

Cubé resin has been known since the 1850s[1] to contain active ingredients useful as pesticides and insecticides.[2] Fractionation of the active ingredients has led to the isolation of biosynthetically related flavonoids and rotenoids which have been reisolated from various botanical sources.[3] The most prominent and active ingredients are rotenone, deguelin and tephrosin (Figure 1). The toxic properties of rotenone were originally ascribed to its inhibition of the NADH/ubiquinone oxidoreductase machinery. Since the mid 90s, several lines of evidence emerged regarding the anticancer and chemopreventive properties of these compounds.[4,5] It was indeed shown by Pezzuto and co-workers that deguelin most potently inhibited the induction of ornithine decarboxylase[6] which is associated with tumor progression.[7,8] It was then shown that the reduction of ornithine decarboxylase activity is coupled to the NADH/ubiquinone inhibition[9] and that rotenone binds to the PSST subunit of this large multi-protein complex.[10] More recently, Lee and co-workers demonstrated that deguelin disrupted the interaction between Hsp90 and HIF1α and suggested that deguelin binds to the nucleotide binding pocket of Hsp90[11] as radicicol and geldanamycin derivatives.[12] Most recently, deguelin and related rotenoids were identified in a screening for modulators of oncogene-regulated hematopoietic differentiation using a zebrafish model supporting the Hsp90 inhibition mechanism of deguelin.[13] Hsp90 has emerged as an important therapeutic target.[14–16] Despite the seemingly ubiquitous functions of this highly expressed chaperone, its role in stabilizing conformationally labile proteins has implications in diverse pathologies ranging from cancer indications,[17,18] neurodegenerative diseases,[19–23] infectious diseases,[24] and inflammation-related disorders.[25] Our interest in Hsp90 inhibition[26,27] instigated the development of synthetic technologies which could be used to broaden the diversity of the rotenoids.
Figure 1.
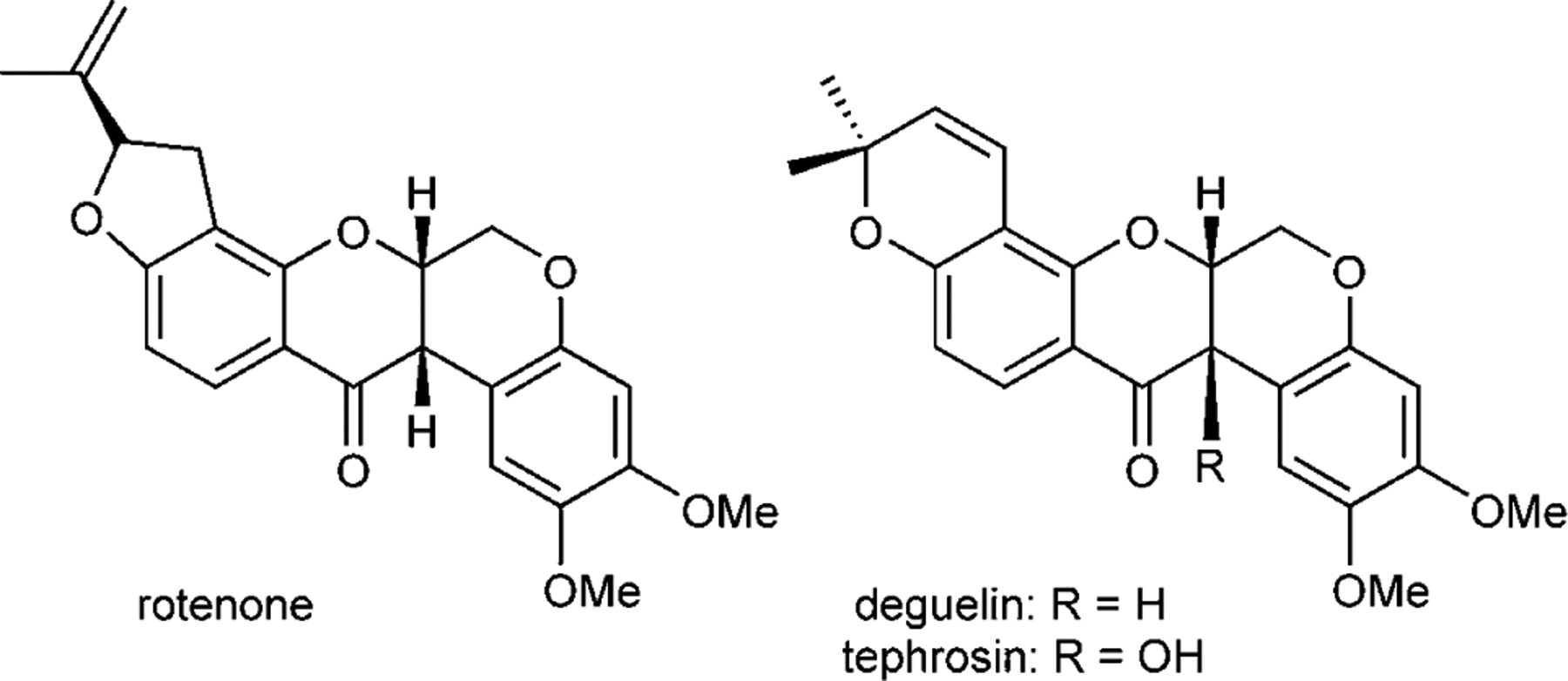
Structure of rotenone, deguelin and tephrosin.
While the first synthesis of rotenone dates back to the 1960s,[28,29] the ready availability of the natural product itself has not stimulated significant synthetic efforts towards the rotenoids and there is no asymmetric synthesis of deguelin (only three total synthesis of racemic deguelin and one formal synthesis).[30–33] Even more surprising considering its potent biological activity, there is no reported synthesis of tephrosin. Structure–activity studies regarding the rotenoids has been largely confined to related natural products extracted from different sources and manipulation by semisyn-thesis.[3] A recent study comparing the activity of deguelin, tephrosin, rotenone and related natural products showed tephrosin to be the most potent inhibitor of HT1080 proliferation amongst the family of rotenoids and related flavins.[34]
We reasoned that deguelin and tephrosin could arise synthetically from a common intermediate 1 via different forms of oxidation (dihydroxylation or epoxidation) of the conjugated alkene 1 which would in turn provide efficient disconnections of the fused ring system (Scheme 1). Further disconnections through a combination of a metathesis and Heck cyclization leads to two readily available intermediates 2 and 3 which could be coupled via Mitsunobu reaction. Indeed, some precedents for the dihydroxylation and the Heck reaction already existed, however, the question remained whether this dihydroxylation could be performed in the presence of the other pyran unsaturation.[35]
Scheme 1.
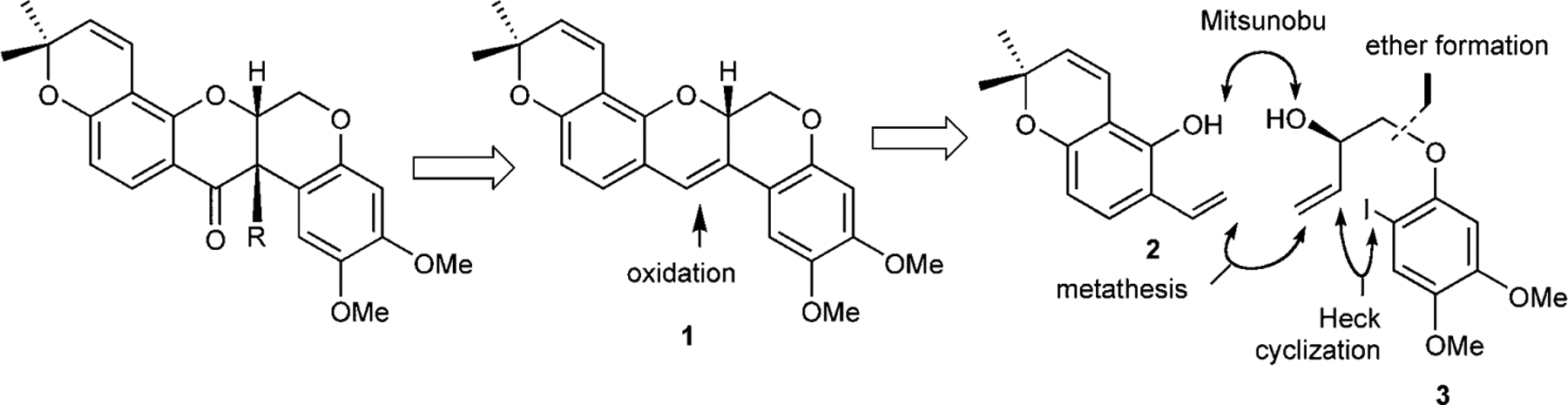
Retrosynthetic disconnections of deguelin and tephrosin.
The synthesis commenced with the derivatization of 2,4-dihydroxybenzaldehyde (4) with 3-chloro-3-methylbutyne (Scheme 2). A very clean conversion to the desired para-alkylated product was obtained due to its greater acidity using potassium carbonate as a base. It turned out to be crucial to include catalytic amounts of copper[36] in the reaction to accentuate the reactivity of this otherwise sterically encumbered electrophile. The alkyne was cyclized under thermal conditions (165°C) to obtain 5.[37] While this reaction can be done at lower temperature in the presence of platinum dichloride catalyst, the thermal reaction proceeded in higher yield and could be achieved quantitatively in only 30 min under microwave irradiation. The aldehyde was then olefinated with an excess of methylenetriphenylphosphorane to afford intermediate 2 in 87% overall yield (three steps). Fragment 9 was obtained in three steps from commercially available 3,4-dimethoxyphenol (6) by nucleophilic epoxide opening of (2R)-vinyloxirane followed by iodination (NIS, TFA)[38] and Heck cyclization in 34% overall yield. The iodination proved to be the most problematic step. While the crude NMR of the reaction showed a quantitative transformation, the product was colored and a quick filtration through a pad of silica always led to loss of material. Other iodination methods and alternative work-up procedures did not improve the outcome of the reaction. The key intermediates 2 and 9 were coupled under Mitsunobu conditions. The cyclization using Grubbs second-generation catalyst[39–41] afforded a quantitative transformation based on NMR and LC/MS analysis, however, purification by silica gel column led to loss of material. Sharpless asymmetric dihydroxylation[42] using the ADmix-α afforded the desired product 10 in excellent yield as a 5:1 diastereomeric ratio (separable by silica gel) while the ADmix-β afforded the opposite diastereoisomer exclusively; this suggests that the existing stereochemical information is mismatched relatively to the asymmetric induction of the ADmix-α. It is noteworthy that no product arising from dihydroxylation of the pyran system was detected. Treatment of the product 10 with IBX resin[43] afforded tephrosin along with a byproduct originating from the cleavage of the diol in a ratio of 8:1 (measured by LC-MS). While these products were not separable by silica gel, tephrosin could be isolated by HPLC albeit with a loss of material affording a 33% isolated yield. It is noteworthy that treatment of 10 with Dess–Martin reagent led exclusively to the cleavage of the diol. Treatment of 10 with acids such as sulfonic acid resin yielded a pinacol rearrangement to give the furan 11 related to glyceollidin (Scheme 3). No trace of proton transfer which would have led to deguelin was detected under a variety conditions. Attempts to achieve this transformation via the epoxide were unsuccessful. Nevertheless, product 11 provides an interesting diverging point into a different ring geometry and could be readily elaborated into diverse oximes such as 12 and 13. Tephrosin could be cleanly reduced to deguelin under the action of zinc with acetic acid albeit without reaching a complete conversion. Attempts to push the reaction beyond 50% conversion led to the emergence of side products. Synthetic tephrosin and deguelin had identical spectral and chromatographic properties to the natural samples. Noting that several isolated yields were low despite excellent conversion, the reaction sequence starting from intermediates 2 and racemic 9 was carried out without any purification. Thus, the crude Mitsunobu product was subjected to a metathesis followed by dihydroxylation and IBX oxidation. To our satisfaction, tephrosine and its diastereoisomers emanating from the S enantiomer of 9 were the only significant products observed by LC/MS (Figure 2) and could be isolated in 23% overall yield.
Scheme 2.
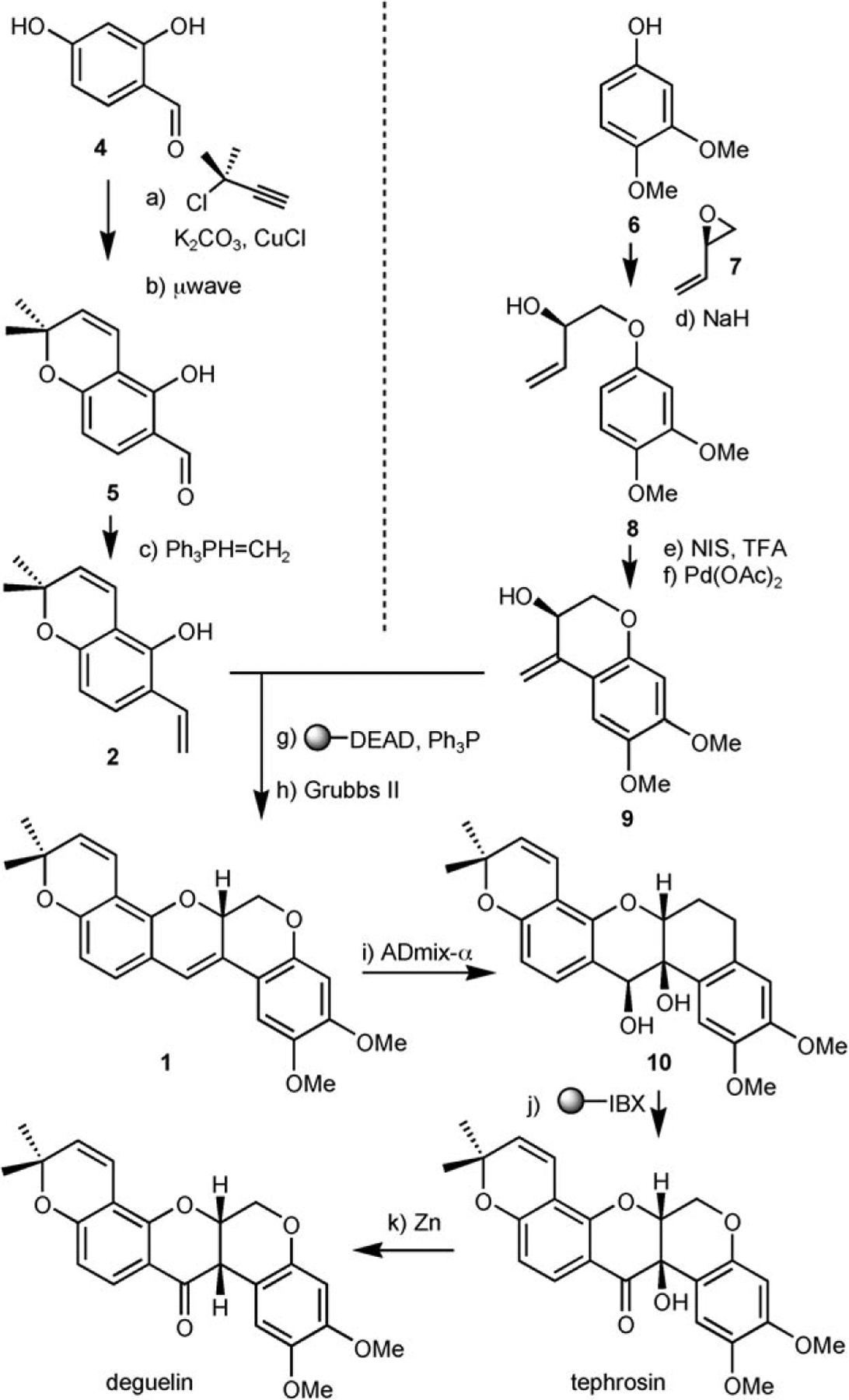
Synthesis of tephrosin and deguelin. a) 3-Chloro-3-methylbut-1-yne (1.1 equiv), K2CO3 (1.1 equiv), KI (0.10 equiv), Cul (0.05 equiv), CH3CN, 23°C, 12 h, 90%; b) m-xylene, μw, 180°C, 30 min, quant; c) PPh3MeBr (3.3 equiv), nBuLi (3.3 equiv), THF, 0°C, 1 h, 97%; d) 7 (6.0 equiv), NaH (1.5 equiv), DMF, μw, 80°C, 20 min, 70%; e) NIS (1.1 equiv), TFA (0.30 equiv), CH3CN, 23 °C, 15 min, 60%; f) KOAc (5.0 equiv), Pd(OAc)2 (0.1 equiv), dppe (0.20 equiv), DMF, 30 min, 100°C, 81 %; g) PS-DEAD (2.0 equiv), PPh3 (2.0 equiv), NEt3.(2.0 equiv), CH2Cl2, 23 °C, 24 h, 63%; h) Grubbs II (0.10 equiv), CH2Cl2, 23 °C, 4h, 78%; i) ADmix-α (3.0 equiv), MeSO2NH2 (3.0 equiv), tBuOH/H2O 1:1, 23°C, 12 h, 97% (5:1 d.r.); j) PS-IBX (10 equiv), CH2Cl2, 23°C, 14 h, 33%; k) Zn, AcOH, μw, 88 °C, 10 min, 50%. PS-DEAD = ethoxycarbonylazocarboxymethyl polystyrene; dppe = 1,2-bis(diphenylphosphino)-ethane; NIS = N-iodosuccinimide; IBX = 2-iodoxybenzoic acid; ADmix-α = 1.6mm (DHQ)2PHAL, 500mm potassium carbonate, 500mm potassium ferricyanide, and 0.7 mm potassium osmate dihydrate.
Scheme 3.
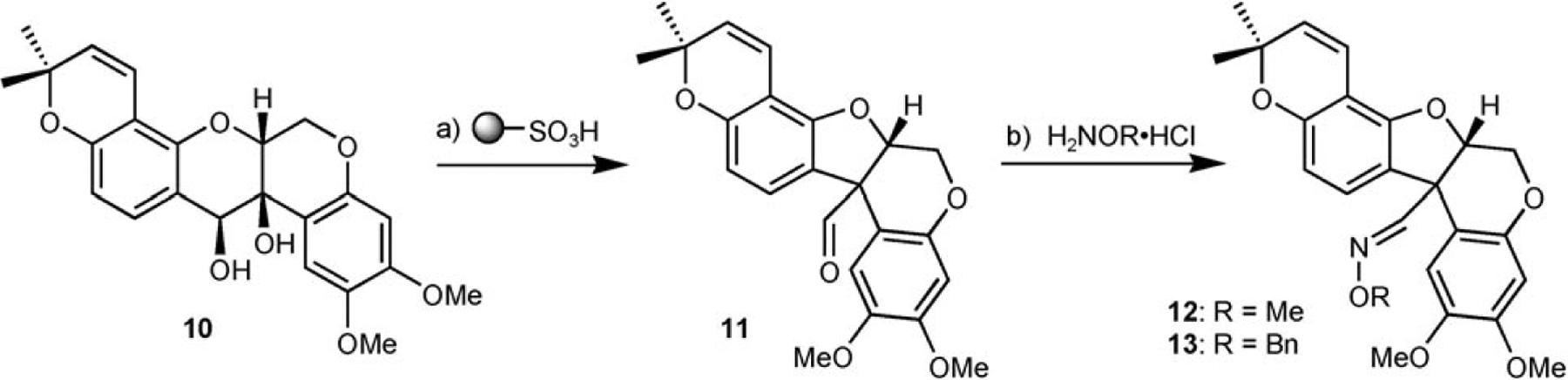
Pinacol rearrangement of intermediate 10. a) PS-SO3H (20 equiv), CH2Cl2, 23°C, 2 h, quant; b) H2NOR·HCl (10 equiv), EtOH, 40°C, 4 h, (12, 90%; 13, 92%).
Figure 2.
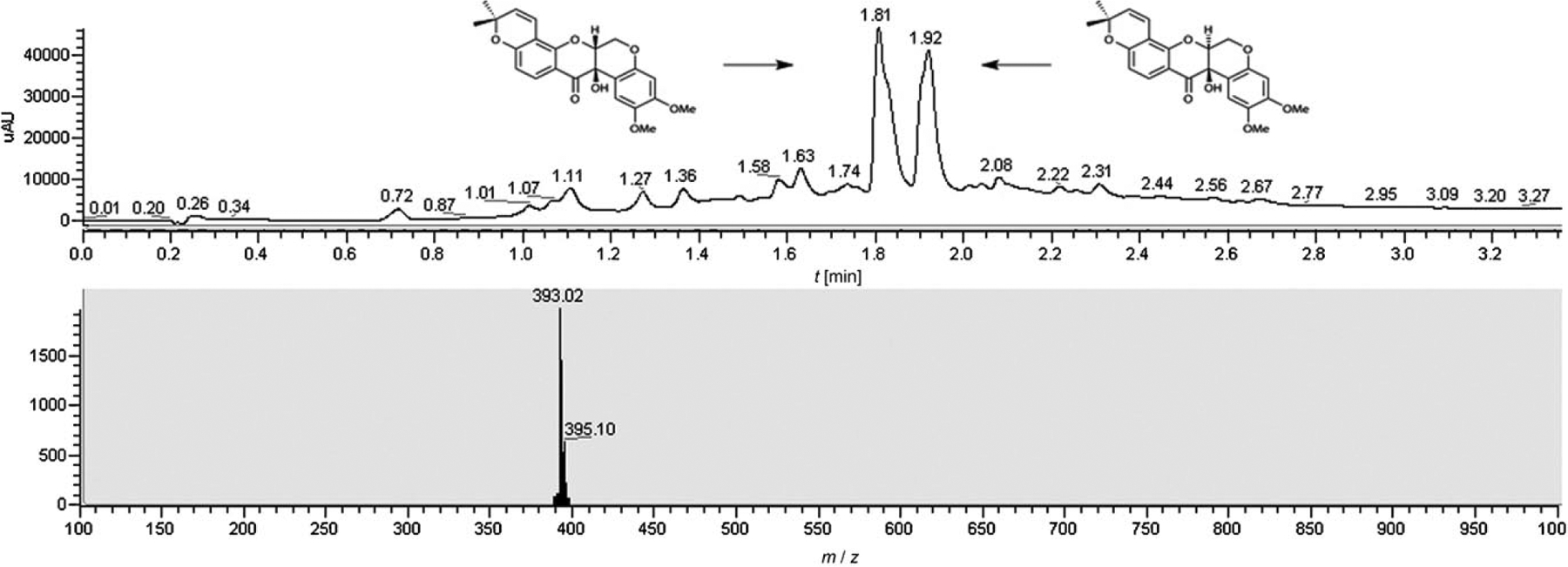
LC-MS chromatogram of the crude product obtained from the reaction sequence starting with 2 and racemic 9 without any silica purification.
With these products in hand we assayed their affinity for Hsp90’s nucleotide-binding pocket using a competition assay[44] with fluorescently labeled 17-AAG known to target the N-terminal nucleotide binding pocket. Contrarily to the previous speculations based on docking studies,[11] deguelin, tephrosin and rotenone showed no notable affinity in this assay (>50 μm). To further investigate potential pharmacological similarities between deguelin and geldanamycin derivative 17-AAG, the three rotenoids (deguelin, tephrosin and rotenone) were assayed for their ability to promote client depletion in SkBr3, a breast cancer cell line known to be responsive to Hsp90 inhibitors such as 17-AAG.[45] The level of ErbB2, a client known to be sensitive to pharmacological inhibition of Hsp90 with 17-AAG, and phosphoAkt (pAkt), a client previously reported to be sensitive to deguelin treatment[11] were analyzed. As shown in Figure 3, 17-AAG caused the expected depletion of both ErbB2 and pAkt. None of the rotenoids caused significant depletion of ErbB2 while all rotenoids caused moderate depletion of pAkt with rotenone being the most effective compound (Figure 3). While deguelin does not bind the N-terminal nucleotide pocket of Hsp90, these results do not exclude that deguelin and related rotenoids modulate the function of Hsp90 by binding to the C-terminal domain in analogy to novobiocin.[46–48] Indeed, it has been shown that C-terminal domain binders affect a different subset of the Hsp90 clients than N-terminal binders.[49,50] To address this question, the impact of the three rotenoids (deguelin, tephrosin and rotenone) on the Hsp90-IP6K2 association was evaluated as it is known to be sensitive to C-terminal binders. To this end, 293T cells cotransfected with Flag-Hsp90 and myc-IP6K2 were incubated with 10 μm of each compound for 18 h and, upon lysis of the cell, the integrity of the Hsp90-IP6K2 association was assessed by immunoprecipitation of Hsp90 (anti-Flag antibody) and blotted with anti-myc antibody. None of the rotenoids showed any disruption (data not shown).
Figure 3.
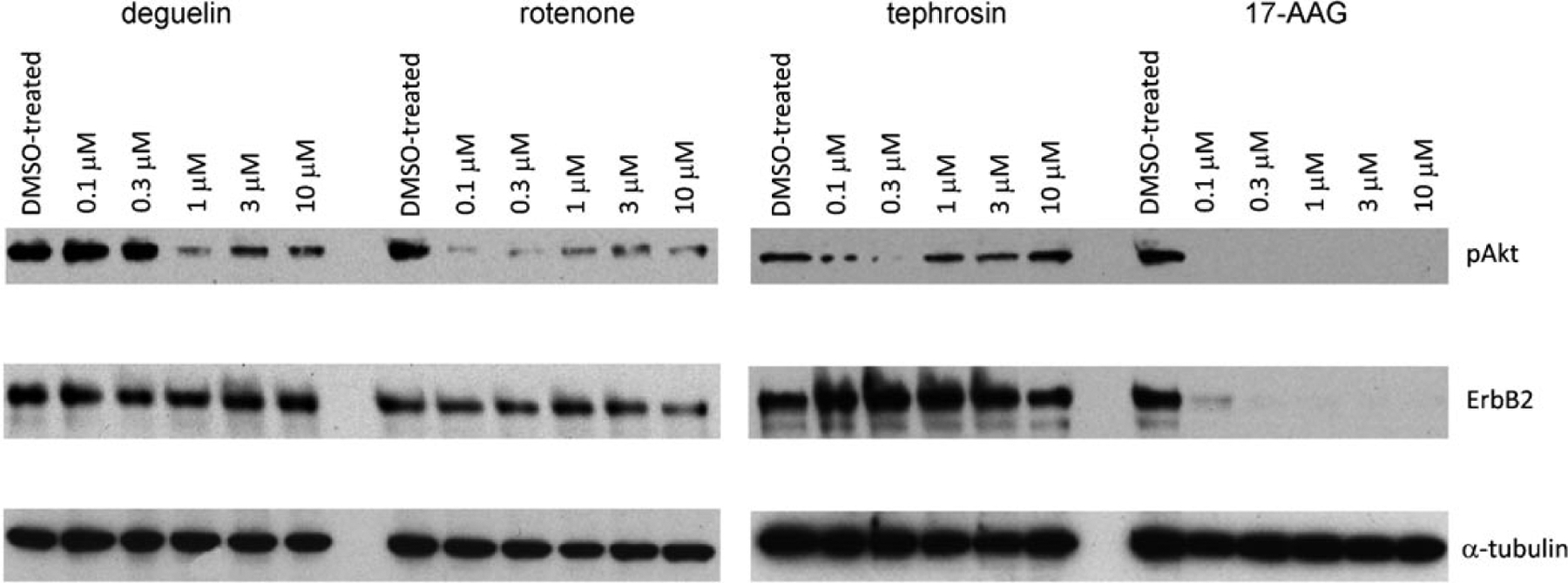
Depletion of ErbB2 and pAkt cells that were treated with the inhibitor for 18 h.
Deguelin and related rotenoids are attractive pharmacophores for chemoprevention showing in vivo activity against breast, colon and skin cancer[51–53] and preventing growth in a mouse xenograft bearing a lung tumor (H1299).[11] While deguelin has been shown to disrupt some of Hsp90’s function, it does not bind to the N-terminal domain nor C-terminal domain as other known pharmacophores. The concise nature and modularity of the synthesis described herein should facilitate access to labeled analogs to dissect the mechanism of action of this important pharmacophore.
Supplementary Material
Acknowledgements
This work was supported in part by a grant from Institut Universitaire de France (IUF). We thank Sylvain Bianchi for preliminary work.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/chem.201001080.
References
- [1].Geoffroy E, Ann Inst. Colon. Marseille 1895, 2, 1–86. [Google Scholar]
- [2].Fukami H, Nakajima M, Naturally Occuring Insecticides, Dekker, New York, 1971. [Google Scholar]
- [3].Botta B, Menendez P, Zappia G, de Lima RA, Torge R, Monachea GD, Curr. Med. Chem 2009, 16, 3414–3468. [DOI] [PubMed] [Google Scholar]
- [4].Cunningham ML, Soliman MS, Badr MZ, Matthews HB, Cancer Lett. 1995, 95, 93–97. [DOI] [PubMed] [Google Scholar]
- [5].Gerhäuser C, Mar W, Lee SK, Suh N, Luo Y, Kosmeder J, Luyengi L, Fong HHS, Kinghorn AD, Nat. Med 1995, 1, 260–266. [DOI] [PubMed] [Google Scholar]
- [6].Gerhauser C, Lee SK, Kosmeder JW, Moriarty RM, Hamel E, Mehta RG, Moon RC, Pezzuto JM, Cancer Res. 1997, 57, 3429–3435. [PubMed] [Google Scholar]
- [7].Lu YP, Chang RL, Huang MT, Conney AH, Carcinogenesis 1993, 14, 293–297. [DOI] [PubMed] [Google Scholar]
- [8].Seiler N, Atanassov C, Raul F, Int. J. Oncology 1998, 13, 993–1006. [DOI] [PubMed] [Google Scholar]
- [9].Fang N, Casida JE, Proc. Natl. Acad. Sci. USA 1998, 95, 3380–3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Schuler F, Yano T, Di Bernardo S, Yagi T, Yankovskaya V, Singer TP, Casida JE, Proc. Natl. Acad. Sci. USA 1999, 96, 4149–4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Oh SH, Woo JK, Yazici YD, Myers JN, Kim W-Y, Jin Q, Hong SS, Park H-J, Suh Y-G, Kim K-W, Hong WK, Lee H-Y, Natl J. Cancer Inst. 2007, 99, 949–961. [DOI] [PubMed] [Google Scholar]
- [12].Schulte TW, Akinaga S, Soga S, Sullivan W, Stensgard B, Toft D, Neckers LM, Cell Stress Chaperones 1998, 3, 100–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yeh J-RJ, Munson KM, Elagib KE, Goldfarb AN, Sweetser DA, Peterson RT, Nat. Chem. Biol 2009, 5, 236–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Workman P, Burrows F, Neckers L, Rosen N, Ann. N. Y. Acad. Sci 2007, 1113, 202–216. [DOI] [PubMed] [Google Scholar]
- [15].Neckers L, Neckers K, Expert Opin. Emerging Drugs 2005, 10, 137–149. [DOI] [PubMed] [Google Scholar]
- [16].Taldone T, Sun W, Chiosis G, Bioorg. Med. Chem. Lett 2009, 19, 2225–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Whitesell L, Lindquist SL, Nat. Rev. Cancer 2005, 5, 761–772. [DOI] [PubMed] [Google Scholar]
- [18].Taldone T, Gozman A, Maharaj R, Chiosis G, Curr. Opin. Pharmacol 2008, 8, 370–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sittler A, Lurz R, Lueder G, Priller J, Lehrach H, Hayer-Hartl MK, Hartl FU, Wanker EE, Hum. Mol. Genet 2001, 10, 1307–1315. [DOI] [PubMed] [Google Scholar]
- [20].Waza M, Adachi H, Katsuno M, Minamiyama M, Sang C, Tanaka F, Inukai A, Doyu M, Sobue G, Nat. Med 2005, 11, 1088–1095. [DOI] [PubMed] [Google Scholar]
- [21].Luo W, Dou F, Rodina A, Chip S, Kim J, Zhao Q, Moulick K, Aguirre J, Wu N, Greengard P, Chiosis G, Proc. Natl. Acad. Sci. USA 2007, 104, 9511–9516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Auluck PK, Bonini NM, Nat. Med 2002, 8, 1185–1186. [DOI] [PubMed] [Google Scholar]
- [23].Luo W, Rodina A, Chiosis G, BMC Neurosci. 2008, 9 Suppl. 2, S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Geller R, Vignuzzi M, Andino R, Frydman J, Genes Dev. 2007, 21, 195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Rice JW, Veal JM, Fadden RP, Barabasz AF, Partridge JM, Barta TE, Dubois LG, Huang KH, Mabbett SR, Silinski MA, Steed PM, Hall SE, Arthritis Rheum. 2008, 58, 3765–3775. [DOI] [PubMed] [Google Scholar]
- [26].Barluenga S, Fontaine JG, Wang C, Aouadi K, Chen R, Beebe K, Neckers L, Winssinger N, ChemBioChem 2009, 10, 2753–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Barluenga S, Wang C, Fontaine JG, Aouadi K, Beebe K, Tsutsumi S, Neckers L, Winssinger N, Angew. Chem 2008, 120, 4504–4507; Angew. Chem. Int. Ed. 2008, 47, 4432–4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Miyano M, J. Am. Chem. Soc 1965, 87, 3958–3962. [DOI] [PubMed] [Google Scholar]
- [29].Miyano M, Kobayashi A, Matsui M, Agric. Biol. Chem 1961, 25, 673–677. [Google Scholar]
- [30].Pastine SJ, Sames D, Org. Lett 2003, 5, 4053–4055. [DOI] [PubMed] [Google Scholar]
- [31].Fukami H, Oda J, Sakata G, Nakajima M, Agric. Biol. Chem 1961, 25, 252–256. [Google Scholar]
- [32].Fukami H, Oda J, Sakata G, Nakajima M, Bull. Agric. Chem. Soc. Jpn 1960, 24, 327–328. [Google Scholar]
- [33].Omokawa H, Yamashita K, Agric. Biol. Chem 1974, 38, 1731–1734. [Google Scholar]
- [34].Matsuda H, Yoshida K, Miyagawa K, Asao Y, Takayama S, Nakashima S, Xu F, Yoshikawa M, Bioorg. Med. Chem 2007, 15, 1539–1546. [DOI] [PubMed] [Google Scholar]
- [35].Ahmad-Junan S, Amos P, Whiting D, J. Chem. Soc. Perkin Trans 1 1992, 539–545. [Google Scholar]
- [36].Bell D, Davies M, Geen G, Mann I, Synthesis 1995, 707–712. [Google Scholar]
- [37].Thomas P, Whiting DA, Tetrahedron Lett. 1984, 25, 1099–1102. [Google Scholar]
- [38].Castanet A, Colobert F, Broutin P, Tetrahedron Lett. 2002, 43, 5047–5048. [Google Scholar]
- [39].Chang S, Grubbs RH, J. Org. Chem 1998, 63, 864–866. [DOI] [PubMed] [Google Scholar]
- [40].Trnka TM, Grubbs RH, Acc. Chem. Res 2001, 34, 18–29. [DOI] [PubMed] [Google Scholar]
- [41].Nicolaou KC, Bulger PG, Sarlah D, Angew. Chem 2005, 117, 4564–4601; Angew. Chem. Int. Ed. 2005, 44, 4490–4527. [DOI] [PubMed] [Google Scholar]
- [42].Kolb H, Van Nieuwenhze M, Sharpless K, Chem. Rev 1994, 94, 2483–2547. [Google Scholar]
- [43].Sorg G, Mengei A, Jung G, Rademann J, Angew. Chem 2001, 113, 4532–4535; Angew. Chem. Int. Ed. 2001, 40, 4395–4397. [Google Scholar]
- [44].Kim J, Felts S, Llauger L, He H, Huezo H, Rosen N, Chiosis G, Biomol J. Screening 2004, 9, 375–381. [DOI] [PubMed] [Google Scholar]
- [45].Xu W, Mimnaugh E, Rosser MF, Nicchitta C, Marcu M, Yarden Y, Neckers L, J. Biol. Chem 2001, 276, 3702–3708. [DOI] [PubMed] [Google Scholar]
- [46].Burlison JA, Neckers L, Smith AB, Maxwell A, Blagg BS, J. Am. Chem. Soc 2006, 128, 15529–15536. [DOI] [PubMed] [Google Scholar]
- [47].Marcu MG, Schulte TW, Neckers L, J. Natl. Cancer Inst 2000, 92, 242–248. [DOI] [PubMed] [Google Scholar]
- [48].Donnelly A, Blagg BS, Curr. Med. Chem 2008, 15, 2702–2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Rosenhagen MC, Soti C, Schmidt U, Wochnik GM, Hartl FU, Holsboer F, Young JC, Rein T, Mol. Endocrinol 2003, 17, 1991–2001. [DOI] [PubMed] [Google Scholar]
- [50].Chakraborty A, Koldobskiy MA, Sixt KM, Juluri KR, Mustafa AK, Snowman AM, van Rossum DB, Patterson RL, Snyder SH, Proc. Natl. Acad. Sci. USA 2008, 105, 1134–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lee H-Y, Biochem. Pharmacol 2004, 68, 1119–1124. [DOI] [PubMed] [Google Scholar]
- [52].Udeani GO, Gerhauser C, Thomas CF, Moon RC, Kosmeder JW, Kinghorn AD, Moriarty RM, Pezzuto JM, Cancer Res. 1997, 57, 3424–3428. [PubMed] [Google Scholar]
- [53].Udeani GO, Zhao G-M, Shin YG, KosmederII JW, Beecher CWW, Kinghorn AD, Moriarty RM, Moon RC, Pezzuto JM, Cancer Chemother. Pharmacol 2001, 47, 263–268. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.