms, such as secretion of soluble factors, cell-tocell contact, and release of extracellular vesicles (Figure 2).29,30
Environmental or self-antigens and homotypic interactions trigger BCR and Toll-like receptor (TLR) signaling, amplifying the response of CLL cells to other signals from the microenvironment and increasing the activation of anti-apoptotic and proliferation pathways.31,32 Genomic studies have identified recurrent mutations in genes regulating tumor cell-microenvironment interactions, which are already required for tumor cell growth. Thus, NOTCH1 mutations are dependent on the presence of Notch ligands in the microenvironment and activate processes such as cell migration, invasion and angiogenesis. 33,34 BCR and NOTCH1 pathways are functionally linked, mutually enhancing their activation.35 MYD88 mutations activate the NF-κB pathway in response to TLR ligands, increasing the cytokine release involved in recruiting stromal and T cells.36 Tumor cells also reconfigure the function of T- and myeloid-derived cells towards a leukemia-supportive and immunosuppressive microenvironment. 30,37 Thus, tumor cells reduce T-cell motility and the effector function of CD4+ cells while inducing CD8+-cell exhaustion38–41 and monocyte differentiation towards macrophages with protumoral functions (M2- like) and nurse-like cells.37
Many studies have confirmed the fundamental role of BCR activation for CLL pathogenesis.42 Several proteins, including phosphatidylinositol 3 kinase (PI3K), Bruton tyrosine kinase (BTK) and spleen tyrosine kinase (SYK) are essential for BCR signal transduction.43 The effect of BCR-mediated signaling varies according to IGHV mutation status: M-CLL cells are generally driven towards anergy, whereas U-CLL cells are more directed towards cell growth and proliferation.44 Moreover, anergic cells normally retain a higher susceptibility to apoptosis unless anti-apoptotic proteins such as BCL2 are overexpressed, as is the case for CLL cells.45 Indeed, most major therapeutic advances occurring in the last decade are related to the inhibition of BCR and BCL2-mediated signaling.
Structural genomic aberrations
Initial chromosome banding analysis revealed that deletions or trisomies were relatively common but only observed in fewer than half of the patients.46 With the advent of fluorescent in situ hybridization (FISH), genomic aberrations were identified in more than 80% of patients, the more relevant being trisomy 12, 13q deletion [del(13q)], 11q deletion [del(11q)] and 17p deletion [del(17p)];47 and FISH became the gold standard for genomic evaluation in CLL. Later, the introduction of more effective mitogens expanded the use of chromosome banding analysis in CLL and revealed other aberrations that could not be detected by FISH, including chromosome translocations in 20-35% of the cases.48 These translocations may occur in the context of complex karyo types. The most common rearrangements involve 13q14, with multiple partners, and the IGH locus. The genes most commonly rearranged with IGH are BCL2 [t(14;18)(q32;q21)] (2% of cases, usually M-CLL);3,49 and BCL3 [t(14;19)(q32;q13)] or BCL11A [t(2;14)(p16;q32)] (<1% of cases, usually U-CLL with atypical features).50,51 Chromosomal microarray analysis has identified novel copy number alterations and also copy number neutral loss of heterozygosity.3,52 This latter is observed in 5% of patients, typically affects deleted regions such as 11q, 17p and 13q, and is associated with mutations of the target gene, particularly TP53.52,53 A novel use of both chromosome banding analysis and chromosomal microarray analysis is the identification of complex karyotypes, observed in up to 20% of patients with CLL. A complex karyotype appears not only prognostic but also predictive in the context of treatment with both conventional and novel agents.54,55 Intriguingly, a subset of patients with complex karyotypes carrying trisomy 12, trisomy 19, and additional trisomies seem to correspond to a particular genetic subgroup with favorable outcome.54–56 In contrast to patients with other hematologic malignancies, patients with five or more aberrations have a worse prognosis compared to those with “less complex” karyotypes (3 or 4 aberrations).55
Table 1.
Clinical and biological characteristics of the three epigenetic subtypes in chronic lymphocytic leukemia.20,27,137
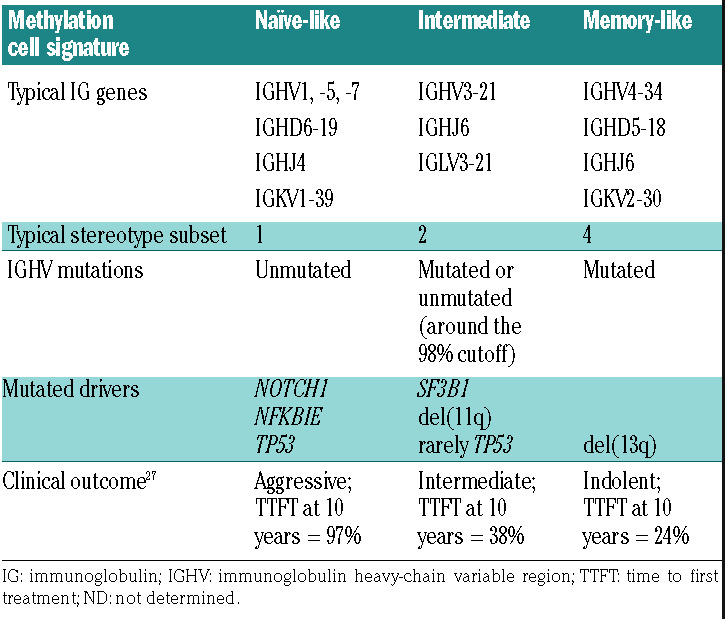
Figure 2.

The chronic lymphocytic leukemia microenvironment. Communication between chronic lymphocytic leukemia (CLL) cells and stromal cells, T cells and nurse-like cells (NLC) is established and maintained by direct contacts, chemokine/cytokine receptors, adhesion molecules and ligand-receptor interactions. CLL cells migrate to tissues attracted by the chemokines CXCL12 secreted by NLC and stromal cells, CXCL13 by follicular dendritic cells (FDC), and CCL19/CCL21 by highendothelial venules, which interact with the CLL receptors CXCR4, CXCR5 and CCR7, respectively. Adhesion molecules (e.g., α4β1 integrin, LFA-1) and their ligands (VCAM1, ICAM, among others) facilitate tumor cell migration and homing. Environmental or auto-/self-antigens and homotypic IG interactions trigger B-cell receptor (BCR) activation capable of driving CLL proliferation.22,138 Interactions between CD40 and CD40 ligand (CD40L) on activated CD4+ T cells are critical in the context of antigen presentation and induction of normal B-cell responses. Activated CLL cells secrete chemokines (CCL2, CCL3, and CCL4) and angiogenic factors that attract T cells and different stromal cells.30,139 Suppressive factors (IL-10)140 and immune inhibitory molecules (PD-L1 among others)141 facilitate tumor cells to evade immuneresponse and maintain tolerance. Anti-tumor CD8+ T cells become exhausted by constant exposure to tumor-derived antigens leading to cell exhaustion.40 Regulatory T cells (Treg) exert an inhibitory effect on CD4+ and CD8+ cells through secretion of suppressive cytokines.142 Tumor-released extracellular vesicles carrying noncoding RNA and proteins induce an inflammatory phenotype in T cells, monocytes, and stromal cells.143
Mutational landscape
On average, CLL tumors accumulate around 2,500 somatic mutations with a clear difference between MCLL and U-CLL (3,000 vs. 2,000 somatic mutations on average, respectively). This increased mutation burden observed in M-CLL has limited transformation potential as patients with M-CLL have fewer mutated drivers and better clinical outcomes than patients with U-CLL.3 The mutational landscape of the disease is remarkably heterogeneous, with only a handful of genes mutated in more than 5% of patients at diagnosis (NOTCH1, SF3B1, TP53, ATM) followed by a long tail of genes mutated at lower frequencies.3,4 Despite this diversity, most mutated drivers cluster in a number of cell pathways (Figure 3), such as NOTCH1 signaling (NOTCH1, FBXW7); BCR and TLR signaling (EGR2, BCOR, MYD88, TLR2, IKZF3); the MAPK-ERK pathway (KRAS, NRAS); NF-κB signaling (BIRC3, NFKB2, NFKBIE, TRAF2, TRAF3); chromatin modifiers (CHD2, SETD2, KMT2D, ASXL1); cell cycle (ATM, TP53, CCND2, CDKN1B, CDKN2A); DNA damage response (ATM, TP53, POT1); and RNA splicing and metabolism (SF3B1, U1, XPO1, DDX3X, RPS15). This clustering suggests that mutations belonging to the same cell process may have similar functional and clinical impacts,57,58 but this hypothesis requires further confirmation. A detailed descriptions of these cell processes can be found elsewhere.51
This accumulation of low-frequency driver alterations highlights the striking interpatient heterogeneity of CLL, which is partly determined by three major factors: (i) the cell of origin: M-CLL have fewer driver mutations than UCLL and some mutated genes are almost exclusively or predominantly seen in one of the two subtypes (e.g MYD88 and PAX5 in M-CLL and U1, NOTCH1, and POT1 in U-CLL), whereas others are seen in both subtypes; (ii) age of the patients: MYD88 mutations seem to be more frequent in younger patients; (iii) disease evolution: some mutations (SF3B1, POT1, ATM) are more frequent in patients requiring therapy compared to those with stable disease, and some others (TP53, BIRC3, MAP2K1, NOTCH1) are more frequent in patients with progressive disease after chemoimmunotherapy (CIT).3,4,59 The co-occurrence of many of these driver alterations within the same tumor complicates the analysis of their relative clinical relevance. For instance, mutations in SF3B1, POT1 or XPO1 are generally associated with poor prognosis, but they rarely appear on their own.13 This has led some investigators to propose a multi-hit model in which the accumulation of driver mutations, regardless of the individual genes targeted by each of these mutations, gradually impairs patients’ outcome.3,13 Indeed, the survival of patients in whom no driver aberrations are identified is comparable to that of individuals in the general population, further reinforcing this concept.3
Figure 3.

Recurrently mutated genes and pathways in chronic lymphocytic leukemia. The main molecular pathways affected by mutations in chronic lymphocytic leukemia are depicted. Genes mutated at higher frequencies (>5%) in newly diagnosed patients are highlighted in bold.
Deep, targeted next-generation sequencing has revealed that subclonal mutations (i.e., those present in only a fraction of tumor cells) can be detected for all driver genes and are associated with rapid disease progression and poor outcome.11–13 This is particularly relevant for TP53 mutations given the fact that, as explained below, CLL therapy is based on the presence or absence of these mutations. The current consensus is that, apart from clonal mutations, subclonal mutations with a variant allelic frequency ranging from 5 to 10% (and therefore below the threshold of detection by conventional molecular techniques) could also be reported, whereas those with a variant allelic frequency lower than 5% should not, but there is much controversy around these issues and this recommendation may well change in the future.60,61 Furthermore, the analysis of clonal and subclonal aberrations has also allowed the reconstruction of each tumor’s phylogeny. Thus, clonal aberrations, which are mostly structural abnormalities [e.g. trisomy 12, del(13q)] generally correspond to earlier driving events, while subclonal mutations in driver genes (e.g., SF3B1, POT1, NOTCH1) are acquired later over the course of the disease.13
Moreover, some genes appear to be specifically selected at relapse. For instance, small clones harboring TP53 mutations typically expand and dominate the disease after CIT, which explains the poor prognosis associated with these subclonal mutations.12,62 Apart from TP53, mutations in IKZF3 and SAMHD1 have also been recurrently selected in small cohorts of patients after CIT.63,64 Clonal evolution plays an important role not only in resistance to CIT, but also to novel agents. Indeed, different point mutations have been identified in the BTK and PLCG2 genes in patients previously treated with the BTK inhibitor ibrutinib,65 and in the BCL2 gene in patients relapsing after treatment with the BCL2 antagonist venetoclax. 66 Resistance to these agents has been associated with these mutations in around 70% of cases, although they are usually subclonal and their specific role causing resistance needs to be proven.67 Other resistance mechanisms involve upregulation of BCL-XL and MEK/ERK, and cell reprogramming and transdifferentiation to cell subtypes that do not require BCR signaling.65,67–70
Epigenomic landscape
The genome of CLL features widespread hypomethylation, and a large fraction of the differences between UCLL and M-CLL can be attributed to their different cell of origin in germinal center-independent or -experienced B cells, respectively.5 Major hypomethylation changes occur at transcription factor binding sites such as TCF3, PU.1/SPIB, NFAT and EGR, and enhancers that modulate genes relevant for CLL pathogenesis involved in B-cell function, BCR signaling, and NF-κB activation among others. This methylation profile is already acquired at the MBL stage3 and remains relatively stable over time. However, some CLL have intratumor variability in certain regions, which may alter the expression of several genes and facilitate tumor evolution.71 Of note, this variability is greater in U-CLL than in M-CLL and is associated with increasing number of subclones.7,71
The genome of CLL features widespread hypomethylation, and a large fraction of the differences between UCLL and M-CLL can be attributed to their different cell of origin in germinal center-independent or -experienced B cells, respectively.5 Major hypomethylation changes occur at transcription factor binding sites such as TCF3, PU.1/SPIB, NFAT and EGR, and enhancers that modulate genes relevant for CLL pathogenesis involved in B-cell function, BCR signaling, and NF-κB activation among others. This methylation profile is already acquired at the MBL stage3 and remains relatively stable over time. However, some CLL have intratumor variability in certain regions, which may alter the expression of several genes and facilitate tumor evolution.71 Of note, this variability is greater in U-CLL than in M-CLL and is associated with increasing number of subclones.7,71
Somatic mutations in chromatin remodeler genes could modify the epigenomic landscape of CLL, but they are uncommon in this malignancy compared to other lymphoid neoplasms. CHD2 is mutated in 5% of CLL and 7% of MBL.75 The histone methyltransferase SETD2 and ARID1A are also mutated in a small proportion of patients. Of note, MYD88 mutations and trisomy 12 are associated with specific remodeling of chromatin activation and accessibility regions. More specifically, the epigenomic profile induced by MYD88 mutations targets regulatory regions related to NF-κB signaling,6 whereas the epigenetic configuration of trisomy 12 CLL is characterized by a subtype-specific hypomethylation signature associated with increased H3K27 acetylation, which leads to the overexpression of 25 target genes including RUNX3.76
Pathogenic mechanisms in the evolution of the disease
CLL is always preceded by an often unnoticed premalignant state known as high-count MBL.77 Low-count MBL may persist for a long time but the risk of progression is negligible.78 Yearly, 1% of cases of high-count MBL evolve into CLL requiring therapy,79 and 2-10% of patients with symptomatic CLL eventually develop Richter transformation.80 At the other end of the spectrum, around 30% of patients with CLL never require any CLL-specific therapy and die of other causes, and 1-2% of them even experience spontaneous regression of their disease.81 It is therefore evident that the rate and pattern of growth (or even decline) of the disease can vary greatly among patients.
Patients with high-count MBL carry mutations in driver genes which may be detected at a median of 41 months prior to progression to CLL.3,82 The mutation rates for the most common drivers (e.g., SF3B1, DDX3X, BIRC3, ATM) are comparable between MBL and CLL, with only a few genes being more commonly mutated in CLL (NOTCH1, TP53, XPO1).82,83 Patients with MBL with mutated drivers have a shorter time to first treatment compared to cases without mutations. Once CLL is established, the growth dynamics of tumor cells is heterogeneous. Some patients exhibit a logistic-like behavior in which the clone stabilizes over time, whereas some others show an exponential- like growth pattern.84 This exponential growth, clinically defined as “short lymphocyte doubling time” is still considered an adverse prognostic parameter in CLL.85,86 As expected, the median number of driver mutations (both clonal and subclonal) is higher in patients with exponential growth, and this patient population also displays unmutated IGHV genes more frequently. In addition, the rate of clonal evolution after therapy (i.e., with a significant shift in at least one subclone) is also higher in patients with exponential growth (Figure 4).
Transformation of CLL into an aggressive lymphoma occurs in 2-10% of patients and in most of them (>90%) corresponds to a DLBCL, but Hodgkin lymphoma may also occur.87 The DLBCL usually emerges as a linear evolution of the same CLL clone with only rare cases deriving from a branching divergent subclone.88 CLL carrying stereotyped subset 8 (IGHV4-39), NOTCH1 or TP53 mutations and complex karyotypes are at higher risk of transformation after CIT. Transformed DLBCL frequently add CDKN2A deletions and MYC translocations or amplifications on top of the genomic alterations already present in the original CLL, but lack the common mutations observed in primary DLBCL indicating that they may correspond to a different biological category.80 Richter transformation also occurs in patients treated with BTK inhibitors. These tumors do not usually acquire BTK or PLCG2 mutations but, if these were present in the original CLL, subclones may emerge with additional independent mutations.89,90
Transformation of CLL into an aggressive lymphoma occurs in 2-10% of patients and in most of them (>90%) corresponds to a DLBCL, but Hodgkin lymphoma may also occur.87 The DLBCL usually emerges as a linear evolution of the same CLL clone with only rare cases deriving from a branching divergent subclone.88 CLL carrying stereotyped subset 8 (IGHV4-39), NOTCH1 or TP53 mutations and complex karyotypes are at higher risk of transformation after CIT. Transformed DLBCL frequently add CDKN2A deletions and MYC translocations or amplifications on top of the genomic alterations already present in the original CLL, but lack the common mutations observed in primary DLBCL indicating that they may correspond to a different biological category.80 Richter transformation also occurs in patients treated with BTK inhibitors. These tumors do not usually acquire BTK or PLCG2 mutations but, if these were present in the original CLL, subclones may emerge with additional independent mutations.89,90
Figure 4.

Evolutionary steps and growth dynamics of chronic lymphocytic leukemia. (Left) The progression of monoclonal B-cell lymphocytosis (MBL) to chronic lymphocytic leukemia (CLL) is a linear process discriminated by the total number of lymphocytes. The presence of driver alterations is associated with rapid progression. Although a few alterations are enriched in CLL compared to MBL, both phases share a similar driver composition. (Middle) The main growth dynamics during the pre-treatment phase of the CLL are shown, including spontaneous regression, logistic growth and exponential growth. The main characteristics associated with each pattern are specified. WBC; peripheral white blood cell count. (Right) Richter transformation to diffuse large B-cell lymphoma (DLBCL) is associated with subset #8, NOTCH1 or TP53 mutations and complex kar yotype. It follows a linear evolution from the CLL clone through the recurrent acquisition of CDKN2A and MYC alterations.
Clinical and prognostic implications of novel discoveries
The clinical course of CLL is rather heterogeneous, ranging from a fairly asymptomatic disease that may even regress spontaneously to a progressive disease that eventually leads to the patient’s death, so there has always been remarkable interest in determining the prognosis of individual patients. Even though many prognostic markers have been identified over the past decades, only a few prevail. For many years, the prognosis of patients with CLL was defined using purely clinical parameters, such as those included in the Rai and Binet staging systems,92,93 the IGHV mutational status,17,18 and numerical aberrations as determined by FISH.47 With the advent of next-generation sequencing, novel drivers were discovered (NOTCH1, SF3B1, BIRC3) and incorporated into these prognostic systems, but none of these attempts succeeded in becoming standard of care.94–96 Indeed, the International Workshop on CLL (iwCLL) guidelines only recommend evaluating the IGHV status and presence/absence of TP53 aberrations in routine practice.86 The recent CLL International Prognostic Index (CLL-IPI) incorporates both clinical and cytogenetic/genomic data (age, clinical staging, β2- microglobulin serum concentration, IGHV mutation status and TP53 aberrations) into one prognostic score.97 The CLL-IPI was developed in cohorts of patients treated with CIT and has been validated in retrospective series.98–100 Among the five items, both TP53 and IGHV have the strongest impact on a patient’s outcome, and it is therefore not surprising that simplified versions of the CLL-IPI incorporating only these two markers have been proposed. 101 A recent study has determined that a score based on the presence of unmutated IGHV, absolute lymphocyte count >15 x109/L, and palpable lymph nodes predicts for a shorter time to first treatment in patients with early, asymptomatic disease.102 On the other hand, several groups are advocating for the incorporation of novel markers, such as a complex karyotype55 or epigenetic subsets, 27,28 into clinical practice. All these novel prognostic and/or predictive models will need to be validated in cohorts of patients treated with targeted agents.
Treatment
Treatment for CLL has changed remarkably in the last decade (Figure 5). The mainstay of therapy used to be CIT - a combination of conventional chemotherapeutic agents plus a monoclonal antibody, such as rituximab or obinutuzumab, - although this is no longer the case, at least for most patients. Novel, targeted agents are now the preferred option, and among them, the drugs currently approved by both the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) are the BTK inhibitor ibrutinib, the BCL2 inhibitor venetoclax and the PI3K inhibitor idelalisib, while the second-generation BTK inhibitor acalabrutinib and PI3K inhibitor duvelisib have already been approved by the FDA and are under evaluation by the EMA.
Frontline therapy
Not all patients with CLL require therapy. Despite all recent advances, the iwCLL still recommends watchful observation for patients with asymptomatic disease.86 This recommendation is based on at least two randomized trials comparing observation to either chlorambucil monotherapy or fludarabine, cyclophosphamide and rituximab (FCR).103,104 Both trials concluded that early therapy in asymptomatic patients was not associated with a prolonged overall survival. Very recently, preliminary results from a third trial comparing ibrutinib versus observation were presented.105 Patients receiving ibrutinib had a longer event-free survival, but no overall survival advantage, although the results were still immature. Moreover, although severe adverse events rates were comparable between groups, patients receiving ibrutinib had a higher incidence of some specific adverse events such as bleeding, hypertension and atrial fibrillation.
For patients with symptomatic disease requiring therapy, ibrutinib is often recommended based on four phase III randomized clinical trials comparing ibrutinib with chlorambucil monotherapy106 and other commonly used CIT combinations, namely FCR, bendamustine plus rituximab and chlorambucil plus obinutuzumab (ClbO).107–109 Ibrutinib was superior to chlorambucil and all CIT combinations in terms of response rate and progression-free survival, and even conferred a longer overall survival compared to that provided by chlorambucil monotherapy and FCR.106,107 In these trials, ibrutinib was sometimes combined with a monoclonal antibody, either rituximab or obinutuzumab, and sometimes given as monotherapy, but the true added value of the monoclonal antibody in this context is unknown.108,110 In terms of toxicity, ibrutinib was less toxic than CIT combinations when severe adverse events or toxic deaths were considered.107–109
Figure 5.

Recommended therapy for patients with symptomatic chronic lymphocytic leukemia. M-CLL, mutated IGHV; U-CLL, unmutated IGHV; FCR: fludarabine + cyclophosphamide + rituximab; BR: bendamustine + rituximab; V: venetoclax; VR: venetoclax + rituximab; VO: venetoclax + obinutuzumab; I: ibrutinib; IO: ibrutinib + obinutuzumab; A: acalabrutinib; ClbO: chlorambucil + obinutuzumab; R: rituximab; D: duvelisib; AlloHCT: allogeneic hematopoietic cell transplantation; R-CHOP, rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone; IGHV, immunoglobulin heavy-chain variable region. *Acalabrutinib (A) is approved for both treatment- naïve and relapsed disease by the FDA but not the EMA. #Venetoclax (V) monotherapy is approved for patients with TP53 aberrations who are refractor y or intolerant to ibrutinib or patients without TP53 aberrations who are refractory or intolerant to both chemoimmunotherapy and ibrutinib. Venetoclax plus rituximab (VR) is approved for any patient with relapsed disease. †Duvelisib (D) monotherapy is approved for relapsed disease (minimum of two prior therapies) by the FDA but not the EMA. ‡AlloHCT is recommended for appropriate patients with high-risk disease, defined by TP53 aberrations and/or complex karyotype in whom ibrutinib and/or venetoclax has failed. Allogeneic HCT is also recommended for appropriate patients with transformed disease who have responded to salvage chemotherapy (e.g., R-CHOP).
Apart from ibrutinib, patients with M-CLL, devoid of TP53 aberrations and fit enough to tolerate FCR therapy, may still be good candidates for the latter, with the benefit being that this treatment can be completed in 6 months while ibrutinib must be taken indefinitely. This option would be particularly valuable for non-compliant patients or those in whom ibrutinib is contraindicated. If FCR is the treatment of choice, caution must be taken in patients with NOTCH1 mutations, in whom rituximab appears to have little added value.59 Other genomic subgroups, such as patients with BIRC3 mutations appear to derive little benefit from CIT,111,112 but these results should be further validated.
Unfit patients also have the alternative of venetoclax plus obinutuzumab (VO) as frontline therapy. This is based on a phase III trial that compared VO with ClbO in elderly/unfit patients.113 VO was superior in terms of response rate and progression-free survival, and had a comparable safety profile. In this trial VO was administered for a definite period of time (2 years), which is quite appealing for older/unfit patients. Moreover, many well established adverse prognostic markers, including U-CLL, ATM aberrations or NOTCH1/BIRC3 mutations, lost their negative effect in patients treated with VO. The only factor that remained predictive of a shorter progression-free survival in this cohort of patients was TP53 aberrations.112 Finally, the alternative BTK inhibitor acalabrutinib was recently approved by the FDA (not by the EMA yet) as frontline therapy in view of the results of a phase III trial comparing acalabrutinib versus ClbO.114
Relapsed/refractory disease
Treatment for relapsed/refractory disease must be decided depending on prior therapy and also the reason why the original treatment was no longer appropriate (e.g., refractoriness vs. intolerance). Ibrutinib is the current gold standard therapy for patients with relapsed/refractory disease, based on the results of several phase I-III trials, 115–119 but this is also changing for two main reasons: (i) an increasing proportion of patients currently receive ibrutinib as frontline therapy; and (ii) a few serious contenders have appeared in the last year.
Venetoclax is one of the best alternatives in this situation, including patients with high-risk genomic aberrations. The drug was already proven effective and safe in several phase I-II trials, in patients who had previously received either CIT or BTK/PI3K inhibitors.120–123 The formal confirmation of this promising activity came with a phase III trial in which venetoclax combined with rituximab was superior to bendamustine plus rituximab in terms of response rate, progression-free survival and overall survival, leading to its full approval for patients with relapsed/refractory CLL.124 Other possibilities are PI3K inhibitors and alternative BTK inhibitors. Idelalisib, in combination with rituximab, was the first PI3K inhibitor approved for the treatment of relapsed/refractory CLL based on the results of a phase III trial,125,126 and yet it is infrequently used because of its less favorable adverseevent profile. It may have a role in patients with complex karyotypes,127who have a higher risk of progression and/or transformation when treated with ibrutinib or venetoclax, 90,128 or in older patients who also tend not to tolerate ibrutinib well,129 but there are no randomized data to substantiate this potential superiority. Duvelisib was the second PI3K inhibitor approved by the FDA, also based on a phase III randomized trial.130 The efficacy and safety profile of the drug appear comparable with those of idelalisib, if not slightly advantageous. Regarding alternative BTK inhibitors, there are several products in development, but only acalabrutinib is approved by the FDA for the treatment of relapsed/refractory CLL. This is based on a phase III trial in which acalabrutinib was superior to either bendamustine plus rituximab or idelalisib plus rituximab.131 In this trial, prior ibrutinib therapy was not allowed, but a separate trial has shown that 85% of patients who were intolerant to ibrutinib were subsequently able to take acalabrutinib, with a 76% response rate.132
Despite all recent therapeutic advances, a proportion of patients will still fail to respond and should be considered for curative therapy. Currently, only allogeneic hematopoietic cell transplantation can be considered potentially curative, but it is also associated with considerable morbidity and mortality. Over the past decades, the number of patients referred for allogeneic hematopoietic cell transplantation has dropped significantly,133 but the procedure should be recommended to young/fit patients in whom BCR/BCL2 inhibitor treatment fails, particularly in those with TP53 aberrations, or in the case of Richter transformation.134,135 Moreover, although chimeric antigen receptor T cells could also be appropriate in this situation and the results are promising,136 none of the commercially available products is yet approved for this indication.
Disease transformation
Richter transformation remains an ominous event for patients with CLL, particularly when it is clonally related to the original CLL, because none of the recently approved novel agents is truly effective. Indeed, disease transformation is a relatively common cause of failure to benefit from these drugs.90,128,129 Histological confirmation is always recommended since it can guide prognosis (i.e., Hodgkin lymphoma and clonally unrelated tumors have more favorable prognosis). Patients with transformed disease should be offered conventional CIT (e.g., RCHOP: rituximab plus cyclophosphamide, doxorubicin, vincristine, prednisone) followed by allogeneic hematopoietic cell transplantation in the case of response. Autologous hematopoietic cell transplantation remains an option if allogeneic transplantation is considered inappropriate.134 Chimeric antigen receptor T cells may also be effective but, unfortunately, none of the approved products is current available for patients with Richter transformation.
Conclusions and perspectives
Recent molecular studies have provided many insights into the processes that govern the development and progression of CLL, including many novel mutated genes clustered in different functional pathways. The CLL epigenome is reprogrammed through the modulation of regulatory regions that appear de novo in the disease, whereas other regions maintain functions already present in different stages of B-cell differentiation. Analysis of the CLL microenvironment has provided clues to understand the survival of tumor cells and resistance to therapy. All this knowledge has offered new perspectives that are being exploited therapeutically with novel agents and strategies. However, these studies are also raising new questions. The relationship between the remarkable molecular heterogeneity of the disease and the clinical diversity is not well understood. The disease is always preceded by a premalignant state (MBL) which shares most molecular drivers with overt CLL. In many cases, these molecular drivers remain constant over time. However, clonal evolution is also possible and is usually associated with exponential tumor growth, progressive disease and, in some cases, disease transformation. Most studies have been performed in pretreated patients and it is not fully understood how the genome and epigenomic alterations and microenvironmental interactions influence the evolution of the disease. Translating new knowledge into clinical practice will require an effort to obtain an integrated view of all these factors in order to understand the disease better and design effective treatments and management strategies.
Acknowledgments
The authors would like to thank Silvia Beà, Jose Ignacio Martín-Subero, and Armando Lopez-Guillermo (Hospital Clinic of Barcelona and IDIBAPS) for their helpful comments on the manuscript, and Neus Giménez (IDIBAPS) for her assistance with the figure design. JD is supported by a grant from Generalitat de Catalunya (PERIS IPFE SLT006/17/301). FN is supported by a predoctoral fellowship from the Ministerio de Ciencia e Innovación (MCI) (BES- 2016-076372). DC is supported by MCI (RTI2018–094584- B-I00), Generalitat de Catalunya Suport Grups de Recerca (AGAUR, grant 2017-SGR-1009) and CIBERONC (CB16/12/00334). EC is supported by grants from “La Caixa” Foundation (CLLEvolution-LCF/PR/HR17/ 52150017), the Instituto de Salud Carlos III and the European Regional Development Fund (FEDER – “Una Manera de Hacer Europa”) (PMP15/00007), MCI (grants RTI2018-094274-B-I00 and SAF2016-81860-REDT), CIBERONC (CB16/12/00225) and AGAUR (2017-SGR-1142). EC is an Academia Researcher of the Institució Catalana de Recerca i Estudis Avançats (ICREA) of the Generalitat de Catalunya. This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (BCLLatlas - 810287). We apologize to authors whose work was not included due to space limitations.
References
- 1.Campo E, Ghia P, Montserrat E, Müller-Hermelink HK, Stein H, Swerdlow SH. Chronic lymphocytic leukaemia/small lymphocytic lymphoma. WHO Classification of Tumours of Haemato - poietic and Lymphoid Tissues. Rev 4th Ed. Lyon: WHO Press; 2017:216-221. [Google Scholar]
- 2.Howlader N, Noone A, Krapcho M, et al. SEER Cancer Statistics Review, 1975-2016, National Cancer Institute. Bethesda, MD, USA. [Google Scholar]
- 3.Puente XS, Beà S, Valdés-Mas R, et al. Noncoding recurrent mutations in chronic lymphocytic leukaemia. Nature. 2015;526 (7574):519-524. [DOI] [PubMed] [Google Scholar]
- 4.Landau DA, Tausch E, Taylor-Weiner AN, et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526(7574):525-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kulis M, Heath S, Bibikova M, et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat Genet. 2012;44(11):1236-1242. [DOI] [PubMed] [Google Scholar]
- 6.Beekman R, Chapaprieta V, Russiñol N, et al. The reference epigenome and regulatory chromatin landscape of chronic lymphocytic leukemia. Nat Med. 2018;24(6):868-880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oakes CC, Claus R, Gu L, et al. Evolution of DNA methylation is linked to genetic aberrations in chronic lymphocytic leukemia. Cancer Discov. 2014;4(3):348-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berndt SI, Camp NJ, Skibola CF, et al. Metaanalysis of genome-wide association studies discovers multiple loci for chronic lymphocytic leukemia. Nat Commun. 2016;7(1): 10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Speedy HE, Beekman R, Chapaprieta V, et al. Insight into genetic predisposition to chronic lymphocytic leukemia from integrative epigenomics. Nat Commun. 2019;10(1): 3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kikushige Y, Ishikawa F, Miyamoto T, et al. Self-renewing hematopoietic stem cell is the primary target in pathogenesis of human chronic lymphocytic leukemia. Cancer Cell. 2011;20(2):246-259. [DOI] [PubMed] [Google Scholar]
- 11.Landau DA, Carter SL, Stojanov P, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152(4):714-726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nadeu F, Delgado J, Royo C, et al. Clinical impact of clonal and subclonal TP53, SF3B1, BIRC3, NOTCH1, and ATM mutations in chronic lymphocytic leukemia. Blood. 2016;127(17):2122-2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nadeu F, Clot G, Delgado J, et al. Clinical impact of the subclonal architecture and mutational complexity in chronic lymphocytic leukemia. Leukemia. 2018;32(3):645-653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agathangelidis A, Ljungström V, Scarfò L, et al. Highly similar genomic landscapes in monoclonal b-cell lymphocytosis and ultrastable chronic lymphocytic leukemia with low frequency of driver mutations. Haematologica. 2018;103(5):865-873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brazdilova K, Plevova K, Skuhrova Francova H, et al. Multiple productive IGH rearrangements denote oligoclonality even in immunophenotypically monoclonal CLL. Leukemia. 2018;32(1):234-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Plevova K, Francova HS, Burckova K, et al. Multiple productive immunoglobulin heavy chain gene rearrangements in chronic lymphocytic leukemia are mostly derived from independent clones. Haematologica. 2014;99(2):329-338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. 1999;94(6):1848-1854. [PubMed] [Google Scholar]
- 18.Damle RN, Wasil T, Fais F, et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood. 1999;94(6):1840-1847. [PubMed] [Google Scholar]
- 19.Seifert M, Sellmann L, Bloehdorn J, et al. Cellular origin and pathophysiology of chronic lymphocytic leukemia. J Exp Med. 2012;209(12):2183-2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stamatopoulos K, Agathangelidis A, Rosenquist R, Ghia P. Antigen receptor stereotypy in chronic lymphocytic leukemia. Leukemia. 2017;31(2):282-291. [DOI] [PubMed] [Google Scholar]
- 21.Jaramillo S, Agathangelidis A, Schneider C, et al. Prognostic impact of prevalent chronic lymphocytic leukemia stereotyped subsets: analysis within prospective clinical trials of the German CLL Study Group (GCLLSG). Haematologica. 2019. Dec 26. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dühren-von Minden M, Übelhart R, Schneider D, et al. Chronic lymphocytic leukaemia is driven by antigen-independent cell-autonomous signalling. Nature. 2012;489(7415):309-312. [DOI] [PubMed] [Google Scholar]
- 23.Minici C, Gounari M, Übelhart R, et al. Distinct homotypic B-cell receptor interactions shape the outcome of chronic lymphocytic leukaemia. Nat Commun. 2017;8(1): 15746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maity PC, Bilal M, Koning MT, et al. IGLV3-21∗01 is an inherited risk factor for CLL through the acquisition of a single-point mutation enabling autonomous BCR signaling. Proc Natl Acad Sci U S A. 2020;117(8):4320-4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stamatopoulos B, Smith T, Crompot E, et al. The light chain IgLV3-21 defines a new poor prognostic subgroup in chronic lymphocytic leukemia: results of a multicenter study. Clin Cancer Res. 2018;24(20):5048-5057. [DOI] [PubMed] [Google Scholar]
- 26.Oakes CC, Seifert M, Assenov Y, et al. DNA methylation dynamics during B cell maturation underlie a continuum of disease phenotypes in chronic lymphocytic leukemia. Nat Genet. 2016;48(3):253-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Queirós AC, Villamor N, Clot G, et al. A Bcell epigenetic signature defines three biologic subgroups of chronic lymphocytic leukemia with clinical impact. Leukemia. 2015;29(3):598-605. [DOI] [PubMed] [Google Scholar]
- 28.Wojdacz TK, Amarasinghe HE, Kadalayil L, et al. Clinical significance of DNA methylation in chronic lymphocytic leukemia patients: results from 3 UK clinical trials. Blood Adv. 2019;3(16):2474-2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herndon TM, Chen SS, Saba NS, et al. Direct in vivo evidence for increased proliferation of CLL cells in lymph nodes compared to bone marrow and peripheral blood. Leukemia. 2017;31(6):1340-1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.ten Hacken E, Burger JA. Microenvironment interactions and B-cell receptor signaling in chronic lymphocytic leukemia: Implications for disease pathogenesis and treatment. Biochim Biophys Acta. 2016;1863(3):401-413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herreros B, Rodríguez-Pinilla SM, Pajares R, et al. Proliferation centers in chronic lymphocytic leukemia: the niche where NF-κB activation takes place. Leukemia. 2010;24(4): 872-876. [DOI] [PubMed] [Google Scholar]
- 32.Herishanu Y, Pérez-Galán P, Liu D, et al. The lymph node microenvironment promotes Bcell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood. 2011;117(2):563-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.López-Guerra M, Xargay-Torrent S, Rosich L, et al. The γ-secretase inhibitor PF- 03084014 combined with fludarabine antagonizes migration, invasion and angiogenesis in NOTCH1-mutated CLL cells. Leukemia. 2015;29(1):96-106. [DOI] [PubMed] [Google Scholar]
- 34.López-Guerra M, Xargay-Torrent S, Fuentes P, et al. Specific NOTCH1 antibody targets DLL4-induced proliferation, migration, and angiogenesis in NOTCH1-mutated CLL cells. Oncogene. 2020;39(6):1185-1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arruga F, Bracciamà V, Vitale N, et al. Bidirectional linkage between the B-cell receptor and NOTCH1 in chronic lymphocytic leukemia and in Richter’s syndrome: therapeutic implications. Leukemia. 2020;34(2):462-477. [DOI] [PubMed] [Google Scholar]
- 36.Puente XS, Pinyol M, Quesada V, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475(7354):101-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hanna BS, Öztürk S, Seiffert M. Beyond bystanders: myeloid cells in chronic lymphocytic leukemia. Mol Immunol. 2019;110(1):77-87. [DOI] [PubMed] [Google Scholar]
- 38.Riches JC, Davies JK, McClanahan F, et al. T cells from CLL patients exhibit features of Tcell exhaustion but retain capacity for cytokine production. Blood. 2013;121(9): 1612-1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramsay AG, Evans R, Kiaii S, Svensson L, Hogg N, Gribben JG. Chronic lymphocytic leukemia cells induce defective LFA-1-directed T-cell motility by altering Rho GTPase signaling that is reversible with lenalidomide. Blood. 2013;121(14):2704-2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hanna BS, Roessner PM, Yazdanparast H, et al. Control of chronic lymphocytic leukemia development by clonally-expanded CD8+ T-cells that undergo functional exhaustion in secondary lymphoid tissues. Leukemia. 2019;33(3):625-637. [DOI] [PubMed] [Google Scholar]
- 41.Llaó Cid L, Hanna BS, Iskar M, et al. CD8+ T-cells of CLL-bearing mice acquire a transcriptional program of T-cell activation and exhaustion. Leuk Lymphoma. 2020;61(2): 351-356. [DOI] [PubMed] [Google Scholar]
- 42.Kipps TJ, Stevenson FK, Wu CJ, et al. Chronic lymphocytic leukaemia. Nat Rev Dis Prim. 2017;316096. [Google Scholar]
- 43.Wiestner A. The role of B-cell receptor inhibitors in the treatment of patients with chronic lymphocytic leukemia. Haematologica. 2015;100(12):1495-1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mockridge CI, Potter KN, Wheatley I, Neville LA, Packham G, Stevenson FK. Reversible anergy of sIgM-mediated signaling in the two subsets of CLL defined by VH-gene mutational status. Blood. 2007;109(10):4424-4431. [DOI] [PubMed] [Google Scholar]
- 45.Moore VDG, Brown JR, Certo M, Love TM, Novina CD, Letai A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest. 2007;117(1):112-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Juliusson G, Oscier DG, Fitchett M, et al. Prognostic subgroups in B-cell chronic lymphocytic leukemia defined by specific chromosomal abnormalities. N Engl J Med. 1990;323(11):720-724. [DOI] [PubMed] [Google Scholar]
- 47.Döhner H, Stilgenbauer S, Benner A, et al. Genomic aberrations and survival in chronic lymphocytic leukemia. N Engl J Med. 2000;343(26):1910-1916. [DOI] [PubMed] [Google Scholar]
- 48.Haferlach C, Dicker F, Schnittger S, Kern W, Haferlach T. Comprehensive genetic characterization of CLL: a study on 506 cases analysed with chromosome banding analysis, interphase FISH, IgV(H) status and immunophenotyping. Leukemia. 2007;21(12):2442-2451. [DOI] [PubMed] [Google Scholar]
- 49.Baliakas P, Iskas M, Gardiner A, et al. Chromosomal translocations and karyotype complexity in chronic lymphocytic leukemia: a systematic reappraisal of classic cytogenetic data. Am J Hematol. 2014;89(3):249-255. [DOI] [PubMed] [Google Scholar]
- 50.Huh YO, Abruzzo L V., Rassidakis GZ, et al. The t(14;19)(q32;q13)-positive small B-cell leukaemia: a clinicopathologic and cytogenetic study of seven cases. Br J Haematol. 2007;136(2):220-228. [DOI] [PubMed] [Google Scholar]
- 51.Nadeu F, Diaz-Navarro A, Delgado J, Puente XS, Campo E. Genomic and epigenomic alterations in chronic lymphocytic leukemia. Annu Rev Pathol Mech Dis. 2020;15(1):149-177. [DOI] [PubMed] [Google Scholar]
- 52.Edelmann J, Holzmann K, Miller F, et al. High-resolution genomic profiling of chronic lymphocytic leukemia reveals new recurrent genomic alterations. Blood. 2012;120(24):4783-4794. [DOI] [PubMed] [Google Scholar]
- 53.Delgado J, Salaverria I, Baumann T, et al. Genomic complexity and IGHV mutational status are key predictors of outcome of chronic lymphocytic leukemia patients with TP53 disruption. Haematologica. 2014;99(11):e231-e234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thompson PA, Stingo F, Keating MJ, et al. Outcomes of patients with chronic lymphocytic leukemia treated with first-line idelalisib plus rituximab after cessation of treatment for toxicity. Cancer. 2016;122(16): 2505-2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baliakas P, Jeromin S, Iskas M, et al. Cytogenetic complexity in chronic lymphocytic leukemia: definitions, associations, and clinical impact. Blood. 2019;133(11): 1205-1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Baliakas P, Puiggros A, Xochelli A, et al. Additional trisomies amongst patients with chronic lymphocytic leukemia carrying trisomy 12: the accompanying chromosome makes a difference. Haematologica. 2016;101(7):299-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Martínez-Trillos A, Pinyol M, Navarro A, et al. Mutations in TLR/MYD88 pathway identify a subset of young chronic lymphocytic leukemia patients with favorable outcome. Blood. 2014;123(24):3790-3796. [DOI] [PubMed] [Google Scholar]
- 58.Giménez N, Martínez-Trillos A, Montraveta A, et al. Mutations in the RAS-BRAF-MAPKERK pathway define a specific subgroup of patients with adverse clinical features and provide new therapeutic options in chronic lymphocytic leukemia. Haematologica. 2019;104(3):576-586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stilgenbauer S, Schnaiter A, Paschka P, et al. Gene mutations and treatment outcome in chronic lymphocytic leukemia: results from the CLL8 trial. Blood. 2014;123(21):3247-3254. [DOI] [PubMed] [Google Scholar]
- 60.Malcikova J, Tausch E, Rossi D, et al. ERIC recommendations for TP53 mutation analysis in chronic lymphocytic leukemia - update on methodological approaches and results interpretation. Leukemia. 2018;32(5): 1070-1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sutton L-A, Ljungström V, Enjuanes A, et al. Comparative analysis of targeted next-generation sequencing panels for the detection of gene mutations in chronic lymphocytic leukemia: an ERIC multi-center study. Haematologica. 2020. Apr 9. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rossi D, Khiabanian H, Spina V, et al. Clinical impact of small TP53 mutated subclones in chronic lymphocytic leukemia. Blood. 2014;123(14):2139-2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ojha J, Ayres J, Secreto C, et al. Deep sequencing identifies genetic heterogeneity and recurrent convergent evolution in chronic lymphocytic leukemia. Blood. 2015;125(3):492-498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Amin NA, Seymour E, Saiya-Cork K, Parkin B, Shedden K, Malek SN. A quantitative analysis of subclonal and clonal gene mutations before and after therapy in chronic lymphocytic leukemia. Clin Cancer Res. 2016;22(17):4525-4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Landau DA, Sun C, Rosebrock D, et al. The evolutionary landscape of chronic lymphocytic leukemia treated with ibrutinib targeted therapy. Nat Commun. 2017;8(1):2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Blombery P, Anderson MA, Gong JN, et al. Acquisition of the recurrent Gly101Val mutation in BCL2 confers resistance to venetoclax in patients with progressive chronic lymphocytic leukemia. Cancer Discov. 2019;9(3):342-353. [DOI] [PubMed] [Google Scholar]
- 67.Lampson BL, Brown JR. Are BTK and PLCG2 mutations necessary and sufficient for ibrutinib resistance in chronic lymphocytic leukemia? Expert Rev Hematol. 2018;11(3):185-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhao X, Lwin T, Silva A, et al. Unification of de novo and acquired ibrutinib resistance in mantle cell lymphoma. Nat Commun. 2017;8(1):14920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Agarwal R, Chan YC, Tam CS, et al. Dynamic molecular monitoring reveals that SWI–SNF mutations mediate resistance to ibrutinib plus venetoclax in mantle cell lymphoma. Nat Med. 2019;25(1):119-129. [DOI] [PubMed] [Google Scholar]
- 70.Pan R, Ruvolo V, Mu H, et al. Synthetic lethality of combined Bcl-2 inhibition and p53 activation in AML: mechanisms and superior antileukemic efficacy. Cancer Cell. 2017;32(6):748-760.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Landau DA, Clement K, Ziller MJ, et al. Locally disordered methylation forms the basis of intratumor methylome variation in chronic lymphocytic leukemia. Cancer Cell. 2014;26(6):813-825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ott CJ, Federation AJ, Schwartz LS, et al. Enhancer architecture and essential core regulatory circuitry of chronic lymphocytic leukemia. Cancer Cell. 2018;34(6):982-995.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mallm J, Iskar M, Ishaque N, et al. Linking aberrant chromatin features in chronic lymphocytic leukemia to transcription factor networks. Mol Syst Biol. 2019;15(5):e8339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pastore A, Gaiti F, Lu SX, et al. Corrupted coordination of epigenetic modifications leads to diverging chromatin states and transcriptional heterogeneity in CLL. Nat Commun. 2019;10(1):1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rodríguez D, Bretones G, Quesada V, et al. Mutations in CHD2 cause defective association with active chromatin in chronic lymphocytic leukemia. Blood. 2015;126(2):195-202. [DOI] [PubMed] [Google Scholar]
- 76.Tsagiopoulou M, Chapaprieta V, Duran-Ferrer M, et al. Chronic lymphocytic leukemias with trisomy 12 show a distinct DNA methylation profile linked to altered chromatin activation. Haematologica. 2020. Feb 27. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Landgren O, Albitar M, Ma W, et al. B-cell clones as early markers for chronic lymphocytic leukemia. N Engl J Med. 2009;360(7):659-667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Criado I, Rodríguez-Caballero A, Gutiérrez ML, et al. Low-count monoclonal B-cell lymphocytosis persists after seven years of follow up and is associated with a poorer outcome. Haematologica. 2018;103(7):1198-1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rawstron AC, Bennett FL, O’Connor SJM, et al. Monoclonal B-cell lymphocytosis and chronic lymphocytic leukemia. N Engl J Med. 2008;359(6):575-583. [DOI] [PubMed] [Google Scholar]
- 80.Rossi D, Spina V, Gaidano G. Biology and treatment of Richter syndrome. Blood. 2018;131(25):2761-2772. [DOI] [PubMed] [Google Scholar]
- 81.Del Giudice I, Chiaretti S, Tavolaro S, et al. Spontaneous regression of chronic lymphocytic leukemia: clinical and biologic features of 9 cases. Blood. 2009;114(3):638-646. [DOI] [PubMed] [Google Scholar]
- 82.Barrio S, Shanafelt TD, Ojha J, et al. Genomic characterization of high-count MBL cases indicates that early detection of driver mutations and subclonal expansion are predictors of adverse clinical outcome. Leukemia. 2017;31(1):170-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Winkelmann N, Rose-Zerilli M, Forster J, et al. Low frequency mutations independently predict poor treatment-free survival in early stage chronic lymphocytic leukemia and monoclonal B-cell lymphocytosis. Haematologica. 2015;100(6):e237-e239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gruber M, Bozic I, Leshchiner I, et al. Growth dynamics in naturally progressing chronic lymphocytic leukaemia. Nature. 2019;570(7762):474-479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Montserrat E, Sanchez‐Bisono J, Viñolas N, Rozman C. Lymphocyte doubling time in chronic lymphocytic leukaemia: analysis of its prognostic significance. Br J Haematol. 1986;62(3):567-575. [DOI] [PubMed] [Google Scholar]
- 86.Hallek M, Cheson BD, Catovsky D, et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood. 2018;131(25):2745-2760. [DOI] [PubMed] [Google Scholar]
- 87.Al-Sawaf O, Robrecht S, Bahlo J, et al. Richter transformation in chronic lymphocytic leukemia (CLL)—a pooled analysis of German CLL Study Group (GCLLSG) front line treatment trials. Leukemia. 2020. Mar 17. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 88.Fabbri G, Khiabanian H, Holmes AB, et al. Genetic lesions associated with chronic lymphocytic leukemia transformation to Richter syndrome. J Exp Med. 2013;210(11):2273-2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kadri S, Lee J, Fitzpatrick C, et al. Clonal evolution underlying leukemia progression and Richter transformation in patients with ibrutinib-relapsed CLL. Blood Adv. 2017;1(12):715-727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Miller CR, Ruppert AS, Heerema NA, et al. Near-tetraploidy is associated with Richter transformation in chronic lymphocytic leukemia patients receiving ibrutinib. Blood Adv. 2017;1(19):1584-1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kwok M, Oldreive C, Rawstron AC, et al. Integrative analysis of spontaneous CLL regression highlights genetic and microenvironmental interdependency in CLL. Blood. 2020;135(6):411-428. [DOI] [PubMed] [Google Scholar]
- 92.Rai KR, Sawitsky A, Cronkite EP, Chanana AD, Levy RN, Pasternack BS. Clinical staging of chronic lymphocytic leukemia. Blood. 1975;46(2):219-234. [DOI] [PubMed] [Google Scholar]
- 93.Binet JL, Auquier A, Dighiero G, et al. A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer. 1981;48(1):198-206. [DOI] [PubMed] [Google Scholar]
- 94.Baliakas P, Hadzidimitriou A, Sutton L-A, et al. Recurrent mutations refine prognosis in chronic lymphocytic leukemia. Leukemia. 2015;29(2):329-336. [DOI] [PubMed] [Google Scholar]
- 95.Rossi D, Rasi S, Spina V, et al. Integrated mutational and cytogenetic analysis identifies new prognostic subgroups in chronic lymphocytic leukemia. Blood. 2013;121(8): 1403-1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Baliakas P, Moysiadis T, Hadzidimitriou A, et al. Tailored approaches grounded on immunogenetic features for refined prognostication in chronic lymphocytic leukemia. Haematologica. 2019;104(2):360-369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.International CLL-IPI working groups. An international prognostic index for patients with chronic lymphocytic leukaemia (CLLIPI): a meta-analysis of individual patient data. Lancet Oncol. 2016;17(6):779-790. [DOI] [PubMed] [Google Scholar]
- 98.da Cunha-Bang C, Christiansen I, Niemann CU. The CLL-IPI applied in a populationbased cohort. Blood. 2016;128(17):2181-2183. [DOI] [PubMed] [Google Scholar]
- 99.Molica S, Shanafelt TD, Giannarelli D, et al. The chronic lymphocytic leukemia international prognostic index predicts time to first treatment in early CLL: Independent validation in a prospective cohort of early stage patients. Am J Hematol. 2016;91(11):1090-1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gentile M, Shanafelt TD, Rossi D, et al. Validation of the CLL-IPI and comparison with the MDACC prognostic index in newly diagnosed patients. Blood. 2016;128(16):2093-2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Delgado J, Doubek M, Baumann T, et al. Chronic lymphocytic leukemia: a prognostic model comprising only two biomarkers (IGHV mutational status and FISH cytogenetics) separates patients with different outcome and simplifies the CLL-IPI. Am J Hematol. 2017;92(4):375-380. [DOI] [PubMed] [Google Scholar]
- 102.Condoluci A, Terzi di Bergamo L, Langerbeins P, et al. International prognostic score for asymptomatic early-stage chronic lymphocytic leukemia. Blood. 2020;135(21): 1859-1869. [DOI] [PubMed] [Google Scholar]
- 103.Dighiero G, Maloum K, Desablens B, et al. Chlorambucil in indolent chronic lymphocytic leukemia. French Cooperative Group on Chronic Lymphocytic Leukemia. N Engl J Med. 1998;338(21):1506-1514. [DOI] [PubMed] [Google Scholar]
- 104.Herling CD, Cymbalista F, Groß-Ophoff-Müller C, et al. Early treatment with FCR versus watch and wait in patients with stage Binet A high-risk chronic lymphocytic leukemia (CLL): a randomized phase 3 trial. Leukemia. 2020. Feb 18. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Langerbeins P, Bahlo J, Rhein C, et al. Ibrutinib versus placebo in patients with asymptomatic treatment-naïve early stage CLL: primary endpoint results of the phase 3 double-blind randomized CLL12 trial. Hematol Oncol. 2019;37(Suppl 2):38-40.31187520 [Google Scholar]
- 106.Burger JA, Tedeschi A, Barr PM, et al. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med. 2015;373(25):2425-2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Shanafelt TD, Wang X V., Kay NE, et al. Ibrutinib–rituximab or chemoimmunotherapy for chronic lymphocytic leukemia. N Engl J Med. 2019;381(5):432-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Woyach JA, Ruppert AS, Heerema NA, et al. Ibrutinib regimens versus chemoimmunotherapy in older patients with untreated CLL. N Engl J Med. 2018;379(26):2517-2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Moreno C, Greil R, Demirkan F, et al. Ibrutinib plus obinutuzumab versus chlorambucil plus obinutuzumab in first-line treatment of chronic lymphocytic leukaemia (iLLUMINATE): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019;20(1):43-56. [DOI] [PubMed] [Google Scholar]
- 110.Burger JA, Sivina M, Jain N, et al. Randomized trial of ibrutinib vs ibrutinib plus rituximab in patients with chronic lymphocytic leukemia. Blood. 2019;133(10): 1011-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Diop F, Moia R, Favini C, et al. Biological and clinical implications of BIRC3 mutations in chronic lymphocytic leukemia. Haematologica. 2020;105(2):448-456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tausch E, Schneider C, Robrecht S, et al. Prognostic and predictive impact of genetic markers in patients with CLL treated with obinutuzumab and venetoclax. Blood. 2020. Mar 23. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 113.Fischer K, Al-Sawaf O, Bahlo J, et al. Venetoclax and obinutuzumab in patients with CLL and coexisting conditions. N Engl J Med. 2019;380(23):2225-2236. [DOI] [PubMed] [Google Scholar]
- 114.Sharman JP, Egyed M, Jurczak W, et al. Acalabrutinib with or without obinutuzumab versus chlorambucil and obinutuzmab for treatment-naive chronic lymphocytic leukaemia (ELEVATE TN): a randomised, controlled, phase 3 trial. Lancet. 2020;395(10232):1278-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(1):32-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Farooqui MZH, Valdez J, Martyr S, et al. Ibrutinib for previously untreated and relapsed or refractory chronic lymphocytic leukaemia with TP53 aberrations: a phase 2, single-arm trial. Lancet Oncol. 2015;16(2): 169-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.O’Brien S, Jones JA, Coutre SE, et al. Ibrutinib for patients with relapsed or refractory chronic lymphocytic leukaemia with 17p deletion (RESONATE-17): a phase 2, open-label, multicentre study. Lancet Oncol. 2016;17(10):1409-1418. [DOI] [PubMed] [Google Scholar]
- 118.Byrd JC, Brown JR, O’Brien S, et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014;371(3):213-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chanan-Khan A, Cramer P, Demirkan F, et al. Ibrutinib combined with bendamustine and rituximab compared with placebo, bendamustine, and rituximab for previously treated chronic lymphocytic leukaemia or small lymphocytic lymphoma (HELIOS): a randomised, double-blind, phase 3 study. Lancet Oncol. 2016;17(2):200-211. [DOI] [PubMed] [Google Scholar]
- 120.Roberts AW, Davids MS, Pagel JM, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374(4):311-322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Stilgenbauer S, Eichhorst B, Schetelig J, et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: a multicentre, open-label, phase 2 study. Lancet Oncol. 2016;17(6):768-778. [DOI] [PubMed] [Google Scholar]
- 122.Jones JA, Mato AR, Wierda WG, et al. Venetoclax for chronic lymphocytic leukaemia progressing after ibrutinib: an interim analysis of a multicentre, open-label, phase 2 trial. Lancet Oncol. 2018;19(1):65-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Coutre S, Choi M, Furman RR, et al. Venetoclax for patients with chronic lymphocytic leukemia who progressed during or after idelalisib therapy. Blood. 2018;131(15):1704-1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Seymour JF, Kipps TJ, Eichhorst B, et al. Venetoclax-rituximab in relapsed or refractory chronic lymphocytic leukemia. N Engl J Med. 2018;378(12):1107-1120. [DOI] [PubMed] [Google Scholar]
- 125.Furman RR, Sharman JP, Coutre SE, et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014;370(11):997-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sharman JP, Coutre SE, Furman RR, et al. Final results of a randomized, phase III study of rituximab with or without idelalisib followed by open-label idelalisib in patients with relapsed chronic lymphocytic leukemia. J Clin Oncol. 2019;37(16):1391-1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kreuzer KA, Furman RR, Stilgenbauer S, et al. The impact of complex karyotype on the overall survival of patients with relapsed chronic lymphocytic leukemia treated with idelalisib plus rituximab. Leukemia. 2020;34(1):296-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Anderson MA, Tam C, Lew TE, et al. Clinicopathological features and outcomes of progression of CLL on the BCL2 inhibitor venetoclax. Blood. 2017;129(25):3362-3370. [DOI] [PubMed] [Google Scholar]
- 129.Maddocks KJ, Ruppert AS, Lozanski G, et al. Etiology of ibrutinib therapy discontinuation and outcomes in patients with chronic lymphocytic leukemia. JAMA. Oncol 2015;1(1):80-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Flinn IW, Hillmen P, Montillo M, et al. The phase 3 DUO trial: duvelisib vs ofatumumab in relapsed and refractory CLL/SLL. Blood. 2018;132(23):2446-2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ghia P, Pluta A, Wach M, et al. ASCEND: phase III, randomized trial of acalabrutinib versus idelalisib plus rituximab or bendamustine plus rituximab in relapsed or refractory chronic lymphocytic leukemia. J Clin Oncol. 2020. May 27. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 132.Awan FT, Schuh A, Brown JR, et al. Acalabrutinib monotherapy in patients with chronic lymphocytic leukemia who are intolerant to ibrutinib. Blood Adv 2019; 3(9):1553-1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gribben JG. How and when I do allogeneic transplant in CLL. Blood. 2018;132(1):31-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kharfan-Dabaja MA, Kumar A, Hamadani M, et al. Clinical practice recommendations for use of allogeneic hematopoietic cell transplantation in chronic lymphocytic leukemia on behalf of the Guidelines Committee of the American Society for Blood and Marrow Transplantation. Biol Blood Marrow Transplant. 2016;22(12): 2117-2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Dreger P, Ghia P, Schetelig J, et al. High-risk chronic lymphocytic leukemia in the era of pathway inhibitors: integrating molecular and cellular therapies. Blood. 2018;132(9): 892-902. [DOI] [PubMed] [Google Scholar]
- 136.Bair SM, Porter DL. Accelerating chimeric antigen receptor therapy in chronic lymphocytic leukemia: the development and challenges of chimeric antigen receptor T-cell therapy for chronic lymphocytic leukemia. Am J Hematol. 2019;94(S1):S10-S17. [DOI] [PubMed] [Google Scholar]
- 137.Sutton LA, Young E, Baliakas P, et al. Different spectra of recurrent gene mutations in subsets of chronic lymphocytic leukemia harboring stereotyped B-cell receptors. Haematologica. 2016;101(8):959-967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Burger JA, Chiorazzi N. B cell receptor signaling in chronic lymphocytic leukemia. Trends Immunol. 2013;34(12):592-601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Burger JA, Quiroga MP, Hartmann E, et al. High-level expression of the T-cell chemokines CCL3 and CCL4 by chronic lymphocytic leukemia B cells in nurselike cell cocultures and after BCR stimulation. Blood. 2009;113(13):3050-3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Dilillo DJ, Weinberg JB, Yoshizaki A, et al. Chronic lymphocytic leukemia and regulatory B cells share IL-10 competence and immunosuppressive function. Leukemia. 2013;27(1):170-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lewinsky H, Barak AF, Huber V, et al. CD84 regulates PD-1/PD-L1 expression and function in chronic lymphocytic leukemia. J Clin Invest. 2018;128(12):5479-5488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.De Matteis S, Molinari C, Abbati G, et al. Immunosuppressive Treg cells acquire the phenotype of effector-T cells in chronic lymphocytic leukemia patients. J Transl Med. 2018;16(1):172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Paggetti J, Haderk F, Seiffert M, et al. Exosomes released by chronic lymphocytic leukemia cells induce the transition of stromal cells into cancer-associated fibroblasts. Blood. 2015;126(9):1106-1117. [DOI] [PMC free article] [PubMed] [Google Scholar]