

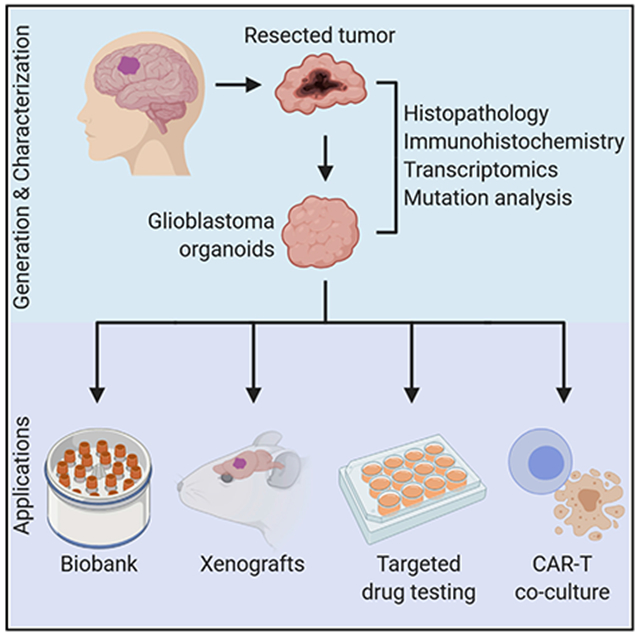
SUMMARY
Glioblastomas exhibit vast inter- and intra-tumoral heterogeneity, complicating the development of effective therapeutic strategies. Current in vitro models are limited in preserving the cellular and mutational diversity of parental tumors and require a prolonged generation time. Here, we report methods for generating and biobanking patient-derived glioblastoma organoids (GBOs) that recapitulate the histological features, cellular diversity, gene expression, and mutational profiles of their corresponding parental tumors. GBOs can be generated quickly with high reliability and exhibit rapid, aggressive infiltration when transplanted into adult rodent brains. We further demonstrate the utility of GBOs to test personalized therapies by correlating GBO mutational profiles with responses to specific drugs and by modeling chimeric antigen receptor T cell immunotherapy. Our studies show that GBOs maintain many key features of glioblastomas and can be rapidly deployed to investigate patient-specific treatment strategies. Additionally, our live biobank establishes a rich resource for basic and translational glioblastoma research.
Graphical Abstract
In Brief
A rapid and reliable method to generate patient-derived glioblastoma organoids captures the features and diversity of their respective parental tumors to allow for testing personalized therapies correlated to organoid profile and the establishment of a biobank for further basic and translational glioblastoma research.
INTRODUCTION
Glioblastoma is the most prevalent primary malignant brain tumor in adults (Ostrom et al., 2018) and remains almost invariably lethal due to its aggressive and invasive nature. Despite many clinical trials (Paolillo et al., 2018), the standard of care therapy for over a decade has been maximal surgical resection followed by temozolomide chemotherapy and radiation treatment, which improves the median survival duration to 14.6 months when compared to 12.1 months with surgery and radiation alone (Stupp et al., 2005). It has been increasingly appreciated that molecular heterogeneity among tumors (Brennan et al., 2013) and within tumors (Darmanis et al., 2017; Neftel et al., 2019; Patel et al., 2014) likely contributes to poor outcomes of numerous clinical trials (Mandel et al., 2018). Characterizing this heterogeneity and developing new models for timely empirical testing of personalized treatment strategies for glioblastoma remain both pre-clinical and clinical challenges.
Several model systems have contributed tremendously to our current understanding of biological mechanisms underlying glioblastoma pathogenesis, but have their limitations. Traditional in vitro culture models, both monolayer and tumor sphere cultures, can require a substantial amount of time to establish and use exogenous EGF, bFGF, and/or serum to propagate tumor cells over serial passages with clonal expansion, which are not favorable to maintain various cellular subtypes and key driver gene expression of parental tumors (Ledur et al., 2017; Lee et al., 2006). Patient-derived xenograft (PDX) models, in which primary dissociated tumor cells are directly injected into mice, are thought to better retain these important features of glioblastomas (Giannini et al., 2005). However, these PDX models exhibit variable engraftment efficiency and host infiltration by tumor cells, have very limited throughput, and are subject to a long latency in tumor generation ranging from 2 to 11 months (Patrizii et al., 2018). The protracted time required to establish clonal tumor cell cultures and PDX models hinders their clinical applicability for testing personalized therapy as current treatment regimens are typically initiated 1 month following surgery, and the median survival time of patients upon diagnosis is 14.6 months.
Recently, 3D organoid culture systems have been developed that capture the phenotypic and molecular heterogeneity found in various organs (Clevers, 2016), including cerebral organoids (Kadoshima et al., 2013; Lancaster et al., 2013; Paşca et al., 2015; Qian et al., 2016). Organoids have since been applied to model various cancers, including pancreatic, prostate, liver, breast, bladder, ovarian, and gastrointestinal cancers (Boj et al., 2015; Broutier et al., 2017; Gao et al., 2014; Kopper et al., 2019; Lee et al., 2018; Sachs et al., 2018; Yan et al., 2018). In most cases, dissociated tumor cells of epithelial origin are cultured within Matrigel in the presence of exogenous growth factors to form 3D structures, and various cancer organoid biobanks have been established as valuable resources (Bleijs et al., 2019). To study glioblastoma, cerebral organoids have been genetically manipulated to develop oncogenic properties (Bian et al., 2018; Ogawa et al., 2018) or co-cultured with tumor spheres to model tumor cell invasion (da Silva et al., 2018; Linkous et al., 2019). Furthermore, patient-derived glioblastoma organoids generated with Matrigel and exogenous EGF/bFGF over several weeks have demonstrated stem cell heterogeneity and a hypoxic gradient (Hubert et al., 2016). The degree to which these glioblastoma organoid systems recapitulate key molecular features of patient tumors remains unclear.
Here, we report a robust method to rapidly generate glioblastoma organoids (named GBOs) in a defined culture medium directly from fresh tumor specimens without single-cell dissociation. We generated a live biobank of GBOs and performed comprehensive histological, molecular, and genomic analyses to show that GBOs recapitulate inter- and intra-tumoral heterogeneity and retain many key features of their corresponding parental tumors. These GBOs can be efficiently xenografted into the adult mouse brain, displaying rapid and aggressive infiltration and maintaining key driver mutation expression. We further show that GBOs can be employed to test responses to standard of care therapy as well as targeted treatments, including drugs from clinical trials and chimeric antigen receptor T (CAR-T) cell immunotherapy, on a clinically relevant timescale. Together, these results highlight the potential utility of our patient-derived glioblastoma organoid model and biobank for basic and translational research and for testing personalized therapies.
RESULTS
Culture and Banking of Glioblastoma Organoids from Patient Tumors
To preserve the local cytoarchitecture and native cell-cell interactions of original tumors, and to avoid clonal selection of specific cell populations in culture, we developed a protocol to generate glioblastoma organoids (GBOs) without mechanical or enzymatic dissociation of the resected tumor tissue into single cells (Figure 1A). Furthermore, the optimized medium to establish and maintain GBOs is fully defined, serum-free, and with no added EGF/bFGF or extracellular matrix that may contribute to further selection. We obtained fresh surgically resected glioblastoma tumor tissue from patients after informed consent (Table S1). Optimal GBOs were generated from tissue along the tumor margin with minimal necrosis and little surrounding brain tissue. The resected tissue was cut into ~1 mm diameter pieces using fine dissection scissors (Figure 1A). Debris and red blood cells were removed and tumor pieces were cultured in the GBO medium on an orbital shaker to facilitate organoid formation and increase nutrient and oxygen diffusion. Tumor pieces generally formed round organoids within 1–2 weeks (Figure 1A). GBOs were propagated by cutting them into ~0.5 mm diameter pieces to avoid necrotic cell death in the inner core (Figure 1A).
Figure 1. Generation of GBOs that Retain Histologic Features of Parental Tumors.
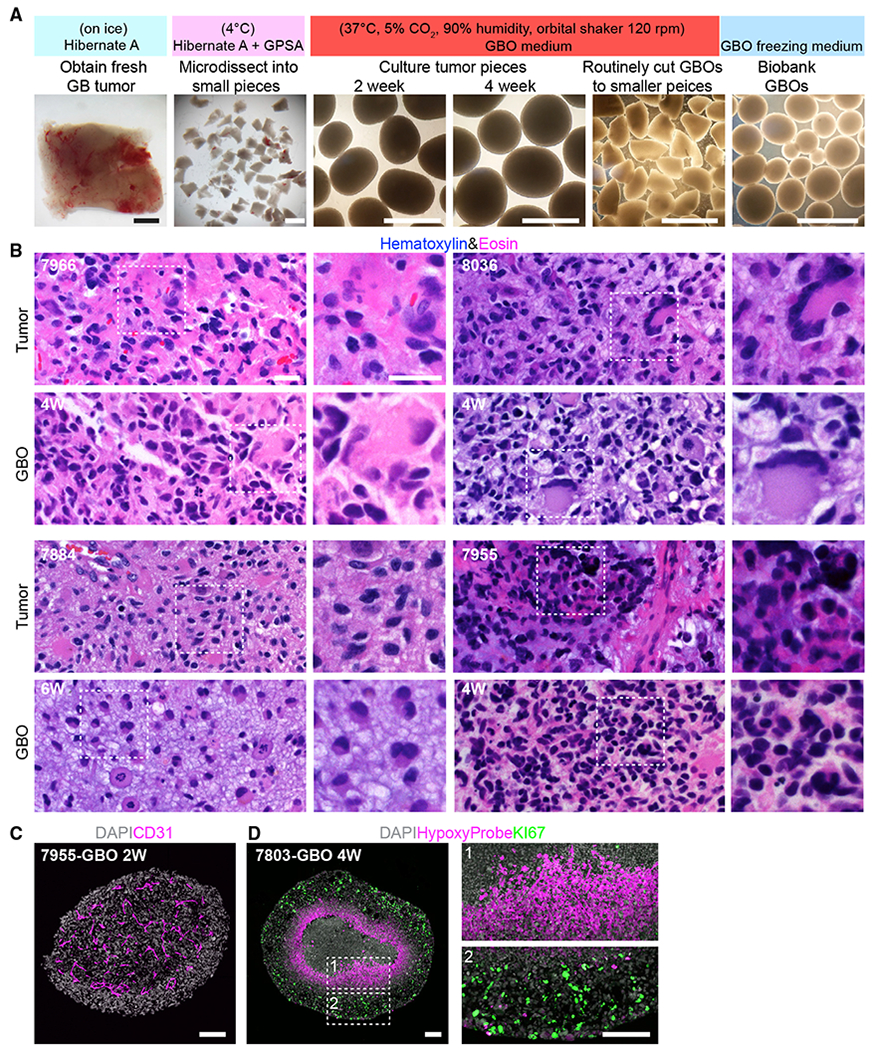
(A) A schematic of the procedure with sample bright-field images. Scale bar, 1 mm.
(B) Sample H&E staining images of parental tumors and corresponding GBOs. Age of GBOs in weeks (W) is listed. Scale bars, 20 μm.
(C) Sample confocal image of micro-vasculature retained in GBO with immunostaining for CD31. Scale bar, 100 μm.
(D) Sample confocal images showing the hypoxia gradient present in a large GBO with immunostaining for HypoxyProbe and KI67. Insets highlight KI67+ proliferating cells at the periphery (box 2), but not in the hypoxic core (box 1). Scale bars, 100 μm.
To assess whether GBOs resemble their corresponding parental tumors, we first performed histological analyses. Each GBO was independently confirmed by a neuropathologist to retain features of high-grade gliomas using H&E staining (Figure 1B). These GBOs displayed the cellular and nuclear atypia of patient tumors, often containing abundant mitotic figures and pleomorphic nuclei (Figure 1B). In particular, GBOs exhibited many characteristic cellular morphologies of their parental tumors, such as gemistocytic cells (UP-7966), multinucleated giant cells (UP-8036 and UP-7955), and cells with abundant vacuoles (UP-7884). Many GBOs also retained CD31+ vasculature (Figure 1C). GBOs allowed to grow larger developed hypoxia gradients (Figure 1D), a hallmark of glioblastomas (Hubert et al., 2016). Hypoxia increased substantially around 300 μm from the surface, correlating with absence of KI67+ proliferating cells (Figure 1D).
To further characterize cellular identities, we performed immunohistological analyses using a panel of neurodevelopmental markers, including glial markers GFAP and S100B, immature neuronal marker DCX, and neural progenitor and glioma stem cell markers NESTIN, BLBP, HOPX, SOX2, and OLIG2. We observed robust heterogeneity in cell identity and morphology in GBOs with close resemblance to the cellular composition of corresponding parental tumors, which exhibited marked inter-tumoral heterogeneity (Figures 2A and S1A). Quantitative analysis of 8 tumor samples showed similar percentages of cells expressing SOX2 and OLIG2 between parental tumors and corresponding GBOs for up to 4 weeks in the majority of cases (Figure 2B). GBOs largely maintained the proliferation rate of corresponding parental tumors as quantified by KI67 immunostaining (Figures 2A and 2B). We observed small numbers of dying cells in non-necrotic regions for some parental tumors and derived GBOs, but not for others (Figure S1B). To examine GBO expansion, we pulsed GBOs with 5-ethynyl-2′-deoxyuridine (EdU). At 1 h after labeling, the majority of EdU+ cells were NESTIN+, S100B+, DCX+, or OLIG2+, revealing progenitor subtypes that were actively dividing in these GBOs (Figures 2C and 2D). Two weeks later, EdU-retaining cells included NESTIN+, HOPX+, DCX+, S100B+, OLIG2+, and GFAP+ cells, indicating continuous generation of diverse cell types that were typically present in parental tumors (Figures 2A and 2D). We have also monitored GBO growth by measuring the size of individual GBOs over time (Figures S2A and S2B).
Figure 2. GBOs Retain and Continuously Generate Heterogeneous Cell Populations.
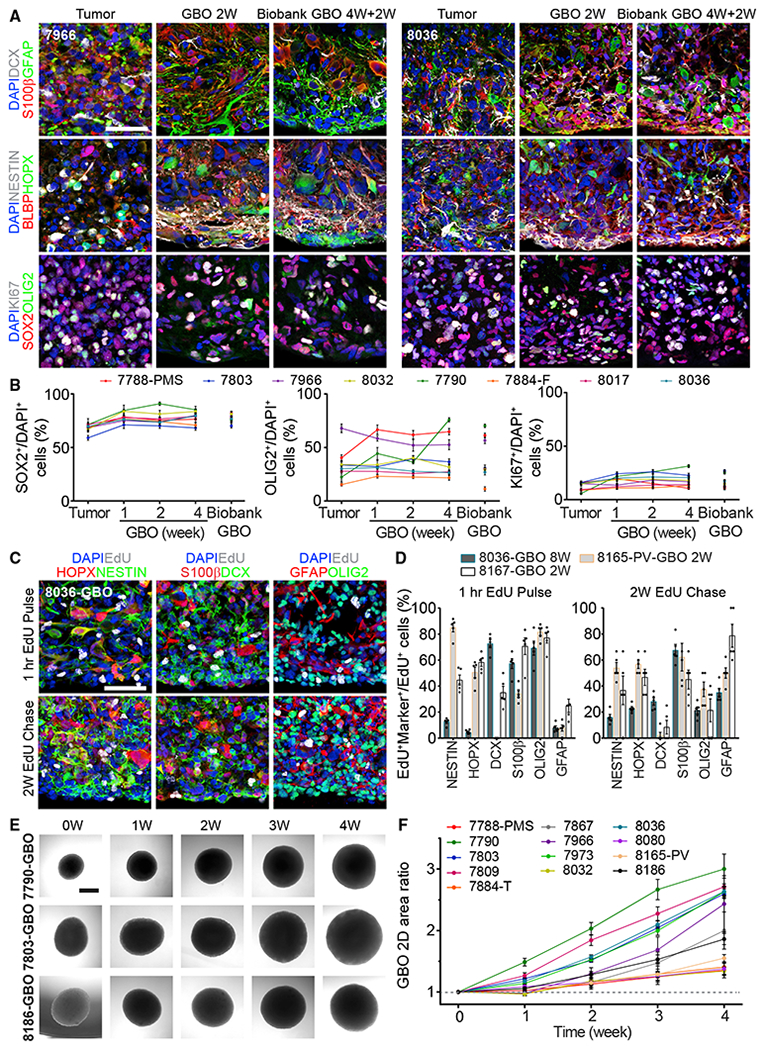
(A) Sample confocal images of immunostaining for different markers showing the maintenance of parental tumor cell populations in cultured GBOs and GBOs recovered from the biobank for two patients. See Figure S1A for additional samples. Scale bar, 50 μm.
(B) Quantifications of SOX2+, OLIG2+, and KI67+ cells in 8 parental tumors, corresponding GBOs for different culture periods, and those recovered from the biobank (4W+2W). Values represent mean ± SEM (n = 5).
(C and D) EdU pulse-chase experiments. GBOs were incubated with 1 μM EdU for 1 h and immunohistology for different markers was performed 1 h and 2 weeks later. Shown are sample confocal images (C; scale bar, 50 μm) and quantifications of EdU+Marker+ cells among EdU+ cells after 1-h EdU pulse and 2-week EdU chase (D). Dots represent data from individual GBOs and bar values represent mean ± SEM (n = 5).
(E and F) Growth of biobanked GBOs after recovery. Shown are sample bright-field images of individual GBOs recovered from the biobank (E; scale bar, 500 μm) and quantification of the ratio of the measured 2D area at each time point to the 2D area at time point 0 for the same GBOs recovered from the biobank (F). Values represent mean ± SEM (n = 10 GBOs per sample).
See also Figures S1 and S2 and Tables S1 and S2.
We have generated GBOs with high reliability from glioblastomas of 53 patient cases, including from anatomical subregions of the same tumors and from recurrent tumors (Figure S2C; Table S1). We defined successful GBO generation as microdissected tumor pieces that could survive, develop a spherical morphology, and continuously grow in culture for 2 weeks. Using these criteria, our overall success rate for generating GBOs was 91.4%, with 66.7% for IDH1 mutant tumors and 75% for recurrent tumors (Table S1). GBOs could be cultured for over 48 weeks and maintained similar expression of markers examined (Figures S2D and S2E). We also developed protocols for freezing cultured GBOs and recovering them from cryopreservation. These recovered GBOs exhibited continuous growth (Figures 2E and 2F) and similar expression of various markers to their corresponding parental tumors (Figures 2A, 2B, S1, and S2D). So far, we have established a biobank of 70 GBOs from 53 patient cases (including subregional samples) carrying a variety of genomic alterations commonly found in glioblastomas (Brennan et al., 2013) (Figure S2F; Table S1) and performed additional characterizations for a subset of these GBOs (Table S2).
Maintenance of Molecular and Mutation Inter- and Intra-tumoral Heterogeneity by GBOs
We next assessed whether GBOs also maintained gene expression signatures and genomic landscapes similar to their corresponding parental tumors. We performed transcriptome analysis by bulk RNA sequencing (RNA-seq) across 12 patients, including 2 patients with subregional samples, for a total of 17 parental tumor samples and 64 derived GBOs in culture for 1 to 12 weeks (Table S3). Overall, transcriptome-wide comparisons of GBOs with their corresponding parental tumors demonstrated high similarity (Figures 3A and S3A). Given the large inter-tumoral heterogeneity for glioblastomas, we identified unique gene expression signatures for individual parental tumors and found that corresponding GBOs maintained these patterns of gene expression in culture over time (Figure 3B). For example, EGFR expression was maintained at high levels in UP-7788-ANT-GBOs and at low levels in UP-7790-GBOs, as shown by both RNA-seq and immunohistology (Figures S3B and S3C). Systematic comparison between parental tumors and derived GBOs showed that the major difference was downregulation of blood- and immune-related genes in GBOs (Figures S3D–S3F), likely reflecting the elimination of blood cells and a lack of immune cell expansion in GBOs overtime. Few genes were upregulated in GBOs compared to parental tumors (Figures S3D and S3F).
Figure 3. GBOs Maintain Inter- and Intra-tumoral Heterogeneity of Gene Expression and Mutational Profiles of Corresponding Parental Tumors.
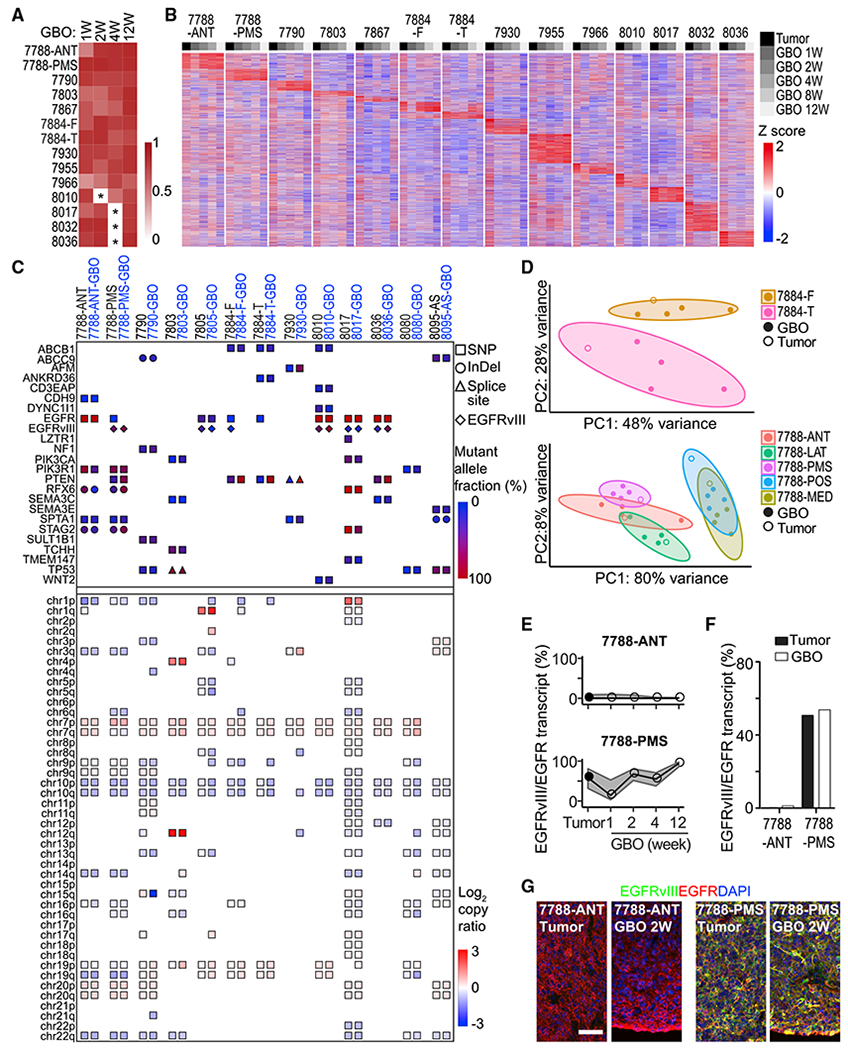
(A) Heatmap of transcriptome-wide gene expression Pearson correlations between parental tumors and corresponding GBOs as determined by RNA-seq. *Unsampled time points.
(B) Gene expression heatmap of the top 10,000 most variably expressed genes in parental tumors.
(C) Somatic variants in glioblastoma-associated genes (top panels) and copy number variations in autosomal chromosomal arms (bottom panels) identified by whole-exome sequencing of parental tumors, derived GBOs at 2 weeks and corresponding blood samples. The types of variants and allele frequencies are listed (see Table S3). The EGFRvIII mutation was determined by RNA-seq.
(D) RNA-seq gene expression PCA plots of subregional samples for parental tumors and corresponding GBOs for different culture periods for two patients with 90% confidence ellipses.
(E) EGFRvIII transcript abundance relative to EGFR as determined by RNA-seq in parental tumors and corresponding GBOs with 95% credible intervals.
(F) EGFRvIII transcript abundance relative to EGFR as determined by digital PCR in parental tumors and corresponding GBOs at 2 weeks.
(G) Confocal images of EGFRvIII and EGFR immunostaining and DAPI for parental tumors and corresponding GBOs. Scale bar, 50 μm.
To determine whether GBOs maintain genomic alterations found in parental tumors, we performed exome sequencing of 13 parental tumor samples, their corresponding GBOs at 2 weeks, and matched blood samples (Table S3). We focused on somatic variants listed in a recent comprehensive study of glioblastoma genomics (Brennan et al., 2013). The majority of somatic variants identified in parental tumors was found in corresponding GBOs at similar allele frequencies (Figure 3C; Table S3). Copy number variants (CNVs) detected in parental tumors were also identified in corresponding GBOs at similar copy number ratios (Figures 3C and S3G). Notably, inter-tumoral heterogeneity was largely retained in corresponding GBOs (Figure 3C).
To assess the maintenance of intra-tumoral heterogeneity in GBOs, we examined subregion samples. Genomics analyses revealed that subregion-specific genomic variants, such as a PTEN missense mutation and copy number loss of 6q and 16q in the UP-7788-PMS subregion, but not in the UP-7788-ANT subregion, were maintained in corresponding GBOs (Figure 3C). Gene expression analysis also showed that GBOs maintained signatures of subregion tumor samples (Figures 3B and 3D). As an example, RNA-seq and digital PCR detected differential expression of the gain-of-function EGFR variant III (EGFRvIII) in different subregions (ANT and PMS) of the UP-7788 tumor and corresponding GBOs (Figures 3E and 3F), which was further confirmed by immunohistology (Figure 3G).
Together, these results demonstrate that GBOs largely maintain molecular signatures of corresponding parental tumors, including inter- and intra-tumoral transcriptomic and genomic heterogeneity.
Maintenance of Cell-Type Heterogeneity and Molecular Signatures by GBOs
To further investigate cell-type heterogeneity and its molecular signatures, we performed single-cell transcriptome analysis of parental tumors from 3 patients and corresponding GBOs at 2 weeks and later time points, including two subregion samples from 1 patient (Table S4). At the single cell level, neoplastic cells from both parental tumors and derived GBOs, as determined by copy number alteration (CNA) status (Tirosh et al., 2016) and marker gene expression (Figure S4A), exhibited patient-specific clustering, whereas non-neoplastic cell types such as macrophages/microglia, T cells, stromal cells, and myelinating oligodendrocytes from different patients clustered together (Figures 4A and 4B). Macrophage/microglia cells showed similar expression of many immune-related genes in the parental tumors and GBOs at 2 weeks, including cytokines such as TNF, IL1B, and TGFB1, suggesting that certain features of the tumor microenvironment were maintained within GBOs (Figure 4C). Some of these cells exhibited microglia-associated gene signatures and others exhibited macrophage-like gene signatures with an overall enrichment of macrophage-like cell states in culture (Figure 4D). We confirmed the presence of macrophage/microglia and T cells within tumors and derived GBOs by immunohistology for IBA1 and CD3, respectively (Figure 4E). We also performed single-cell RNA-seq for UP-8036-GBOs at 8 weeks and UP-8165 and UP-8167 GBOs at 24 weeks (Table S4). We found similar distributions of cells at later time points as for the parental tumor and corresponding GBOs at 2 weeks (Figures 4F, S4B, and S4G), suggesting the maintenance of cell types and molecular signatures of parental tumors by GBOs over extended time in culture.
Figure 4. Single-Cell RNA-Seq Analyses of Parental Tumors and Corresponding GBOs.
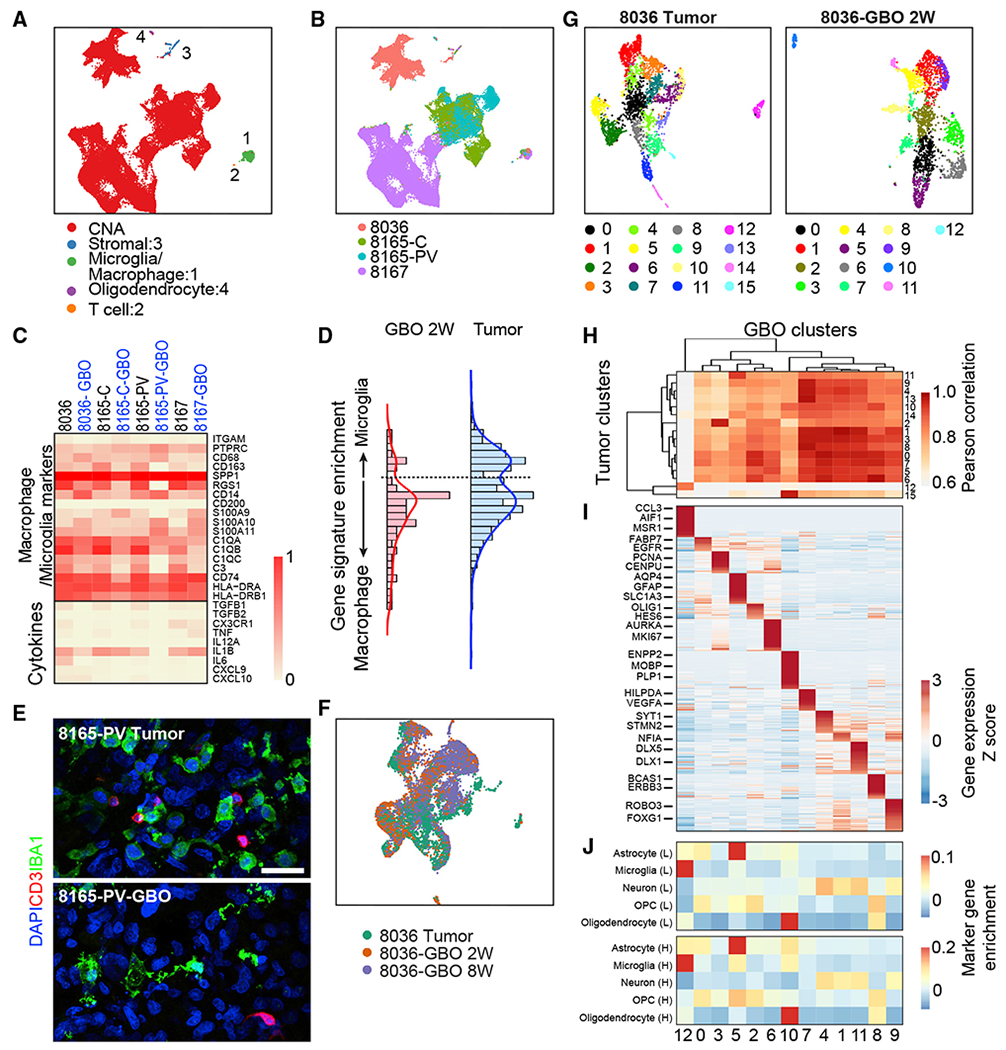
(A) UMAP plot of single-cell RNA expression from UP-8036, UP-8165-C, UP-8165-PV, and UP-8167 parental tumors and corresponding GBOs. Neoplastic cells are identified and colored by the presence of CNAs (see Figure S4A). Non-neoplastic cell clusters shared by cells from different patients and corresponding GBOs are colored and marked: 1 (microphage/microglia cluster), 2 (T cell cluster), 3 (stromal cell cluster), and 4 (mature oligodendrocyte cluster).
(B) UMAP plot of single-cell RNA expression from four parental tumors and corresponding GBOs. Cells are colored by patients and subregions.
(C) Heatmap of gene expression of selected macrophage/microglia marker genes and cytokines in the macrophage/microglia cell cluster (1 in A).
(D) Histogram of microglia versus macrophage gene signature expression in cells from the macrophage/microglia cell population from all parental tumors and all GBOs at 2 weeks.
(E) Confocal images of immunostaining for microphage/microglia marker IBA1 and T cell marker CD3 in the parental tumor and corresponding GBO at 2 weeks. Scale bar, 50 μm.
(F) UMAP plot of UP-8036 parental tumor and GBOs at 2 and 8 weeks colored by samples.
(G) UMAP plots of UP-8036 parental tumor and GBOs at 2 weeks colored by cluster. The same cluster number is listed in (H) and (J).
(H) Heatmap of gene expression Pearson correlation of clusters identified in the UP-8036 parental tumor (rows) and GBOs at 2 weeks (columns) with hierarchical clustering by Euclidian distance.
(I) Heatmap of gene expression of cluster-specific markers in UP-8036-GBOs with columns corresponding to (H). See Table S4 for the detailed gene list.
(J) Comparison of cell clusters in UP-8036 GBOs at 2 weeks (corresponding to that in H) with normal adult brain cells identified by single-nuclei RNA-seq of human adult brains in Lake et al. (2018) (L, top panel) and Habib et al. (2017) (H, bottom panel) with marker gene enrichment analysis. OPC, oligodendrocyte precursor cell.
Many different cell clusters were identified in parental tumors and corresponding GBOs at 2 weeks (Figures 4G, S4C, and S4H), reflecting the diversity of cell types and cellular states. GBO cell clusters were mapped to the parental tumor cell clusters by pairwise comparisons of whole transcriptome gene expression with a high degree of similarity, indicating that GBOs largely maintain the cellular heterogeneity of parental tumors (Figures 4H, S4D, and S4I). To further explore this heterogeneity, marker genes were identified for each GBO cell cluster (Table S4). For UP-8036-GBOs, we identified neoplastic populations of proliferating cells, oligodendrocyte precursor cell (OPC)-like, astrocyte-like, oligodendrocyte-like, and neuron-like cells (Figure 4I), similar to results recently reported in primary tumors (Neftel et al., 2019). Comparing their transcriptome profiles to two independent single-nuclei RNA-seq datasets for adult human non-tumor brain cells (Habib et al., 2017; Lake et al., 2018) revealed that these neoplastic cells share similarities with different cell types in the adult human brain (Figure 4J). Similarly, UP-8165 and UP-8167 exhibited multiple populations of neoplastic cells with distinct transcriptomic features in parental tumors and corresponding GBOs (Figures S4E and S4J). Notably, UP-8165-C contained a subregion-specific and distinct population of cells not found in UP-8165-PV in both parental tumors and corresponding GBOs (Figure S4B). This cell population was identified by high expression levels of GPNMB (Figure S4B), which has been linked to worse prognoses in glioblastomas (Kuan et al., 2006).
Together, single-cell RNA-seq analyses highlight marked cellular heterogeneity in GBOs and further support that GBOs recapitulate cell-type heterogeneity and molecular properties of corresponding parental tumors.
Robust Engraftment and Aggressive Infiltration by GBOs upon Xenograft
We next asked whether GBOs reproduce glioblastoma properties in vivo with murine xenografts. To minimize the survival bias of different cell types following dissociation and to maintain the integrity and cytoarchitecture of GBOs, we transplanted intact GBOs into adult immunodeficient mouse brains using an established brain organoid transplantation protocol (Mansour et al., 2018) (Figure S5A). We transplanted a total of 8 GBO samples from 7 patients into 5–7 animals for each GBO sample, all of which exhibited engraftment when examined at 1–3 months, indicating a very efficient xenograft model (Table S5).
One major hallmark of glioblastomas is infiltration of tumor cells into the surrounding brain tissue, which is often associated with a FLAIR signal on the patient MRI scan (Kelly et al., 1987) (Figure 5A). At 2 months post-transplantation, analyses of xenografted UP-7788-PMS and UP-7790 GBOs revealed similar tissue architecture at original xenograft sites and infiltrated areas as compared to those of corresponding parental tumors by H&E staining (Figure 5B). We confirmed extensive ipsilateral and contralateral infiltration of GBO-derived cells by immunohistology of human-specific antigens HuNu and STEM121 (Figures 5C–5F). Quantification of infiltrated cells within one brain section of 35 μm thickness showed over 10,000 and 20,200 cells migrated out from original xenograft sites, respectively (Figures 5C and 5E). The majority of infiltrating cells migrated in the white matter, including a subset that crossed the corpus callosum and into the cortex, while a few invaded subcortical areas (Figures 5C and 5E). Reconstruction of the UP-7790 GBO xenograft using serial brain sections showed aggressive tumor growth and infiltration within 3 months (Video S1). Among all xenografted GBOs derived from different tumors, 92% (48 out 52) displayed various degrees of infiltration into the surrounding mouse brain tissue (Table S5). Interestingly, we noted a resemblance of a satellite tumor phenotype in 3 out of 6 UP-7803-GBO xenografted animals and the corresponding original tumor as seen in the patient MRI scan (Figures S5B and S5C). We did not observe such satellite tumor phenotypes in any other GBO xenografts or patient MRI scans, suggesting maintenance of tumor-specific features in these xenografts.
Figure 5. Orthotopic Transplantation of GBOs into Adult Immunodeficient Mice Displays Efficient Engraftment and Extensive Infiltration into the Brain Parenchyma.
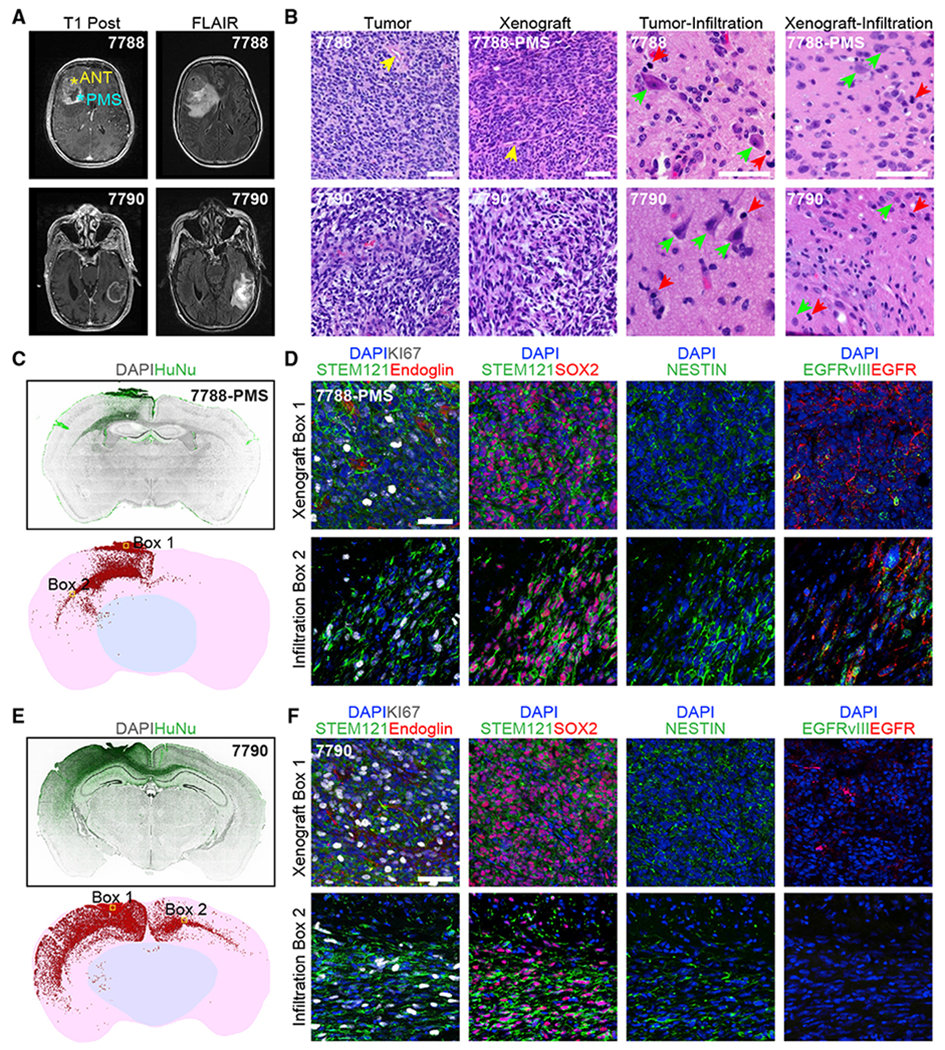
(A) MRI T1 post-contrast (left) and FLAIR (right) patient brain images.
(B) Sample H&E staining images of the tumor bulk versus infiltrated areas of parental patient tissues and the original xenograft sites and infiltrated areas for corresponding GBOs at 2 months post-transplantation. Prominent blood vessels (yellow arrow heads) are observed in both the tumor bulk in patients and original GBO xenograft sites in mice. Infiltrated areas are shown with human neurons and mouse neurons (green arrow heads) and tumor cells (red arrow heads), respectively. Scale bar, 100 μm.
(C and D) Xenograft of UP-7788-PMS-GBO at 2 months post-transplantation. (C) Coronal section views of human nuclear antigen (HuNu) immunostaining (top) and reconstruction for quantification of infiltrated cells (bottom). Each red dot represents a HuNu+ cell. (D) Sample confocal images of areas in box 1 and 2 in (C, bottom panel) for immunostaining of human-specific cytoplasmic antigen (STEM121) showing the extensive vascularization from the host (endoglin immunostaining) in the original xenograft site, and proliferation (KI67 immunostaining), progenitor (SOX2, NESTIN immunostaining), and EGFR/mutant EGFRvIII expression status of tumor cells in the original xenograft site (box 1) and the infiltrated area (box 2). Scale bar, 50 μm.
(E and F) Xenograft of UP-7790-GBO at 2 months post-transplantation. Cornal section image and reconstruction (E) and sample confocal images in box 1 and 2 (F) are similar as in (C) and (D).
Consistent with the high angiogenic hallmark of glioblastomas, all xenografted GBOs with the exception of those from one patient were extensively vascularized by host Endoglin+ endothelial cells at 2 months after transplantation (Figures 5D, 5F, and S5D; Table S5). Populations of KI67+ proliferative cells and SOX2+ or NESTIN+ progenitors were found at both initial xenograft sites and distant infiltrated areas (Figures 5D, 5F, and S5D). It has been reported that maintenance of EGFR amplification or EGFRvIII mutation can be challenging in tumor cell cultures and exogenous overexpression of wild-type EGFR or mutant EGFRvIII is often required for in vitro and in vivo modeling (Bigner et al., 1990; Pandita et al., 2004). Importantly, in our model, both EGFR amplifications and EGFRvIII mutant expression were retained in xenograft sites and infiltrated areas by GBOs derived from EGFR amplified and EGFRvIII+ tumors (Figures 5D and S5D).
Our xenografts also exhibited rapid infiltration within 1 month in vivo (Figure S5E). In addition, GBOs recovered from the biobank were successfully engrafted and exhibited prominent infiltration within 1 month and retained EGFRvIII mutant expression (Figure S5E; Table S5).
Taken together, these results demonstrate that orthotopic transplantation of intact GBOs offers a timing advantage over existing PDX models with high efficiency of engraftment, robust infiltration, and retention of key driver mutation expression.
Modeling Targeted Drug Treatments Using GBOs
We next applied our GBO model for testing treatment responses in vitro. To mimic the post-surgical standard of care treatment, we subjected 8 GBO samples from 7 patients (Table S6) to a single exposure of 10 Gy radiation with concurrent temozolomide (TMZ, 50 μM) treatment for a week. The therapeutic response was evaluated by quantifying the percentage of cells expressing KI67, which has previously been clinically associated with overall patient survival in treated tumor specimens (Bagley et al., 2019). GBOs from 3 out of 7 patients exhibited decreased percentages of KI67+ cells with temozolomide and radiation treatment (Figures 6A and 6B). One patient (UP-7788) with reduced KI67+ cells in GBOs had a radiographic reduction of tumor volume following treatment of recurrence at 1 month (Figure S6A). Another patient (UP-7884), with reduced KI67+ cells in GBOs from 2 subregions, had no recurrence in the temporal region and exhibited a pattern favoring pseudo-progression in the frontal region (Figure S6B), which has been associated with extended survival (Brandes et al., 2008; Roldán et al., 2009). Meanwhile, 3 patients (UP-7790, UP-7803, and UP-7966) with no significant changes in KI67+ cells in GBOs (Figure 6B) had a below median survival after treatment (1, 3, and 8 months, respectively). Future studies with a larger sample size will be necessary to better establish the correlation between patient and GBO treatment responses. O6-methylguanine-DNA-methyltransferase (MGMT) promoter methylation has been reported to be a predictive marker for response to temozolomide and radiation treatment (Brandes et al., 2008). We did not find a clear association between GBO responses and MGMT methylation from this sample set (Figures 6A and 6B). To determine whether the treatment responses had any underlying molecular basis, we performed gene set enrichment analysis (GSEA) on pre-treated GBO and parental tumor transcriptomes stratified by KI67 responses. The radiation-sensitive group enriched for gene sets associated with response to radiation therapy and tumor necrosis factor (TNF) signaling, while the resistant group enriched for gene sets related to neural stem cells (Figures 6C and S6C; Table S6).
Figure 6. Therapeutic Testing of GBOs In Vitro.
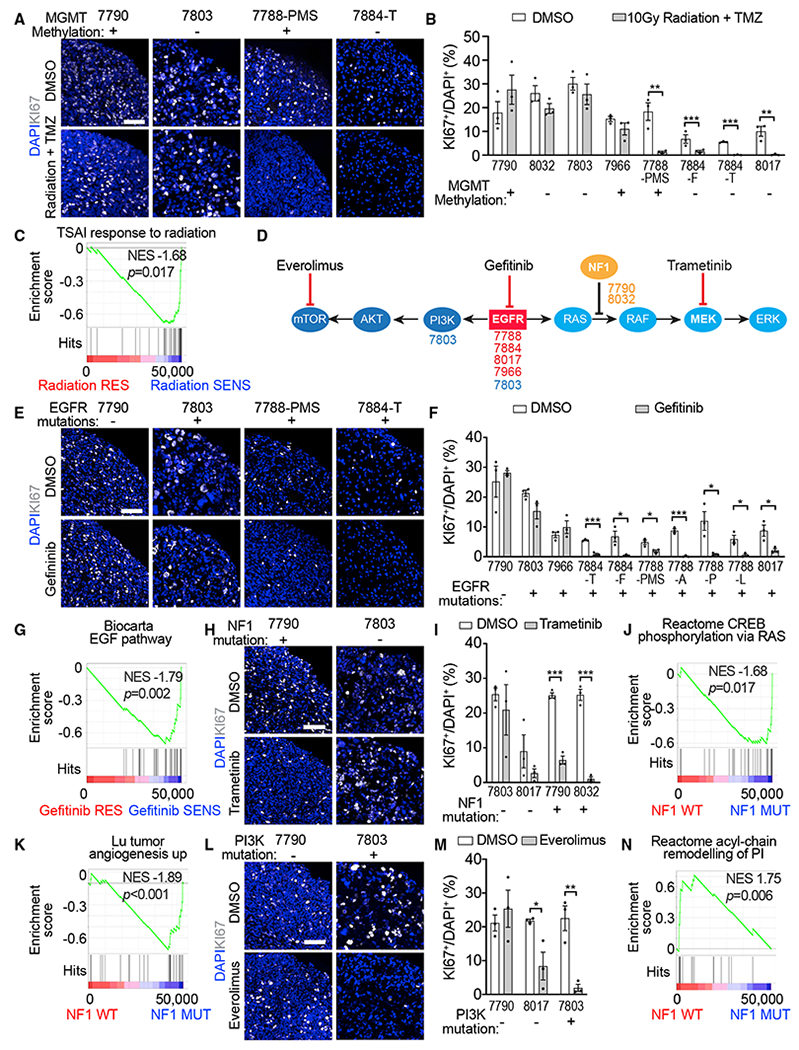
(A–C) Treatment of GBOs with 10 Gy radiation and temozolomide (TMZ; 50 μM). (A) Sample confocal images of KI67 immunostaining and DAPI staining. Scale bar, 100 μm. The MGMT methylation status identified in each parental tumor is listed. (B) Quantification of percentages of KI67+ cells among DAPI+ cells. See Table S6 for the age of GBOs used in the analysis. Dots represent individual data points and bar values represent mean ± SEM (n = 3; *p < 0.05; **p < 0.01; ***p < 0.001; Student’s t test). (C) Gene set enrichment in GBOs with significant reduction of KI67+ cells following radiation and temozolomide treatment.
(D) Schematic of targeted treatment strategy showing genetic pathways, location of tumor-specific mutations, and mechanism of action of targeted treatments. The mutations were based on identifications in patient tumor samples via clinical sequencing (see Table S1).
(E–G) Treatment of GBOs with gefitinib (5 μM). Sample images (E) and quantification (F) are similar as in (A) and (B). Shown in (G) is gene set enrichment in samples with significant reduction of KI67+ cells following gefitinib treatment.
(H–K) Treatment of GBOs with trametinib (1 μM). Sample image (H) and quantification (J) are similar as in (A) and (B). Also shown are gene set enrichment in samples with NF1 mutations (J and K).
(L–N) Treatment of GBOs with everolimus (1 μM). Sample images (L) and quantification (M) are similar as in (A) and (B). Shown in (N) is gene set enrichment in samples without NF1 mutations.
We next explored more targeted drug treatments for specific signaling pathways based on somatic mutations identified in parental tumors from a routinely performed clinical sequencing panel (Figure 6D; Table S1). EGFR tyrosine kinase inhibition did not improve the overall survival of patients with glioblastomas (Rich et al., 2004; Uhm et al., 2011), but showed a survival benefit in patients with mutated EGFR in the absence of downstream PTEN mutation (Arif et al., 2018; Mellinghoff et al., 2005). We hypothesized that the clinical benefit could be mutation-specific. We therefore treated 10 GBO samples derived from 6 patients with the EGFR inhibitor gefitinib (5 μM) for a week. Reduction of KI67+ cells was observed in 7 GBO samples from 3 patients (Figures 6E and 6F), all of which had EGFR alterations (Table S1). These results provide functional evidence suggesting that these EGFR alterations drive cell proliferation in these tumors. Two tumors had EGFR alterations (UP-7966 with a copy number gain and UP-7803 with clinical detection of EGFRvIII) but did not show a reduction of KI67+ cells in GBOs (Figure 6F), indicating that mutation analysis alone without functional testing is not sufficient to predict treatment responses. GSEA for the gefitinib-responders enriched for gene sets associated with EGF signaling and immune-related CCR5 signaling, while the gefitinib-resistant group enriched for gene sets associated with stem cell-associated neural development (Figures 6G and S6D; Table S6).
Mutations downstream of targeted tyrosine kinases are known to contribute to therapeutic resistance (de Bruin et al., 2014; Huang and Fu, 2015). Two tumors had downstream NF-1 mutations. We therefore tested the MEK inhibitor trametinib (1 μM) to inhibit signaling downstream of NF-1 (Figure 6D). There was a significant reduction of KI67+ cells in NF-1 mutant GBOs, but not in the EGFR-mutated or PI3K-mutated GBOs (Figures 6H and 6I). GSEA for NF1 mutants enriched for gene sets associated with RAS signaling and angiogenesis (Figures 6J and 6K; Table S6), while the NF1 WT group enriched for a gene set associated with acyl chain remodeling of phosphoinositol, the target of PI3K (Figure 6N), and glutathione conjugation (Figure S6E; Table S6), which is associated with drug resistance (Backos et al., 2012). We next tested the effect of inhibiting PI3K-induced mTOR activation for NF1 WT tumors (Figure 6D). Everolimus is an mTOR inhibitor with known brain penetration and is currently used to treat subependymal giant cell astrocytoma (Krueger et al., 2010). Treatment with everolimus (1 μM) in UP-7803-GBOs, which had a PI3K mutation, exhibited a near complete reduction of KI67+ cells (Figures 6L and 6M). Meanwhile, treatment of UP-8017-GBOs, which had an EGFR alteration upstream to both the RAS and PI3K pathways, had a partial reduction of KI67+ cells (Figure 6M). In contrast, UP-7790-GBOs, which had a mutation in the parallel RAS pathway, did not show any reduction (Figures 6L and 6M). The dichotomous treatment effect between PI3K and RAS pathway mutations was further explored with measurement of individual GBO growth. Following an initial 1 week of drug exposure and subsequent 2 weeks in the normal GBO media, mutation-specific resistance and treatment response were also observed when measuring GBO size over time (Figures S6F and S6G). Dose-response analysis with trametinib exposure to UP-7790-GBOs demonstrated growth inhibition at ~100 nM, while everolimus exposure diminished the size of UP-7803-GBOsat ~10 nM (Figure S6FH). We further confirmed the in vivo efficacy of trametinib treatment on reducing tumor cell proliferation upon transplantation of UP-7790-GBOs recovered from the biobank into the adult immunodeficient mouse brain (Figures S6I–S6K).
Together, these results show that responses of GBOs derived from different tumors to various drug treatments are heterogeneous, and the efficacy of targeted treatments is largely consistent with the mutational status and pathway enrichment in tumors. These results demonstrate the value of GBOs for rapid, functional testing of personalized drug treatment responses.
Modeling Personalized CAR-T Immunotherapy Using GBOs
Despite success in many blood cancers (June et al., 2018), CAR-T cell immunotherapy in solid tumors has not been as effective and remains to be further developed (Newick et al., 2017). Recently, the EGFRvIII variant commonly found in glioblastomas has been targeted by CAR-T cells in clinical trials (Goff et al., 2019; O’Rourke et al., 2017). Despite the ability of these EGFRvIII-specific CAR-T cells to penetrate tumors, treatment efficacy was unclear. Current in vitro models testing CAR-T cell therapy for glioblastomas generally lack the cellular heterogeneity and maintenance of specific mutant antigens, such as EGFRvIII, in prolonged cultures and often rely on overexpression of antigens in tumor cells for testing (Johnson et al., 2015). To assess the utility of our GBO model, which maintains cellular heterogeneity and endogenous EGFRvIII expression, in testing emerging immunotherapies in glioblastomas, we co-cultured GBOs with 2173BBz CAR-T cells designed to react specifically with cells expressing EGFRvIII (O’Rourke et al., 2017). CAR-T cells targeting CD19 for the B cell blood lineage were used as a control (Porter et al., 2011). We tested CAR-T cell co-cultures with 6 GBO samples, including GBOs containing high (UP-8036) and low (UP-8017) percentages of EGFRvIII+ cells and two pairs of GBOs from subregional sampling of tumors in which one subregion contained a high percentage of EGFRvIII+ cells (UP-7788-PMS and UP-7884-F) and the other subregion did not (UP-7788-ANT and UP-7884-T) as confirmed by immunostaining (Figures 7A and S7A). GBOs were analyzed at 0, 24, and 72 h after CAR-T cell addition for invasion and proliferation of T cells, tumor cell death, and EGFRvIII antigen loss. Both CD19 and 2173BBz CAR-T cells invaded all GBOs, but marked expansion of CAR-T cells was observed only when 2173BBz CAR-T cells were incubated with EGFRvIII+ GBOs (Figures 7A and S7A). This was evidenced by an increased presence of CD3+T cells, many of which were KI67+ within GBOs (Figures 7B and S7B). This increased CAR-T cell expansion was accompanied by increased cleaved-caspase-3 signal and a decreased ratio of EGFRvIII/EGFR signal intensity in GBOs (Figures 7A, 7C, and 7D), suggesting that EGFRvIII+ cells were being targeted and killed by 2173BBz CAR-T cells. To further support this notion, immunostaining for granzyme B, an effector of T cell killing (Shi et al., 2000), revealed T cells filled with granules near apoptotic EGFRvIII+ cells (Figure S7C). These granules often localized on the side of the T cell closest to the EGFRvIII+ tumor cell. ELISA for cytokines interleukin (IL)-2, TNF-α, and interferon (IFN)-γ revealed increases in their levels only in conditions where 2173BBz CAR-T cells were incubated with EGFRvIII+ GBOs, suggesting antigen recognition and subsequent T cell activation (Figure S7D). Many EGFR+ EGFRvIII− tumor cells persisted after 3 days (Figures 7A and S7A), suggesting that the 2173Bbz CAR-T cells are fairly specific to their target and are unable to completely eradicate all tumor cells under our conditions. Together, these results demonstrate the utility of GBOs for rapid testing of antigen-specific CAR-T cell treatment responses with the endogenous target in culture.
Figure 7. Modeling Immunotherapy with Co-cultures of CAR-T Cells and GBOs.
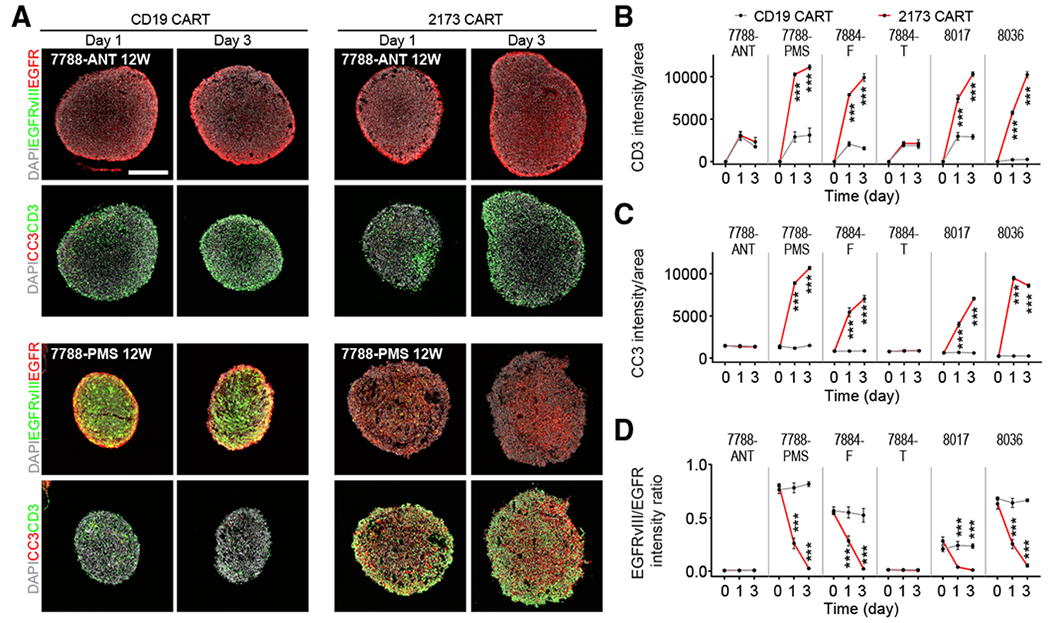
(A) Sample confocal images of immunostaining of EGFR, EGFRvIII, cleaved-caspase-3 (CC3), and CD3 after 1 and 3 days of co-culture with either CD19 or 2173BBz CAR-T cells. Scale bar, 200 μm.
(B–D) Summary of quantifications of averaged signal intensity of CD3 (B), CC3 (C), and averaged EGFRvIII/EGFR signal intensity ratio (D) in GBOs after co-culture with either CD19 or 2173BBz CAR-T cells. Values represent mean ± SEM (n = 3; ***p < 0.001; two-way ANOVA with uncorrected Fisher LSD test).
DISCUSSION
In contrast to previously reported cancer organoids that have been generated from dissociated tumor cells mostly of epithelial origin with added mitogens and Matrigel, our GBOs retain native cell-cell interactions and are cultured in a defined medium without exogenous EGF/bFGF or extracellular matrix. These GBOs recapitulate the heterogeneity of their corresponding parental tumors as evidenced by (1) histology illustrating similar tissue architecture and cellular morphologies, (2) immunohistology displaying the presence and continual generation of a similar spectrum of diverse cell types, (3) RNA-seq showing maintenance of similar transcriptomic signatures, (4) whole-exome sequencing confirming the preservation of somatic variants and CNVs at similar frequencies, and (5) single-cell RNA-seq revealing the maintenance of different cell populations and their gene expression profiles. Additionally, many GBOs recapitulate specific elements of the tumor microenvironment, such as hypoxia gradients, microvasculature, and immune cell populations. By orthotopic transplantation of intact GBOs into mice, our xenografts displayed efficient engraftment with rapid and aggressive infiltration. We further demonstrate the utility of GBOs by showing that drug treatments and engineered CAR-T cells elicit differential responses depending on tumor-specific mutations. Our robust and rapid method for GBO generation with sufficient throughput for targeted testing makes it possible to accelerate personalized medicine efforts and influence clinical decisions. We have also generated a live biobank of GBOs with diverse mutational profiles as a resource for due future biological studies and therapeutic testing.
A Live Culture and Biobank of Organoids Recapitulating Heterogeneity of Patient Tumors
One important feature of our organoid generation method is that by avoiding single-cell dissociation, we preserve native cell-cell interactions to enable GBO formation and expansion in the absence of exogenous EGF/bFGF, serum and extracellular matrix, which may help GBOs maintain properties similar to the parental tumors. The use of a fully defined culture medium enhances the reproducibility of cultures (Gjorevski et al., 2016) and facilitates its use in future clinical applications. Our GBOs maintain relatively similar proportions of actively proliferating cells as the corresponding parental tumors. Traditional tumor cultures clonally select for highly proliferative cells in growth factor-rich media, reducing the proportion of the more slowly proliferating and non-proliferating cells originally present within parental tumors. These populations may play an important role in glioblastoma pathogenesis and treatment resistance.
Organoid biobanks have been previously established for a number of cancers, including pancreatic, liver, prostate, breast, bladder, ovarian, and gastrointestinal cancers, but not yet for glioblastomas (Bleijs et al., 2019). We provide a resource of 70 biobanked GBOs from different patients that captures major genomic alterations associated with glioblastoma pathogenesis (Figure S2; Table S1). Our optimized freeze-and-thaw methods allow efficient recovery and continuous growth of GBOs that maintain their resemblance to parental tumors and can efficiently engraft and rapidly infiltrate upon transplantation into the adult mouse brain. We provide detailed characterization for many of these biobanked GBOs, including histology, RNA-seq, whole-exome sequencing, and responses to different drugs and CAR-T therapies (Table S2). As tumor collection and GBO generation is ongoing, this biobank will be a useful resource for future biological studies and testing therapeutics for glioblastomas.
Clinically Relevant Timing
Given the short survival period after diagnosis for the majority of glioblastoma patients, the timing of testing personalized treatment strategies is critical. Compared to the variable efficacy and prolonged generation time for tumor cell lines and PDX models, our methodology is robust for generating patient-derived GBOs from a wide range of glioblastomas within 1–2 weeks from the initial surgical resection. This provides a timely platform with sufficient throughput to test personalized treatment strategies based on individual patient tumor characteristics. Future studies can test the effectiveness of several therapeutic strategies with GBOs from multiple tumor subregions before treatment initiation. Given the heterogeneity of glioblastomas, in vitro testing of various therapeutic options may also help refine patient enrollment in clinical trials. Our GBO transplantation model exhibits rapid and aggressive infiltration phenotypes within one month and can be used to test in vivo treatment responses in a timely fashion.
Practical Limitations and Future Applications
While our GBOs resemble many features of their corresponding tumors, the model is not without its limitations. Optimal tumor tissue acquisition relies on close coordination with the neurosurgeon intra-operatively to ensure that viable tissue is resected en bloc without cauterization. Close coordination with neuropathology is also important to confirm the diagnosis and limit the time span between resection and tissue processing, which is critical for maximum reliability of GBO generation. Our ability to maintain and expand GBOs over very long periods has been variable, likely owing to both tissue quality and the diverse composition and growth characteristics of tumors themselves. In keeping with its more aggressive growth phenotype, the vast majority (96.4%) of IDH1-WT tumors have resulted in plentiful GBOs, but our limited experience with IDH1 mutant tumors and recurrent tumors showed reduced success rates (66.7% and 75.0%, respectively). Further optimization is likely required to establish and propagate these cultures more efficiently. Likewise, our methodology could be adapted and optimized to culture organoids from other brain tumors, such as medulloblastoma or ependymoma. Routine in-depth characterization of all tumors and GBOs as described in this study may be challenging due to the variation in the initial tumor volume obtained and GBO expansion rate, but ample tissue is usually available for basic characterization and testing targeted therapies.
We report the partial preservation of microvasculature and immune cells in some GBOs, which may prove useful in better understanding the tumor microenvironment. However, we found evidence of divergence from primary tumors over time with a decreased abundance of vasculature and macrophage/microglia populations and lower expression of immune-related genes in GBOs. This is not unexpected as our culture conditions were optimized to preserve tumor cell viability and growth, and resident immune cells have a limited lifespan and become diluted without expansion. Future studies involving the immune microenvironment would ideally be performed early after GBO establishment or after exogenous immune cell reconstitution (Neal et al., 2018). Given the differential proliferation and death of different cell populations in GBOs, the composition of cell types could also drift overtime and it will be best to analyze soon after GBOs are established.
Access to ongoing cultures of live glioblastoma tissue that resemble the parental tumor provides a unique avenue for many future applications. Viral barcoding labeling can be used to trace the division behavior and clonal lineage of specific cell subtypes within GBOs, which can be combined with single-cell RNA-seq and bioinformatic approaches to infer lineage trajectories. Our system should also enable the study of genetic and transcriptional alterations by CRISPR-Cas9 genome editing, small interfering RNA (siRNA) knockdowns, and overexpression vectors, to gain mechanistic insight into glioblastoma pathogenesis. Our platform for drug testing may lead to new therapeutic strategies. Our 3D GBO model additionally provides a platform to test and optimize CAR-T therapies for solid tumors in vitro before in vivo testing. Importantly, our GBOs recapitulate the endogenous expression of antigens, allowing for a more accurate assessment of CAR-T cell target reactivity, threshold for responses, and specificity, as compared to models engineered to overexpress specific antigens. Therapeutic testing can be extended to GBO xenografts, which recapitulate tumor infiltration into the surrounding brain tissue. The effects of specific drugs, immunotherapy, and/or radiation treatments on GBO composition and regrowth capacity may provide insight into treatment-resistant and recurrence-initiating tumor cell populations. Moreover, matched primary and recurrent parental/GBO pairs may provide additional insight into more invasive and treatment-resistant tumor cell populations.
In summary, our patient-derived GBO model recapitulates the heterogeneity and key features of glioblastomas and has the potential for timely testing of personalized treatment responses and broad applications in basic and translational research of glioblastomas.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Hongjun Song (shongjun@pennmedicine.upenn.edu). There are restrictions to the availability of biobanked glioblastoma organoids due to the lack of an external centralized repository for their distribution and our need to maintain the stock; however, biobanked organoids generated in this study will be made available upon reasonable request following approval by an internal review board and completion of a Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Subjects
The use of human brain tissue and peripheral blood samples was coordinated by the University of Pennsylvania Tumor Tissue/Biospecimen Bank following ethical and technical guidelines on the use of human samples for biomedical research purposes. Patient glioblastoma tissue and peripheral blood samples were collected at the Hospital of the University of Pennsylvania after informed patient consent under a protocol approved by the University of Pennsylvania’s Institutional Review Board. All patient samples were de-identified before processing. A total of 58 patient cases (including recurrent cases) from both male and female subjects between the ages of 21-90 years old were included in the present study. Table S1 summarizes detailed epidemiological data for each subject and histological data for each tumor provided by the Neurosurgery Clinical Research Division (NCRD) at the University of Pennsylvania, and testing results for a panel of disease-associated genomic alterations (Agilent Haloplex assay, Illumina HiSeq2500), fusion transcripts (Illumina HiSeq2500), and MGMT promoter methylation (PyroMark Q24, QIAGEN) provided by the University of Pennsylvania Center for Personalized Diagnostics.
Animal Models
All animal procedures used in this study were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania. Animals were housed at a maximum of five per cage with a 14-hour light/10-hour dark cycle with food and water ad libitum. Female 4-8-week-old athymic nude (NU/J) mice (The Jackson Laboratory, RRID: IMSR_JAX:002019) were used for all experiments and were randomly assigned to treatment groups. Animals were monitored at a minimum of twice weekly for weight loss and were examined routinely for physical and/or neurological abnormalities.
METHOD DETAILS
Collection, Dissection, and Processing of Patient Glioblastoma Samples
Fresh surgically resected glioblastoma tissue was placed in sterile phosphate buffered saline and taken immediately to the University of Pennsylvania Department of Pathology to confirm a preliminary diagnosis of high-grade glioma by the attending neuropathologist (M.N.). In cases where a large amount of en bloc tissue was available, the tissue was sub-divided into anatomically distinct subregions for analysis of intra-tumoral heterogeneity. After preliminary diagnosis of glioblastoma was confirmed, the tissue was distributed and placed in Hibernate A medium (BrainBits) kept at 4°C. For reliable organoid generation it was imperative that the tissue was processed immediately as a prolonged time between surgical removal and tissue processing reduced the reliability of GBO generation. The tissue was transferred to a sterile glass dish with H+GPSA medium containing Hibernate A, 1X GlutaMax (Thermo Fisher Scientific), 1X PenStrep (Thermo Fisher Scientific), and 1X Amphotericin B (Thermo Fisher Scientific) for dissection under a stereomicroscope (Zeiss) within a laminar flow biosafety cabinet. The amount of glioblastoma tissue received ranged from 0.5 to 2 mL in volume. The resected tumors were minced into approximately 0.5 to 1 mm diameter pieces using fine dissection scissors (Fine Science Tools) and washed with H+GPSA medium to remove cellular debris. Pieces containing substantial amounts of necrosis or surrounding brain tissue were removed. Tumor pieces were incubated in 1X RBC lysis buffer (Thermo Fisher Scientific) under gentle rotation for 10 minutes at room temperature to lyse the majority of contaminating red blood cells. RBC lysis buffer was aspirated, and tumor pieces were washed with H+GPSA medium. Several tumor pieces were snap frozen for bulk RNA sequencing and whole exome sequencing. For histological studies, several tumor pieces were placed directly in 4% methanol-free formaldehyde (Polysciences) diluted in DPBS (Thermo Fisher Scientific) for 1 hour at room temperature under gentle rotation. After fixation, the tumor pieces were washed in DPBS and cryoprotected by overnight incubation in 30% sucrose (Sigma-Aldrich) in DPBS at 4°C. Tumor pieces were placed in a plastic cryomold (Electron Microscopy Sciences) and snap frozen in tissue freezing medium (General Data) on dry ice. Frozen tissue was stored at −80°C until processing.
Generation of GBOs from Resected Patient Glioblastoma Tissue
The remaining tumor pieces not set aside for RNA sequencing, whole exome sequencing, or histology were distributed in ultra-low attachment 6-well culture plates (Corning) with 4 mL of GBO medium containing 50% DMEM:F12 (Thermo Fisher Scientific), 50% Neurobasal (Thermo Fisher Scientific), 1X GlutaMax (Thermo Fisher Scientific), 1X NEAAs (Thermo Fisher Scientific), 1X PenStrep (Thermo Fisher Scientific), 1X N2 supplement (Thermo Fisher Scientific), 1XB27 w/o vitamin A supplement (Thermo Fisher Scientific), 1X 2-mercaptoethanol (Thermo Fisher Scientific), and 2.5 μg/ml human insulin (Sigma) per well and placed on an orbital shaker rotating at 120 rpm within a 37°C, 5% CO2, and 90% humidity sterile incubator. Roughly 75% of the medium was changed every 48 hours by tilting the plates at a 45° angle and aspirating the medium above the sunken GBOs. Within the first week of culture, the tumor pieces often shed cellular and blood debris making the medium slightly cloudy. The shedding soon ceased, and the tumor pieces generally formed rounded organoids within 1-2 weeks, depending on tissue quality and patient-specific tumor growth characteristics. The criteria for successful establishment of GBOs from a given patient’s tumor was that the micro-dissected tumor pieces survived for 2 weeks, developed a spherical morphology, and continuously grew in culture. GBOs cultured for prolonged periods of time (> 1 month) were routinely cut to ~200-500 μm diameter pieces using fine dissection scissors to prevent substantial necrosis within the center due to limited nutrient and oxygen diffusion. GBOs were sampled for RNA sequencing, whole exome sequencing, and histology by the same methods as the corresponding parental tumor pieces.
GBO Growth Analysis
To measure the growth of GBOs over time, similarly sized GBOs (0.5 −1 mm diameter) were placed into individual wells of a 48-well tissue culture plate with 300 μL of GBO medium per well. Images of individual GBOs were taken every week using a brightfield microscope and Zen software. The 2D projected area of each GBO was quantified in ImageJ by carefully outlining each GBO and measuring the area within the outlined region. The 2D area at each time point was divided by the 2D area at time 0 to calculate a growth ratio for each time point. Ten individual GBOs were measured for each GBO sample. GBOs recovered from the biobank were cultured for 3 days before the start of analysis of GBO growth.
Tissue Processing and Immunohistology
Serial tissue sections (20 μm for GBOs and 35 μm for xenografted rodent brains) were sliced using a cryostat (Leica, CM3050S), and melted onto charged slides (Thermo Fisher Scientific). Slides were dried at room temperature and stored at −20°C until ready for immunohistology. For immunofluorescence staining, the tissue sections were outlined with a hydrophobic pen (Vector Laboratories) and washed with TBS containing 0.1% Tween-20 (v/v). Tissue sections were permeabilized and non-specific binding was blocked using a solution containing 10% donkey serum (v/v), 0.5% Triton X-100 (v/v), 1% BSA (w/v), 0.1% gelatin (w/v), and 22.52 mg/ml glycine in TBST for 1 hour at room temperature. For rodent brain sections, mouse-on-mouse blocking reagent (Vector Laboratories) was added to the blocking solution. The tissue sections were incubated with primary antibodies (see the Key Resources Table) diluted in TBST with 5% donkey serum (v/v) and 0.1% Triton X-100 (v/v) overnight at 4°C. After washing in TBST, the tissue sections were incubated with secondary antibodies (see the Key Resources Table) diluted in TBST with 5% donkey serum (v/v) and 0.1% Triton X-100 (v/v) for 1.5 hours at room temperature. After washing with TBST, sections were incubated with TrueBlack reagent (Biotium) diluted 1:20 in 70% ethanol for 1 minute to block autofluorescence due to lipofuscin and blood components. After washing with DPBS, slides were mounted in mounting solution (Vector Laboratories), coverslipped, and sealed with nail polish.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat polyclonal anti-Doublecortin (C-18) | Santa Cruz Biotechnology | Cat# sc-8066; RRID: AB_2088494 |
| Goat polyclonal anti-Endoglin/CD105 | R&D Systems | Cat# AF1320; RRID: AB_354735 |
| Goat polyclonal anti-Nestin (C-20) | Santa Cruz Biotechnology | Cat# sc-21247; RRID: AB_650014 |
| Goat polyclonal anti-Sox2 (Y-17) | Santa Cruz Biotechnology | Cat# sc-17320; RRID: AB_2286684 |
| Mouse monoclonal anti-BLBP (AT1D1) | Abcam | Cat# ab131137; RRID: AB_11157091 |
| Mouse monoclonal anti-CD3 | BioLegend | Cat# 344802; RRID: AB_2043995 |
| Mouse monoclonal anti-EGFR (H11) | Thermo Fisher Scientific | Cat# MA1-12693; RRID: AB_1074165 |
| Mouse monoclonal anti-GFAP (GA5) | Millipore | Cat# MAB360; RRID: AB_11212597 |
| Mouse monoclonal anti-Granzyme B | R&D Systems | Cat# MAB2906; RRID: AB_2263752 |
| Mouse monoclonal anti-Nuclei, human (235-1) | Millipore | Cat# MAB1281; RRID: AB_94090 |
| Mouse monoclonal anti-Ki67 (B56) | BD Biosciences | Cat# 550609; RRID: AB_39377 |
| Mouse monoclonal anti-STEM121 | Takara Bio | Cat# Y40410; RRID: AB_2801314 |
| Mouse monoclonal anti-VWF (C-12) | Santa Cruz Biotechnology | Cat# sc-365712; RRID: AB_10842026 |
| Rabbit polyclonal anti-CD31 | Abcam | Cat# ab28364; RRID: AB_726362 |
| Rabbit polyclonal anti-Cleaved Caspase-3 (Asp175) | Cell Signaling Technology | Cat# 9661; RRID: AB_2341188 |
| Rabbit monoclonal anti-EGF Receptor vIII (D6T2Q) | Cell Signaling Technology | Cat# 64952; RRID: AB_2773018 |
| Rabbit polyclonal anti-Hopx (FL-73) | Santa Cruz Biotechnology | Cat# sc-30216; RRID: AB_2120833 |
| Rabbit polyclonal anti-Iba1 | Wako | Cat# 019-19741; RRID: AB_839504 |
| Rabbit polyclonal anti-Ki67 | Abcam | Cat# ab15580; RRID: AB_443209 |
| Rabbit monoclonal anti-Olig2 | Abcam | Cat# ab109186; RRID: AB_10861310 |
| Rabbit polyclonal anti-S100b | Sigma-Aldrich | Cat# S2644; RRID: AB_477501 |
| Rat monoclonal anti-CD3 (CD3-12) | GeneTex | Cat# GTX11089; RRID: AB_369097 |
| Donkey polyclonal anti-Goat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11055; RRID: AB_2534102 |
| Donkey polyclonal anti-Goat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | Thermo Fisher Scientific | Cat# A-21432; RRID: AB_2535853 |
| Donkey polyclonal anti-Goat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-21447; RRID: AB_2535864 |
| Donkey polyclonal anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-21202; RRID: AB_141607 |
| Donkey polyclonal anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | Thermo Fisher Scientific | Cat# A-31570; RRID: AB_2536180 |
| Donkey polyclonal anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-31571; RRID: AB_162542 |
| Donkey polyclonal anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-21206; RRID: AB_2535792 |
| Donkey polyclonal anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | Thermo Fisher Scientific | Cat# A-31572; RRID: AB_162543 |
| Donkey polyclonal anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Thermo Fisher Scientific | Cat# A-31573; RRID: AB_2536183 |
| Donkey polyclonal anti-Rat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-21208; RRID: AB_2535794 |
| Biological Samples | ||
| Human glioblastoma tissue | Hospital of the University of Pennsylvania | https://www.pennmedicine.org |
| Human blood samples | Hospital of the University of Pennsylvania | https://www.pennmedicine.org |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 1X RBC lysis buffer | Thermo Fisher Scientific | 00433357 |
| 2-Mercaptoethanol | Thermo Fisher Scientific | 21985023 |
| Advantage 2 PCR Kit | Takara Bio | 639206 |
| Advantage UltraPure PCR deoxynucleotide mix (10mM each dNTP) | Takara Bio | 639125 |
| Amphotericin B | Thermo Fisher Scientific | 15290026 |
| B-27 Supplement (50X), minus vitamin A | Thermo Fisher Scientific | 12587010 |
| Bovine serum albumin (BSA) | Sigma-Aldrich | Cat# B6917; CAS# 9048-46-8 |
| Corn oil | Sigma-Aldrich | Cat# C8267; CAS# 8001-30-7 |
| DAPI | Sigma-Aldrich | Cat# 10236276001; CAS# 28718-90-3 |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | Cat# D2650; CAS# 67-68-5 |
| DL-Dithiothreitol solution (1 M) | Sigma-Aldrich | Cat# 43816; CAS# 3483-12-3 |
| Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F-12) | Thermo Fisher Scientific | 11320033 |
| Dulbecco’s phosphate-buffered saline (DPBS), calcium, magnesium | Thermo Fisher Scientific | 14040133 |
| Dulbecco’s phosphate-buffered saline (DPBS), no calcium, no magnesium | Thermo Fisher Scientific | 14190144 |
| Dynabeads Human T-Activator CD3/CD28 for T Cell Expansion and Activation | Thermo Fisher Scientific | 11131D |
| EdU | Abcam | ab146186 |
| Everolimus | Santa Cruz Biotechnology | Cat# sc-218452; CAS# 159351-69-6 |
| EZ-Tn5 Transposase | Lucigen | TNP92110 |
| Formaldehyde, 16%, methanol free, Ultra Pure | Polysciences | Cat# 18814-10; CAS#: 50-00-0 |
| Gefitinib | Santa Cruz Biotechnology | Cat# sc-202166; CAS# 184475-35-2 |
| Gelfoam Sponge | Pfizer | 031508 |
| GlutaMAX supplement | GIBCO | 35050061 |
| Hibernate A medium | BrainBits | HA |
| Human insulin solution | Sigma-Aldrich | Cat# I9278; CAS# 11061-68-0 |
| IDT for Illumina Nextera DNA UD Indexes Set A | Illumina | 20027213 |
| Illumina Exome Panel - Enrichment Oligos | Illumina | 20020183 |
| ImmunoCult-XF T Cell Expansion Medium | StemCell Technologies | 10981 |
| KAPA HiFi DNA Polymerase with dNTPs | Kapa Biosystems | KK2102 |
| Maxi-Cure Super Glue | Bob Smith Industries | BSI-113 |
| MEM Non-Essential Amino Acids Solution (100X) | Thermo Fisher Scientific | 11140050 |
| MgCl2 (1M) | Thermo Fisher Scientific | AM9530G |
| Mouse on Mouse (M.O.M.) Blocking Reagent | Vector Laboratories | MKB-2213 |
| N-2 Supplement (100X) | Thermo Fisher Scientific | 17502048 |
| Neurobasal medium | Thermo Fisher Scientific | 21103049 |
| Nextera DNA Flex Pre-Enrichment Library Prep and Enrichment Reagents | Illumina | 20025523 |
| Nuclease-Free Water (not DEPC-Treated) | Thermo Fisher Scientific | AM9937 |
| Penicillin-Streptomycin (5,000 U/mL) | Thermo Fisher Scientific | 15070063 |
| Polyethylene glycol solution, 40% | Sigma-Aldrich | P1458 |
| Human Recombinant IL-2 | StemCell Technologies | 78036 |
| RNA Clean & Concentrator 5 | Zymo Research | R1013 |
| RNase Inhibitor, Murine | New England Biolabs | M0314S |
| RPMI 1640 medium | Thermo Fisher Scientific | 11875093 |
| SDS (10% w/v) | Fisher Scientific | 50-751-7490 |
| SMARTScribe Reverse Transcriptase | Takara | 639537 |
| Sodium hydroxide solution (1 N) | Sigma-Aldrich | 1091371000 |
| SPRIselect Reagent | Beckman Coulter | B23318 |
| Sterile saline solution injection | Midwest Veterinary Supply | 193.74500.3 |
| Sucrose | Sigma-Aldrich | Cat# S0389; CAS# 57-50-1 |
| Temozolamide | Santa Cruz Biotechnology | Cat# sc-203292; CAS# 85622-93-1 |
| Tissue Freezing Medium | General Data | 1518313 |
| Trametinib | Santa Cruz Biotechnology | Cat# sc-364639A; CAS# 871700-17-3 |
| Tris (1 M), pH 7.0, RNase-free | Thermo Fisher Scientific | AM9850G |
| Tris (1 M), pH 8.0, RNase-free | Thermo Fisher Scientific | AM9855G |
| Triton X-100 | Sigma-Aldrich | Cat# T9284; CAS# 9002-93-1 |
| TRIzol reagent | Thermo Fisher Scientific | 15596026 |
| TrueBlack Lipofuscin Autofluorescence Quencher | Biotium | 23007 |
| Trypan blue stain, 0.4% | Thermo Fisher Scientific | T10282 |
| TWEEN 20 | Sigma-Aldrich | Cat# P1379; CAS# 9005-64-5 |
| VECTASHIELD Vibrance Antifade Mounting Medium | Vector Laboratories | H170010 |
| Y-27632 | StemCell Technologies | Cat# 72304; CAS# 129830-38-2 |
| Critical Commercial Assays | ||
| Bioanalyzer high sensitivity DNA analysis | Agilent | 5067-4626 |
| Click-iT EdU Alexa Fluor 647 Imaging Kit | Thermo Fisher Scientific | C10340 |
| DuoSet ELISA Ancillary Reagent Kit 1 | R&D Systems | DY007 |
| EZ-PCR Mycoplasma test kit | Biological Industries | 2070020 |
| Human IFN-gamma DuoSet ELISA | R&D Systems | DY285B-05 |
| Human IL-2 DuoSet ELISA | R&D Systems | DY202-05 |
| Human TNF-alpha DuoSet ELISA | R&D Systems | DY210-05 |
| Hypoxyprobe Kit | Hypoxyprobe | HP1-100Kit |
| KAPA Library Quantification Kit for Illumina NGS | Kapa Biosystems | KK4835 |
| Neural Tissue Dissociation Kit - Postnatal Neurons | Miltenyi Biotech | 130094802 |
| NextSeq High Output v2 150 Cycles | Illumina | TG-160-2002 |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | Q33231 |
| Quick-DNA Microprep Kit | Zymo Research | D3020 |
| Deposited Data | ||
| Bulk RNA sequencing | This paper | GEO: GSE141947 |
| Whole exome sequencing | This paper | GEO: GSE141947 |
| Single-cell RNA sequencing | This paper | GEO: GSE141947 |
| Single nuclei RNA sequencing of adult human brain | Habib et al., 2017 | https://doi.org/10.1038/nmeth.4407 |
| Single nuclei RNA sequencing of adult human brain | Lake et al., 2018 | https://doi.org/10.1038/nbt.4038 |
| Experimental Models: Cell Lines | ||
| Glioblastoma organoid samples | This paper | Tables S1 and S2 |
| Experimental Models: Organisms/Strains | ||
| NU/J Mus musculus, female | The Jackson Laboratory | Cat# 002019; RRID: IMSR_JAX:002019 |
| Oligonucleotides | ||
| CDS Primer sequence: 5′-AAGCAGTGGTATCAACGCAGAGTACT30VN-3′ | IDT | N/A |
| TSO Primer sequence: 5′-AAGCAGTGGTATCAACGCAGAGTACATrGrGrG-3′ | IDT | N/A |
| LS PCR Primer sequence: 5′-AAGCAGTGGTATCAACGCAGAGT-3′ | IDT | N/A |
| Software and Algorithms | ||
| Adobe Illustrator CC | Adobe | https://www.adobe.com/products/illustrator.html; RRID:SCR_010279 |
| Adobe Photoshop CC | Adobe | https://www.adobe.com/products/photoshop.html; RRID:SCR_014199 |
| ANNOVAR | Wang et al., 2010 | http://www.openbioinformatics.org/annovar/; RRID:SCR_012821 |
| avsnp150 | Annovar | http://annovar.openbioinformatics.org/en/latest/ |
| bcl2fastq v.2.19.0.316 | Illumina | https://support.illumina.com/sequencing/sequencing_software/bcl2fastq-conversion-software.html; RRID:SCR_015058 |
| BWA v.0.7.10 | Li and Durbin, 2009 | http://bio-bwa.sourceforge.net/; RRID:SCR_010910 |
| COSMIC v88 | Sanger | https://cancer.sanger.ac.uk/cancergenome/projects/cosmic/; RRID:SCR_002260 |
| dbNSFP35a | Liu et al., 2016 | https://sites.google.com/site/jpopgen/dbNSFP; RRID:SCR_005178 |
| dbSNP b146 | Broad Institute | https://www.ncbi.nlm.nih.gov/SNP/; RRID:SCR_002338 |
| DESeq2 v.1.22.2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html; RRID:SCR_015687 |
| Drop-seq tools v.2.1.0 | Saunders et al., 2018 | https://github.com/broadinstitute/Drop-seq |
| Enrichr | Kuleshov et al., 2016 | http://amp.pharm.mssm.edu/Enrichr/; RRID:SCR_001575 |
| Freebayes v1.2.0-4-gd15209e | Garrison and Math, 2012 | https://github.com/ekg/freebayes; RRID:SCR_010761 |
| GATK v.4.1.0.0 | Broad Institute | https://software.broadinstitute.org/gatk/; RRID:SCR_001876 |
| gnomAD | Broad Institute | http://gnomad.broadinstitute.org/; RRID:SCR_014964 |
| GraphPad Prism | GraphPad Software | https://www.graphpad.com/scientific-software/prism/; RRID:SCR_002798 |
| GRCh38 v 28 | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.38 |
| GRCh38 (exome) | Broad Institute | https://software.broadinstitute.org/gatk/documentation/article?id=11010 |
| IGV v2.4.14 | Broad Institute | https://www.broadinstitute.org/igv/; RRID:SCR_011793 |
| ImageJ | NIH | https://imagej.nih.gov/ij/; RRID:SCR_003070 |
| Imaris | Bitplane | https://imaris.oxinst.com/packages; RRID:SCR_007370 |
| inferCNV | Broad Institute | https://github.com/broadinstitute/infercnv |
| Manta v 1.5.0 illumina github | Chen et al., 2016 | https://github.com/Illumina/manta |
| MuTect V4.1.0.0 | Broad Institute | https://www.broadinstitute.org/cancer/cga/mutect; RRID:SCR_000559 |
| Picard v. 1.141 | Broad Institute | http://broadinstitute.github.io/picard/; RRID:SCR_006525 |
| R Project v.3.5.1 | Open source | https://www.r-project.org/; RRID:SCR_001905 |
| RefGenes | Annovar | https://www.refgenes.org/rg/; RRID:SCR_003372 |
| RStudio | Open source | https://rstudio.com/; RRID:SCR_000432 |
| Rsubread v.1.32.2 | Liao et al., 2019 | https://bioconductor.org/packages/release/bioc/html/Rsubread.html; RRID:SCR_016945 |
| SAMtools/BCFtools v1.9 | Li et al., 2009; Li, 2011 | http://samtools.sourceforge.net/mpileup.shtml; RRID:SCR_005227 |
| Seurat | Stuart et al., 2019 | https://github.com/satijalab/seurat; RRID:SCR_007322 |
| Small ExAX common variants | Broad Institute | http://exac.broadinstitute.org/ |
| STAR v.2.6.1d | Dobin et al., 2013 | https://github.com/alexdobin/STAR; RRID:SCR_015899 |
| Strelka v.2.9.10 | Kim et al., 2018 | https://academic.oup.com/bioinformatics/article-pdf/28/14/1811/16904379/bts271.pdf; RRID:SCR_005109 |
| SVA v.3.30.1 | Leek et al., 2012 | https://bioconductor.org/packages/release/bioc/html/sva.html RRID:SCR_002155 |
| VarScan v2.3.9 | Koboldt et al., 2012 | https://sourceforge.net/projects/varscan/files/ RRID:SCR_006849 |
| Vcflib | GitHub | https://github.com/vcflib/vcflib; RRID:SCR_001231 |
| VCFtools v 0.1.13 | Danecek et al., 2011 | http://vcftools.sourceforge.net/; RRID:SCR_001235 |
| Zen 2Blue | Carl Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen-lite.html; RRID:SCR_013672 |
| Other | ||
| Bioanalyzer 2100 | Agilent | G2939BA |
| C&B Metabond Quick Adhesive Cement System Kit | Benco Dental | 1681-343 |
| Cell counting slides | Thermo Fisher Scientific | C10228 |
| Countess II Automated Cell Counter | Thermo Fisher Scientific | AMQAX1000 |
| Disposable pellet pestle | Fisher | 12-141-368 |
| Fine Forceps - Curved/Serrated | Fine Science Tools | 11065-07 |
| Forma Steri-Cult CO2 Incubator | Thermo Fisher Scientific | 3310 |
| MACS SmartStrainer, 70 μM | Miltenyi Biotech | 130-098-462 |
| MaxQ CO2 Plus Shaker | Thermo Fisher Scientific | 88881102 |
| Microfluidic chips for drop-seq | FlowJEM | Custom order |
| NanoDrop 2000 | Thermo Fisher Scientific | ND-2000 |
| NextSeq550 | Illumina | SY-415-1002 |
| Pellet pestle cordless motor | Fisher | 12-141-361 |
| Qubit 3 Fluorimeter | Thermo Fisher Scientific | Q33216 |
| T100 Thermal Cycler | Bio-rad | 1861096EDU |
| TB Syringe (26 G x 3/8 in, 1 ml) | BD Biosciences | 309625 |
| Vannas Spring Scissors - Curved/3mm Cutting Edge | Fine Science Tools | 15000-10 |
Tissue dehydration, paraffin embedding, microtome sectioning, and standard H&E staining was performed by the University of Pennsylvania Pathology Clinical Service Center. GBOs were fixed overnight in 4% methanol-free formaldehyde and stored in 70% ethanol at 4°C until ready for processing. GBOs and parental tumor samples were independently analyzed by an attending neuropathologist (M.N.).
Confocal Microscopy and Image Processing
Tumor tissue, GBOs, and brain sections were imaged as z stacks using a Zeiss LSM 810 confocal microscope or a Zeiss LSM 710 confocal microscope (Zeiss) using a 10X, 20X, 40X, or 63X objective with Zen 2 software (Zeiss). Images were analyzed using either Imaris 7.6 or ImageJ software. Images were cropped and edited using Adobe Photoshop (Adobe) and Adobe Illustrator (Adobe).
Detection of Hypoxia Gradients in GBOs
GBOs were incubated in GBO medium containing 200 μM pimonidazole-HCl (Hypoxyprobe) for three hours on an orbital shaker rotating at 120 rpm within a 37°C, 5% CO2, 90% humidity sterile incubator and processed for immunohistology as described (Varia et al., 1998). Mouse anti-pimonidazole monoclonal antibody (Hypoxyprobe) was used to detect bound pimonidazole.
EdU Pulse-Chase in GBOs
GBOs were incubated in GBO medium containing 1 μM EdU (Thermo Fisher Scientific) for one hour on an orbital shaker rotating at 120 rpm within a 37°C, 5% CO2, 90% humidity sterile incubator. GBOs were fixed and processed for immunohistology at 1-hourand 2-week time points. EdU incorporation was detected using Click-iT EdU Alexa Fluor 647 kit (Thermo Fisher Scientific) (Berg et al., 2019).
Biobanking and Recovery of GBOs
Within one month of culture, GBOs were cut into approximately 100 μm diameter pieces, washed with GBO medium to remove cell debris, and incubated with GBO medium supplemented with 10 μM Y-27632 (StemCell Technologies) for 1 hour. GBOs were resuspended in GBO freezing medium comprised of GBO medium supplemented with 10 μM Y-27632 and 10% DMSO (Sigma) and placed in cryovials (Thermo Fisher Scientific) with 20 GBOs per vial. GBOs were kept in the cryovials with GBO freezing medium for 15 minutes at room temperature to allow the DMSO to better penetrate the core before slowly being cooled to −80°C using a CoolCell freezing container (Thermo Fisher Scientific) placed in a −80°C freezer. Frozen GBOs were placed in a liquid nitrogen tank for long-term storage. For recovery, vials were quickly thawed in a 37°C water bath, and GBOs were placed in a 50 mL conical tube. Ten ml of GBO medium containing 10 μM Y-27632 was added dropwise while vigorously swirling the tube to slowly dilute the DMSO. The medium was aspirated and GBOs were cultured overnight in GBO medium supplemented with 10 μM Y-27632. The medium was replaced with GBO medium without Y-27632 the next day. GBO pieces generally rounded up and were ready for experimentation within 1-2 weeks. All biobanked GBOs were confirmed free of Mycoplasma, Acholeplasma, and Spiroplasma with a detection limit of 10 CFU/ml by targeted PCR (Biological Industries).
Solid Tumor Sequencing Panel
The solid tumor sequencing panel was performed by the University of Pennsylvania Center for Personalized Diagnostics. Genomic DNA was extracted from fresh solid tumor specimens. Sequence analysis was performed on a panel of 152 genes: ABL1, AKT1*, AKT2, AKT3*, ALK*, APC, AR*, ARAF*, ARID1A, ARID2, ATM, ATRX, AURKA, BAP1, BRAF*, BRCA1*, BRCA2, BRIP, BTK*, CREBBP*, CCND1, CCND2, CCND3, CCNE1,CDH1, CDK4, CDK6, CDKN2A, CHEK2*, CIC, CRKL, CSF1R, CTNNB1*, DAXX, DDR2*, DNMT3A, EIF1AX, EGFR*, EP300, EPHA3, ERBB2*, ERBB3, ERBB4, ERCC2, ERG, ESR1*, ESR2, EZH2, FBXW7, FGF3, FGFR1, FGFR2, FGFR3, FGFR4, FLT3, FUBP1, GATA3, GNA11*, GNAQ*, GNAS, HRAS*, H3F3A, IDH1*, IDH2*, IGF1R, JAK1, JAK2, JAK3, KDM5A, KDM5C, KDM6A, KDR*, KIT*, KMT2C, KRAS*, MAP2K1*, MAP2K2, MAP2K4, MAPK1, MAPK3, MAX, MCL1, MDM2, MDM4, MED12, MEN1, MET*, MITF, MLH1, MRE11A, MSH2, MSH6, MTOR, MYC, MYCN, NBN, NF1, NF2, NKRT1, NKRT2, NKRT3, NKX2-1, NOTCH1, NOTCH2, NOTCH3, NRAS*, PAK1, PALB2, PBRM1, PDGFRA*, PIK3CA*, PIK3CB, PIK3R1, PTCH1, PTEN, PTPN11, RAB35, RAC1, RAD50, RAD51, RAD51B, RAD51C, RAD51D, RAF1, RB1, RET*, RHOA, RNF43, SETD2, SF3B1, SLIT2, SMAD4, SMARCA4, SMO, SPOP, SRC, STAG2, STK11, SUFU, SUZ12, SYK, TET2, TGFBR2, TP53, TRAF7, TSC1, TSHR, TSC2, U2AF1, VHL, WT1, and XRCC2. Targeted analysis for variants in the regions specified in this testing panel was achieved by enrichment of those genomic loci using a custom Agilent Haloplex assay. The library preparation included unique molecular identifiers to identify duplicate reads. Sequencing of enriched libraries was performed on the Illumina HiSeq 2500 platform using multiplexed, paired-end reads. Analyses and interpretation were performed using a customized bioinformatics pipeline, Halo_v1.2. All variants were annotated with reference to the hg19 Genome build. Variants were reported according to HGVS nomenclature and classified into 3 categories: disease-associated variants, variants of uncertain significance, and indeterminate. Variants internally classified as benign were not listed in the report. Variants determined to be clinically relevant but failing to meet technical reporting criteria were categorized as indeterminate. The lower limit of detection for single nucleotide variants (SNVs) and small insertions or deletions (indels) was established for this assay at 2% variable allele frequency. Categorization of variants was dependent upon multiple criteria, including (but not limited to) literature review and the presence of the variant in publicly available databases including dbSNP, COSMIC, gnomAD, and the 1000 genome project. Copy number gain was inferred from this assay by increased read depth of the targeted regions which are designated above with an asterisk. Because analysis was conducted on a single piece of tissue, the results of this assay may not be representative of the entire tumor.
Fusion Transcript Panel
The fusion transcript panel was performed by the University of Pennsylvania Center for Personalized Diagnostics. RNA was extracted from formalin fixed paraffin embedded (FFPE) tumor tissue and reverse transcribed into cDNA allowing direct detection of normal and variant gene expression. PCR primers were designed to capture exons within transcripts of the following genes: AKT1, ALK, AXL, BCOR, BRAF, CALCA, CAMTA1, CCNB3, CCND1, CIC, EGFR, EML4, EPC1, ERBB2, ERG, ESR1, EWSR1, FGFR1, FGFR2, FGFR3, FOXO1, FUS, GLI1, HMGA2, JAZF1, KRT20, KRT7, MEAF6, MET, MKL2, NCOA2, NRG1, NTRK1, NTRK2, NTRK3, PDGFB, PIK3CA, PLAG1, PMS2, PPARG, PTH, RAF1, RET, ROS1, SLC5A5, SS18, STAT6, TAF15, TCF12, TERT, TFE3, TFG, THADA, TMPRSS2, USP6, and YWHAE. In addition, exons in internal housekeeping genes CHMP2A, GPI, RAB7A, and VCP were included as quality controls. The design of the panel was based on the literature at the time of development and the target regions were selected to include the most common fusion points of the specified genes. Sequencing of PCR-enriched libraries was performed on the Illumina HiSeq platform using multiplexed, paired-end reads. Analysis and interpretation utilized a customized bioinformatics pipeline, Archer_v6.0.3.2. All variants listed are with reference to the hg19 Genome build. The Archer Analysis 6.0.3.2 Software (ArcherDX, Inc.), was used to process the data and identify normal and abnormal reads. Because analysis was conducted on a single piece of tissue, the results of this assay may not be representative of the entire tumor.
MGMT Promoter Methylation Assay
The MGMT promoter methylation assay was performed by the University of Pennsylvania Center for Personalized Diagnostics. Genomic DNA was extracted from FFPE tumor tissue and underwent bisulfite conversion followed by amplification with primers to target DMR2 of the MGMT (O-6-methylguanine-DNA methyltransferase) promoter, including 4 CpG sites (chr10:131,265,519-131,265,537; hg19 Assembly). Using pyrosequencing (PyroMark Q24, QIAGEN), the PCR product was evaluated to assess for percent methylation across the 4 CpG sites. The result was considered positive when the mean and median percent methylation across the 4 interrogated CpG sites was greater than or equal to 10%, considered low positive when the mean and median level of DNA methylation seen were either relatively low (above the limit of detection but below 10%) or highly variable across the CpG sites, and considered not detected when the mean and median percent methylation across the 4 CpG sites were below the limit of detection (4.5%). Appropriate positive and negative controls were included in each assay. Because analysis was conducted on a single piece of tissue, the results of this assay may not be representative of the entire tumor.
RNA Sequencing of Parental Tumors and Derived GBOs
In an effort to minimize variability due to sampling, we pooled 5-10 pieces of parental tumor tissue or GBOs into one sample for analyses. They were snap-frozen on dry ice, and stored at −80°C until processing. Samples were homogenized in TRIzol (Thermo Fisher Scientific) using a disposable pestle and handheld motor. RNA clean-up was performed using a Zymo Research Clean & Concentrator kit after TRIzol (ThermoFisher) phase separation according to the manufacturers’ protocol. RNA concentration and quality were assessed using a Nanodrop 2000 (Thermo Fisher Scientific).
Library preparation was performed as previously described with some minor modifications (Weng et al., 2017). About 100 ng RNA in 3.2 μL was combined with 0.25 μL RNase inhibitor (NEB) and 1 μL CDS primer (5′-AAGCAGTGGTATCAACGCAGAGTACT30VN-3′) in an 8-well PCR tube strip, heated to 70°C, and immediately placed on ice. 5.55 μL RT mix, containing 2 μL of 5X SMARTScribe RT buffer (Takara), 0.5 μL of 100 mM DTT (Millipore Sigma), 0.3 μL of 200 mM MgCl2 (Thermo Fisher Scientific), 1 μL of 10 mM dNTPs (Takara), 1 μL of 10 μM TSO primer (5′-AAGCAGTGGTATCAACGCAGAGTACATrGrGrG-3′), 0.25 μL of RNase inhibitor (NEB), and 0.5 μL SMARTScribe reverse transcriptase (Takara) was added to the reaction. RT was performed under the following conditions: 42°C for 90 minutes, 10 cycles of 50°C for 2 minutes and 42°C for 2 minutes, 70°C for 15 minutes, and 4°C indefinitely. For cDNA amplification, 2 μL of the RT reaction was combined with 2.5 μL of 10X Advantage 2 buffer (Takara), 0.5 μL of 10 mM dNTPs (Takara), 0.25 μL of 10 mM IS PCR primer (5′-AAGCAGTGGTATCAACGCAGAGT-3′), 19.25 μL nuclease free water (ThermoFisher), and 0.5 μL Advantage 2 DNA Polymerase (Takara). Thermocycling conditions were as follows: 94°C for 3 minutes, 8 cycles of 94°C for 15 s, 65°C for 30 s, and 68°C for 6 minutes, 72°C for 10 minutes, and 4°C indefinitely. Amplified cDNA was purified using 0.8X SPRI beads (Beckman Coulter), eluted in 10 μL nuclease-free water, and quantified using Qubit dsDNA HS assay kit (Thermo Fisher Scientific). cDNA was fragmented by combining 100 pg cDNA in 1 μL with 1 μL nuclease free water, 2X TD buffer (20 mM Tris, pH 8.0 (Thermo Fisher Scientific), 10 mM MgCl2, and 16% PEG 8000 (MilliporeSigma), and 0.5 μL Tn5 (Lucigen). The mixture was heated to 55°C for 10 minutes, and the reaction was terminated upon the addition of 1.25 μL of 0.2% SDS (Fisher) and incubation at room temperature for 10 minutes. Fragments were amplified by adding 27.75 μL nuclease free water (Thermo Fisher Scientific), 10 μL of 5X KAPA HiFi Fidelity Buffer (Roche), 10 mM dNTPs (Roche), 1 μL of 10 mM Nextera i7 primer, 1 μL of 10 mM Nextera i5 primer, and 2 μL KAPA HiFi Polymerase (Roche). Thermocycling conditions were as follows: 72°C for 5 minutes, 95°C for 1 minute, 14 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 30 s, 72°C for 1 minute, and 4°C indefinitely. DNA was purified twice with 0.8X SPRI beads (Beckman Coulter) and eluted in 10 μL of 10 mM Tris, pH 8 (Thermo Fisher Scientific). Samples were quantified by qPCR (KAPA) and pooled at equal molar amounts. Final sequencing library fragment sizes were quantified by bioanalyzer (Agilent) with an average size of ~350 bp, and concentrations were determined by qPCR (KAPA). Samples were loaded at concentrations of 2.2 pM, and sequenced on a NextSeq 550 (Illumina) using 2x76 bp reads to an average depth of 25 million reads per sample.
Raw sequencing data were demultiplexed with bcl2fastq2 v.2.19.0.316 (Illumina) with adaptor trimming turned off. Alignment was performed using STAR v.2.6.1d (Dobin et al., 2013) with the following additional parameters: –outFilterMatchNminOverLread 0.33–outFilterScoreMinOverLread 0.33–twopassMode Basic–chimSegmentMin 12–chimJunctionOverhangMin 12–alignSJDBoverhangMin 1–alignMatesGapMax 100000–alignIntronMax 200000–chimSegmentReadGapMax 3–alignSJstitchMismatchNmax 5 –1 5 5–outSAMstrandField intronMotif–chimOutJunctionFormat 1. GRCh38 (gencode) was used as the reference genome, and gencode v.28 GTF was used as the annotation file. Multimapping and chimeric alignments were discarded, and uniquely mapped reads were quantified at the exon level and summarized to gene counts using Rsubread (Liao et al., 2019). Counts were converted to units of TPM, and batch correction was performed using SVA (Leek et al., 2012). Further analyses were performed in R v.3.5.1.
Pearson’s correlation coefficient was calculated across all genes that were detected in at least one sample in the final dataset. To identify highly variable genes in the primary tumors, the coefficient of variation of the expression of each gene was modeled against the mean expression in log10 scale using a LOESS regression. The top 10,000 genes with the greatest positive deviation from the fit and a mean expression of greater than 1 TPM were selected.
Differential gene expression analysis (Figures S3D–S3F) was performed using the DESeq2 suite of analysis tools (Love et al., 2014). Time course analysis was performed using the likelihood ratio test where the full model design was ~Batch + Tumor + Time point and the reduced model design was ~Batch + Tumor. Cutoffs for significance were set at Benjamini-Hochberg corrected p value < 0.01 and log2 fold change > 2. Gene ontology enrichment analysis was performed using Enrichr (Kuleshov et al., 2016). Regional sample PCA analysis was also performed using the DESeq2 suite of tools, including VST transformation of raw counts and selection of the top 100 most variably expressed genes for PCA analysis.
EGFRvIII transcripts were detected by RNA-seq and quantified by examining the number of exon 1 to exon 8 spanning reads versus the number of exon 7 to exon 8 spanning reads (Brennan et al., 2013). 95% credible intervals were determined using a beta distribution with a uniform prior.
Whole Exome Sequencing of Parental Tumors and Derived GBOs
At least 5-10 pieces of parent tumor or GBOs at 2 weeks were combined into one sample, snap-frozen on dry ice, and stored at −80°C until processing. A 50 μL aliquot of whole blood obtained at the time of surgery was also snap-frozen on dry ice and stored at −80°C until processing. DNA was extracted using the Zymo Quick-DNA Microprep Kit. DNA quality were assessed using a Nanodrop 2000 (Thermo Fisher Scientific) and quantified using the Qubit high sensitivity dsDNA assay (Thermo Fisher Scientific). Exomeseq libraries were prepared using the Nextera Flex for Enrichment kit (Illumina), Illumina exome panel oligos (Illumina), and IDT for Illumina DNA UD set A index primers (Illumina) according to the manufacturer’s instructions. Genomic DNA inputs varied from 25 to 300 μg, and pre-enrichment samples were combined at equal amounts by mass in 9 or 10 plex pools for enrichment. Sequencing library fragment sizes were quantified by bioanalyzer (Agilent), and concentrations were quantified by qPCR (KAPA). Samples were combined into 18- or 19-plex equimolar pools with whole blood, corresponding parental tumor, and corresponding GBO combined in the same sequencing run, loaded at a concentration of 2.2 pM, and sequenced on a NextSeq 550 (Illumina) using 2x74 bp reads for an average of 32 million reads per sample and an average target coverage of 47x.
Raw sequencing data were demultiplexed with bcl2fastq2 with adaptor trimming turned off. Alignment was performed using BWA-MEM with default parameters and a GRCh38 reference genome kindly provided by the Broad Institute on a public GATK resource FTP server (Li and Durbin, 2009). Pre-processing according to GATK best practices, including marking PCR duplicates and base quality recalibration, was performed using Picard tools v.1.141 and GATK v.4.1.0.0. (DePristo et al., 2011; McKenna et al., 2010; Van der Auwera et al., 2013). A database of known polymorphic sites was used from the GATK resource bundle based on dbSNP build 146.
Somatic variants were identified using four different variant calling tools: Mutect2, Varscan2, Freebayes, and Manta/Strelka2. Mutect2 was run with a panel of normals composed of the 11 samples used in this study, a population germline variant resource from gnomAD (Broad Institute), and allele frequencies of variants not included in the germline resource set to 0.0000025 (Cibulskis et al., 2013). GATK contamination detection was performed using a small ExAC reference provided by the GATK resource bundle, and filtering was performed using default parameters. Varscan2 was run using default parameters, and somatic variants were filtered for a p value < 0.05 and alternate allele frequency > 0.05 (Koboldt et al., 2012; Li et al., 2009). Freebayes was run in somatic mode with the following additional parameters (Garrison and Math, 2012): -F 0.05–pooled-continuous–pooled-discrete. Variants were filtered using vcflib by breaking multiallelic records into separate lines, identifying putative somatic variants by comparison with the paired whole blood sample, filtering for variant calls where the QUAL field > 20, and filtering for variant calls where the coverage was > 20. Prior to running Strelka2, Manta was used to call candidate somatic indels in exome mode across only the primary GRCh38 contigs (Chen et al., 2016). Then, Strelka2 was run in exome mode using identified candidate indels also across the primary GRCh38 contigs to identify somatic variants (Kim et al., 2018). Somatic variants were filtered using default settings. VCFtools was used for variant manipulations throughout these analyses (Danecek et al., 2011).
Identified somatic variants in the individual samples were called if identified by two or more variant calling tools using the above described workflow. Variants identified in the parental tumor and corresponding GBOs were pooled to yield a final variant list. Allele frequencies were queried at these sites in the parental tumors and corresponding GBOs using bcftools mpileup accepting anomalous read pairs and disabling probabilistic realignment for base alignment quality calculation (Li, 2011). This approach of “recovering” variants is inspired by previous work in cancer organoids (Kopper et al., 2019). Annovar was used to annotate variants with the following databases (Wang et al., 2010): RefGene (2017), dbNSFP35a (Liu et al., 2011, 2016), COSMIC v88 (Forbes et al., 2017), Clin-Var (Landrum et al., 2016), and avsnp (based on dbsnp 150). Variants highlighted in Figure 3C come from a list of significantly mutated genes from work by Brennan et al. (2013).
Copy number analysis was performed using the GATK4 pipeline. A target manifest was obtained from the exome panel supplier (Illumina) in hg19 coordinates and lifted over to GRCh38 coordinates using Picard tools and a chain file from UCSC genome browser. Allosomal chromosomes were excluded and all whole blood samples were included in the construction of a panel of normals. Read counts were quantified and denoised using GATK. Allele counts were quantified over a set of genomic intervals defined by a subset of gnomad snps provided in the GATK resource bundle. Genomic segments were modeled with the following additional parameters: –number-of-smoothing-iterations-per-fit 1–number-of-changepoints-penalty-factor 5. Copy ratios were called and visualized using IGV (Robinson et al., 2011). Copy ratios with a greater than 10% change and with the most number of independent probes supporting the call were summarized to chromosomal arms and shown in Figure 3C.
Tissue Dissociation and Single-cell RNA Sequencing
Multiple tumor pieces and GBOs were dissociated using a papain-based neural tissue dissociation kit (Miltenyi Biotech). Crude dissociates from parental tumor samples were treated with ammonium chloride based RBC lysis buffer (Thermo Fisher Scientific) for 5 minutes. All samples were washed three times by centrifugation at 200 g for 5 minutes and resuspension in 10 mL of calcium-free, magnesium-free DPBS (Thermo Fisher Scientific). Cells were strained through a 70 μm filter (Miltenyi Biotech), analyzed for viability by trypan blue staining, and counted using an automatic cell counter (Thermo Fisher Scientific). Samples had a viability of >80% and were diluted to a final concentration of 100 viable cells/μL in DPBS with 0.01% BSA(w/v). Single cell droplet encapsulation and library preparation was performed using the drop-seq method with minor modifications (Macosko et al., 2015). Microfluidic chips were obtained from FlowJEM with plasma bonding and aquapel treatment using the same design as previously published. Each droplet co-encapsulation run was performed with 50% additional reagent volume to account for syringe and tubing dead volume as well as occasional microfluidic channel clogging and re-starts. Each sample was loaded onto 2-4 droplet generation runs, and cell suspensions and droplets were kept on ice prior to droplet encapsulation and droplet breakage. In the cDNA PCR amplification stage, between 4,000-8,000 beads were combined in a single PCR tube reaction and purified individually using 0.6x SPRI beads (Beckman Coulter). cDNA across multiple runs of the same sample were pooled together and used as input for tagmentation, where the tagment reaction time was increased to 8 minutes. Final samples were purified once with 0.6x SPRI beads and a second time with 1.0x SPRI beads (Beckman Coulter). Sequencing library fragment sizes were quantified by bioanalyzer (Agilent), and concentrations were quantified by qPCR (KAPA). Samples were pooled, loaded at 2.2 pM, and sequenced on a NextSeq 550 (Illumina) with a 20 bp Read 1 and 64 bp Read 2. The custom Read 1 primer was spiked into the usual Illumina sequencing primer well (#20) at the manufacturer’s recommended concentration.
Raw sequencing data was demultiplexed with bcl2fastq2 (Illumina) with adaptor trimming turned off. Additional processing was performed using drop-seq tools v.2.1.0 with GRCh38 as the reference genome, and gencode v.28 GTF was used as the annotation file (Saunders et al., 2018). Seurat v3.1 was used to analyze the scRNA-seq data (Stuart et al., 2019). Cells with > 400 unique genes detected, < 5,000 genes detected, and < 25% mitochondrial reads were retained for further analysis. Standard pre-processing pipelines, including normalization and scaling, were followed without additional modifications. Clustering and UMAP dimensionality reduction were performed based on empirically selected PCs.
For CNA analysis, inferCNV was used (Patel et al., 2014; Tirosh et al., 2016; Venteicher et al., 2017) on each patient sample individually with microglia/macrophages and T cells set as references (Figure S4A). Non-neoplastic cells were identified by the expression of classical markers. Microglia/macrophages were further sub-setted for detailed analysis. Expression of genes associated with microglia or macrophages (Darmanis et al., 2017) was calculated using the AddModuleScore function in Seurat with bin size set to 10. Enrichment of macrophage versus microglia gene expression was taken as the difference between the gene signature expressions.
To compare the parental tumor and derived GBOs at 2 weeks, gene expression within clusters were averaged and compared by calculating the transcriptome-wide Pearson correlation. Hierarchical clustering was performed using Euclidian distances of correlations to the primary tumor. Cluster specific marker expression was determined using the Wilcoxon rank sum test. For comparison with normal brain cell types, two single-nuclei RNA-seq datasets of normal adult human cerebral cortex were retrieved from public repositories (Habib et al., 2017; Lake et al., 2018). These datasets were selected because they were generated using adaptations of the drop-seq platform and were expected to best match the technical characteristics of our single-cell RNA-seq data. Annotated granule, CA1, and CA3 neurons were excluded, and all other neuron types were combined as one large neuronal population in the downstream analysis. Differential gene expression was performed using the FindAllMarkers function in Seurat with a log fold-change threshold of 0.1 and a minimum percent expressed threshold of 0.1. The top 100 marker genes based on adjusted p value were selected for each cell type. Gene enrichment was calculated from these marker gene lists in the GBO single-cell RNA-seq data using the AddModuleScore function with the number of bins set to 10 and averaged across all cells in each GBO cluster.
Digital PCR for EGFR and EGFRvIII Transcripts
Primers and minor groove binder (MGB) probes for WT EGFR and EGFRvIII were designed. The WT EGFR MGB probe was fluorescently labeled with 6-FAM and the EGFRvIII MGB probe was fluorescently labeled with 6-FAM or VIC. Gene copies per μl were normalized with the VIC labeled internal reference control RNase P (Thermo Fisher Scientific) by running a parallel reaction on the same chip with either FAM labeled WT EGFR or EGFRvIII reaction mix. The Digital PCR reactions were carried out using Quantstudio 3D digital PCR master mix V2 (Applied Biosystems) following the manufacturer’s instructions. The chips were loaded onto a Thermal cycler (ProFlex PCR system, Applied Biosystems) and each individual chip was read on a chip reader (Quantstudio 3D Digital PCR Instrument, Applied Biosystems). For each chip, the instrument generated a single.eds file that contained the processed imaged data and the results from preliminary analysis on the instrument. The data were then transferred onto the Thermo Fisher Scientific cloud for further analysis by the software. Tumor samples and GBOs were analyzed for WT EGFR and EGFRvIII. For every tumor and GBO sample, two chips were run separately with the same concentration of cDNA containing EGFRvIII primers with RNaseP (VIC labeled) and probe for WT EGFR (FAM labeled), respectively. WT EGFR and EGFRvIII copies per μl were normalized with RNaseP (internal control). Amplified EGFR was considered positive if the ratio of WT EGFR to RNaseP was ≥ 1.5. EGFRvIII is considered positive if vIII % is ≥ 10%.
Animals and Orthotopic Transplantation of GBOs
Orthotopic transplantation of GBOs was performed using a method described for brain organoid transplantation into immunodeficient mice (Mansour et al., 2018). Mice were induced into anesthesia with 5% and maintained with 2% isoflurane in oxygen. An approximately 1 mm2 craniotomy above the right cerebral cortex at the intersection between the sagittal and lambdoid sutures was performed using a micromotor drill (Stoelting). After removing the meninges, the underlying brain tissue was aspirated using a 23G blunt needle (Neta Scientific) to create a 1 mm3 cavity. Bleeding was controlled using Gelfoam (Pfizer) and sterile saline. A single GBO was transferred into the cavity and sealed by 3-mm coverslipand super glue (Bob Smith Industries), followed by application of dental cement (Benco Dental) onto the surrounding skull area. Each transplanted GBO was approximately 1 mm in diameter and consisted of roughly 1 million cells as determined by automated cell counting (Countess II, Thermo Fisher Scientific) of similarly sized GBOs. Animals were weighed twice a week and euthanized immediately after weight loss and/or the onset of neurological symptoms. Anesthesia by intraperitoneal injection of a lethal dose of ketamine, xylazine and acepromazine cocktail was performed. Mice were transcardially perfused with cold 0.1 M phosphate buffer followed by cold 4% formaldehyde in 0.1 M phosphate buffer (pH 7.4). Brains were carefully removed from the skull and fixed in 4% formaldehyde at 4°C overnight, washed with DPBS, and cryoprotected in 30% sucrose (w/v) overnight at 4°C. Brains were placed in plastic cryomolds and snap frozen in tissue freezing medium (General Data) on dry ice. Frozen brains were stored at −80°C until processing.
Radiation and Drug Treatment of GBOs
For radiation experiments, GBOs were irradiated at a total dose of 10 Gray (Gy) at a dose rate of 2.65 Gy/min using a Precision XRAD320iX cabinet irradiator with 320 kV voltage, 12.5 mA current, and 50 source to surface distance (SSD). For drug experiments, GBOs were cultured for a week in GBO medium containing either 50 μM temozolomide (Santa Cruz Biotechnology), 5 μM gefitinib (Santa Cruz Biotechnology), 1 μM trametinib (Santa Cruz Biotechnology), 1 μM everolimus (Santa Cruz Biotechnology), or DMSO vehicle on an orbital shaker rotating at 120 rpm within a 37°C, 5% CO2, and 90% humidity sterile incubator. Media containing fresh drug was replaced every 48 hours. Different batches of GBOs were used for different treatment experiments (See Table S6 for their ages in culture). For a given treatment, GBOs were treated in triplicate and quantified for DAPI and KI67 immunohistology. Samples were subsequently stratified based on a statistically significant reduction in the percentage of KI67+ nuclei for gene expression analyses. Gene Set Enrichment Analysis (Subramanian et al., 2005), based on normalized expression value (TPM) from RNA-seq of pretreatment GBOs and parental tumors, was performed using GenePattern from the publicly available Broad Institute Servers (cloud.genepattern.org).
GBO size experiments were performed utilizing biobanked samples that were recovered for a minimum of 2 weeks prior to use. GBOs of similar size were placed in individual wells of a 24-well plate in 1 mL of GBO media containing drug or vehicle control. Similar to GBO growth analyses, images of individual GBOs were taken on a weekly basis using a brightfield microscope and Zen software. The 2D projected area of each GBO was quantified in ImageJ by manually outlining each GBO and measuring the area within the outlined region. GBO size was calculated as the ratio of the measured 2D projected area at each time point to the measured 2D area at time point 0. Analysis was done in the form of growth over time following 1 week of drug exposure of 1 μM trametinib (Santa Cruz Biotechnology), or 1 μM everolimus (Santa Cruz Biotechnology) and subsequent growth for 2 additional weeks in normal GBO media. GBO growth was also analyzed in the form of dose response with serial dilutions of trametinib and everolimus ranging from 0.1 nM to 1000 nM with GBO size measurement after 1 week of drug exposure.
For in vivo drug treatment experiments, UP-7790-GBOs were recovered from the biobank and cultured in vitro for 14 days before xenografting individual organoids into female adult athymic nude (NU/J) mice. At 14 days after xenograft, mice were randomized into treatment groups and given daily intraperitoneal injections of either vehicle (10% DMSO, 90% corn oil) or trametinib (3 mg/kg) for 12 days. Mice were weighed every other day to monitor animal health. Mice were given a single injection of EdU (10 mg/kg) 2 hours before sacrificing. Xenografts were analyzed using immunohistology for proliferation (KI67 and EdU) and cell death (Cleaved-Caspase-3). Quantifications were performed blind to the treatment groups.
Generation of CD19 and 2173BBz CAR-T Cells and GBO-CAR-T Co-culture
T cells were isolated from leukapheresis products obtained from de-identified healthy donors under a protocol approved by the University of Pennsylvania’s Institutional Review Board. T cells were stimulated with Dynabeads Human T-Activator CD3/CD28 (Thermo Fisher Scientific) at a bead to cell ratio of 3:1 (first stimulation) and transduced with lentiviral vector coding CAR transgene. T cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum, 20 mM HEPES, and PenStrep (Thermo Fisher Scientific) and medium was replaced every 48 hours. The end of the first stimulation was determined by decreased log-phase growth and reduced mean lymphocytic volume to 300-330 fl as measured on a Coulter Multisizer. This was usually reached 10 days after stimulation, and the T cells were frozen down for functional assays or used for re-stimulation. The percentage of T cells expressing CARs was determined by flow cytometry (LSR II, BD Biosciences) using biotinylated anti-human and anti-mouse antibodies for 2173BBz and CD19 CAR-T cells, respectively, with streptavidin-coupled PE. Data were analyzed using FlowJo software. In the current study, T cells transduced with CD19 and 2173BBz CARs were 30% and 32% CAR+, respectively.
Vials containing frozen CD19 and 2173BBz CAR-T cells were recovered in ImmunoCult-XF T Cell Expansion Medium (StemCell Technologies) with 100 units/ml of recombinant human IL-2 (StemCell Technologies). After 24 hours, the medium was replaced with ImmunoCult-XF T Cell Expansion Medium without IL-2 and the T cells were cultured for an additional 48 hours. T cells were incubated with GBOs in ultra-low attachment 24-well culture plates (Corning) at a ratio of approximately one T cell for every ten GBO cells (30,000 T cells and a single organoid in one well) with 1 mL of ImmunoCult-XF T Cell Expansion Medium per well. Cell counts within GBOs were estimated by dissociating similarly sized GBOs from each patient followed by automated cell counting (Countess II, Thermo Fisher Scientific Scientific). The plates were placed on an orbital shaker rotating at 120 rpm within a 37°C, 5% CO2, 90% humidity sterile incubator. Co-culture experiments were performed for six GBO samples with triplicates for each condition. The GBO-CAR-T co-cultures were monitored by brightfield microscopy at 0,1,2, and 3 day time points with samples taken for immunohistological analysis at 0, 1 and 3 days.
Cytokine ELISA
For each experimental condition, 200 μL of medium was sampled at 0,1, and 3-day time points after GBO-CAR-T co-culture from the same well, snap frozen on dry ice, and stored at −80°C until ready for analysis. Displaced medium was replaced with 200 μL of ImmunoCult-XF T Cell Expansion Medium to prevent concentration of cytokines. An ELISA was performed in triplicates on 10-fold diluted medium using human IL-2, IFN-γ, and TNF-α DuoSet ELISA kits (R&D) and absorbances were recorded at 450 nm using a microplate absorbance reader (BioRad). The absorbance value at 450 nm for the blank condition was subtracted from the experimental values. Seven-point standard curves were used to calculate cytokine concentrations from absorbance values.
QUANTIFICATION AND STATISTICAL ANALYSIS
Studies were not blinded with the exception for analysis of in vivo drug treatments. All statistical tests and sample sizes are included in the Figure Legends and text. All data are shown as mean ± SEM. In all cases, the p values are represented as follows: ***p < 0.001, ** p < 0.01, *p < 0.05, and not statistically significant when p > 0.05. In all cases, the stated “n” value is either parental tumor pieces, GBOs, or mice with multiple independent images used to obtain data points for each. No statistical methods were used to pre-determine sample sizes. For all quantifications of immunohistology, the samples being compared were processed in parallel and imaged using the same settings and laser power for confocal microscopy. Immuno-positive cells were quantified manually using the cell counter function in ImageJ (NIH). Images from the outer region of five organoids were used for quantification and compared to healthy, non-necrotic regions from five different pieces of the parental tumors. Fluorescence intensity was quantified by outlining the region of interest and using the measure function in ImageJ (NIH) to obtain the mean gray value for the marker of interest. To quantify the number of migrated tumor cells, we created “spots” for each individual cell based on immunofluorescent staining for human nuclear antigen, then drew “surfaces” by outlining the contour of the coronal brain section as well as initial graft site in Imaris (Bitplane) based on aligned image stacks as previously described (Sun et al., 2015). For 3-D reconstruction, optical stacks of images were serially aligned along the rostro-caudal axis using Reconstruct 1.1.0 (J.C. Fiala, NIH). The extent of infiltration was quantified using STEM121 immunofluorescence. Concentric rings were drawn using ImageJ to measure the distance of the furthest infiltrated cells from the margin of the initial graft site, and the scores were assigned as 0 (no infiltration), 1 (< 2 mm), 2 (2-4 mm) and 3 (> 4 mm). All quantifications of cell counts were statistically analyzed using a Student’s t test performed using GraphPad Prism (GraphPad Software Inc). Statistical analysis of CD3, Cleaved-caspase-3, and EGFRvIII/EGFR fluorescent intensities and cytokine concentrations by ELISAs was performed using two-way-ANOVA with uncorrected Fisher’s LSD test using GraphPad Prism.
DATA AND CODE AVAILABILITY
The access number for the RNA-seq, exome-seq and single-cell RNA-seq data reported in this study is NCBI GEO: GSE141947.
Supplementary Material
Figure S1. GBOs and Biobanked Samples Retain Cellular Diversity of Parental Tumors as Shown by immunostaining, Related to Figure 2.
Shown are sample confocal images of immunostaining for neural progenitor, glial, and glioma stem cell markers (A) and cell death marker (Cleaved-Caspase-3/CC3; B) for six additional parental tumors, corresponding GBOs and GBOs recovered from the biobank.
Biobanked GBOs (cryopreserved at 4 weeks after culture) were all examined at 2 weeks after recovery. Similar as in Figure 2A. See Figure 2B for quantification. Scale bar, 50 μm.
Figure S2. GBO Expansion, Efficacy of Generation, Long-Term Culturing, and Mutational Diversity, Related to Figures 1 and 2
(A-B) Continuous growth of GBOs over time. Shown in (A) are sample brightfield images of individual GBOs over a 4-week period. Scale bar, 500 μm. Shown in (B) is the quantification of the ratio of the measured 2D area at each time point to the 2D area at time point 0 of the same GBO for 5 different samples. Values represent mean ± SEM (n = 10 GBOs per sample).
(C) Quantification of percentages of micro-dissected tumor pieces that were successfully generated GBOs after 2 weeks of culture for different tumors. Values represent mean ± SEM (n = 3 wells).
(D-E) Sample confocal images of immunostaining for neural progenitor, glial, and glioma stem cell markers showing the maintenance of parental tumor cell populations in cultured GBOs up to 48 weeks and GBOs recovered from the biobank for UP-7788-PMS (D) and quantification of SOX2+, OLIG2+, and KI67+ cells in three GBOs over long-term cultures (E; the same data for tumors, GBOs at 1, 2 and 4 weeks as in Figure 2B are replotted for comparison). Similar as in Figures 2A and 2B. Scale bar, 50 μm.
(F) Bar graph comparing the incidence of major mutations in glioblastomas from the TCGA dataset (Brennan et. al, 2013) to samples in our GBO biobank. Also see Table S1 for a summary of mutations detected in our patient cases.
Figure S3. Bulk RNA-Seq and Exome Sequencing Analyses of Parental Tumors and Corresponding GBOs, Related to Figure 3
(A) Scatterplots comparing gene expression in UP-7788-PMS parental tumor and corresponding GBOs at 1, 2, 4, and 12 weeks in culture.
(B-C) Expression of EGFR in UP-7788-ANT and UP-7790 parental tumors and corresponding GBOs shown by RNA-seq (B) and immunohistology (C; Scale bar, 50 μm).
(D-F) Differential expression between parental tumors and derived GBOs. Shown in (D) is a Volcano plot of differentially expressed genes (red) in the GBOs versus in the corresponding parental tumors. The fold changes in gene expression compare the parental tumors and the corresponding GBOs at 12 weeks. Shown in (E) are boxplots of expression of sample genes in the parental tumors and corresponding GBOs. Shown in (F) are bubble plots of enriched gene ontology biological process terms in the down- and upregulated genes in GBOs compared to corresponding parental tumors.
(G) Sample copy number tracks of UP-7788-ANT and UP-8017 parental tumors and corresponding GBOs at 2 weeks from exome-sequencing. Ratio was normalized to the corresponding blood sample.
Figure S4. Single-Cell RNA-Seq Analyses of GBOs and Corresponding Parental Tumors, Related to Figure 4
(A) Plots of CNA analysis of parental tumors and derived GBOs. Macrophage/microglia and T cells in each sample were used as the non-neoplastic reference.
(B) UMAP plot of UP-8165-C and UP-8165-PV parental tumors and GBOs at 2 and 24 weeks colored by sample (left panel). The right panel shows the expression of GPNMB. Note cells with high levels of GPNMB expression were only present in UP-8165-C tumor and derived GBOs, but not in UP-8165-PV tumor and derived GBOs.
(C) UMAP plots of UP-8165-C and UP-8166-PV parental tumors and GBOs at 2 weeks colored by cluster. The same cluster number is listed in (D) and (F).
(D) Heatmap of gene expression Pearson correlation of clusters identified in UP-8165-C and UP-8165-PV parental tumors (rows) and GBOs at 2 weeks (columns) with hierarchical clustering by euclidian distance.
(E) Heatmap of gene expression of cluster-specific markers in UP-8165-C and UP-8165-PV GBOs with columns corresponding to that of (D). See Table S4 for the detailed list.
(F) Comparison of cell clusters in UP-8165C-GBOs and UP-8165PV-GBOs at 2 weeks (corresponding to that in D) with normal adult brain cells identified by single-nuclei RNA-seq of human adult brains in Lake et al. (2018) (L, top panel) and Habib et al. (2017) (H, bottom panel) with marker gene enrichment analysis. OPC: oligodendrocyte precursor cell.
(G) UMAP plot of UP-8167 parental tumors and corresponding GBOs at 2 and 24 weeks colored by sample.
(H) UMAP plots of UP-8167 parental tumors and corresponding GBOs at 2 weeks colored by cluster. The same cluster number is listed in (I) and (K).
(I) Heatmap of gene expression Pearson correlation of clusters identified in UP-8167 parental tumors (rows) and GBOs at 2 weeks (columns) with hierarchical clustering by euclidian distance.
(J) Heatmap of gene expression of cluster-specific markers in UP-8167-GBOs with columns corresponding to that of (I). See Table S4 for the detailed list.
(K) Comparison of cell clusters in UP-8167 GBOs at 2 weeks (corresponding to that in I) with normal adult brain cells identified by single-nuclei RNA-seq of human adult brains in Lake et al. (2018) (L, top panel) and Habib et al. (2017) (H, bottom panel) with marker gene enrichment analysis.
Figure S5. Consistent Engraftment, Infiltration, and Maintenance of EGFRvIII Mutation Expression of Xenografted GBOs, Related to Figure 5
(A) Sample view of GBO transplantation into the adult mouse cortex with craniotomy followed by placement of an approximately 1-mm diameter GBO into the aspirated cavity (white dashed square).
(B) MRI T1 post-contrast image with tumor satellites (arrow heads) from patient UP-7803.
(C) Sample confocal images of immunostaining for human-specific cytoplasmic antigen (STEM121) showing similar satellite phenotype in the whole brain section (left) and zoomed-in inset (right) of UP-7803-GBO xenograft at 2 months post-transplantation. Arrows point to satellite-like infiltration. Scale bar, 100 μm.
(D) Sample confocal images of immunostaining of tumor cells in the original xenograft sites and the infiltrated areas of UP-7788-ANT, UP-7803, UP-8010 and UP-8036 GBOs showing extensive vascularization from the host (Endoglin immunostaining) in the original xenograft sites, proliferation (KI67 immunostaining), progenitor marker (SOX2, NESTIN immunostaining) and EGFR/EGFRvIII expression status at 2 months post-transplantation. Scale bar, 50 um. See Table S5 for the age of GBOs used.
(E) Sample confocal images of immunostaining for STEM121 in xenografts of UP-7788-PMS and UP-7790 GBOs as well as their biobanked samples after recovery at 1 month post-transplantation displaying robust engraftment and maintenance of EGFRvIII mutation expression (inset). Scale bars, 100 um.
Figure S6. Patient Treatment Responses, Gene Expression Pathway Enrichment Stratified by GBO Treatment Responses or Mutation Status, Drug Treatment Responses Measured by GBO Size, and In Vivo Drug Treatment Responses, Related to Figure 6
(A-B) MRI T1 post-contrast images of the patient (UP-7788, A) treated with temozolomide and radiation following one month post-operative MRI scan. Note reduction in volume of tumor recurrence following the treatment. Shown in (B) are MRI T1 post-contrast images of a patient (UP-7884) with two distinct tumor regions treated with temozolomide and radiation following 1 month post-operative MRI scan. Note pseudoprogression in the frontal region and no recurrence in the temporal region.
(C) Gene set enrichment analysis stratified by reduction in KI67+ cells following radiation and temozolomide treatment. Also see Table S6.
(D) Gene set enrichment analysis stratified by reduction in KI67+ cells following gefitinib treatment. Also see Table S6.
(E) Gene set enrichment analysis stratified by reduction in KI67+ cells following trametinib treatment. Also see Table S6.
(F) Sample brightfield images of individual GBO growth over time following 1 week of exposure of drug treatment (1 μM trametinib, 1 μM everolimus, or vehicle) for UP-7790-GBOs and UP-7803-GBOs and 2 subsequent weeks in the normal GBO media. Scale bars, 200 μm.
(G) Quantification of relative GBO size over time. Similar as in Figure 2F. Each line represents single GBO growth over time. Statistical significance noted on legend compared to DMSO at equivalent time points by Student’s t test with different colors indicating different drug treatments (n = 6; **p < 0.01; ***p < 0.001; n.s.: p > 0.05).
(H) Dose response analysis of UP-7790-GBOs to trametinib (top panel) and UP-7803-GBOs to everolimus (bottom panel) treatments. Low dose and High dose of drug treatments were performed as separate experiments and separated by color with overlap in the DMSO control and 10 nM concentration. Each dot represents quantification of the ratio of the measured 2D area following 1 week of exposure of varying drug concentrations to the 2D area at time point 0 of the same single GBO. Bar values represent mean ± SEM and statistical significance measured separately for each experiment in comparison to DMSO by Student’s t test (n = 6; **p < 0.01; ***p < 0.001).
(I-K) In vivo trametinib treatment results in reduced cell proliferation within GBO xenografts. Shown in (I) is the schematic diagram of experimental procedure. UP-7790-GBOs were recovered from the biobank and cultured in vitro for 14 days before xenografting individual organoids into the adult athymic nude mouse brain as in Figure 5. At 14 days after xenograft, mice were randomized into treatment groups and were given daily intraperitoneal injections of either vehicle (10% DMSO, 90% corn oil) or 3 mg/kg trametinib for 12 days. Mice were given a single injection of EdU (10 mg/kg) 2 hours before sacrificing for analysis. Shown in (J) are sample immunostaining images of GBO xenografts from vehicle and trametinib treated mice for human nuclear antigen (huNA), KI67, EdU, and Cleavedcaspase-3 (CC3). Scale bar: 50 μm. Shown in (K) are summary bar graphs displaying the percentage of huNA+ cells that were KI67+, EdU+, or CC3+ within GBO xenografts. Values represent mean ± SEM (n = 5 animals per treatment group, **p < 0.01, ns: p > 0.05; Student’s t test).
Figure S7. Modeling Immunotherapy with Co-culture of GBOs and CAR-T Cells, Related to Figure 7
(A) Sample confocal images of immunostaining of EGFR, EGFRvIII, Cleaved-caspase-3 (CC3) and CD3 in UP-8036 and UP-8017 GBOs after 1 and 3 days of co-culture with either CD19 or 2173BBz CAR-T cells. Scale bar, 200 μm.
(B) Sample confocal images of immunostaining for CD3 and KI67 in UP-7788-PMS-GBOs after 24 hours of co-culture with either CD19 or 2173BBz CAR-T cells. Scale bar, 100 μm.
(C) Sample confocal images of immunostaining for CD3, EGFRvIII, and Granzyme B in UP-7788-PMS-GBOs after 24 hours of co-culture with 2173BBz CAR-T cells. Note the presence of activated and proliferating T cells near apoptotic EGFRvIII+ cells. Red arrows point to de-granulating T cells, blue arrows point to mitotic T cells, and yellow arrows point to apoptotic EGFRvIII+ cells. Scale bar, 50 μm.
(D) Quantification of IL2, TNF-α, and IFN-γ concentrations in media collected at 0, 1, and 3 days after co-culture of GBOs with either CD19 or 2173BBz CAR-T cells by ELISA. Values represent mean ± SEM (n = 3; *p < 0.05; **p < 0.01; ***p < 0.001; two-way ANOVA with uncorrected Fisher LSD test).
Highlights.
Rapid establishment of glioblastoma organoids (GBOs) in a defined medium with biobank
GBOs maintain parental tumor cellular heterogeneity, gene expression, and mutations
GBO transplantation exhibits efficient engraftment and aggressive infiltration
Tumor-specific treatment responses in GBOs to drugs and CAR-T cells
ACKNOWLEDGMENTS
We would like to thank all patients who generously donated tissue. We thank members of the Ming and Song laboratories, the Glioblastoma Translational Center of Excellence, and D. Lim for comments and suggestions; A.A. Mansour and F.H. Gage for initial guidance of the transplantation procedure; B. Temsamrit and E. LaNoce for technical support; W. Sarchiapone for assistance with clinical information; and K. Abdullah, E. Hudgins, and M. Piazza for assistance with tissue acquisition. The graphical abstract was created using illustrations from https://www.biorender.com. This work was partially supported by Glioblastoma Translational Center of Excellence at the Abramson Cancer Center, NIH (R37NS047344 to H.S. and R35NS097370 and U19AI131130 to G.-l.M.), and the Sheldon G.Adelson Medical Research Foundation (to G.-l.M.). D.Y.Z. was partially supported by the Blavatnik Family Fellowship in Biomedical Research. P.T.T.N. was partially supported by the Hearst Foundation Fellowship.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cell.2019.11.036.
DECLARATION OF INTERESTS
J.F.D. has ownership interest (including equity, patents, etc.) in Liquid Biotech USA, Inc., and PolyAurum, LLC. The University of Pennsylvania has submitted a patent application based on the EGFRvIII digital PCR assay utilized. Other authors declare no competing interests.
REFERENCES
- Arif SH, Pandith AA, Tabasum R, Ramzan AU, Singh S, Siddiqi MA, and Bhat AR (2018). Significant Effect of Anti-tyrosine Kinase Inhibitor (Gefitinib) on Overall Survival of the Glioblastoma Multiforme Patients in the Backdrop of Mutational Status of Epidermal Growth Factor Receptor and PTEN Genes. Asian J. Neurosurg 13, 46–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backos DS, Franklin CC, and Reigan P (2012). The role of glutathione in brain tumor drug resistance. Biochem. Pharmacol 83, 1005–1012. [DOI] [PubMed] [Google Scholar]
- Bagley SJ, Schwab RD, Nelson E,Viaene AN, Binder ZA, Lustig RA, O’Rourke DM, Brem S, Desai AS, and Nasrallah MP (2019). Histopathologic quantification of viable tumor versus treatment effect in surgically resected recurrent glioblastoma. J. Neurooncol 141, 421–429. [DOI] [PubMed] [Google Scholar]
- Berg DA, Su Y, Jimenez-Cyrus D, Patel A, Huang N, Morizet D, Lee S, Shah R, Ringeling FR, Jain R, et al. (2019). A Common Embryonic Origin of Stem Cells Drives Developmental and Adult Neurogenesis. Cell 177, 654–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian S, Repic M, Guo Z, Kavirayani A, Burkard T, Bagley JA, Krauditsch C, and Knoblich JA (2018). Genetically engineered cerebral organoids model brain tumor formation. Nat. Methods 15, 631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigner SH, Humphrey PA, Wong AJ, Vogelstein B, Mark J, Friedman HS, and Bigner DD (1990). Characterization of the epidermal growth factor receptor in human glioma cell lines and xenografts. Cancer Res. 50, 8017–8022. [PubMed] [Google Scholar]
- Bleijs M, van de Wetering M, Clevers H, and Drost J (2019). Xenograft and organoid model systems in cancer research. EMBO J. 38, e101654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, Jager M, Ponz-Sarvise M, Tiriac H, Spector MS, et al. (2015). Organoid models of human and mouse ductal pancreatic cancer. Cell 160, 324–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandes AA, Franceschi E, Tosoni A, Blatt V, Pession A, Tallini G, Bertorelle R, Bartolini S, Calbucci F, Andreoli A, et al. (2008). MGMT promoter methylation status can predict the incidence and outcome of pseudoprogression after concomitant radiochemotherapy in newly diagnosed glioblastoma patients. J. Clin. Oncol 26, 2192–2197. [DOI] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. TCGA Research Network (2013). The somatic genomic landscape of glioblastoma. Cell 155, 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broutier L, Mastrogiovanni G, Verstegen MM, Francies HE, Gavarró LM, Bradshaw CR, Allen GE, Arnes-Benito R, Sidorova O, Gaspersz MP, et al. (2017). Human primary liver cancer-derived organoid cultures for disease modeling and drug screening. Nat. Med 23, 1424–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, Cox AJ, Kruglyak S, and Saunders CT (2016). Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222. [DOI] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, and Getz G (2013). Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol 31, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H (2016). Modeling Development and Disease with Organoids. Cell 165, 1586–1597. [DOI] [PubMed] [Google Scholar]
- da Silva B, Mathew RK, Polson ES, Williams J, and Wurdak H (2018). Spontaneous Glioblastoma Spheroid Infiltration of Early-Stage Cerebral Organoids Models Brain Tumor Invasion. SLAS Discov. 23, 862–868. [DOI] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, et al. 1000 Genomes Project Analysis Group (2011). The variant call format and VCFtools. Bioinformatics 27, 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmanis S, Sloan SA, Croote D, Mignardi M, Chernikova S, Samghababi P, Zhang Y, Neff N, Kowarsky M, Caneda C, et al. (2017). Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 21, 1399–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruin EC, Cowell C, Warne PH, Jiang M, Saunders RE, Melnick MA, Gettinger S, Walther Z, Wurtz A, Heynen GJ, et al. (2014). Reduced NF1 expression confers resistance to EGFR inhibition in lung cancer. Cancer Discov. 4, 606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 20, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes SA, Beare D, Boutselakis H, Bamford S, Bindal N, Tate J, Cole CG, Ward S, Dawson E, Ponting L, et al. (2017). COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 45 (D1), D777–D783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, Dowling C, Wanjala JN, Undvall EA, Arora VK, et al. (2014). Organoid cultures derived from patients with advanced prostate cancer. Cell 159, 176–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison E, and Math G (2012). Haplotype-based variant detection from short-read sequencing. arXiv, arXiv:1207.3907. [Google Scholar]
- Giannini C, Sarkaria JN, Saito A, Uhm JH, Galanis E, Carlson BL, Schroeder MA, and James CD (2005). Patient tumor EGFR and PDGFRA gene amplifications retained in an invasive intracranial xenograft model of glioblastoma multiforme. Neuro-oncol. 7, 164–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjorevski N, Sachs N, Manfrin A, Giger S, Bragina ME, Ordóñez-Morán P, Clevers H, and Lutolf MP (2016). Designer matrices for intestinal stem cell and organoid culture. Nature 539, 560–564. [DOI] [PubMed] [Google Scholar]
- Goff SL, Morgan RA, Yang JC, Sherry RM, Robbins PF, Restifo NP, Feldman SA, Lu YC, Lu L, Zheng Z, et al. (2019). Pilot Trial of Adoptive Transfer of Chimeric Antigen Receptor-transduced T Cells Targeting EGFRvIII in Patients With Glioblastoma. J. Immunother 42, 126–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib N, Avraham-Davidi I, Basu A, Burks T, Shekhar K, Hofree M, Choudhury SR, Aguet F, Gelfand E, Ardlie K, et al. (2017). Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods 14, 955–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, and Fu L (2015). Mechanisms of resistance to EGFR tyrosine kinase inhibitors. Acta Pharm. Sin. B 5, 390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubert CG, Rivera M, Spangler LC, Wu Q, Mack SC, Prager BC, Couce M, McLendon RE, Sloan AE, and Rich JN (2016). A Three-Dimensional Organoid Culture System Derived from Human Glioblastomas Recapitulates the Hypoxic Gradients and Cancer Stem Cell Heterogeneity of Tumors Found In Vivo. Cancer Res. 76, 2465–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, Nace AK, Dentchev T, Thekkat P, Loew A, et al. (2015). Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci. Transl. Med 7, 275ra22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- June CH, O’Connor RS, Kawalekar OU, Ghassemi S, and Milone MC (2018). CAR T cell immunotherapy for human cancer. Science 359, 1361–1365. [DOI] [PubMed] [Google Scholar]
- Kadoshima T, Sakaguchi H, Nakano T, Soen M, Ando S, Eiraku M, and Sasai Y (2013). Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc. Natl. Acad. Sci. USA 110, 20284–20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly PJ, Daumas-Duport C, Scheithauer BW, Kall BA, and Kispert DB (1987). Stereotactic histologic correlations of computed tomography- and magnetic resonance imaging-defined abnormalities in patients with glial neoplasms. Mayo Clin. Proc 62, 450–459. [DOI] [PubMed] [Google Scholar]
- Kim S, Scheffler K, Halpern AL, Bekritsky MA, Noh E, Kallberg M, Chen X, Kim Y, Beyter D, Krusche P, and Saunders CT (2018). Strelka2: fast and accurate calling of germline and somatic variants. Nat. Methods 15, 591–594. [DOI] [PubMed] [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, and Wilson RK (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopper O, de Witte CJ, Lõhmussaar K,Valle-Inclan JE, Hami N, Kester L, Balgobind AV, Korving J, Proost N, Begthel H, et al. (2019). An organoid platform for ovarian cancer captures intra-and interpatient heterogeneity. Nat. Med 25, 838–849. [DOI] [PubMed] [Google Scholar]
- Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, Wilson KA, Byars A, Sahmoud T, and Franz DN (2010). Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N. Engl. J. Med 363, 1801–1811. [DOI] [PubMed] [Google Scholar]
- Kuan CT, Wakiya K, Dowell JM, Herndon JE 2nd, Reardon DA, Graner MW, Riggins GJ, Wikstrand CJ, and Bigner DD (2006). Glycoprotein nonmetastatic melanoma protein B, a potential molecular therapeutic target in patients with glioblastoma multiforme. Clin. Cancer Res 12, 1970–1982. [DOI] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44 (W1), W90–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake BB, Chen S, Sos BC, Fan J, Kaeser GE, Yung YC, Duong TE, Gao D, Chun J, Kharchenko PV, and Zhang K (2018). Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol 36, 70–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, and Knoblich JA (2013). Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, et al. (2016). ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 44 (D1), D862–D868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledur PF, Onzi GR, Zong H, and Lenz G (2017). Culture conditions defining glioblastoma cells behavior: what is the impact for novel discoveries? Oncotarget 8, 69185–69197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, Pastorino S, Purow BW, Christopher N, Zhang W, et al. (2006). Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 9, 391–403. [DOI] [PubMed] [Google Scholar]
- Lee SH, Hu W, Matulay JT, Silva MV, Owczarek TB, Kim K, Chua CW, Barlow LJ, Kandoth C, Williams AB, et al. (2018). Tumor Evolution and Drug Response in Patient-Derived Organoid Models of Bladder Cancer. Cell 173, 515–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek JT, Johnson WE, Parker HS, Jaffe AE, and Storey JD (2012). The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2011).A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2019). The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 47, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkous A, Balamatsias D, Snuderl M, Edwards L, Miyaguchi K, Milner T, Reich B, Cohen-Gould L, Storaska A, Nakayama Y, et al. (2019). Modeling Patient-Derived Glioblastoma with Cerebral Organoids. Cell Rep. 26, 3203–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Jian X, and Boerwinkle E (2011). dbNSFP: a lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat 32, 894–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wu C, Li C, and Boerwinkle E (2016). dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat 37, 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. (2015). Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel JJ, Yust-Katz S, Patel AJ, Cachia D, Liu D, Park M, Yuan Y, Kent TA, and de Groot JF (2018). Inability of positive phase II clinical trials of investigational treatments to subsequently predict positive phase III clinical trials in glioblastoma. Neuro-oncol. 20, 113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour AA, Goncalves JT, Bloyd CW, Li H, Fernandes S, Quang D, Johnston S, Parylak SL, Jin X, and Gage FH (2018). An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol 36, 432–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, and DePristo MA (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, Lu KV, Yoshimoto K, Huang JH, Chute DJ, et al. (2005). Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N. Engl. J. Med 353, 2012–2024. [DOI] [PubMed] [Google Scholar]
- Neal JT, Li X, Zhu J, Giangarra V, Grzeskowiak CL, Ju J, Liu IH, Chiou SH, Salahudeen AA, Smith AR, et al. (2018). Organoid Modeling of the Tumor Immune Microenvironment. Cell 175, 1972–1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, Richman AR, Silverbush D, Shaw ML, Hebert CM, et al. (2019). An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 178, 835–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newick K, O’Brien S, Moon E, and Albelda SM (2017). CAR T Cell Therapy for Solid Tumors. Annu. Rev. Med 68, 139–152. [DOI] [PubMed] [Google Scholar]
- O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, Martinez-Lage M, Brem S, Maloney E, Shen A, et al. (2017). A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med 9, eaaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa J, Pao GM, Shokhirev MN, and Verma IM (2018). Glioblastoma Model Using Human Cerebral Organoids. Cell Rep. 23, 1220–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, and Barnholtz-Sloan JS (2018). CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2011-2015. Neuro-oncol. 20 (suppl_4), iv1–iv86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandita A, Aldape KD, Zadeh G, Guha A, and James CD (2004). Contrasting in vivo and in vitro fates of glioblastoma cell subpopulations with amplified EGFR. Genes Chromosomes Cancer 39, 29–36. [DOI] [PubMed] [Google Scholar]
- Paolillo M, Boselli C, and Schinelli S (2018). Glioblastoma under Siege: An Overview of Current Therapeutic Strategies. Brain Sci. 8, E15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paşca AM, Sloan SA, Clarke LE,Tian Y, Makinson CD, Huber N, Kim CH, Park JY, O’Rourke NA, Nguyen KD, et al. (2015). Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods 12, 671–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. (2014). Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrizii M, Bartucci M, Pine SR, and Sabaawy HE (2018). Utility of Glioblastoma Patient-Derived Orthotopic Xenografts in Drug Discovery and Personalized Therapy. Front. Oncol 8, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter DL, Levine BL, Kalos M, Bagg A, and June CH (2011). Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med 365, 725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Nguyen HN, Song MM, Hadiono C, Ogden SC, Hammack C, Yao B, Hamersky GR, Jacob F, Zhong C, et al. (2016). Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 165, 1238–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rich JN, Reardon DA, Peery T, Dowell JM, Quinn JA, Penne KL, Wikstrand CJ, Van Duyn LB, Dancey JE, McLendon RE, et al. (2004). Phase II trial of gefitinib in recurrent glioblastoma. J. Clin. Oncol 22, 133–142. [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roldán GB, Scott JN, McIntyre JB, Dharmawardene M, de Robles PA, Magliocco AM, Yan ES, Parney IF, Forsyth PA, Cairncross JG, et al. (2009). Population-based study of pseudoprogression after chemoradiotherapy in GBM. Can. J. Neurol. Sci 36, 617–622. [DOI] [PubMed] [Google Scholar]
- Sachs N, de Ligt J, Kopper O, Gogola E, Bounova G, Weeber F, Balgobind AV, Wind K, Gracanin A, Begthel H, et al. (2018). A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 172, 373–386. [DOI] [PubMed] [Google Scholar]
- Saunders A, Macosko EZ, Wysoker A, Goldman M, Krienen FM, de Rivera H, Bien E, Baum M, Bortolin L, Wang S, et al. (2018). Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 174, 1015–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Yang X, Froelich CJ, and Greenberg AH (2000). Purification and use of granzyme B. Methods Enzymol. 322, 125–143. [DOI] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive integration of single cell data. Cell 177, 1888–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group (2005). Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med 352, 987–996. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun GJ, Zhou Y, Stadel RP, Moss J, Yong JH, Ito S, Kawasaki NK, Phan AT, Oh JH, Modak N, et al. (2015). Tangential migration of neuronal precursors of glutamatergic neurons in the adult mammalian brain. Proc. Natl. Acad. Sci. USA 112, 9484–9489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Venteicher AS, Hebert C, Escalante LE, Patel AP, Yizhak K, Fisher JM, Rodman C, Mount C, Filbin MG, et al. (2016). Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature 539, 309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhm JH, Ballman KV, Wu W, Giannini C, Krauss JC, Buckner JC, James CD, Scheithauer BW, Behrens RJ, Flynn PJ, et al. (2011). Phase II evaluation of gefitinib in patients with newly diagnosed Grade 4 astrocytoma: Mayo/North Central Cancer Treatment Group Study N0074. Int. J. Radiat. Oncol. Biol. Phys 80, 347–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. (2013). From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics 43, 11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varia MA, Calkins-Adams DP, Rinker LH, Kennedy AS, Novotny DB, Fowler WC Jr., and Raleigh JA (1998). Pimonidazole: a novel hypoxia marker for complementary study of tumor hypoxia and cell proliferation in cervical carcinoma. Gynecol. Oncol 71, 270–277. [DOI] [PubMed] [Google Scholar]
- Venteicher AS, Tirosh I, Hebert C, Yizhak K, Neftel C, Filbin MG, Hovestadt V, Escalante LE, Shaw ML, Rodman C, et al. (2017). Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science 355, eaai8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, and Hakonarson H (2010). ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng YL, An R, Cassin J, Joseph J, Mi R, Wang C, Zhong C, Jin SG, Pfeifer GP, Bellacosa A, et al. (2017). An Intrinsic Epigenetic Barrier for Functional Axon Regeneration. Neuron 94, 337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan HHN, Siu HC, Law S, Ho SL, Yue SSK, Tsui WY, Chan D, Chan AS, Ma S, Lam KO, et al. (2018). A Comprehensive Human Gastric Cancer Organoid Biobank Captures Tumor Subtype Heterogeneity and Enables Therapeutic Screening. Cell Stem Cell 23, 882–897. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. GBOs and Biobanked Samples Retain Cellular Diversity of Parental Tumors as Shown by immunostaining, Related to Figure 2.
Shown are sample confocal images of immunostaining for neural progenitor, glial, and glioma stem cell markers (A) and cell death marker (Cleaved-Caspase-3/CC3; B) for six additional parental tumors, corresponding GBOs and GBOs recovered from the biobank.
Biobanked GBOs (cryopreserved at 4 weeks after culture) were all examined at 2 weeks after recovery. Similar as in Figure 2A. See Figure 2B for quantification. Scale bar, 50 μm.
Figure S2. GBO Expansion, Efficacy of Generation, Long-Term Culturing, and Mutational Diversity, Related to Figures 1 and 2
(A-B) Continuous growth of GBOs over time. Shown in (A) are sample brightfield images of individual GBOs over a 4-week period. Scale bar, 500 μm. Shown in (B) is the quantification of the ratio of the measured 2D area at each time point to the 2D area at time point 0 of the same GBO for 5 different samples. Values represent mean ± SEM (n = 10 GBOs per sample).
(C) Quantification of percentages of micro-dissected tumor pieces that were successfully generated GBOs after 2 weeks of culture for different tumors. Values represent mean ± SEM (n = 3 wells).
(D-E) Sample confocal images of immunostaining for neural progenitor, glial, and glioma stem cell markers showing the maintenance of parental tumor cell populations in cultured GBOs up to 48 weeks and GBOs recovered from the biobank for UP-7788-PMS (D) and quantification of SOX2+, OLIG2+, and KI67+ cells in three GBOs over long-term cultures (E; the same data for tumors, GBOs at 1, 2 and 4 weeks as in Figure 2B are replotted for comparison). Similar as in Figures 2A and 2B. Scale bar, 50 μm.
(F) Bar graph comparing the incidence of major mutations in glioblastomas from the TCGA dataset (Brennan et. al, 2013) to samples in our GBO biobank. Also see Table S1 for a summary of mutations detected in our patient cases.
Figure S3. Bulk RNA-Seq and Exome Sequencing Analyses of Parental Tumors and Corresponding GBOs, Related to Figure 3
(A) Scatterplots comparing gene expression in UP-7788-PMS parental tumor and corresponding GBOs at 1, 2, 4, and 12 weeks in culture.
(B-C) Expression of EGFR in UP-7788-ANT and UP-7790 parental tumors and corresponding GBOs shown by RNA-seq (B) and immunohistology (C; Scale bar, 50 μm).
(D-F) Differential expression between parental tumors and derived GBOs. Shown in (D) is a Volcano plot of differentially expressed genes (red) in the GBOs versus in the corresponding parental tumors. The fold changes in gene expression compare the parental tumors and the corresponding GBOs at 12 weeks. Shown in (E) are boxplots of expression of sample genes in the parental tumors and corresponding GBOs. Shown in (F) are bubble plots of enriched gene ontology biological process terms in the down- and upregulated genes in GBOs compared to corresponding parental tumors.
(G) Sample copy number tracks of UP-7788-ANT and UP-8017 parental tumors and corresponding GBOs at 2 weeks from exome-sequencing. Ratio was normalized to the corresponding blood sample.
Figure S4. Single-Cell RNA-Seq Analyses of GBOs and Corresponding Parental Tumors, Related to Figure 4
(A) Plots of CNA analysis of parental tumors and derived GBOs. Macrophage/microglia and T cells in each sample were used as the non-neoplastic reference.
(B) UMAP plot of UP-8165-C and UP-8165-PV parental tumors and GBOs at 2 and 24 weeks colored by sample (left panel). The right panel shows the expression of GPNMB. Note cells with high levels of GPNMB expression were only present in UP-8165-C tumor and derived GBOs, but not in UP-8165-PV tumor and derived GBOs.
(C) UMAP plots of UP-8165-C and UP-8166-PV parental tumors and GBOs at 2 weeks colored by cluster. The same cluster number is listed in (D) and (F).
(D) Heatmap of gene expression Pearson correlation of clusters identified in UP-8165-C and UP-8165-PV parental tumors (rows) and GBOs at 2 weeks (columns) with hierarchical clustering by euclidian distance.
(E) Heatmap of gene expression of cluster-specific markers in UP-8165-C and UP-8165-PV GBOs with columns corresponding to that of (D). See Table S4 for the detailed list.
(F) Comparison of cell clusters in UP-8165C-GBOs and UP-8165PV-GBOs at 2 weeks (corresponding to that in D) with normal adult brain cells identified by single-nuclei RNA-seq of human adult brains in Lake et al. (2018) (L, top panel) and Habib et al. (2017) (H, bottom panel) with marker gene enrichment analysis. OPC: oligodendrocyte precursor cell.
(G) UMAP plot of UP-8167 parental tumors and corresponding GBOs at 2 and 24 weeks colored by sample.
(H) UMAP plots of UP-8167 parental tumors and corresponding GBOs at 2 weeks colored by cluster. The same cluster number is listed in (I) and (K).
(I) Heatmap of gene expression Pearson correlation of clusters identified in UP-8167 parental tumors (rows) and GBOs at 2 weeks (columns) with hierarchical clustering by euclidian distance.
(J) Heatmap of gene expression of cluster-specific markers in UP-8167-GBOs with columns corresponding to that of (I). See Table S4 for the detailed list.
(K) Comparison of cell clusters in UP-8167 GBOs at 2 weeks (corresponding to that in I) with normal adult brain cells identified by single-nuclei RNA-seq of human adult brains in Lake et al. (2018) (L, top panel) and Habib et al. (2017) (H, bottom panel) with marker gene enrichment analysis.
Figure S5. Consistent Engraftment, Infiltration, and Maintenance of EGFRvIII Mutation Expression of Xenografted GBOs, Related to Figure 5
(A) Sample view of GBO transplantation into the adult mouse cortex with craniotomy followed by placement of an approximately 1-mm diameter GBO into the aspirated cavity (white dashed square).
(B) MRI T1 post-contrast image with tumor satellites (arrow heads) from patient UP-7803.
(C) Sample confocal images of immunostaining for human-specific cytoplasmic antigen (STEM121) showing similar satellite phenotype in the whole brain section (left) and zoomed-in inset (right) of UP-7803-GBO xenograft at 2 months post-transplantation. Arrows point to satellite-like infiltration. Scale bar, 100 μm.
(D) Sample confocal images of immunostaining of tumor cells in the original xenograft sites and the infiltrated areas of UP-7788-ANT, UP-7803, UP-8010 and UP-8036 GBOs showing extensive vascularization from the host (Endoglin immunostaining) in the original xenograft sites, proliferation (KI67 immunostaining), progenitor marker (SOX2, NESTIN immunostaining) and EGFR/EGFRvIII expression status at 2 months post-transplantation. Scale bar, 50 um. See Table S5 for the age of GBOs used.
(E) Sample confocal images of immunostaining for STEM121 in xenografts of UP-7788-PMS and UP-7790 GBOs as well as their biobanked samples after recovery at 1 month post-transplantation displaying robust engraftment and maintenance of EGFRvIII mutation expression (inset). Scale bars, 100 um.
Figure S6. Patient Treatment Responses, Gene Expression Pathway Enrichment Stratified by GBO Treatment Responses or Mutation Status, Drug Treatment Responses Measured by GBO Size, and In Vivo Drug Treatment Responses, Related to Figure 6
(A-B) MRI T1 post-contrast images of the patient (UP-7788, A) treated with temozolomide and radiation following one month post-operative MRI scan. Note reduction in volume of tumor recurrence following the treatment. Shown in (B) are MRI T1 post-contrast images of a patient (UP-7884) with two distinct tumor regions treated with temozolomide and radiation following 1 month post-operative MRI scan. Note pseudoprogression in the frontal region and no recurrence in the temporal region.
(C) Gene set enrichment analysis stratified by reduction in KI67+ cells following radiation and temozolomide treatment. Also see Table S6.
(D) Gene set enrichment analysis stratified by reduction in KI67+ cells following gefitinib treatment. Also see Table S6.
(E) Gene set enrichment analysis stratified by reduction in KI67+ cells following trametinib treatment. Also see Table S6.
(F) Sample brightfield images of individual GBO growth over time following 1 week of exposure of drug treatment (1 μM trametinib, 1 μM everolimus, or vehicle) for UP-7790-GBOs and UP-7803-GBOs and 2 subsequent weeks in the normal GBO media. Scale bars, 200 μm.
(G) Quantification of relative GBO size over time. Similar as in Figure 2F. Each line represents single GBO growth over time. Statistical significance noted on legend compared to DMSO at equivalent time points by Student’s t test with different colors indicating different drug treatments (n = 6; **p < 0.01; ***p < 0.001; n.s.: p > 0.05).
(H) Dose response analysis of UP-7790-GBOs to trametinib (top panel) and UP-7803-GBOs to everolimus (bottom panel) treatments. Low dose and High dose of drug treatments were performed as separate experiments and separated by color with overlap in the DMSO control and 10 nM concentration. Each dot represents quantification of the ratio of the measured 2D area following 1 week of exposure of varying drug concentrations to the 2D area at time point 0 of the same single GBO. Bar values represent mean ± SEM and statistical significance measured separately for each experiment in comparison to DMSO by Student’s t test (n = 6; **p < 0.01; ***p < 0.001).
(I-K) In vivo trametinib treatment results in reduced cell proliferation within GBO xenografts. Shown in (I) is the schematic diagram of experimental procedure. UP-7790-GBOs were recovered from the biobank and cultured in vitro for 14 days before xenografting individual organoids into the adult athymic nude mouse brain as in Figure 5. At 14 days after xenograft, mice were randomized into treatment groups and were given daily intraperitoneal injections of either vehicle (10% DMSO, 90% corn oil) or 3 mg/kg trametinib for 12 days. Mice were given a single injection of EdU (10 mg/kg) 2 hours before sacrificing for analysis. Shown in (J) are sample immunostaining images of GBO xenografts from vehicle and trametinib treated mice for human nuclear antigen (huNA), KI67, EdU, and Cleavedcaspase-3 (CC3). Scale bar: 50 μm. Shown in (K) are summary bar graphs displaying the percentage of huNA+ cells that were KI67+, EdU+, or CC3+ within GBO xenografts. Values represent mean ± SEM (n = 5 animals per treatment group, **p < 0.01, ns: p > 0.05; Student’s t test).
Figure S7. Modeling Immunotherapy with Co-culture of GBOs and CAR-T Cells, Related to Figure 7
(A) Sample confocal images of immunostaining of EGFR, EGFRvIII, Cleaved-caspase-3 (CC3) and CD3 in UP-8036 and UP-8017 GBOs after 1 and 3 days of co-culture with either CD19 or 2173BBz CAR-T cells. Scale bar, 200 μm.
(B) Sample confocal images of immunostaining for CD3 and KI67 in UP-7788-PMS-GBOs after 24 hours of co-culture with either CD19 or 2173BBz CAR-T cells. Scale bar, 100 μm.
(C) Sample confocal images of immunostaining for CD3, EGFRvIII, and Granzyme B in UP-7788-PMS-GBOs after 24 hours of co-culture with 2173BBz CAR-T cells. Note the presence of activated and proliferating T cells near apoptotic EGFRvIII+ cells. Red arrows point to de-granulating T cells, blue arrows point to mitotic T cells, and yellow arrows point to apoptotic EGFRvIII+ cells. Scale bar, 50 μm.
(D) Quantification of IL2, TNF-α, and IFN-γ concentrations in media collected at 0, 1, and 3 days after co-culture of GBOs with either CD19 or 2173BBz CAR-T cells by ELISA. Values represent mean ± SEM (n = 3; *p < 0.05; **p < 0.01; ***p < 0.001; two-way ANOVA with uncorrected Fisher LSD test).
Data Availability Statement
The access number for the RNA-seq, exome-seq and single-cell RNA-seq data reported in this study is NCBI GEO: GSE141947.