

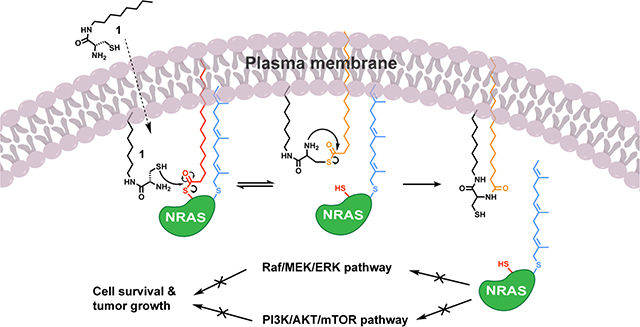
Abstract
Activating mutations in the small GTPase NRAS are responsible for driving tumor growth in several cancers. Unfortunately, the development of NRAS inhibitors has proven difficult due to the lack of hydrophobic binding pockets on the protein’s surface. To overcome this limitation, we chose to target the posttranslational S-palmitoyl modification of NRAS, which is required for its signaling activity. Utilizing an amphiphile-mediated depalmitoylation (AMD) strategy, we demonstrate the ability to directly cleave S-palmitoyl groups from NRAS and inhibit its function. C8 alkyl cysteine causes a dose-dependent decrease in NRAS palmitoylation and inhibits downstream signaling in melanoma cells with an activating mutation in NRAS. This compound reduces cell growth in NRAS-driven versus non-NRAS-driven melanoma lines and inhibits tumor progression in an NRAS-mutated melanoma xenograft mouse model. Our work demonstrates that AMD can effectively suppress NRAS activity and could represent a promising new avenue for discovering lead compounds for treatment of NRAS-driven cancers.
Graphical Abstract
INTRODUCTION
Ras proteins are small GTPases that regulate many cellular events including proliferation1, differentiation2, adhesion3, apoptosis4, and migration5. There are four known isoforms of the Ras family of proteins – NRAS, HRAS, KRAS4a, and KRAS4b – that are mutated in numerous diseases ranging from neurodevelopmental disorders to various cancers6. Activating mutations in the RAS oncogenes are found in approximately one third of all cancers6, making it the most frequently mutated gene family in human cancer.
Ras proteins are very difficult to target and have even been deemed “undruggable”7. Small-molecule inhibitors that directly bind to Ras proteins with high affinity are extremely challenging to develop due to the lack of deep binding pockets on their surfaces6, though recent breakthroughs have been made for the G12C mutant through use of covalent inhibitors8,9. Because of this complexity, current treatments and therapeutic approaches for targeting Ras-related diseases rely on targeting upstream proteins and downstream effectors. However, these approaches often come with significant challenges such as feedback loop activation or acquired resistance10.
Membrane anchoring post-translational modifications (PTMs) on Ras occur at the C-terminal hypervariable region (HVR)11. These PTMs are involved in regulation of cell signaling pathways as well as membrane localization, cellular trafficking, and activation of Ras proteins6,12,13. Membrane localization of Ras proteins is necessary for their downstream signaling events to occur6. The membrane localization of NRAS, HRAS, and KRAS4a is regulated by prenylation and palmitoylation at the CAAX terminus, which creates a hydrophobic lipid domain on the C-terminal cysteine6. On the other hand, KRAS4b is not palmitoylated but rather contains a polybasic region in its HVR allowing for its passive diffusion to the plasma membrane6,12,13. NRAS and HRAS are palmitoylated by the palmitoyl acyltransferase (PAT) enzyme DHHC9-GPC16 complex and depalmitoylated by acyl-protein thioesterases, which allow for their release from the plasma membrane and diffusion back to the Golgi14,15.
A potential therapeutic strategy to target oncogenic Ras isoforms in cancers would be to prevent membrane association by direct targeting of lipid PTMs, such as prenylation or palmitoylation. The first small-molecule inhibitors developed to target PTMs of Ras proteins were farnesyltransferase inhibitors (FTIs), which directly blocked the farnesyl transferase enzyme by competitively binding to the CAAX binding site16. These FTIs inhibited HRAS farnesylation and membrane localization in cells, which decreased cell growth in various human cancer cell lines6. However, although FTIs seem to have promising effects on certain HRAS mutant cancers in recent clinical trials, they failed to work for all of the Ras isoforms17,18. This result was explained by the fact that NRAS, KRAS4a, and KRAS4b were found to undergo geranylgeranylation as an alternative prenylation modification when farnesylation was blocked by FTIs6,12.
Due to the ineffectiveness of FTIs for NRAS, a good alternative approach would be to target palmitoylation, since there are no known compensatory mechanisms for lipidating S-palmitoylation sites if PATs are blocked. Currently, there are no selective inhibitors for palmitoylation because of the difficulty to target only one of the 23 PAT enzymes and their overlapping substrate specificity15,16,19. However, the irreversible inhibitor 2-bromopalmitate (2-BP) has been used as a pharmacological tool to study protein palmitoylation for many years11,19. Because 2-BP is a promiscuous electrophile, it targets multiple enzymes involved in lipid metabolism as well as many other proteins and non-CoA-dependent enzymes, which limits its potential as a therapeutic candidate for depalmitoylation due to off-target side effects and associated toxicity issues20. Thus, it would be beneficial to take an alternative approach to traditional enzymatic inhibitors and use a chemoselective chemical agent to directly target palmitoylation of NRAS as a means to suppress its signaling capabilities that are responsible for tumor progression. Recently, our group has developed an amphiphile-mediated depalmitoylation (AMD) strategy using a compound, termed C8 alkyl cysteine (1), to cleave S-palmitoyl groups from endogenous membrane-associated proteins21. We sought to apply the AMD approach to depalmitoylate NRAS and inhibit its function in the context of an NRAS-driven cancer.
Here we demonstrate that C8 alkyl cysteine (1) is able to depalmitoylate NRAS in NRAS-mutated melanoma, leading to significant phenotypic effects in vitro and in vivo. In turn, depalmitoylation of NRAS decreased phosphorylation of downstream proteins such as AKT and ERK in NRAS-mutated melanoma cells. Fluorescence microscopy studies showed that treatment with the depalmitoylating agent caused delocalization of NRAS from the plasma membrane. We also determined that as concentrations of 1 increased, the cell viability of NRAS-mutated melanoma cell lines decreased, while the cell viability of a BRAF-mutated melanoma cell line was unaffected until much higher concentrations. Cell death was partially rescued by addition of an NRAS mutant bearing a non-palmitoylated membrane binding tail derived from KRAS4b, which is not dependent on palmitoylation for plasma membrane localization. In an NRAS-mutated melanoma xenograft mouse model, treatment with 1 caused a decrease in p-ERK, increase in apoptosis, and decrease in tumor growth. These results suggest that directly depalmitoylating NRAS with 1 can inhibit its downstream signaling pathways involved in NRAS-driven cancers.
RESULTS AND DISCUSSION
Depalmitoylation of NRAS in NRAS-Driven Melanoma
Based on our group’s initial findings that C8 alkyl cysteine (1) is able to depalmitoylate membrane-associated proteins in cells21, we sought to determine if this compound would have similar effects on the palmitoylation state of mutant NRAS. For these initial tests, we used the WM3000 human melanoma cell line, which has an activating Q61R mutation in NRAS22,23. We found that 2 hour incubation with increasing concentrations of 1 (Figure 1a) resulted in a dose-dependent decrease in the levels of NRAS(Q61R) palmitoylation in cells, with an IC50 of 7 μM, as determined by acyl-resin assisted capture (acyl-RAC)24,25 (Figure 1b,c and Supplementary Figure 1a). Controls were performed in which WM3000 cells were treated with vehicle or C8 alkyl serine (2), a structurally similar molecule which is not capable of S-palmitoyl thioester cleavage because it lacks a nucleophilic thiol (Figure 1a)21. As expected, there was no observable thioester cleavage under these conditions, indicating that the observed reduction in NRAS palmitoylation is due to direct cleavage of the NRAS S-palmitoyl group by 1 and not a non-specific effect of 1 (Figure 1d,e). As a positive control, cells treated with the non-specific, irreversible palmitoylation inhibitor 2-BP11,19,20 also caused a decrease in NRAS palmitoylation (Figure 1d,e). However, total protein palmitoylation is significantly decreased when treated with 2-BP compared to 1 as observed by silver staining (Supplementary Figure 1b), which suggests that there is less off-target depalmitoylation with 1. Additionally, a small panel of known S-palmitoylated proteins were studied after treatment with 1, 2, or 2-BP to determine if there were significant decreases in protein palmitoylation levels.21 Compound 1 showed a preference for NRAS over other tested palmitoylated proteins in this panel. On the other hand, compound 2 did not have observable effects on the palmitoylation of the various proteins, while 2-BP non-selectively caused an overall decrease in protein palmitoylation (Supplementary Figure 2a,b). With this information, we confirmed biochemically that compound 1 is able to depalmitoylate NRAS in NRAS-mutated melanoma cells through direct nucleophilic cleavage of the S-palmitoyl thioester.
Figure 1.

C8 Alkyl Cysteine Causes Depalmitoylation of NRAS in NRAS-Driven Melanoma Cells. (a) Chemical structures of C8 alkyl cysteine (1) and control compound C8 alkyl serine (2). (b) Western blot detection of NRAS Q61R in acyl resin-assisted capture fractions of WM3000 melanoma cells after treatment with increasing concentrations (0–25 μM) of C8 alkyl cysteine (1). Input fractions (IF) contain total cellular protein and bound fractions (BF) contain only S-palmitoylated proteins. (c) Western blot lanes were analyzed and quantified using ImageJ. Ratios of BF to IF for each of the treatment conditions were calculated and normalized to the 0 μM control to determine the amount of S-palmitoylated NRAS in each sample. Results are the mean ± s.d. of three independent experiments (one-way ANOVA). (d) WM3000 melanoma cells were treated with different conditions, including the untreated control (C), vehicle control (20 μM TCEP in DMSO) (V), 10 μM C8 alkyl serine (2), 10 μM C8 alkyl cysteine (1), and 10 μM 2-bromopalmitate (2-BP) for 2 hours. (e) Ratios of BF to IF for each of the treatment conditions were calculated and normalized to vehicle control to determine the amount of S-palmitoylated NRAS in each sample. Results are the mean ± s.d. of three independent experiments (one-way ANOVA).
Delocalization of NRAS from the Plasma Membrane
To understand the biological effects of compound 1 on NRAS after depalmitoylation, we investigated the change in subcellular localization that occurred after treatment using fluorescence microscopy. Previous studies have shown that S-palmitoylation is required for NRAS anchorage to the plasma membrane26,27. Due to their ease of transfection and clear localization of RAS to the plasma membrane, we chose to use HeLa S3 cells as a model to visualize changes in NRAS localization. HeLa S3 cells stably expressing enhanced green fluorescent protein (EGFP)-tagged NRAS were treated with 20 μM of 1. We observed the delocalization of EGFP-NRAS from the plasma membrane within 30 minutes of treatment with 1 (Figure 2a,b and Supplementary Video 1), whereas there was no change in localization after treatment with the control 2 (Figure 2c,d). We also treated cells with 20 μM of 2-BP, which was expected to non-specifically inhibit palmitoylation. Although the results showed a partial delocalization of the EGFP-NRAS from the plasma membrane, there was no observation of an increase in localization at the Golgi28 (Figure 2e,f). This result may be attributed to the fact that 2-BP irreversibly inhibits palmitoylation and targets multiple other metabolic enzymes20. Additionally, 2-BP would not remove already palmitoylated NRAS from the plasma membrane, but rather it would only inhibit NRAS from being palmitoylated at the Golgi. This highlights the difference between direct depalmitoylation using 1 and inhibition of palmitoylation using 2-BP. The localization experiments confirmed that 1 causes NRAS to delocalize from the plasma membrane after depalmitoylation.
Figure 2.

C8 Alkyl Cysteine Causes Rapid Delocalization of NRAS from the Plasma Membrane. (a) EGFP-NRAS plasmid was stably transfected in HeLa S3 cells and treated with 20 μM C8 alkyl cysteine (1) for 30 min to visualize delocalization of NRAS from the plasma membrane. (b) Profile plots of cross sections (blue line) show EGFP-NRAS delocalization before (0 min) and after (30 min) treatment with 1. (c) EGFP-NRAS HeLa S3 cells were treated with 20 μM of the control C8 alkyl serine (2) for 30 min. (d) Profile plots of cross sections show EGFP-NRAS delocalization before (0 min) and after (30 min) treatment with 2. (e) EGFP-NRAS HeLa S3 cells were treated with 20 μM 2-bromopalmitate (2-BP) for 30 min. (f) Profile plots of cross sections show EGFP-NRAS delocalization before (0 min) and after (30 min) treatment with 2-BP. NRAS was fused to EGFP (green) and the Golgi apparatus was stained with BODIPY TR Ceramide (red). Scale bars denote 10 μm.
Inhibition of RAS Signaling in NRAS-Mutated Melanoma
Since there was an observable decrease in NRAS palmitoylation and a delocalization of NRAS from the plasma membrane after treatment with 1, we wanted to study the changes that occur in downstream RAS signaling. It is well known that NRAS activation occurs at the plasma membrane29. Therefore, it would be expected that NRAS would no longer be able to interact with its downstream effector proteins if it was not at the membrane. We treated both NRAS-driven and non-NRAS-driven melanoma cell lines with 1 as well as different controls to assess the differences in phosphorylated-AKT and phosphorylated-ERK. The phosphorylation of both of these proteins leads to activation of the oncogenic signaling pathways related to cancer cell growth and survival30. In the NRAS-driven melanoma cells, there was a dose-dependent decrease in p-AKT and p-ERK after 2-hour treatment with 1, (Supplementary Figure 3a,b). Single dose studies showed a significant decrease in p-AKT after 2-hour treatment with 10 μM of 1 compared to that of the untreated control, vehicle control, and compound 2 (Figure 3a,b and Supplementary Figure 4a,c). A decrease in p-ERK after treatment with 1 was also observed. (Figure 3a,c and Supplementary Figure 4e).
Figure 3.

C8 Alkyl Cysteine Inhibits Oncogenic RAS Signaling in NRAS-Mutated Melanoma Cells Compared to BRAF-Mutated Melanoma Cells. (a) Western blot of six different treatment conditions in the WM3000 melanoma cell line with the NRAS Q61R mutation. Treatment conditions include the untreated control (C), vehicle control (20 μM TCEP in DMSO) (V), 10 μM C8 alkyl serine (2), 10 μM C8 alkyl cysteine (1), 10 μM 2-bromopalmitate (2-BP), and 12 nM binimetinib (BM). (b,c) Western blot lanes for p-AKT and p-ERK were analyzed and quantified using ImageJ. Each treatment condition was normalized to the vehicle control. Results are the mean ± s.d. of three independent experiments. *p ≤ 0.05, **p ≤ 0.005, ***p ≤ 0.0005 (one-way ANOVA). (d) Western blot of the six different treatment conditions in the Sk-Mel-28 melanoma cell line with the BRAF V600E mutation. (e,f) Western blot lanes for p-AKT and p-ERK were analyzed and quantified using ImageJ. Each treatment condition was normalized to the vehicle control. Results are the mean ± s.d. of three independent experiments. *p ≤ 0.05 (one-way ANOVA).
Next, we treated the Sk-Mel-28 BRAF-driven melanoma cell line with the same six treatment conditions to determine if the effects on downstream phosphorylation were dependent on NRAS palmitoylation in a non-NRAS-dependent cell line. This particular melanoma cell line served as a control because it is dependent on mutated BRAF V600E for cancer survival and has wildtype NRAS31,32, suggesting that NRAS depalmitoylation should not affect its oncogenic downstream signaling. In the BRAF-mutated melanoma cell line, there was no significant decrease in either p-AKT or p-ERK after 2-hour treatment with 10 μM of 1, while there was an expected significant decrease after treatment with 12 nM of the MEK inhibitor binimetinib33,34 (Figure 3d-f). The total protein levels of β-actin, AKT, and ERK were similar for all treatment conditions (Supplementary Figure 4b,d,f), except for the ERK level in the binimetinib-treated BRAF-mutated melanoma cells. This particular ERK level was much higher than the rest of the treatments, which could be due to feedback loop activation when MEK is blocked by binimetinib33. These results confirmed that compound 1 causes an inhibition of downstream RAS signaling by the depalmitoylation of NRAS in an NRAS-dependent cell line compared to a non-NRAS-dependent cell line.
Reduction of Cell Viability in NRAS-Driven Melanoma
High levels of p-AKT and p-ERK are associated with an upregulation of cell growth and survival pathways in cancer1,30. Since we determined that 1 inhibits oncogenic RAS signaling by decreasing p-AKT and p-ERK, we wanted to study the direct effects of the depalmitoylating compound on cell viability. Although our amphiphilic compound is expected to depalmitoylate multiple membrane-bound proteins21, we expected that the depalmitoylation of NRAS would have the most significant effects on cell viability of NRAS-driven cell lines because they are dependent on NRAS for survival. In order to assess the importance of AMD chemical structure on growth inhibition activity, both NRAS-dependent and BRAF-dependent melanoma cell lines were treated with a variety of alkyl cysteine derivatives that had different chain lengths (Supplementary Figure 5 and Supplementary Figure 6). We determined that compound 1 was the most potent inhibitor of NRAS-mutated melanoma cell viability compared to the other lipid compounds (2–6), which could be due to the appropriate balance in hydrophobicity and hydrophilicity in the chemical structure and alkyl chain length of 1 (Supplementary Figure 7a). This balance would allow compound 1 to have enough solubility for transmembrane permeability and amphiphilicity to insert itself into the membrane. Additionally, 1 was more selective for inhibiting NRAS-mutated melanoma viability vs. BRAF-mutated melanoma viability compared to 4, which was the only other derivative that had an effect on cell viability of the NRAS-mutated melanoma cell line (Supplementary Figure 7a–c).
Next, the WM3000 and Sk-Mel-2 NRAS-mutated human melanoma cell lines as well as the control Sk-Mel-28 BRAF-mutated human melanoma cell line were all treated with 1 at increasing concentrations of 0, 3.125, 6.25, 12.5, 25, and 50 μM for 24 hours. We found that 1 had over 8-fold more inhibitory activity in NRAS-mutant versus non-NRAS-mutant melanoma cells, with an inhibitory concentration of 50% (IC50) of 9 μM in the WM3000 NRAS-mutated melanoma cell line and 72 μM in the Sk-Mel-28 BRAF-mutated melanoma cell line (Figure 4a). There was similarly high potency in Sk-Mel-2 melanoma cell line, which showed an IC50 of 15 μM. The dose-dependent decrease in cell viability correlated well with the dose-dependent decrease in the palmitoylation of NRAS (Figure 1c and Figure 4a). These results support that 1 causes a reduction in NRAS palmitoylation leading to a decrease in NRAS-mutated melanoma cell viability.
Figure 4.

C8 Alkyl Cysteine Preferentially Reduces Cell Viability in NRAS-Driven versus BRAF-Driven Melanoma. (a) A WST-1 assay was used to assess the cell viability of NRAS-mutated melanoma cell lines (WM3000 and Sk-Mel-2) vs. a BRAF-mutated melanoma cell line (Sk-Mel-28) after 24-hour incubation with increasing concentrations of C8 alkyl cysteine. Results are the mean ± s.d. of three independent experiments. Each row (concentration) was analyzed individually, without assuming a consistent SD. **p ≤ 0.01, ***p ≤ 0.001 (multiple t-tests). (b) The last 24 amino acids of the hypervariable region (HVR) of NRAS and KRAS4b are marked in red. The final protein construct of the NRAS-KRAS4bHVR fusion that was used for experimentation is at the bottom. (c) A WST-1 assay was used to assess the cell viability of the original WM3000 NRAS-mutated melanoma cell line and the WM3000 NRAS-mutated melanoma cell line with KRAS4b HVR replacing the original NRAS C-terminal HVR. Both cell lines were treated with increasing concentrations of C8 alkyl cysteine for 24 hours. Results are the mean ± s.d. of three independent experiments. Each row (concentration) was analyzed individually, without assuming a consistent SD. ***p ≤ 0.001 (multiple t-tests).
To determine if the inhibitory effects of compound 1 were mediated by the release of NRAS from the plasma membrane, a cell viability rescue experiment was conducted using a fusion construct of NRAS(Q61R) with its C-terminal HVR replaced with the KRAS4b HVR (Figure 4b). The KRAS4b HVR anchors RAS to the plasma membrane via a polylysine motif instead of S-palmitoylation, and its localization is not affected by AMD21. Furthermore, while NRAS is dependent on S-palmitoylation for cell growth and survival26,27, KRAS4b is not13. The original WM3000 NRAS(Q61R) melanoma cell line and the generated fusion WM3000 NRAS(Q61R)-KRAS4bHVR melanoma cell line both were treated with increasing concentrations of 1 for 24 hours. The IC50 of 1 was 9 μM in the WM3000 NRAS-mutated melanoma cell line and 16 μM in the stable WM3000 NRAS(Q61R)-KRAS4bHVR melanoma cell line (Figure 4c). Almost twice the concentration of 1 was necessary to have a similar decrease in cell viability in the NRAS-KRAS4bHVR cell line compared to the original NRAS-mutated melanoma cell line. It is possible that since the NRAS-KRAS4bHVR cell line still has endogenous NRAS(Q61R) mutant protein, there could be a threshold at which the NRAS-KRAS4bHVR fusion protein is unable to completely rescue cell viability if endogenous NRAS(Q61R) mutant protein is targeted by 1. However, these results support that the reduction in cell viability is mediated by the depalmitoylation and release of NRAS from the plasma membrane.
Apoptosis Induction in NRAS-Driven Melanoma Tumors
Having demonstrated the inhibitory effect of NRAS depalmitoylation in vitro, we sought to explore the activity of 1 in melanoma xenograft mouse models. Mice implanted with either WM3000 (NRAS-mutant) or Sk-Mel-28 (BRAF-mutant) melanoma cells were intratumorally injected with either 50 mg/kg compound 1 or vehicle control (Figure 5a).
Figure 5.

C8 Alkyl Cysteine Reduces Proliferation and Induces Apoptosis in NRAS-Driven versus BRAF-Driven Melanoma Tumors. (a) Schematic and timeline of in vivo subcutaneous melanoma xenograft implantation and intratumoral (IT) treatments. (b) Histology was performed for BRAF-mutated melanoma tumors (Sk-Mel-28) and NRAS-mutated melanoma tumors (WM3000) for both vehicle control (123.51 mg/kg TCEP in saline) and C8 alkyl cysteine (1) treatment groups. Consecutive tissue sections were stained with hematoxylin and eosin (H&E) for tumor cell morphology, anti-Ki-67 for cell proliferation, anti-cleaved caspase-3 (CC3) for cell apoptosis, and anti-phosphorylation of ERK (p-ERK) for RAS signaling.
To evaluate tumor cell survival, apoptosis, and p-ERK activity, consecutive tumor sections were stained for Ki-67, cleaved-caspase-3 (CC3), and p-ERK 24-hours after treatment. The Ki-67 nuclear antigen is highly expressed in cells undergoing active phases of the cell cycle (G1, S, G2, and M) but is absent in the G0 phase when the cells are in a quiescent state35. Hence, the expression of Ki-67 is strongly correlated with cell proliferation. We observed a decrease in Ki-67 in the NRAS tumors treated with compound 1 compared to vehicle control, while the BRAF tumors treated with 1 and vehicle control exhibited similar levels of Ki-67 staining (Figure 5b).
Intratumoral injection of compound 1 in NRAS-mutated tumors showed strong staining for CC3 activity, which is a common and reliable marker to assess apoptosis in various tissues36,37, compared to vehicle control, and minimal CC3 activity was observed in BRAF-mutated tumors. This confirmed that compound 1 was capable of inducing programmed cell death in NRAS, but not BRAF-mutated tumors (Figure 5b).
Finally, to determine if intratumoral injection of compound 1 can downregulate RAS signaling in NRAS tumors, tissue sections were stained for p-ERK activity. We observed p-ERK staining in the BRAF-mutated tumors treated with 1 as well as vehicle controls of both the BRAF and NRAS-mutated tumors, but there was much less p-ERK staining in the NRAS-mutated tumors treated with 1 (Figure 5b). These results were in line with our in vitro data showing a downregulation of the RAS signaling pathway in an NRAS-mutated melanoma cell line upon treatment with 1 (Figure 3a-c). Together, these studies demonstrated that compound 1 was able to achieve tumor cell killing and downregulate p-ERK signaling in NRAS-driven melanoma tumors, but not in BRAF-driven melanoma tumors.
Tumor Growth Decrease in NRAS Melanoma Xenografts
After confirming the ability of compound 1 to induce cell death in the NRAS-mutated melanoma tumors, we proceeded to examine the effectiveness of treatment with 1 in the NRAS-mutant tumor bearing mice by intraperitoneal (IP) administration. The xenograft mice were randomized into either compound 1 (20 mg/kg) or vehicle control (49.40 mg/kg) treatment groups for a 7-day study (Figure 6a). Daily treatment with 1 resulted in a significant decrease in tumor growth compared to the vehicle control cohort (Figure 6b). Importantly, the body weight of the animals did not change over the course of treatment, suggesting that compound 1 was well tolerated at this treatment regimen (Figure 6c).
Figure 6.

C8 Alkyl Cysteine Causes a Decrease in Tumor Growth in NRAS-Mutated Melanoma Xenograft Mice. (a) Schematic and timeline of in vivo melanoma xenograft intraperitoneal (IP) treatments. (b) WM3000 NRAS-mutated human melanoma xenograft mice were IP injected with either 20 mg/kg/day of 1 or 49.40 mg/kg/day (2 molar equivalents) of vehicle control. Tumor volume was measured every other day for one week for both treatment groups. Results are the mean ± SEM with n = 6. *p ≤ 0.05 (unpaired, two-tailed Student’s t test). (c) Animal weight was measured every other day for the duration of the IP treatments. (d) Liver toxicity was analyzed by assessing the alanine aminotransferase (ALT) activity in the blood samples of the xenograft mice in both treatment groups. The conditions included 49.40 mg/kg/day of vehicle control, 20 mg/kg/day of C8 alkyl cysteine (1), ALT enzyme assay positive control (+), and untreated mouse negative control (−).
Drug-induced hepatotoxicity is a frequent cause of liver injury with the use of many cancer therapies38. To determine if 1 caused liver toxicity in the xenograft mice, alanine aminotransferase (ALT) enzyme was measured from the serum. ALT is found mainly in the liver and kidney, and normally at low levels in the blood. However, when the liver is damaged ALT is released resulting in high levels in the bloodstream. ALT levels after IP treatment were approximately 60 mU/mL for 1 and 90 mU/ml for vehicle control (Figure 6d). These levels were not significantly different from one another and within the normal range for mice, which is between 26–126 U/L39. This strongly suggested that compound 1 did not induce liver toxicity in the mice.
Discussion
NRAS GTPase has been deemed as an undruggable protein because of the challenging nature of targeting its shallow binding pockets. Directly targeting the S-palmitoylation modification on the C-terminal HVR of NRAS using a chemoselective approach can be applied as a unique inhibition strategy. It has been well documented through in vitro and in vivo studies that NRAS palmitoylation is required for its cancer signaling activity26,27. Taking this into account, our depalmitoylation strategy can be used to target NRAS palmitoylation and inhibit its oncogenic signaling capabilities in the context of NRAS-driven cancers.
We have determined that our depalmitoylating compound, C8 alkyl cysteine (1), causes delocalization of NRAS from the plasma membrane by the mechanism of depalmitoylation. Additionally, 1 inhibits downstream signaling associated with cancer cell growth and survival pathways in NRAS-mutated melanoma cells. Specifically, it causes a decrease in p-AKT and p-ERK in an NRAS-driven cell line but not in a BRAF-driven cell line that is not dependent on NRAS signaling. Accordingly, 1 causes an inhibitory effect on cell viability in multiple NRAS-mutated melanoma cell lines, which is rescued by addition of an NRAS mutant with its HVR replaced with the non-palmitoylated HVR of KRAS4b. However, it is important to note that there is not a complete rescue in cell viability of the NRAS-KRAS4bHVR cell line. A possible explanation for this result is that there is still endogenous mutant NRAS that is depalmitoylated by 1, which may contribute to the partial rescuing capability of the NRAS-KRAS4bHVR fusion protein up until a certain concentration. In a xenograft mouse model, tumor histology confirmed that 1 was able to induce apoptosis after intratumoral injection in NRAS-mutated melanoma tumors, but not BRAF-mutated melanoma tumors. We were also able to detect a downregulation of p-ERK signaling in the NRAS-dependent melanoma tumors with direct intratumoral injection. Furthermore, we observed a significant decrease in tumor growth in NRAS-mutated melanoma xenograft mice treated with 1 compared to the vehicle control, with no detectable liver toxicity. Taken together, these results indicate that compound 1 can inhibit NRAS function in vitro and in vivo.
This unique depalmitoylation approach overcomes the issues associated with designing small molecules to bind the active site of NRAS and provides a direct method to inhibit its function by targeting the S-palmitoylation post-translational modification. A depalmitoylation-based strategy has the potential to be transformative for the treatment of melanoma as well as other cancers with high rates of NRAS-mutations, such as leukemias and lymphomas. Our ongoing work includes enhancing the selectivity of the amphiphile-mediated depalmitoylating compounds. We plan to use appropriate large-scale capture techniques and proteomic methods for analyzing the palmitoylated proteins that are targeted by these compounds40,41. In addition, we plan to explore the direct relationship between NRAS palmitoylation and total Ras-GTP levels in future studies42,43. With further optimization of the selectivity and pharmacokinetics, amphiphile-mediated depalmitoylation could become a strategy to uncover novel lead compounds that can be adapted to generate inhibitors of several other S-palmitoylated drug targets across a wide range of disease indications.
Supplementary Material
ACKNOWLEDGMENT
We would like to thank S. Gutkind (UCSD) for his advice and suggestions as well as providing the Sk-Mel-28 melanoma cell line. H.D.V. was supported by the Molecular Biophysics Training Grant, NIH Grant T32 GM008326. This study was supported by grants from the National Institute of Health (DP2DK111801–01S2 and T32CA009523–33). Tissue Technology Shared Resources P30CA23100.
Footnotes
METHODS
Synthetic methods, materials, and methods are included in the Supporting Information.
ASSOCIATED CONTENT
Supporting Information
Supporting Information Available: This material is available free of charge via the Internet.
Detailed methods, chemical schemes, chemical structures, HPLC/ELDS spectra, NMR spectra, additional data and figures for silver staining, depalmitoylation protein panel western blot and analysis, multidose signaling western blot and analysis, total β-actin, AKT, and ERK levels, cell viability assay data, plasmid sequence map, and EGFP-NRAS live-cell imaging video.
University of California San Diego has filed a Patent Cooperation Treaty (PCT) Application that covers the use of amphiphilic thiol compounds for protein depalmitoylation. (International Application no. PCT/US2019/021944; H.D.V., M.J., R.J.B., A.K.R., N.K.D.). A.K.R. and N.K.D. are shareholders of Palm Therapeutics, Inc.
REFERENCES
- (1).Wei Z, and Liu HT (2002) MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 12, 9–18. [DOI] [PubMed] [Google Scholar]
- (2).Crespo P, and León J (2000) Ras proteins in the control of the cell cycle and cell differentiation. Cell. Mol. Life Sci 57, 1613–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Nakada M, Niska JA, Tran NL, McDonough WS, and Berens ME (2005) EphB2/R-ras signaling regulates glioma cell adhesion, growth, and invasion. Am. J. Pathol 167, 565–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Spandidos DA, Sourvinos G, Tsatsanis C, and Zafiropoulos A (2002) Normal ras genes: their onco-suppressor and pro-apoptotic functions (review). Int. J. Oncol 21, 237–241. [DOI] [PubMed] [Google Scholar]
- (5).Giehl K, Skripczynski B, Mansard A, Menke A, and Gierschik P (2000) Growth factor-dependent activation of the Ras-Raf-MEK-MAPK pathway in the human pancreatic carcinoma cell line PANC-1 carrying activated K-ras: Implications for cell proliferation and cell migration. Oncogene 19, 2930–2942. [DOI] [PubMed] [Google Scholar]
- (6).Simanshu DK, Nissley DV, and McCormick F (2017) RAS Proteins and Their Regulators in Human Disease. Cell 170, 17–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Papke B, and Der CJ (2017) Drugging RAS: Know the enemy. Science (80-.). 355, 1158–1163. [DOI] [PubMed] [Google Scholar]
- (8).Ostrem JM, Peters U, Sos ML, Wells JA, and Shokat KM (2013) K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ostrem JML, and Shokat KM (2016) Direct small-molecule inhibitors of KRAS: From structural insights to mechanism-based design. Nat. Rev. Drug Discov 15, 771–785. [DOI] [PubMed] [Google Scholar]
- (10).Samatar AA, and Poulikakos PI (2014) Targeting RAS-ERK signalling in cancer: Promises and challenges. Nat. Rev. Drug Discov 13, 928–942. [DOI] [PubMed] [Google Scholar]
- (11).Webb Y, Hermida-Matsumoto L, and Resh MD (2000) Inhibition of protein palmitoylation, raft localization, and T cell signaling by 2-bromopalmitate and polyunsaturated fatty acids. J. Biol. Chem 275, 261–270. [DOI] [PubMed] [Google Scholar]
- (12).Rajalingam K, Schreck R, Rapp UR, and Albert S (2007) Ras oncogenes and their downstream targets. Biochim. Biophys. Acta - Mol. Cell Res 1773, 1177–1195. [DOI] [PubMed] [Google Scholar]
- (13).Ahearn IM, Haigis K, Bar-Sagi D, and Philips MR (2012) Regulating the regulator: Post-translational modification of RAS. Nat. Rev. Mol. Cell Biol 13, 39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Xiang S, Bai W, Bepler G, and Zhang X (2017) Activation of Ras by Post-Translational Modifications Conqu. RAS Elsevier Inc. [Google Scholar]
- (15).Resh MD (2012) Targeting protein lipidation in disease. Trends Mol. Med 18, 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Yeste-Velasco M, Linder ME, and Lu YJ (2015) Protein S-palmitoylation and cancer. Biochim. Biophys. Acta - Rev. Cancer 1856, 107–120. [DOI] [PubMed] [Google Scholar]
- (17).Gajewski TF, Salama AKS, Niedzwiecki D, Johnson J, Linette G, Bucher C, Blaskovich MA, Sebti SM, and Haluska F (2012) Phase II study of the farnesyltransferase inhibitor R115777 in advanced melanoma (CALGB 500104). J. Transl. Med 10, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Berndt N, Hamilton AD, and Sebti SM (2011) Targeting protein prenylation for cancer therapy. Nat. Rev. Cancer 11, 775–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Mikic I, Planey S, Zhang J, Ceballos C, Seron T, von Massenbach B, Watson R, Callaway S, McDonough PM, Price JH, Hunter E, and Zacharias D (2006) A Live Cell, Image-Based Approach to Understanding the Enzymology and Pharmacology of 2-Bromopalmitate and Palmitoylation. Methods Enzymol. 414, 150–187. [DOI] [PubMed] [Google Scholar]
- (20).Davda D, Azzouny M. A. El, Tom CTMB, Jeannie L, Majmudar JD, Kennedy RT, and Martin BR (2014) Profiling targets of the irreversible palmitoylation inhibitor 2- bromopalmitate. ACS Chem. Biol 8, 1912–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Rudd AK, Brea RJ, and Devaraj NK (2018) Amphiphile-Mediated Depalmitoylation of Proteins in Living Cells. J. Am. Chem. Soc 140, 17374–17378. [DOI] [PubMed] [Google Scholar]
- (22).Ahmed RL, Shaughnessy DP, Knutson TP, Vogel RI, Ahmed K, Kren BT, and Trembley JH (2019) CDK11 loss induces cell cycle dysfunction and death of BRAF and NRAS Melanoma Cells. Pharmaceuticals 12, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Reyes-Uribe P, Adrianzen-Ruesta MP, Deng Z, Echevarria-Vargas I, Mender I, Saheb S, Liu Q, Altieri DC, Murphy ME, Shay JW, Lieberman PM, and Villanueva J (2018) Exploiting TERT dependency as a therapeutic strategy for NRAS-mutant melanoma. Oncogene 37, 4058–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Forrester MT, Hess DT, Thompson JW, Hultman R, Moseley MA, Stamler JS, and Casey PJ (2011) Site-specific analysis of protein S -acylation by resin-assisted capture. J. Lipid Res 52, 393–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Howie J, Reilly L, Fraser NJ, Vlachaki Walker JM, Wypijewski KJ, Ashford MLJ, Calaghan SC, McClafferty H, Tian L, Shipston MJ, Boguslavskyi A, Shattock MJ, and Fuller W (2014) Substrate recognition by the cell surface palmitoyl transferase DHHC5. Proc. Natl. Acad. Sci 111, 17534–17539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Cuiffo B, and Ren R (2010) Palmitoylation of oncogenic NRAS is essential for leukemogenesis. Blood 115, 3598–3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Xu J, Hedberg C, Dekker FJ, Li Q, Haigis KM, Hwang E, Waldmann H, and Shannon K (2012) Inhibiting the palmitoylation/depalmitoylation cycle selectively reduces the growth of hematopoietic cells expressing oncogenic Nras. Blood 119, 1032–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Rocks O, Gerauer M, Vartak N, Koch S, Huang ZP, Pechlivanis M, Kuhlmann J, Brunsveld L, Chandra A, Ellinger B, Waldmann H, and Bastiaens PIH (2010) The palmitoylation machinery is a spatially organizing system for peripheral membrane proteins. Cell 141, 458–471. [DOI] [PubMed] [Google Scholar]
- (29).Song SP, Hennig A, Schubert K, Markwart R, Schmidt P, Prior IA, Böhmer FD, and Rubio I (2013) Ras palmitoylation is necessary for N-Ras activation and signal propagation in growth factor signalling. Biochem. J 454, 323–332. [DOI] [PubMed] [Google Scholar]
- (30).Mendoza MC, Er EE, and Blenis J (2011) The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci 36, 320–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JWC, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Wooster R, Stratton MR, and Futreal PA (2002) Mutations of the BRAF gene in human cancer. Nature 417, 949–954. [DOI] [PubMed] [Google Scholar]
- (32).Ellerhorst JA, Greene VR, Ekmekcioglu S, Warneke CL, Johnson MM, Cooke CP, Wang LE, Prieto VG, Gershenwald JE, Wei Q, and Grimm EA (2011) Clinical correlates of NRAS and BRAF mutations in primary human melanoma. Clin. Cancer Res 17, 229–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Woodfield SE, Zhang L, Scorsone KA, Liu Y, and Zage PE (2016) Binimetinib inhibits MEK and is effective against neuroblastoma tumor cells with low NF1 expression. BMC Cancer 16, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Crassini K, Shen Y, Stevenson WS, Christopherson R, Ward C, Mulligan SP, and Best OG (2018) MEK1/2 inhibition by binimetinib is effective as a single agent and potentiates the actions of Venetoclax and ABT-737 under conditions that mimic the chronic lymphocytic leukaemia (CLL) tumour microenvironment. Br. J. Haematol 182, 360–372. [DOI] [PubMed] [Google Scholar]
- (35).Duchrow M, Schlüter C, Wohlenberg C, Flad HD, and Gerdes J (1996) Molecular characterization of the gene locus of the human cell proliferation-associated nuclear protein defined by monoclonal antibody Ki-67. Cell Prolif. 29, 1–12. [PubMed] [Google Scholar]
- (36).Eckle VS, Buchmann A, Bursch W, Schulte-Hermann R, and Schwarz M (2004) Immunohistochemical Detection of Activated Caspases in Apoptotic Hepatocytes in Rat Liver. Toxicol. Pathol 32, 9–15. [DOI] [PubMed] [Google Scholar]
- (37).Hadjiloucas I, Gilmore AP, Bundred NJ, and Streuli CH (2001) Assessment of apoptosis in human breast tissue using an antibody against the active form of caspase 3: Relation to tumour histopathological characteristics. Br. J. Cancer 85, 1522–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Vincenzi B, Armento G, Spalato Ceruso M, Catania G, Leakos M, Santini D, Minotti G, and Tonini G (2016) Drug-induced hepatotoxicity in cancer patients - implication for treatment. Expert Opin. Drug Saf 15, 1219–1238. [DOI] [PubMed] [Google Scholar]
- (39).Suckow MA, Danneman P, and Brayton C (2001) The Laboratory Mouse. CRC Press. [Google Scholar]
- (40).Draper JM, and Smith CD (2009) Palmitoyl acyltransferase assays and inhibitors (Review). Mol. Membr. Biol 26, 5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Martin BR, and Cravatt BF (2009) Large-scale profiling of protein palmitoylation in mammalian cells. Nat. Methods 6, 135–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Jeng HH, Taylor LJ, and Bar-Sagi D (2012) Sos-mediated cross-activation of wild-type Ras by oncogenic Ras is essential for tumorigenesis. Nat. Commun 3, 1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Margarit SM, Sondermann H, Hall BE, Nagar B, Hoelz A, Pirruccello M, Bar-Sagi D, and Kuriyan J (2003) Structural evidence for feedback activation by Ras·GTP of the Ras-specific nucleotide exchange factor SOS. Cell 112, 685–695. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.