

Introduction
Neurodegeneration with brain iron accumulation (NBIA) comprises a group of disorders with a progressive extrapyramidal syndrome and excessive iron deposition in the brain, particularly globus pallidus and substantia nigra. NBIA is considered to be a very rare disease group, with a prevalence of less than 1/1,000,000 in the general population [1–3]. Ten genes have been identified as associated with different NBIA subtypes. Only two of these genes (FTL and CP) encode proteins that play a direct role in iron metabolism, while the remaining eight (PANK2, PLA2G6, C19orf12, WDR45, FA2H, ATP13A2, DCAF17, and COASY) encode proteins involved in lipid metabolism, mitochondrial function, coenzyme A (CoA) metabolism, and autophagy [4]. The most common forms of NBIA are pantothenate kinase-associated neurodegeneration (PKAN), PLA2G6-associated neurodegeneration (PLAN), and beta propeller protein-associated neurodegeneration (BPAN). Mitochondrial membrane protein-associated neurodegeneration (MPAN) occurs less frequently. It is inherited in an autosomal recessive fashion, and a common Polish mutation has been identified in several cases. MPAN is caused by biallelic mutations in C19orf12, which encodes a protein of the mitochondrial membrane [5, 6]. Herein, we present a patient with progressive neurocognitive decline after normal development during infancy, undiagnosed for 19 years until detailed genetic analysis revealed mutations in C19orf12.
Case report
A previously healthy 14-year-old Turkish girl, the third child of related parents, had normal developmental milestones and presented with a history of frequent falls for 10 years. Her parents reported that she had always been clumsy and displayed persistent difficulty walking upstairs. She learned to walk by 16 months of age and to speak at 2 years. An intellectual decline associated with psychiatric disturbances eventually emerged. An electromyogram showed infrequent, but definite acute denervation potentials in the distal leg muscles bilaterally, suggesting an axonal neuropathy. Muscle biopsy of the right quadriceps muscle showed mild myopathic changes. Evaluation at that time included normal metabolic testing.
There was a history of personality change and learning difficulties. There is no history of neurological disorders in her family. The brain MRI was performed and revealed moderate hypointense signal in the globus pallidus and substantia nigra on T2, which appeared mildly hyperintense on T1, suggestive of pathological iron accumulation (Fig. 1).
Fig. 1.
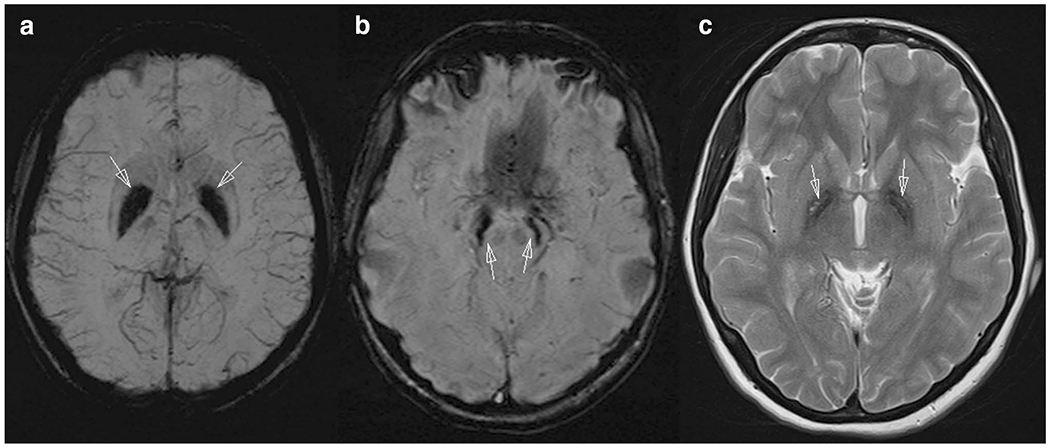
Brain MRI of the patient shows iron accumulation in bilateral globus pallidus (a), substantia nigra, and red nucleus (b) on the susceptibility weighted images (SWI) as dark signal. Axial plane T2-weighted image (c) demonstrates globus pallidus hypointensity with hyperintense streaking in the region of the medial medullary lamina
Upon admission at the age of 19 years, she was wheelchair bound. She presented with progressive cognitive deficits. Neurological examination revealed spastic tetraparesis and severe bilateral optic atrophy. The anterior segment was normal with no corneal or lens involvement.
She was ataxic, dysarthric, and unable to walk. She had bladder and bowel incontinence. She had levadopa-resistant orolingual and limb dystonia. Brain MRI findings (signal reduction in the globus pallidus and substantia nigra) suggested a metabolic disorder, in particular an inborn error of metal metabolism such as NBIA.
A DNA sample was submitted, as part of a cohort of idiopathic NBIA cases, for whole exome sequencing to the University of Washington Center for Mendelian Genomics (University of Washington Center for Mendelian Genomics detailed sequencing methods, http://uwcmg.org/docs/Exome_Genome_Sequencing/uwcmg_Detailed_Methods_Sequencing.docx). Pathogenicity of identified variants was evaluated using multiple bioinformatics tools. For each variant, minor allele frequencies were checked in variant databases including the 1000 Genomes, Exome Variant Server (EVS), and Exome Aggregation Consortium (ExAC) databases. Whole exome studies identified a homozygous 11 bp deletion, c.171_181delCGGGGGGCTGT, predicted to cause a frameshift and premature termination of the C19orf12 protein (p.Gly58Argfs*10).The segregation analysis of the parents showed that they were both heterozygous for the same mutation, consistent with their report of consanguinity. Sanger sequencing confirmed the mutation in the proband.
Discussion
Neurodegeneration with brain iron accumulation (NBIA) constitute a group of neurodegenerative disorders inherited as autosomal dominant, recessive or X-linked traits in which iron accumulates in the brain, resulting in progressive dystonia, spasticity, parkinsonism, neuropsychiatric abnormalities, and optic atrophy or retinal degeneration. In this case, a homozygous 11 bp deletion, c.171_181delCGGGGGGCTGT in C19orf12 identified the diagnosis as MPAN. We have not seen it reported in other patients. There is a different mutation (C19orf12 p.Thr11Met) which is frequent among adult Turkish patients with MPAN [3, 7, 8]. The onset of MPAN is typically between 4 and 20 years of age, and the progression is generally slower than PKAN. Brain MRI of our patient demonstrates iron accumulation in bilateral globus pallidus, substantia nigra, and red nucleus that were seen only on the SWI- and T2-weighted images. In the literature, it was described that, on T2-weighted images MPAN patients have hyperintense streaking of the medial medullary lamina between the globus pallidus interna and externa that could be mistaken for an “eye-of-the-tiger sign”, leading to a wrong radiologic diagnosis of PKAN. Also, in the literature it was said that “hyperintense streaking of the medial medullary lamina” may discriminate MPAN from other NBIA subtypes. Secondly, cortical and cerebellar atrophy may be seen in more advanced disease in MPAN patients. But we did not observe cortical or cerebellar atrophy in our patient [9, 10].
Ophthalmological abnormalities have been described in NBIA patients, and optic atrophy is one of the core symptoms in most MPAN cases. Optic nerve atrophy has not been described in PKAN patients. For this reason, detailed ophthalmological examination can help in the differential diagnosis of NBIA patients. There is no curative treatment, and the management of patients relies on rehabilitation and symptomatic medications. Treatment for NBIA disorders remains symptomatic, but encouraging research offers new perspectives for treatment. Deep brain stimulation (DBS) of the internal globus pallidus may produce some benefit, in particular, in patients with PKAN. Moreover, oral or intrathecal baclofen often provides symptomatic relief but has no known disease-modifying effects [11, 12]. Assuming the role of brain iron accumulation in the pathogenesis of NBIA, the current focus is on the use of iron-chelating agents. Recently, a new therapeutic chelating agent deferiprone (3-hydroxy-1,2-dimethylpyridin-4(1H)-one, DFP), with the potential to cross the blood-brain barrier, has been studied in an attempt to slow down the progression of the disease. This clinical trial showed a consistent reduction of brain iron levels in the globus pallidus [13]. Our patient used deferiprone for 2 years; nonetheless, the clinical benefit was absent in our case. While clinical diagnosis of NBIA is a challenge, analysis of both clinical findings, including age at onset, family history, and presence of optic atrophy, and characteristic imaging abnormalities allows accurate diagnosis of most of the NBIA subtypes and guides the genetic testing [9, 14].
Acknowledgements
Sequencing was provided by the University of Washington Center for Mendelian Genomics (UW-CMG) and was funded by the National Human Genome Research Institute and the National Heart, Lung and Blood Institute Grant 2UM1HG006493 to Drs. Debbie Nickerson, Michael Bamshad, and Suzanne Leal.
Footnotes
Conflict of interest The authors declare that they have no conflict of interest.
Ethical approval All procedures were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments.
Informed consent Informed consent was obtained from our patient’s family included in the study.
References
- 1.Selikhova M, Fedotova E, Wiethoff S, Schottlaender LV, Klyushnikov S, Illarioshkin SN, Houlden H (2017) A 30-year history of MPAN case from Russia. Clin Neurol Neurosurg 159:111–113. 10.1016/j.clineuro.2017.05.025 [DOI] [PubMed] [Google Scholar]
- 2.Deutschländer A, Konno T, Ross OA (2017) Mitochondrial membrane protein-associated neurodegeneration. Parkinsonism Relat Disord 39:1–3. 10.1016/j.parkreldis.2017.03.014 [DOI] [PubMed] [Google Scholar]
- 3.Olgiati S, Doğu O, Tufekcioglu Z, Diler Y, Saka E, Gultekin M, Kaleagasi H, Kuipers D, Graafland J, Breedveld GJ, Quadri M, Sürmeli R, Sunter G, Doğan T, Yalçın AD, Bilgiç B, Elibol B, Emre M, Hanagasi HA, Bonifati V (2017) The p.Thr11Met mutation in c19orf12 is frequent among adult Turkish patients with MPAN. Parkinsonism Relat Disord 39:64–70. 10.1016/j.parkreldis.2017.03.012 [DOI] [PubMed] [Google Scholar]
- 4.Skowronska M, Kmiec T, Jurkiewicz E, Malczyk K, Kurkowska-Jastrzęska I, Czlonkowska A (2017) Evolution and novel radiological changes of neurodegeneration associated with mutations in C19orf12. Parkinsonism Relat Disord 39:71–76. 10.1016/j.parkreldis.2017.03.013 [DOI] [PubMed] [Google Scholar]
- 5.Al Macki N, Al Rashdi I (2017) A novel deletion mutation of exon 2 of the C19orf12 gene in an Omani family with mitochondrial membrane protein-associated neurodegeneration (MPAN). Oman Med J 32(1):66–68. 10.5001/omj.2017.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gore E, Appleby BS, Cohen ML, DeBrosse SD, Leverenz JB, Miller BL, Siedlak SL, Zhu X, Lerner AJ (2016) Clinical and imaging characteristics of late onset mitochondrial membrane protein-associated neurodegeneration (MPAN). Neurocase 22(5):476–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Langwinska-Wosko E, Skowronska M, Kmiec T, Czlonkowska A (2016) Retinal and optic nerve abnormalities in neurodegeneration associated with mutations in C19orf12 (MPAN). J Neurol Sci 370:237–240. 10.1016/j.jns.2016.09.046 [DOI] [PubMed] [Google Scholar]
- 8.Salomão RP, Pedroso JL, Gama MT, Dutra LA, Maciel RH, Godeiro-Junior C, Chien HF, Teive HA, Cardoso F, Barsottini OG (2016) A diagnostic approach for neurodegeneration with brain iron accumulation: clinical features, genetics and brain imaging. Arq Neuropsiquiatr 74(7):587–596. 10.1590/0004-282X20160080 (review) [DOI] [PubMed] [Google Scholar]
- 9.Hogarth P (2015) Neurodegeneration with brain iron accumulation: diagnosis and management. J Mov Disord 8(1):1–13. 10.14802/jmd.14034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoganathan S, Sudhakar SV, Thomas M, Dutta AK, Danda S (2016) “Eye of tiger sign” mimic in an adolescent boy with mitochondrial membrane protein associated neurodegeneration (MPAN). Brain Dev 38(5):516–519. 10.1016/j.braindev.2015.10.017 [DOI] [PubMed] [Google Scholar]
- 11.Meyer E, Kurian MA, Hayflick SJ (2015) Neurodegeneration with brain iron accumulation: genetic diversity and pathophysiological mechanisms. Annu Rev Genom Hum Genet 16:257–279. 10.1146/annurev-genom-090314-025011 [DOI] [PubMed] [Google Scholar]
- 12.Tschentscher A, Dekomien G, Ross S, Cremer K, Kukuk GM, Epplen JT, Hoffjan S (2015) Analysis of the C19orf12 and WDR45 genes in patients with neurodegeneration with brain iron accumulation. J Neurol Sci 349(1–2):105–9. 10.1016/j.jns.2014.12.036 [DOI] [PubMed] [Google Scholar]
- 13.Amaral LL, Gaddikeri S, Chapman PR, Roy R, Gaddikeri RS, Marussi VH, Bag AK (2015) Neurodegeneration with brain iron accumulation: clinicoradiological approach to diagnosis. J Neuroimaging 25(4):539–551. 10.1111/jon.12195 [DOI] [PubMed] [Google Scholar]
- 14.Hartig M, Prokisch H, Meitinger T, Klopstock T (2013) Mitochondrial membrane protein-associated neurodegeneration (MPAN). Int Rev Neurobiol 110:73–84. 10.1016/B978-0-12-410502-7.00004-1 [DOI] [PubMed] [Google Scholar]