

Abstract
A synthetic approach to paxilline indole diterpenes is described. The route to the pentacyclic core relies on a new regioselective alkenylation of ketones and a tandem radical addition−aldol reaction sequence to access vicinal quaternary stereocenters. Emindole SB, the simplest member of the family, is synthesized in 11 steps from commercially available material to demonstrate the application of this approach.
Paxilline indole diterpenes comprise a family of fungal metabolites with a diverse set of biological activities. Notable examples include inhibition of potassium channels by paspalinine, penitrem A, and aflatrem;1 alteration of cellular lipid balance by terpendoles C and D;2 insecticidal activity of nodulisporic acids;3 and inhibition of mitosis by terpendole E and 11-ketopaspaline.4 Other reports detail antimicrobial5 and antiviral6 activities of paxilline indole diterpenes, further broadening the scope of potentially useful biological effects exerted by these natural products. Over 80 congeners identified to date (e.g., 1, 2, and 3, Figure 1) share a common polycyclic core that is believed to arise from an unusual polycyclization of a prenylated indole precursor.7 The resulting structural complexity of paxilline indole diterpenes attracted considerable attention from synthetic organic chemists. In 1985, Smith and Mewshaw reported a pioneering synthesis of paspaline, with over a dozen more syntheses of this and other related natural products appearing in the following years.8 A notable hallmark of many of those investigations has been the challenge associated with the direct assembly of an embedded transhexahydroindene fragment including vicinal quaternary stereocenters.9 Here we demonstrate a solution to this problem that allows an 8-step stereocontrolled assembly of a functionalized pentacyclic core of paxilline indole diterpenes. As a proof of concept, we also report a synthesis of emindole SB (3),6,10 a key biosynthetic precursor to more complex congeners, in 11 steps from commercially available material.
Figure 1.
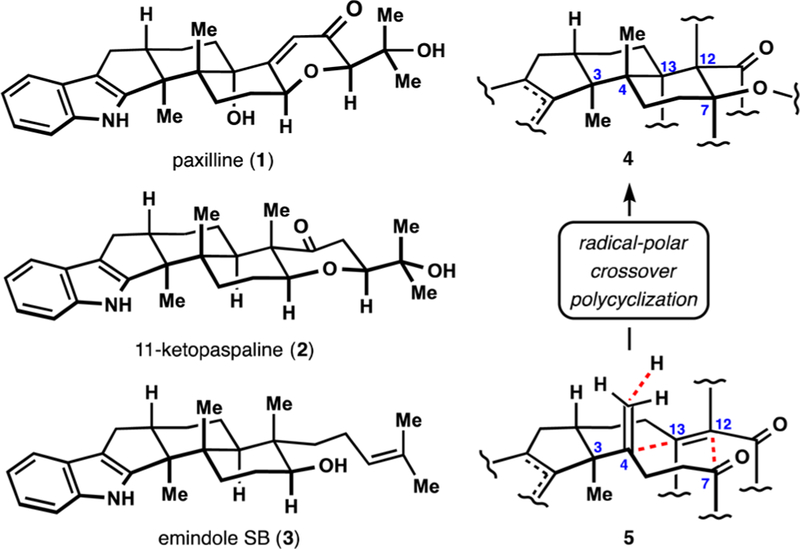
Representative paxilline indole diterpenes.
We believed that rapid access to a functionalized polycyclic terpenoid core (such as 4, Figure 1) would allow the concise assembly of various paxilline indole terpenes. The aldol motif of 4 was expected to provide a convenient handle for diversification of the terpenoid scaffold, and a carbonyl or related functionality of the five-membered ring would serve for installation of the indole moiety. We sought to set the stereochemistry of the crucial trans-juncture of the hexahydroindene motif early in the synthesis and envisioned a direct assembly of the tricyclic core bearing vicinal quaternary stereocenters (at C3 and C4) via a radical−polar crossover polycyclization of a cyclopentanone derivative 5 initiated by chemoselective hydrogen atom transfer (HAT) to the 1,1-disubstitued alkene at C4.11−13 In this process, the key carbon−carbon bond formations would result from intramolecular radical conjugate addition12,14 (C4−C13 bond) and aldol (C12−C7 bond) reactions.15
We reasoned that the brevity of the proposed route would depend in part on effcient installation of the C3 quaternary stereocenter of 5 and we identified alkenylation of the corresponding cyclopentanone derivative as the most direct approach. A literature survey revealed that regioselective intermolecular alkenylation of unsymmetrical ketones bearing two unactivated enolizable positions finds little precedent,16 with examples of formation of quaternary stereocenters being particularly scarce.17 After extensive experimentation, we chose to employ terminal alkynes as electrophiles in the reaction with silyl enol ethers serving as enol surrogates.18,19 The desired alkenylation was eventually achieved by treating enol ether 6 with O-TBS-protected 4-pentyn-1-ol in the presence of indium(III) bromide19,20 (Scheme 1). Subsequent mild acidic workup afforded alcohol 7 in a highly regio- and stereoselective manner. This reaction likely proceeds via the intermediacy of corresponding vinylindium species and requires utilization of substoichiometric amounts of indium(III) bromide.19 Subsequent chemoselective Sharpless allylic oxidation21 of 7 followed by double oxidation of the resulting diol completed the assembly of dialdehyde 8.
Scheme 1.
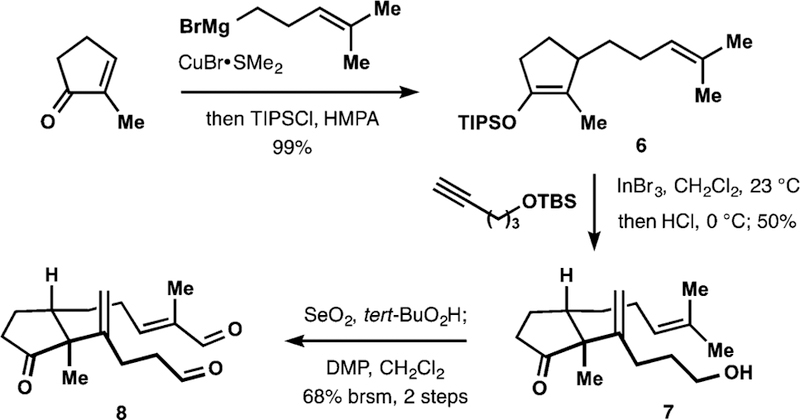
Synthesis of the Polycyclization Precursor
Our initial attempts to perform radical−polar crossover polycyclization of dialdehyde 8 resulted in successful formation of the desired C4−C13 and C12−C7 bonds, but favored the undesired diastereomer and exhibited overall low efficiency of the transformation. Extensive screening of catalysts and reductants revealed that application of iron(III) acetylacetonate12 and (isopropoxy)phenylsilane22 led to substantial increase in the yield of tricyclic products (Scheme 2). Thus, treatment of 8 with (isopropoxy)phenylsilane in a mixture of 1,2-dichloroethane and ethylene glycol at 0 °C in the presence of iron(III) acetylacetonate afforded a nearly equimolar mixture of 9 and 10 in ca. 58% yield (24% isolated yield of 9).23 This transformation likely involves HAT to the 1,1-disubstituted alkene, followed by the conjugate addition of the intermediate tert-alkyl radical 11, reduction to the corresponding enolate 12 (or its diastereomer en route to 10), and subsequent aldol reaction with the aliphatic aldehyde functionality. The structures of 9 and 10 (after conversion to dioxane 13) were confirmed by X-ray crystallographic analysis. It should be noted that in both cases the β-hydroxyaldehyde motif was formed in a highly diastereoselective manner. Surprisingly, changing the nature of the solvent and performing the reaction at lower temperatures led only to the increase in content of the undesired diastereomer, with ratios as high as 2.5:1 favoring ketone 10 being obtained. Application of other iron(III), cobalt(III), and manganese(III)-based catalysts did not favorably affect the ratio of diastereomers 9 and 10, but instead led to increased formation of byproducts resulting from the conjugate reduction of α,β-unsaturated aldehyde 8.
Scheme 2.
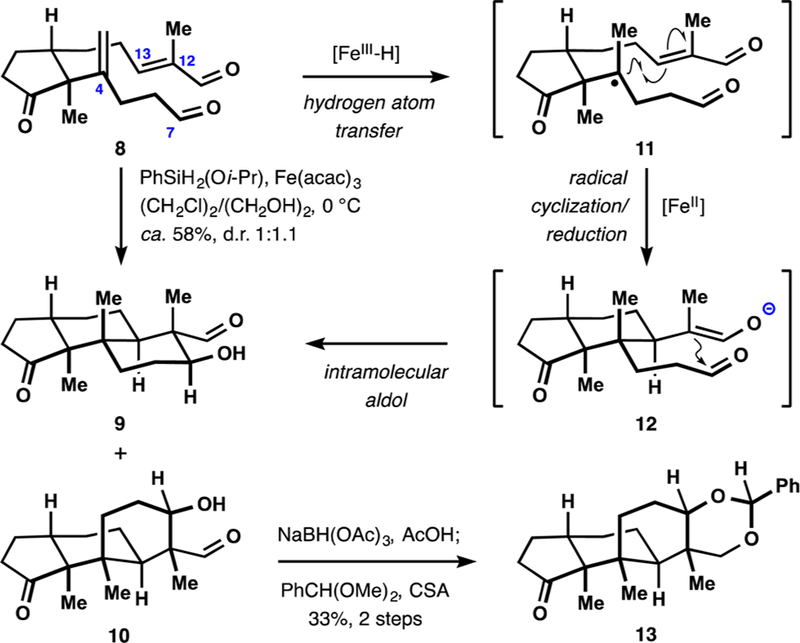
Polycyclization En Route to the Tricyclic Core
During our studies, we observed that aldehyde 8 existed predominantly in the form of the corresponding hemiacetals at C7 in the presence of alcoholic solvents such as deuterated methanol. We reasoned that introduction of a hydroxy group in place of the ketone functionality of 8 could allow for reversible formation of cyclic hemiacetal intermediates. Indeed, secondary alcohol 14 was found to exist as an approximately 1:2 mixture with the corresponding cyclic hemiacetal 15 in a solution of deuterated chloroform (Table 1). We speculated that this transient tether could allow for the desired stereocontrol over the initial radical cyclization. In accord with our predictions, subjection of 14 to the standard polycyclization conditions (see Scheme 2) afforded a 5:1 mixture of diols 16 and 17 (entry 1, Table 1),24 effectively reversing the selectivity exhibited by the parent substrate 8. The ratio of products was temperature dependent with lower temperatures favoring the desired scaffold 16 (entries 2 and 3), thereby reversing the trend observed for the parent substrate 8 (see discussion above). When the solvent was replaced with ethanol, the ratio of 16 to 17 deteriorated (entry 4).25 Methylation of the secondary hydroxy group led to similar deterioration of diastereomeric ratios with ether 18 affording a 1.6:1 mixture of tricycles 19 and 20 (entry 5).26 Effects of solvent and temperature on polycyclization of substrate 18 were similar to those for the parent compound 8 (entries 6 and 7).
Table 1.
Tether-Controlled Polycyclization
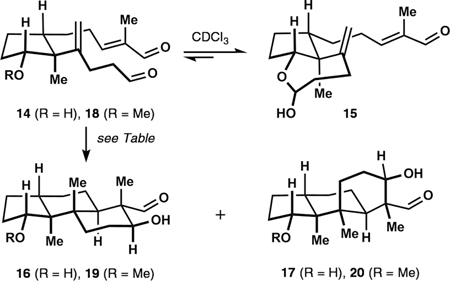 | ||
|---|---|---|
| entry | conditions | ratioa |
| 1 | 14, Fe(acac)3, PhSiH2(Oi-Pr), (CH2CI)2/ (CH2OH)2, 0 °C | 5.0 (16) : 1.0 (17) |
| 2 | 14, Fe(acac)3, PhSiH2(Oi-Pr), (CH2CI)2/ (CH2OH)2, −20 °C | 7.1 (16) : 1.0 (17) |
| 3 | 14, Fe(acac)3, PhSiH2(Oi-Pr), (CH2CI)2/ (CH2OH)2, 23 °C | 3.1 (16) : 1.0 (17) |
| 4 | 14, Fe(acac)3, PhSiH2(Oi-Pr), EtOH, 0 °C | 1.5 (16): 1.0 (17) |
| 5 | 18, Fe(acac)3, PhSiH2(Oi-Pr), (CH2CI)2 / (CH2OH)2, 0 °C | 1.6 (19): 1.0 (20) |
| 6 | 18, Fe(acac)3, PhSiH2(Oi-Pr), (CH2CI)2 / (CH2OH)2, 23 °C | 1.9 (19) : 1.0 (20) |
| 7 | 18, Fe(acac)3, PhSiH2(Oi-Pr), EtOH, 0 °C | 1.0 (19) : 1.4 (20) |
According to integration of aldehyde peaks in 1H NMR spectra of crude reaction mixtures.
Guided by the aforementioned logic, we proposed that an indole N−H in place of a hydroxy group could serve as an anchor to control the stereoselectivity of polycyclization in a similar fashion. Corresponding cyclic hemiaminal motifs have been previously identified in a number of indole-containing alkaloids,27 suggesting viability of similar intermediates. To test our hypothesis, we began with the Fischer synthesis of indole 22 from ketone 21 (Scheme 3). Conversion of the N-phenylhydrazone of 21 into 22 was best achieved by heating with an excess of anhydrous zinc chloride in toluene.28 Parikh− Doering conditions29 proved optimal for oxidation of diol 22 and secured access to hemiaminal 23 after mild basic workup. Hemiaminal 23 was produced as a single diastereomer (stereochemistry of the hemiaminal was not determined) and underwent facile dehydration to the corresponding enamine upon prolonged exposure to acid. Subjection of 23 to the cyclization conditions ((isopropoxy)phenylsilane and iron(III) acetylacetonate in ethanol at 0 °C) triggered facile radical cyclization to pentacyclic intermediate 24.30 However, the stability of the hemiaminal fragment31 required subsequent treatment with strong base in order to complete construction of the desired pentacyclic core, concluding the assembly of four new stereocenters with excellent selectivity. The radical cyclization required rigorous exclusion of oxygen from the reaction mixture due to facile oxidation of intermediates. Hemiaminal 24 proved unstable in the presence of acid, undergoing dehydration to the corresponding enamine. In the end, the key functionalized pentacycle 25 was prepared in a total of eight steps from commercially available 2-methyl-2- cyclopenten-1-one. To demonstrate the application of our strategy, we synthesized (±)-emindole SB (3) in three steps from hydroxyaldehyde 25. Thus, treatment of 25 with a large excess of (Z)-2-ethoxyvinyllithium followed by acidic workup,32 subsequent hydrogenation of the corresponding unsaturated aldehyde, and Wittig olefination of the resulting mixture of lactols afforded emindole SB (3) in 11 steps from the commercially available precursor.
Scheme 3.
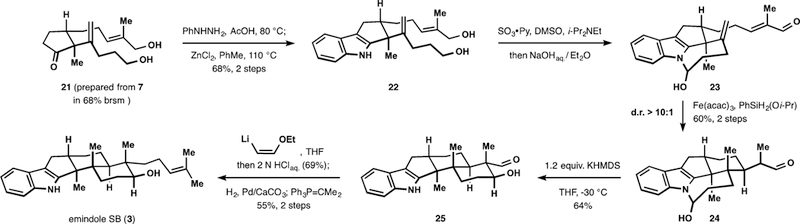
Synthesis of (±)-Emindole SB (3)
In summary, we disclose a concise approach to the functionalized pentacyclic core of paxilline indole diterpenes featuring a new and potentially useful alkenylation of ketones to construct quaternary stereocenters and a tandem radical addition−aldol reaction sequence to access challenging vicinal quaternary stereocenters in a highly stereoselective manner. We also demonstrate a short synthesis of (±)-emindole SB, which serves as a proof of concept for our approach. We anticipate that the strategy reported herein will be applicable to the asymmetric synthesis of a wide variety of paxilline indole diterpenes as well as their unnatural analogues,33 the implementation of which is currently underway.
ACKNOWLEDGMENTS
Funding from the School of Physical Sciences at the University of California, Irvine, and Chao Family Comprehensive Cancer Center is gratefully acknowledged. We thank Prof. Ryan Shenvi (TSRI) for disclosure of (isopropoxy)phenylsilane and helpful discussions, and Dr. Joseph Ziller and Jordan Corbey for X-ray crystallographic analyses. We also thank Profs. Larry Overman and Chris Vanderwal for providing routine access to their instrumentation and helpful discussions.
Footnotes
ASSOCIATED CONTENT
Supporting Information
- Experimental procedures and spectroscopic data for new compounds (PDF)
- X-ray crystallographic data for 9 (CIF)
- X-ray crystallographic data for 13 (CIF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Knaus H-G; McManus OB; Lee SH; Schmalhofer WA; Garcia-Calvo M; Helms LMH; Sanchez M; Giangiacomo K; Reuben JP; Smith AB III; Kaczorowski GJ; Garcia ML Biochemistry 1994, 33, 5819. [DOI] [PubMed] [Google Scholar]
- (2).Huang X-H; Tomoda H; Nisnida H; Masuma R; Ōmura SJ Antibiot 1995, 48, 1. [DOI] [PubMed] [Google Scholar]
- (3).Meinke PT; Smith MM; Shoop WL Curr. Top. Med. Chem 2002, 2, 655 and references therein. [DOI] [PubMed] [Google Scholar]
- (4).Tarui Y; Chinen T; Nagumo Y; Motoyama T; Hayashi T; Hirota H; Muroi M; Ishii Y; Kondo H; Osada H; Usui T ChemBioChem 2014, 15, 934. [DOI] [PubMed] [Google Scholar]
- (5).Ogata M; Ueda J; Hoshi M; Hashimoto J; Nakashima T; Anzai K; Takagi M; Shin-ya KJ Antibiot 2007, 60, 645. [DOI] [PubMed] [Google Scholar]
- (6).Fan Y; Wang Y; Liu P; Fu P; Zhu T; Wang W; Zhu WJ Nat. Prod 2013, 76, 1328. [DOI] [PubMed] [Google Scholar]
- (7).(a) de Jesus AE; Gorst-Allman CP; Steyn PS; van Heerden FR; Vleggaar R; Wessels PL; Hull WEJ Chem. Soc., Perkin Trans 1 1983, 1863. [Google Scholar]; (b) Byrne KM; Smith KS; Ondeyka JG J. Am. Chem. Soc 2002, 124, 7055. [DOI] [PubMed] [Google Scholar]; (c) Uchida R; Tomoda H; Ōmura S J. Antibiot 2006, 59, 298. [DOI] [PubMed] [Google Scholar]
- (8).(a) Smith AB III; Mewshaw RJ Am. Chem. Soc 1985, 107, 1769. [Google Scholar]; (b) Smith AB III; Leenay TL Tetrahedron Lett 1988, 29, 2791. [Google Scholar]; (c) Mewshaw R; Taylor MD; Smith AB III. J Org. Chem 1989, 54, 3449. [Google Scholar]; (d) Smith AB III; Leenay TL J. Am. Chem. Soc 1989, 111, 5761. [Google Scholar]; (e) Smith AB III; Sunazuka T; Leenay TL; Kingery-Wood JJ Am. Chem. Soc 1990, 112, 8197. [Google Scholar]; (f) Smith AB III; Kingery-Wood J; Leenay TL; Nolen EG; Sunazuka TJ Am. Chem. Soc 1992, 114, 1438. [Google Scholar]; (g) Smith AB III; Kanoh N; Ishiyama H; Hartz RA J. Am. Chem. Soc 2000, 122, 11254. [Google Scholar]; (h) Smith AB III; Cui H Org. Lett 2003, 5, 587. [DOI] [PubMed] [Google Scholar]; (i) Smith AB III; Kanoh N; Ishiyama H; Minakawa N; Rainier JD; Hartz RA; Cho YS; Cui H; Moser WM J. Am. Chem. Soc 2003, 125, 8228. [DOI] [PubMed] [Google Scholar]; (j) Smith AB III; Cui H Helv. Chim. Acta 2003, 86, 3908. [Google Scholar]; (k) Smith AB III; Davulcu AH; Kürti L Org. Lett 2006, 8, 1665. [DOI] [PubMed] [Google Scholar]; (l) Smith AB III; Kürti L; Davulcu AH; Cho Y-S; Ishiyama H; Ohmoto K J. Org. Chem 2007, 72, 4596. [DOI] [PubMed] [Google Scholar]; (m) Enomoto M; Morita A; Kuwahara S Angew. Chem., Int. Ed 2012, 51, 12833. [DOI] [PubMed] [Google Scholar]; (n) Asanuma A; Enomoto M; Nagasawa T; Kuwahara S Tetrahedron Lett 2013, 54, 4561. [Google Scholar]; (o) Teranishi T; Murokawa T; Enomoto M; Kuwahara S Biosci., Biotechnol., Biochem 2015, 79, 11. [DOI] [PubMed] [Google Scholar]; (p) Sharpe RJ; Johnson JS J. Am. Chem. Soc 2015, 137, 4968. [DOI] [PMC free article] [PubMed] [Google Scholar]; (q) Zou Y; Melvin JE; Gonzales SS; Spafford MJ; Smith AB III. J. Am. Chem. Soc 2015, 137, 7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9). For recent discussions, see refs 8m, 8p, and 8q.
- (10).Nozawa K; Nakajima S; Kawai K.-i. J. Chem. Soc., Perkin Trans 1 1988, 2607. [Google Scholar]
- (11).For pioneering work on relevant hydrofunctionalization, see: Mukaiyama T; Yamada T Bull. Chem. Soc. Jpn 1995, 68, 17 [Google Scholar]; and references therein. Shenvi and Herzon propose HAT as the initial step in their related hydrogenations: (a) Iwasaki K; Wan KK; Oppedisano A; Crossley SWM; Shenvi RA J. Am. Chem. Soc 2014, 136, 1300. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) King SM; Ma X; Herzon SB J. Am. Chem. Soc 2014, 136, 6884. [DOI] [PubMed] [Google Scholar]; For similar mechanistic considerations, see: (c) Tang L; Papish ET; Abramo GP; Norton JR; Baik M-H; Friesner RA; Rappé AJ Am. Chem. Soc 2003, 125, 10093. [DOI] [PubMed] [Google Scholar]; (d) Ishikawa H; Colby DA; Seto S; Va P; Tam A; Kakei H; Rayl TJ; Hwang I; Boger DL J. Am. Chem. Soc 2009, 131, 4904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).HAT-initiated conjugate addition: (a) Lo JC; Yabe Y; Baran PS J. Am. Chem. Soc 2014, 136, 1304. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lo JC; Gui J; Yabe Y; Pan C-M; Baran PS Nature 2014, 516, 343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).For selected examples of chemoselective HAT to polyenes, see: (a) Magnus P; Payne AH; Waring MJ; Scott DA; Lynch V Tetrahedron Lett 2000, 41, 9725. [Google Scholar]; (b) Waser J; González-Gómez JC; Nambu H; Huber P; Carreira EM Org. Lett 2005, 7, 4249. [DOI] [PubMed] [Google Scholar]; (c) Smith DM; Pulling ME; Norton JR J. Am. Chem. Soc 2007, 129, 770. [DOI] [PubMed] [Google Scholar]; (d) Taniguchi T; Goto N; Nishibata A; Ishibashi H Org. Lett 2010, 12, 112. [DOI] [PubMed] [Google Scholar]; (e) Leggans EK; Barker TJ; Duncan KK; Boger DL Org. Lett 2012, 14, 1428. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Crossley SWM; Barabé F; Shenvi RA J. Am. Chem. Soc 2014, 136, 16788. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Ma X; Herzon SB Chem. Sci 2015, 6, 6250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).For selected examples of radical conjugate additions to form quaternary carbons, see: (a) Barton DHR; Crich D Tetrahedron Lett 1985, 26, 757. [Google Scholar]; (b) Okada K; Okamoto K; Morita N; Okubo K; Oda MJ Am. Chem. Soc 1991, 113, 9401. [Google Scholar]; (c) Schnermann MJ; Overman LE Angew. Chem., Int. Ed 2012, 51, 9576. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Sun Y; Li R; Zhang W; Li A Angew. Chem., Int. Ed 2013, 52, 9201. [DOI] [PubMed] [Google Scholar]; (e) Lackner GL; Quasdorf KW; Overman LE J. Am. Chem. Soc 2013, 135, 15342. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Chu L; Ohta C; Zuo Z; MacMillan DW C. J. Am. Chem. Soc 2014, 136, 10886. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Zhou S; Zhang D; Sun Y; Li R; Zhang W; Li A Adv. Synth. Catal 2014, 356, 2867. [Google Scholar]; (h) Müller DS; Untiedt NL; Dieskau AP; Lackner GL; Overman LE J. Am. Chem. Soc 2015, 137, 660. [DOI] [PubMed] [Google Scholar]; (i) Chinzei T; Miyazawa K; Yasu Y; Koike T; Akita M RSC Adv 2015, 5, 21297. [Google Scholar]; (j) Lackner GL; Quasdorf KW; Pratsch G; Overman LE J. Org. Chem 2015, 80, 6012. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Pratsch G; Lackner GL; Overman LE J. Org. Chem 2015, 80, 6025. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Nawrat CC; Jamison CR; Slutskyy Y; MacMillan DWC; Overman LE J. Am. Chem. Soc 2015, 137, 11270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).For relevant aldol reactions, see: Isayama S; Mukaiyama T Chem. Lett 1989, 2005. [Google Scholar]
- (16).(a) Kosugi M; Hagiwara I; Migita T Chem. Lett 1983, 12, 839. [Google Scholar]; (b) Yamaguchi M; Tsukagoshi T; Arisawa MJ Am. Chem. Soc 1999, 121, 4074. [Google Scholar]; (c) Ooi T; Goto R; Maruoka KJ Am. Chem. Soc 2003, 125, 10494. [DOI] [PubMed] [Google Scholar]; (d) Su W; Raders S; Verkade JG; Liao X; Hartwig JF Angew. Chem., Int. Ed 2006, 45, 5852. [DOI] [PubMed] [Google Scholar]; (e) Datta GK; Larhed M Org. Biomol. Chem 2008, 6, 674. [DOI] [PubMed] [Google Scholar]; (f) Grigalunas M; Ankner T; Norrby P-O; Wiest O; Helquist PJ Am. Chem. Soc 2015, 137, 7019. [DOI] [PubMed] [Google Scholar]
- (17). See refs 16a, 16b, and 16e.
- (18).(a) Staben ST; Kennedy-Smith JJ; Huang D; Corkey BK; LaLonde RL; Toste FD Angew. Chem., Int. Ed 2006, 45, 5991. [DOI] [PubMed] [Google Scholar]; (b) Schäfer C; Miesch M; Miesch L Chem. - Eur. J 2012, 18, 8028. [DOI] [PubMed] [Google Scholar]
- (19).For pioneering work on indium(III)-mediated alkenylation of ketene silyl acetals, see: Nishimoto Y; Moritoh R; Yasuda M; Baba A Angew. Chem., Int. Ed 2009, 48, 4577. [DOI] [PubMed] [Google Scholar]
- (20).For relevant indium(III)-catalyzed alkenylation, see: Nakamura M; Endo K; Nakamura EJ Am. Chem. Soc 2003, 125, 13002. [DOI] [PubMed] [Google Scholar]
- (21).Umbreit MA; Sharpless KB J. Am. Chem. Soc 1977, 99, 5526. [Google Scholar]
- (22).Shenvi RA. personal communication.
- (23). An elegant 19-step synthesis of hydroxyaldehyde (−)-9 from 2-methyl-1,3-cyclohexadione was recently reported. See ref 8q.
- (24). Relative configuration of the decalin fragment of 16 was confirmed by reduction to the corresponding triol, which proved identical by 1H and 13C NMR analysis to the product of double reduction of compound 9. Relative configuration of the cyclopentanol fragment of 16 was established by NOE experiments.
- (25). Ethylene glycol and 1,2-dichloroethane are immiscible; therefore, the concentration of alcohols in the 1,2-dichloroethane phase is expected to be significantly lower than in ethanol, decreasing the extent of intermolecular hemiacetalization. The reagents appear to dissolve preferentially in the 1,2-dichloroethane phase.
- (26). Relative configurations of compounds 19 and 20 were established by NOE experiments.
- (27).Chbani M; Païs M; Delauneux J-M; Debitus C J. Nat. Prod 1993, 56, 99. [DOI] [PubMed] [Google Scholar]
- (28).Vaswani RG; Day JJ; Wood JL Org. Lett 2009, 11, 4532. [DOI] [PubMed] [Google Scholar]
- (29).Parikh JR; Doering W; von E J. Am. Chem. Soc 1967, 89, 5505. [Google Scholar]
- (30). Aldehyde 24 is formed with high diastereoselectivity (dr > 10:1). Complete stereochemical assignment of 24 was not determined.
- (31).(a) Arai E; Tokuyama H; Linsell MS; Fukuyama T Tetrahedron Lett 1998, 39, 71. [Google Scholar]; (b) Evans DA; Borg G; Scheidt KA Angew. Chem., Int. Ed 2002, 41, 3188. [DOI] [PubMed] [Google Scholar]
- (32).Lau KSY; Schlosser MJ Org. Chem 1978, 43, 1595. [Google Scholar]
- (33).For asymmetric cuprate additions to 2-methyl-2-cyclopenten-1-one, see: Calvo BC; Madduri AVR; Harutyunyan SR; Minnaard AJ Adv. Synth. Catal 2014, 356, 2061. [Google Scholar]