

Abstract
Objectives
Metallo-β-lactamases (MBLs) are an emerging class of antimicrobial resistance enzymes that degrade β-lactam antibiotics, including last-resort carbapenems. Infections caused by carbapenemase-producing Enterobacteriaceae (CPE) are increasingly prevalent, but treatment options are limited. While several serine-dependent β-lactamase inhibitors are formulated with commonly prescribed β-lactams, no MBL inhibitors are currently approved for combinatorial therapies. New compounds that target MBLs to restore carbapenem activity against CPE are therefore urgently needed. Herein we identified and characterized novel synthetic peptide inhibitors that bound to and inhibited NDM-1, which is an emerging β-lactam resistance mechanism in CPE.
Methods
We leveraged Surface Localized Antimicrobial displaY (SLAY) to identify and characterize peptides that inhibit NDM-1, which is a primary carbapenem resistance mechanism in CPE. Lead inhibitor sequences were chemically synthesized and MBCs and MICs were calculated in the presence/absence of carbapenems. Kinetic analysis with recombinant NDM-1 and select peptides tested direct binding and supported NDM-1 inhibitor mechanisms of action. Inhibitors were also tested for cytotoxicity.
Results
We identified approximately 1700 sequences that potentiated carbapenem-dependent killing against NDM-1 Escherichia coli. Several also enhanced meropenem-dependent killing of other CPE. Biochemical characterization of a subset indicated the peptides penetrated the bacterial periplasm and directly bound NDM-1 to inhibit enzymatic activity. Additionally, each demonstrated minimal haemolysis and cytotoxicity against mammalian cell lines.
Conclusions
Our approach advances a molecular platform for antimicrobial discovery, which complements the growing need for alternative antimicrobials. We also discovered lead NDM-1 inhibitors, which serve as a starting point for further chemical optimization.
Introduction
β-Lactams are an important class of antibiotics that not only possess activity against Gram-positive bacteria, but also Gram-negatives, which include some of the most difficult-to-treat pathogens.1,2 The defining β-lactam ring inhibits the transpeptidase activity of PBPs, which are essential bacterial enzymes that polymerize the peptidoglycan cell wall.3,4 Disruption of peptidoglycan assembly leads to rapid cell lysis.
Carbapenems are important β-lactam therapeutics because they possess potent broad-spectrum activity and are not susceptible to common resistance mechanisms.5,6 In fact, imipenem and meropenem are last-line carbapenem antibiotics used to treat MDR Gram-negative infections.7–11 Despite restriction of the use of carbapenems, resistance mechanisms have inevitably emerged.
The most common β-lactam resistance mechanism in Gram-negative organisms is production of β-lactamase enzymes.3,12 β-Lactamases hydrolyse the β-lactam ring, which prevents the antibiotic from binding its target PBP.3,5 β-Lactamases are divided into four classes (A, B, C and D) based on structural and sequence similarities, but comprise two distinct groups based on hydrolytic activity.5,13–15 Serine-dependent β-lactamases (SBLs), including classes A, C and D, rely on an active-site serine for β-lactam degradation.5,13–16 Class B β-lactamases rely on active-site Zn2+ ions to coordinate hydrolysis; hence, they are designated metallo-β-lactamases (MBLs).13,17–19 MBLs emerged recently, but have rapidly spread among Gram-negative pathogens, which threatens our current antimicrobial treatment options.12,20–25
New Delhi metallo-β-lactamase (NDM-1) was first reported in 2009 when blaNDM-1 was found in a Klebsiella pneumoniae isolate from a patient in Sweden who had been hospitalized in India.22 NDM-1 is particularly concerning because it quickly spread to all continents, is highly associated with other antibiotic resistance mechanisms, inactivates all bicyclic β-lactam antibiotics, including carbapenems, and is a primary resistance mechanism in many ‘superbugs’.3,5,26,27 Gram-negative pathogens carrying NDM and other MBL subclasses (i.e. IMP, VIM) are considered to be among the most urgent antibiotic resistance problems worldwide.28,29
Class A, C and D β-lactamase inhibitors have been developed and include molecules that covalently modified the conserved serine residue (i.e. avibactam, tazobactam, sulbactam etc.) required for catalytic activity.30–33 Administered in combination with prescription β-lactam antibiotics, inhibitors inactivate serine-dependent β-lactamases to restore antimicrobial activity. In contrast, we currently lack effective MBL inhibitors, including those that target the NDM subclass. ANT431 is an NDM-1 inhibitor in preclinical development, but does not possess broad-spectrum activity against MBLs and is not a candidate for further development.27,34 Development of small-molecule inhibitors has been challenging due to the flexibility of the MBL active site and challenges associated with metallo-enzymes. Alternatively, peptides with activity against MBLs have been reported.35–39
Here, we used a cell-based high-throughput antimicrobial discovery system, termed Surface Localized Antimicrobial displaY (SLAY), to screen for synthetic randomized peptides that potentiate carbapenem-dependent killing of NDM-1 Escherichia coli.40 We discovered thousands of peptides that restored NDM-1 E. coli susceptibility to carbapenems. A subset of leads was genetically validated and four were biochemically tested against recombinant NDM-1 for inhibitory activity. Kinetic analysis was performed to determine the inhibitory mechanism of each lead and cytotoxicity and haemolytic activity were assayed. Our results advanced our basic understanding of the chemical diversity of NDM-1 inhibition and further developed an innovative molecular platform for antimicrobial discovery.
Materials and methods
Bacterial strains and antibiotics
Strains and plasmids used in this study are listed in Table S1 (available as Supplementary data at JAC Online). Primers are listed in Table S2. All strains were grown aerobically from freezer stocks on LB agar at 37°C. Antibiotics were used at the following concentrations: 75 mg/L carbenicillin (Fisher Scientific), 10 mg/L tetracycline (Sigma–Aldrich), 4 or 8 mg/L meropenem (TCI America) and 8 mg/L imipenem (Alfa Aesar).
Peptide library construction
SLAY for the NDM-1 screen was constructed on the broad-host plasmid pMMB67EHTet, which was derived from pMMB67EH, where the bla gene was exchanged with a tetracycline resistance gene.40 Peptide sequences were randomly generated using 30 bp NNB codons to produce peptides comprising 10 amino acids. The sequences were cloned into the KpnI and SalI sites of pMMB67EHTet using primers homologous to the tether sequence on the reverse primer. Plasmid libraries were transformed into C2987 competent cells (NEB). Approximately 1.20 × 106 colonies were pooled. Plasmid DNA was isolated from the C2987 library and re-transformed into E. coli W3110/pNDM-1 to three times coverage.
Screening and sequencing the random peptide library
An aliquot of the frozen E. coli W3110/pNDM-1 peptide library was added to 5 mL of LB supplemented with carbenicillin and tetracycline for growth, shaking at 37°C for 1 h. Duplicate cultures were back-diluted into 5 mL LB with carbenicillin and tetracycline to OD600 0.01 supplemented with IPTG (MP Biomedicals). The remaining culture was used as the ‘input’ sample for the peptide screen. Induced cultures were grown with shaking at 37°C. Cells were harvested at 4 h, which was considered the ‘output’ sample. Plasmids were isolated from input and output groups using the QIAprep Spin Miniprep Kit. Primers with regions homologous to the plasmid were used to simultaneously amplify the peptide-encoding region of the plasmid and add sequencing adapters. Four reactions per sample were amplified for a total of 20 cycles. The reactions were pooled and cleaned using QIAquick PCR Purification Kit. Libraries were gel purified using the QIAquick Gel Extraction Kit. Amplicons were sequenced using 50 bp single-end reads on an Illumina HiSeq supplemented with Phi-X. DNA was sequenced at The University of Texas Genomic Sequencing and Analysis Facility.
Raw read processing
Raw Illumina reads each contained a 30 bp sequence of interest (SOI) coding for the 10 amino acid peptides, flanked on both sides by a library-specific amplicon pattern. For each library, we used custom scripts to identify the conserved flanking amplicon region sequences and raw reads without an exact match to expected flanking sequences were excluded from subsequent analyses. Reads were also excluded if one or more nucleotides within the SOI had a Phred quality score less than 20 (i.e. 1% error rate) or if less than 90% of nucleotides within the SOI had a Phred score less than 30 (i.e. 0.1% error rate). The SOI for reads passing these filtering steps were translated into 10 amino acid peptides.
Differential abundance analysis
Within each library, the number of sequences containing each unique peptide was tallied, and counts were normalized using trimmed mean of M-values normalization in edgeR to control for differences in sequencing depth between experiments.41 Fisher’s exact tests were performed in edgeR to identify significantly differentially abundant peptides between each input and output peptide population and the corresponding no-carbapenem control.41 To control for type I errors resulting from thousands of statistical tests, we corrected P values for each pairwise comparison using the independent hypothesis weighting (IHW) correction procedure with average normalized count per peptide as the covariate.42 Peptides were considered NDM-1 inhibitors when the IHW P value between comparisons was <0.05 and when there was a log2-fold average count depletion in the treatment group relative to the control. Because we were interested in peptides that potentiate carbapenem-dependent killing, peptides depleted only when carbapenems were added to the growth medium were reported. Physiochemical properties of NDM-1-inhibiting peptides were inferred using the Peptides package in R.43,44 Peptide sequence logos and characterization of the physiochemical properties of NDM-1-inhibiting peptides were visualized using pheatmap (https://github.com/raivokolde/pheatmap) and ggseqlogo in R.45
Bacterial growth curves using SLAY
Growth curves were determined as previously described with slight modifications.40 Briefly, strains were grown on LB agar containing carbenicillin, tetracycline and 0.2% glucose overnight at 37°C. The next day, cultures were grown to mid-logarithmic phase (OD600 0.4–0.6). Cultures were back-diluted to OD600 0.01 in 5 mL LB under three different conditions: (−) meropenem/(−) IPTG; (+) meropenem/(+) IPTG; and (+) meropenem/(−) IPTG. Meropenem was used at 8 mg/L. Cultures were grown at 37°C with shaking and the OD600 was collected over a 4 h period using a Fisherbrand accuSkan GO UV/Vis Microplate Spectrophotometer with SkanIt Software 5.0. Each experiment was done twice in triplicate and a representative assay was reported. NDM-1 E. coli carrying the empty SLAY vector was used as a control.
Antimicrobial activity assays
MBC assays were adapted from previously described methods.40,46,47 A small number of NDM-1 bacteria from an overnight plate were resuspended in 1 mL LB for OD600 measurement. Cultures were back-diluted to OD600 0.05 in 5 mL LB containing carbenicillin and grown to logarithmic phase at 37°C with shaking. Cells were collected, washed twice with 5 mL 10 mM Tris (pH 7.4) + 25 mM NaCl + 0.1% glucose and diluted to OD600 0.001 in 5 mL 10 mM Tris (pH 7.4) + 25 mM NaCl + 0.1% glucose, either with or without a final concentration of 4 mg/L meropenem. Fifty microlitres of bacteria was added to each well in a polypropylene 96-well plate (Greiner Bio-One). Chemically synthesized peptides (GenScript) were diluted to 512 μM in water and serial diluted to a volume of 75 μL. Fifty microlitres of each peptide dilution was added to 50 μL of cells to result in final peptide concentrations of 4, 8, 16, 32, 64, 128 and 256 μM. Plates were sealed and incubated at 37°C. At 24 h, each well was spotted on LB agar to assess killing in (+) or (−) meropenem conditions. MBCs were determined as the lowest concentration of peptide that results in at least 99.90% killing of the initial inoculums. Each experiment was performed twice in triplicate and the representative MBC was reported.
MIC assays were performed as previously described with slight modifications.48 A small number of bacteria from an overnight plate were used to inoculate 5 mL LB at OD600 0.05 and grown to logarithmic phase. Cells were washed twice with MOPS media + 0.1% glucose and diluted to OD600 0.001. Fifty microlitres of cells was added to each well of a polypropylene 96-well plate. Peptides were diluted in water to 256 μM and serial diluted. Fifty microlitres of each peptide solution was added to 50 μL of cells at 0, 32, 64 and 128 μM. Increasing concentrations of meropenem (0.1–64 mg/L) were added to the appropriate well. Plates were sealed and incubated at 37°C overnight. MICs were determined by OD600 measurements where cell density was 0. Each experiment was performed twice in triplicate. A representative MIC was reported.
Permeability assays
Assays were performed as previously described.49 A small number of bacteria were scraped from an overnight plate, resuspended in PBS and normalized to OD600 0.2. CCCP (Acros Organics) was added at 200 μM to inhibit efflux pump activity. Ethidium bromide (MP Biomedicals) was added immediately prior to measurement to final concentration of 1.2 μM in 200 μL total volume. Readings were taken, using a BioTek Synergy Neo2 multi-mode reader, every 15 s for 30 min with samples assessed in triplicate in Greiner Bio-One 96-well flat-bottom black plates. Each experiment was performed twice in triplicate. One representative assay was reported.
Kinetic assays
NDM-1 was purified as previously described.50 Kinetic assays were performed as previously reported with slight modifications.35 NDM-1 inhibitory activity was calculated following hydrolysis of increasing concentrations of nitrocefin (BioVision) (2–250 μM) in 10 mM Tris, 50 mM KCl, 5% glycerol, pH 8.0 (P6 and P9), or 10 mM Tris, 50 mM KCl, 5% glycerol 40% DMSO, pH 8.0 (P5 and P7) in the presence (125 μM to 3 mM) of chemically synthesized peptides. Compound dilutions were performed in water (P6 and P9) or DMSO (P5 and P7). GraphPad Prism (8.4.1) was used to calculate the kinetic parameters for NDM-1 inhibition by each peptide using non-linear regression and Michaelis–Menten enzyme kinetics, which determined the maximal velocity (Vmax) and the Michaelis constant (Km). Lineweaver–Burk plots were used to display the data where the slope was Km/Vmax, the y intercept 1/Vmax and the x intercept −1/Km. Inhibitor constant (Ki) values for NDM-1 inhibition were calculated using non-linear regression with global curve fitting in Prism, where parameters were determined by finding the global best-fit value among all datasets for each inhibitor. The datasets were analysed using the following models: competitive inhibition model, KmObserved = Km × (1 + [I]/Ki), Y = Vmax × X/KmObserved + X); non-competitive inhibition model, Vmax inh = Vmax/(1 + I/Ki), Y = Vmax inh × X/(Km + X), where I is the inhibitor concentration. The global fitting function was also used to support inhibitory mechanisms along with Michaelis–Menten kinetic analysis and Lineweaver–Burk plot values, which were reported.
MTT assays
MTT assays were performed as previously described.51 MODE-K and HEK cell lines were used to assess peptide cytotoxicity. Initially, standard curves (Figure S1) were calculated to determine the linear range, which included a concentration of 75000 cells for both cell lines. Cells were seeded in tissue culture-treated 96-well plates at a final volume of 100 μL and incubated for 24 h to allow attachment. After 24 h, media containing increasing concentrations of purified peptide inhibitors were added. Plates were incubated for 24 h. Following incubation, 10 μL of 5 mg/L MTT (Sigma–Aldrich) solution in PBS was added to a final concentration of 0.45 mg/L and the plate was incubated at 37°C for 4 h. Liquid was removed and 100 μL of 100% DMSO was added to each well to dissolve the formazan crystals. The plate was incubated in the dark at room temperature with shaking for 1 h and the absorbance was measured at 570 nm.
Haemolysis assays
Haemolysis assays were performed as described previously.40,52 Briefly, human RBCs (hRBCs) were purchased (Rockland Immunochemicals) and 50 μM solutions of chemically synthesized peptides were mixed in 10 mM PBS at pH 7.4 for a total volume of 0.5 mL. The hRBC solution was made by washing 0.4 mL of the red blood cells twice with 7 mL of PBS. Precipitates were resuspended in 4 mL PBS. Haemolytic activity was measured by mixing the 0.5 mL peptide solutions with 0.4 mL hRBC solution at 37°C for 1 h. The absorbance was measured at 540 nm. Percentage haemolysis was calculated using the equation:
Results
To screen for peptides that restore the antimicrobial activity of carbapenems against carbapenemase-producing Enterobacteriaceae (CPE), we generated a random synthetic library that encoded more than 1 × 106 10 amino acid peptides in the SLAY plasmid system (Figure 1a). Approximately 1700 unique sequences were identified that potentiate carbapenem-dependent killing of NDM-1 E. coli (Figure 1b), of which 37 peptides were found to restrict growth in the presence of both imipenem and meropenem (Figure 1b, orange dots). We prioritized the 37 peptides for analysis because increased susceptibility to multiple carbapenems suggested a conserved mechanism of action. Interestingly, the 37 peptides only modestly increased CPE susceptibility to both meropenem and imipenem.
Figure 1.
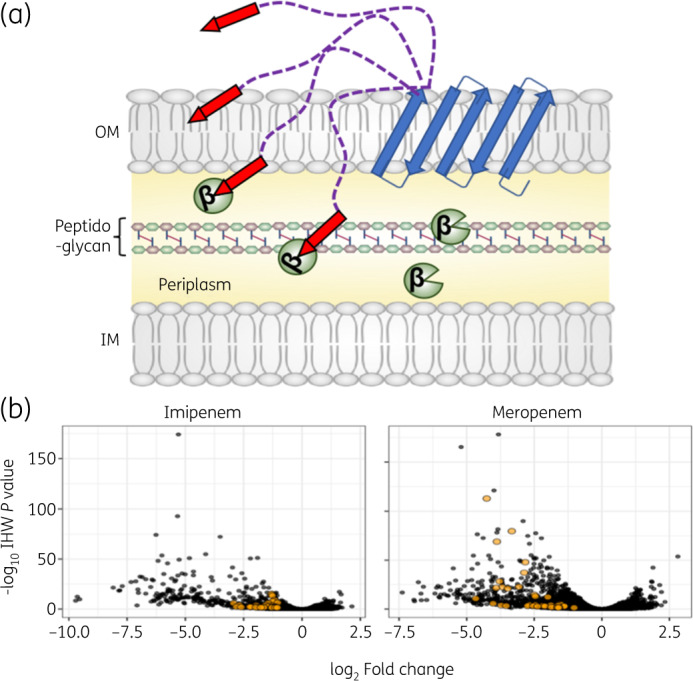
Self-screening of displayed peptides to discover NDM-1 inhibitors that potentiate killing of carbapenemase-producing E. coli. (a) Illustration of SLAY, including the OmpA (46–159) transmembrane protein (blue arrows), flexible tether (purple dashed lines) and C-terminal peptide (red arrows). NDM-1 is localized to the periplasm (yellow). (b) Volcano plot of library sequences depleted in NDM-1 E. coli after 4 h of growth in imipenem (left) or meropenem (right). Sequences depleted in both conditions are labelled orange. This figure appears in colour in the online version of JAC and in black and white in the printed version of JAC.
We computationally analysed the amino acid composition of the 37 peptides at each position to determine specific amino acids over-represented within the pool (Figure 2a). Our analyses indicated enrichment of proline, arginine, leucine and isoleucine at site-specific positions. Furthermore, the physicochemical composition of the peptide pool highlighted a prevalence of basic, hydrophobic and polar amino acids (Figure 2b). Twenty-six of the 37 sequences were genetically validated using the SLAY system (Figures 2c and S2).
Figure 2.
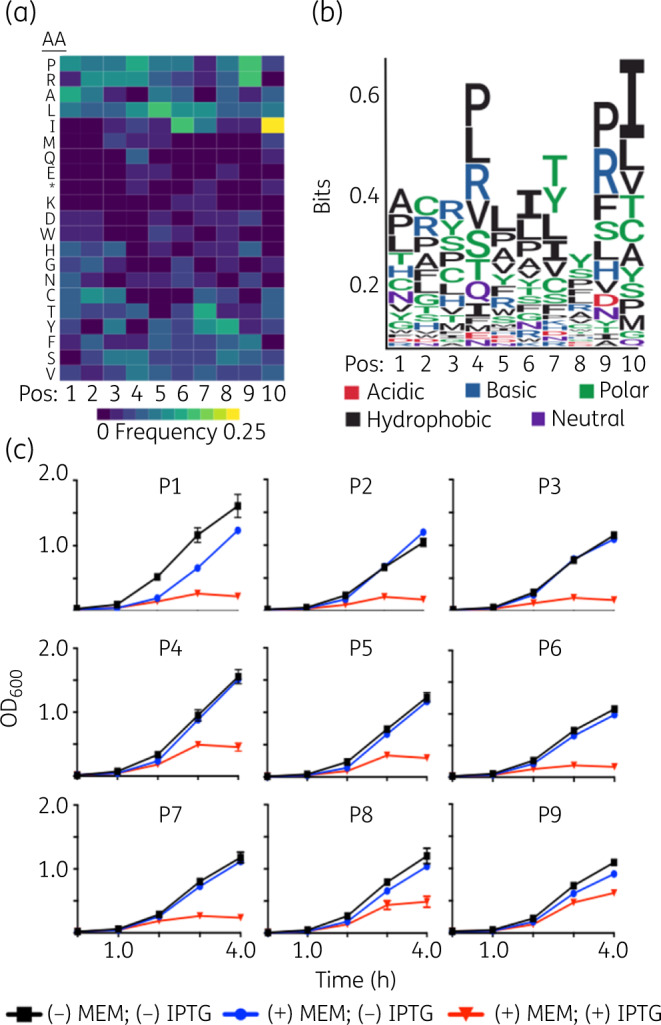
Physicochemical properties and genetic validation of inhibitor peptides that potentiate killing of NDM-1 E. coli. (a) Heat map of amino acids in each position of the depleted sequences in the screen. (b) Depleted sequences were used to build a consensus motif. Larger letters indicate stronger amino acid enrichment at each position. The chemical properties of each amino acid are colour coded. (c) Growth curves of NDM-1 E. coli containing P1 to P9 cloned into the SLAY system. Cultures were grown in 8 mg/L meropenem (MEM) with or without 0.1 mM IPTG for 4 h. This figure appears in colour in the online version of JAC and in black and white in the printed version of JAC.
The top nine validated peptides with the greatest inhibitory effect from the screen were chemically synthesized to measure meropenem activity against NDM-1 E. coli independent of SLAY localization. MBCs for each were determined when NDM-1 E. coli was exposed to 4 mg/L of meropenem (Table 1), which is 2-fold higher than the EUCAST breakpoint.53 The reported MBCs represent the lowest concentration of peptide inhibitor that resulted in at least 99.90% killing of the initial inoculum. MBC assays using increasing concentrations of synthesized peptides showed a correlative response with meropenem susceptibility, which indicated potentiation of meropenem-dependent killing independent of SLAY subcellular localization. Moreover, we tested peptide MBCs in two additional CPE, including NDM-1 K. pneumoniae and Enterobacter cloacae.
Table 1.
Fold change (log2) and NDM-1 inhibitor concentrations of peptides from SLAY
| MBC (μM)a, b |
||||||||
|---|---|---|---|---|---|---|---|---|
| Peptide | Sequence | Charge | Hydrophobicity | Fold change (log2) | P value | Ec W3110 NDM-1 | Kp NIH1 NDM-1 | Ecl ATCC13047 NDM-1 |
| P1 | AFRPIPTHSC | 1.026 | −0.08 | −4.26 | <4.57e−118 | 8 | 32 | 32 |
| P2 | YNYSYIITRS | 0.995 | −0.52 | −3.33 | <1.50e−84 | 128 | 32 | ≥128 |
| P3 | PTTVHIIYRI | 1.088 | 0.57 | −3.88 | <2.24e−73 | 16 | 16 | 64 |
| P4 | ASVTWLLYAM | −0.002 | 1.36 | −2.83 | <3.75e−52 | 128 | 128 | ≥128 |
| P5 | LRCLMLKFPI | 1.935 | 1.31 | −2.87 | <3.62e−41 | 8 | 4 | 4 |
| P6 | CLRPSIISRA | 1.936 | 0.49 | −3.07 | <2.24e−26 | 32 | ≥128 | ≥128 |
| P7 | WRYQWTILFI | 0.997 | 0.38 | −3.91 | <1.81e−25 | 16 | ≥128 | ≥128 |
| P8 | FCIRLATYVI | 0.935 | 1.76 | −2.68 | <2.02e−07 | 8 | ≤4 | 64 |
| P9 | PGHRVSCWLS | 1.026 | −0.17 | −2.17 | <5.36e−06 | ≤4 | ≤4 | 32 |
Ec, E. coli; Kp, Klebsiella pneumoniae; Ecl, Enterobacter cloacae.
MBCs were determined as the lowest concentration of peptide that results in at least 99.90% killing of the initial inoculums. Data are representative of three independent experiments.
All MBCs were determined with 4 mg/L of meropenem.
Porins are the main entryways for carbapenems to penetrate the bacterial periplasm and are known to contribute to resistance.54 Therefore, we first tested if membrane permeability increased when cells were treated with each peptide (Figure S3). Colistin (10 mg/L), a cationic antimicrobial peptide (CAMP) known to perturb the outer membrane of and lyse Gram-negative bacterial cells, was used as a positive control to indicate lysis.48,55,56 Our data showed that high concentrations (256 μM) of select peptides slightly increased cell permeability, whereas other peptides did not. Specifically, exposure to P5 and P9 marginally increased permeability. However, we reasoned the increased permeability was independent of meropenem-dependent killing (Table 1) because other peptides demonstrated a more robust increase in permeability, but the respective MBC values indicated less killing (i.e. P4 and P8). These data suggested an additional mechanism, independent of membrane permeability, to restore carbapenem susceptibility in NDM-1 E. coli. Therefore, we investigated how P5 and P9, which slightly increased permeability, and P6 and P7, which did not increase permeability, potentiate meropenem-dependent killing of NDM-1 E. coli.
Next, we tested if peptides directly bound and inhibited NDM-1 activity using a biochemical assay to calculate NDM-1 hydrolysis of the β-lactam analogue nitrocefin. Hydrolysis of the amide bond in nitrocefin induces a measurable colorimetric shift (from 386 to 482 nm). Recombinant NDM-1, increasing inhibitor (P5, P6, P7 and P9) concentrations and nitrocefin were used to calculate the Michaelis–Menten kinetics of β-lactam hydrolysis.35,57,58 NDM-1 was used at a concentration of 10 nM, nitrocefin concentrations were 0, 2, 10, 25, 50, 100, 200 and 250 μM, and peptide inhibitors were used at the indicated concentrations (Figure 3). Importantly, Lineweaver–Burk plots of Michaelis–Menten kinetic analysis from three inhibitor peptides (P5, P6 and P9) suggested competitive inhibition of NDM-1, whereas P7 demonstrated non-competitive inhibition. We calculated the Ki of each kinetic reaction to determine inhibitor potency; the lowest Ki observed was for P6 (0.11 μM), followed by P9 (1.34 μM), P7 (158.60 μM) and P5 (176.50 μM).
Figure 3.

Lineweaver–Burk plots showing inhibition of NDM-1-dependent nitrocefin hydrolysis activity by inhibitor peptides (a) P5, (b) P6, (c) P7 and (d) P9. Recombinant NDM-1 was used at 10 nM. Circles indicate hydrolysis without inhibitor. Squares, triangles and inverted triangles indicate increasing concentration of the respective inhibitor at the indicated concentrations. The position where the lines intersect indicates the type of inhibition (P5, competitive; P6, competitive; P7, non-competitive; P9, competitive). Ki is indicated above each plot.
Meropenem MICs were calculated using increasing concentrations of the four NDM-1 inhibitors (Table 2). P5 and P9 strongly potentiated meropenem-dependent growth inhibition of NDM-1 E. coli at high and low peptide concentrations (32 μM), whereas P6 and P7 potentiated meropenem activity at higher concentrations (>64 μM). Meropenem MICs were also determined in NDM-1 K. pneumoniae and E. cloacae to establish whether the NDM-1 inhibitors were broadly effective against CPE. While all peptides reduced the meropenem MIC in a concentration-dependent manner, P5 increased meropenem susceptibility to below the EUCAST breakpoint in all CPE tested, which indicated broad-spectrum activity.
Table 2.
Meropenem MICs (mg/L) against NDM-1 Enterobacteriaceae in the presence of peptides
| Organism and peptide concentration |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Peptide |
E. coli W3110 NDM-1 |
K. pneumoniae NIH1 NDM-1 |
E. cloacae ATCC 13047 NDM-1 |
|||||||||
| – | 32 μM | 64 μM | 128 μM | – | 32 μM | 64 μM | 128 μM | – | 32 μM | 64 μM | 128 μM | |
| P5 | 8 | <0.5 | <0.5 | <0.5 | 32 | 8 | <0.5 | <0.5 | 16 | 8 | 2 | <0.5 |
| P6 | 8 | 8 | 4 | 4 | 32 | 16 | 16 | 8 | 16 | 8 | 4 | 4 |
| P7 | 8 | 4 | 4 | 2 | 32 | 8 | 8 | 8 | 16 | 2 | 2 | 2 |
| P9 | 8 | 2 | <0.1 | <0.1 | 32 | 16 | 16 | 16 | 16 | 8 | 8 | 8 |
As with any therapeutic compound, cytotoxicity is a concern when prioritizing lead development. Cell viability in the presence of the NDM-1 inhibitor peptides was assessed by MTT assay in two cell lines, MODE-K and HEK293. None of the peptides exhibited notable cytotoxic activity when exposed to MODE-K cells, with each demonstrating well below 20% lysis. The one exception was P7, when exposed at 256 μM, which was associated with the high concentration (9% of total concentration) of DMSO required to solubilize the peptide. While MODE-K cells showed minimal reduction of cell viability (Figure 4a), HEK293 cells showed a dose-dependent cytotoxic effect (Figure S4).
Figure 4.

Cytotoxicity of NDM-1 inhibitor peptides (a) MTT assays of MODE-K (intestinal epithelial) cells to measure viability after 24 h when exposed to increasing concentrations of NDM-1 inhibitor peptides. Cell viability is reported as the percentage of the no-peptide control (−), which was normalized to 100. Triton X-100 was used as a positive control (+). Values with P value <0.05 are indicated with an asterisk. Dotted line indicates 80% cytotoxicity. (b) Haemolytic activity of selected peptides at 50 μM. Dotted line indicates 20% haemolysis.
Haemolysis is a known off-target effect of antimicrobial peptides, with several CAMPs showing marked haemolysis at therapeutically relevant concentrations.59 We assayed the haemolytic activities of all synthesized peptides at 50 μM. The haemolytic activity of each is summarized in Figure 4(b). None of the peptides tested exhibited notable haemolytic activity, with each demonstrating below 20% lysis.
Discussion
NDM-1 is a primary β-lactam resistance mechanism that has emerged in many Gram-negative ‘superbugs’, which threaten our antimicrobial repertoire. Here we used SLAY to identify and characterize peptides that restore the antimicrobial activity of carbapenems in NDM-1 Enterobacteriaceae. As anticipated, the vast majority of sequences in the random peptide screen did not restore carbapenem antimicrobial activity (99.99%). However, high-throughput sequencing identified approximately 1700 candidate inhibitors that potentiated NDM-1 E. coli killing via a carbapenem-dependent mechanism. Of these, only 37 peptides increased susceptibility to both meropenem and imipenem based on our cut-offs, which suggested a conserved mechanism of action. Interestingly, these overlapped peptides only modestly increased carbapenem susceptibility (Figure 1b, orange dots), whereby the ‘best’ peptides for one carbapenem did not function as well with the other in our screen.
Sequence analysis of the 37 peptides suggested enrichment of specific amino acids, including enrichment of arginine, lysine and histidine residues, which contain positively charged side chains at pH 7.0, and proline residues. Interestingly, each sequence (with the exception of P6) encoded a positively charged residue at the 2, 3 or 4 position (or reverse sequence). The conserved site-specific charge could contribute to NDM-1 inhibition or peptide penetration of the periplasm. Several lytic antimicrobial peptides require a cationic charge, which promotes electrostatic interactions with the negatively charged outer membrane to displace divalent ions to perturb the membrane.60–63 In contrast, selected lead peptides in our study only slightly increased permeability, but strongly potentiated meropenem-dependent killing, which suggested an additional membrane-independent mechanism of action. It is possible that the positive charge enriched NDM-1 inhibitor peptides at the negatively charged cell surface, which distorted the membrane allowing access to the periplasm, where the sequence bound and directly inhibited NDM-1. Consistent with this hypothesis, select peptides did not increase membrane permeability (i.e. P6 and P7) at a level we could detect (Figure S3), but nonetheless potentiated meropenem-dependent killing of CPE at higher concentrations (Table 1), likely through direct inhibition of NDM-1 (Figure 3).
Despite reports of several small-molecule MBL inhibitors, none has been approved for therapeutic use for various reasons, including ineffectiveness across Enterobacteriaceae and poor pharmacokinetics/pharmacodynamics.27,64–69 Several MBL peptide inhibitors have also been discovered with limited therapeutic efficacy.35,36,38,39,57 Peptides offer diverse avenues to sample chemical space and SLAY provides a valuable platform to optimize and quickly test new sequence pools in a high-throughput manner to understand inhibitory motifs. Additionally, identified inhibitor motif combinations provide an opportunity to explore broad- or narrow-spectrum inhibitors in a more directed fashion, which could be useful in future development of pathogen-specific therapeutics.
Supplementary Material
Acknowledgements
We thank Bryan Davies for providing the SLAY plasmids and for helpful insights regarding the peptide display system.
Funding
This work was supported by funding from the National Institutes of Health (Grant AI146829 to J.M.B., Grant GM128885 to M.W.P., Grant AI134673 to T.A.C. and Grants AI111753, AI141101 to D.E.G.) and Cancer Prevention & Research Institute of Texas (RR160053 to M.W.P., a CPRIT Scholar in Cancer Research) are gratefully acknowledged.
Transparency declarations
None to declare.
Author contributions
M.I.K, D.E.G. and J.M.B designed the research; M.I.K, R.D.S., V.C.O, H.B.A, K.N.K., B.W.P., D.C.C., M.S., T.A.C and J.M.B performed research; M.I.K., M.S., B.W.P., D.C.C., T.A.C. and J.M.B contributed new reagents/analytic tools; M.I.K., B.W.P, D.C.C., D.E.G., M.W.P., T.A.C. and J.M.B analysed data; and M.I.K and J.M.B wrote the paper.
References
- 1. Livermore DM, Woodford N.. The β-lactamase threat in Enterobacteriaceae, Pseudomonas and Acinetobacter. Trends Microbiol 2006; 14: 413–20. [DOI] [PubMed] [Google Scholar]
- 2. Livermore DM. Fourteen years in resistance. Int J Antimicrob Agents 2012; 39: 283–94. [DOI] [PubMed] [Google Scholar]
- 3. Palzkill T. Metallo-β-lactamase structure and function. Ann NY Acad Sci 2013; 1277: 91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sauvage E, Kerff F, Terrak M. et al. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol Rev 2008; 32: 234–58. [DOI] [PubMed] [Google Scholar]
- 5. Papp-Wallace KM, Endimiani A, Taracila MA. et al. Carbapenems: past, present, and future. Antimicrob Agents Chemother 2011; 55: 4943–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bassetti M, Nicolini L, Esposito S. et al. Current status of newer carbapenems. Curr Med Chem 2009; 16: 564–75. [DOI] [PubMed] [Google Scholar]
- 7. Torres JA, Villegas MV, Quinn JP.. Current concepts in antibiotic-resistant gram-negative bacteria. Expert Rev Anti Infect Ther 2007; 5: 833–43. [DOI] [PubMed] [Google Scholar]
- 8. Boon JM, Smith BD.. Chemical control of phospholipid distribution across bilayer membranes. Med Res Rev 2002; 22: 251–81. [DOI] [PubMed] [Google Scholar]
- 9. Paterson DL. Recommendation for treatment of severe infections caused by Enterobacteriaceae producing extended-spectrum β-lactamases (ESBLs). Clin Microbiol Infect 2000; 6: 460–3. [DOI] [PubMed] [Google Scholar]
- 10. Paterson DL. Serious infections caused by enteric gram-negative bacilli – mechanisms of antibiotic resistance and implications for therapy of gram-negative sepsis in the transplanted patient. Semin Respir Infect 2002; 17: 260–4. [DOI] [PubMed] [Google Scholar]
- 11. Paterson DL, Bonomo RA.. Extended-spectrum β-lactamases: a clinical update. Clin Microbiol Rev 2005; 18: 657–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Queenan AM, Bush K.. Carbapenemases: the versatile β-lactamases. Clin Microbiol Rev 2007; 20: 440–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zmarlicka MT, Nailor MD, Nicolau DP.. Impact of the New Delhi metallo-β-lactamase on β-lactam antibiotics. Infect Drug Resist 2015; 8: 297–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ambler RP, Coulson AF, Frère JM. et al. A standard numbering scheme for the class A β-lactamases. Biochem J 1991; 276: 269–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bush K, Jacoby GA.. Updated functional classification of β-lactamases. Antimicrob Agents Chemother 2010; 54: 969–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ghuysen JM. Serine β-lactamases and penicillin-binding proteins. Annu Rev Microbiol 1991; 45: 37–67. [DOI] [PubMed] [Google Scholar]
- 17. Bebrone C. Metallo-β-lactamases (classification, activity, genetic organization, structure, zinc coordination) and their superfamily. Biochem Pharmacol 2007; 74: 1686–701. [DOI] [PubMed] [Google Scholar]
- 18. Crowder MW, Spencer J, Vila AJ.. Metallo-β-lactamases: novel weaponry for antibiotic resistance in bacteria. Acc Chem Res 2006; 39: 721–8. [DOI] [PubMed] [Google Scholar]
- 19. Wang Z, Fast W, Valentine AM. et al. Metallo-β-lactamase: structure and mechanism. Curr Opin Chem Biol 1999; 3: 614–22. [DOI] [PubMed] [Google Scholar]
- 20. Laraki N, Galleni M, Thamm I. et al. Structure of In31, a blaIMP-containing Pseudomonas aeruginosa integron phyletically related to In5, which carries an unusual array of gene cassettes. Antimicrob Agents Chemother 1999; 43: 890–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lauretti L, Riccio ML, Mazzariol A. et al. Cloning and characterization of blaVIM, a new integron-borne metallo-β-lactamase gene from a Pseudomonas aeruginosa clinical isolate. Antimicrob Agents Chemother 1999; 43: 1584–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yong D, Toleman MA, Giske CG. et al. Characterization of a new metallo-β-lactamase gene, bla(NDM-1), and a novel erythromycin esterase gene carried on a unique genetic structure in Klebsiella pneumoniae sequence type 14 from India. Antimicrob Agents Chemother 2009; 53: 5046–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Walsh TR. Clinically significant carbapenemases: an update. Curr Opin Infect Dis 2008; 21: 367–71. [DOI] [PubMed] [Google Scholar]
- 24. Walsh TR. Emerging carbapenemases: a global perspective. Int J Antimicrob Agents 2010; 36: S8–14. [DOI] [PubMed] [Google Scholar]
- 25. Poirel L, Naas T, Nicolas D. et al. Characterization of VIM-2, a carbapenem-hydrolyzing metallo-β-lactamase and its plasmid- and integron-borne gene from a Pseudomonas aeruginosa clinical isolate in France. Antimicrob Agents Chemother 2000; 44: 891–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cornaglia G, Giamarellou H, Rossolini GM.. Metallo-β-lactamases: a last frontier for β-lactams? Lancet Infect Dis 2011; 11: 381–93. [DOI] [PubMed] [Google Scholar]
- 27. Everett M, Sprynski N, Coelho A. et al. Discovery of a novel metallo-β-lactamase inhibitor that potentiates meropenem activity against carbapenem-resistant Enterobacteriaceae. Antimicrob Agents Chemother 2018; 62: pii=e00074-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tacconelli E. Global Priority List of Antibiotic-resistant Bacteria to Guide Research, Discovery and Development of New Antibiotics. The World Health Organization. 2017.
- 29. Tacconelli E, Carrara E, Savoldi A. et al. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect Dis 2018; 18: 318–27. [DOI] [PubMed] [Google Scholar]
- 30. Reading C, Cole M.. Clavulanic acid: a β-lactamase-inhibiting β-lactam from Streptomyces clavuligerus. Antimicrob Agents Chemother 1977; 11: 852–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. English AR, Retsema JA, Girard AE. et al. CP-45,899, a β-lactamase inhibitor that extends the antibacterial spectrum of β-lactams: initial bacteriological characterization. Antimicrob Agents Chemother 1978; 14: 414–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fisher J, Belasco JG, Charnas RL. et al. β-Lactamase inactivation by mechanism-based reagents. Philos Trans R Soc Lond B Biol Sci 1980; 289: 309–19. [DOI] [PubMed] [Google Scholar]
- 33. Ehmann DE, Jahić H, Ross PL. et al. Avibactam is a covalent, reversible, non-β-lactam β-lactamase inhibitor. Proc Natl Acad Sci USA 2012; 109: 11663–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bush K, Bradford PA.. Interplay between β-lactamases and new β-lactamase inhibitors. Nat Rev Microbiol 2019; 17: 295–306. [DOI] [PubMed] [Google Scholar]
- 35. Sanschagrin F, Levesque RC.. A specific peptide inhibitor of the class B metallo-β-lactamase L-1 from Stenotrophomonas maltophilia identified using phage display. J Antimicrob Chemother 2005; 55: 252–5. [DOI] [PubMed] [Google Scholar]
- 36. Sun Q, Law A, Crowder MW. et al. Homo-cysteinyl peptide inhibitors of the L1 metallo-β-lactamase, and SAR as determined by combinatorial library synthesis. Bioorg Med Chem Lett 2006; 16: 5169–75. [DOI] [PubMed] [Google Scholar]
- 37. Walter MW, Felici A, Galleni M. et al. Trifluoromethyl alcohol and ketone inhibitors of metallo-β-lactamases. Bioorg Med Chem Lett 1996; 6: 2455–8. [Google Scholar]
- 38. King AM, Reid-Yu SA, Wang W. et al. Aspergillomarasmine A overcomes metallo-β-lactamase antibiotic resistance. Nature 2014; 510: 503–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu X-L, Yang K-W, Zhang Y-J. et al. Optimization of amino acid thioesters as inhibitors of metallo-β-lactamase L1. Bioorg Med Chem Lett 2016; 26: 4698–701. [DOI] [PubMed] [Google Scholar]
- 40. Tucker AT, Leonard SP, DuBois CD. et al. Discovery of next-generation antimicrobials through bacterial self-screening of surface-displayed peptide libraries. Cell 2018; 172: 618–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Robinson MD, McCarthy DJ, Smyth GK.. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010; 26: 139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ignatiadis N, Klaus B, Zaugg JB. et al. Data-driven hypothesis weighting increases detection power in genome-scale multiple testing. Nat Methods 2016; 13: 577–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Osorio D, Rondón-Villarreal P, Torres R.. Peptides: a package for data mining of antimicrobial peptides. R J 2015; 7: 4. [Google Scholar]
- 44.R Core Team. R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing; 2019. http://www.R-project.org/. [Google Scholar]
- 45. Wagih O. ggseqlogo: a versatile R package for drawing sequence logos. Bioinformatics 2017; 33: 3645–7. [DOI] [PubMed] [Google Scholar]
- 46. Mah T-F. Establishing the minimal bactericidal concentration of an antimicrobial agent for planktonic cells (MBC-P) and biofilm cells (MBC-B). J Vis Exp 2014: e50854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Qaiyumi S. Macro- and microdilution methods of antimicrobial susceptibility testing In: Schwalbe GR, Steele-Moore L, eds. Antimicrobial Susceptibility Testing Protocols. CRC Press; 75–79. [Google Scholar]
- 48. Kang KN, Klein DR, Kazi MI. et al. Colistin heteroresistance in Enterobacter cloacae is regulated by PhoPQ-dependent 4-amino-4-deoxy-l-arabinose addition to lipid A. Mol Microbiol 2019; 111: 1604–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kamischke C, Fan J, Bergeron J. et al. The Acinetobacter baumannii Mla system and glycerophospholipid transport to the outer membrane. Elife 2019; 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Docquier J-D, Lamotte-Brasseur J, Galleni M. et al. On functional and structural heterogeneity of VIM-type metallo-β-lactamases. J Antimicrob Chemother 2003; 51: 257–66. [DOI] [PubMed] [Google Scholar]
- 51. Alatrash N, Narh ES, Yadav A. et al. Synthesis, DNA cleavage activity, cytotoxicity, acetylcholinesterase inhibition, and acute murine toxicity of redox-active ruthenium(ii) polypyridyl complexes. ChemMedChem 2017; 12: 1055–69. [DOI] [PubMed] [Google Scholar]
- 52. Zhang S-K, Song J-W, Gong F. et al. Design of an α-helical antimicrobial peptide with improved cell-selective and potent anti-biofilm activity. Sci Rep 2016; 6: 27394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rodloff AC, Goldstein EJC, Torres A.. Two decades of imipenem therapy. J Antimicrob Chemother 2006; 58: 916–29. [DOI] [PubMed] [Google Scholar]
- 54. Pagès J-M, James CE, Winterhalter M.. The porin and the permeating antibiotic: a selective diffusion barrier in Gram-negative bacteria. Nat Rev Microbiol 2008; 6: 893–903. [DOI] [PubMed] [Google Scholar]
- 55. Boll JM, Crofts AA, Peters K. et al. A penicillin-binding protein inhibits selection of colistin-resistant, lipooligosaccharide-deficient Acinetobacter baumannii. Proc Natl Acad Sci USA 2016; 113: E6228–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Boll JM, Tucker AT, Klein DR. et al. Reinforcing lipid A acylation on the cell surface of Acinetobacter baumannii promotes cationic antimicrobial peptide resistance and desiccation survival. MBio 2015; 6: e00478–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Thomas PW, Spicer T, Cammarata M. et al. An altered zinc-binding site confers resistance to a covalent inactivator of New Delhi metallo-β-lactamase-1 (NDM-1) discovered by high-throughput screening. Bioorg Med Chem 2013; 21: 3138–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Makena A, Brem J, Pfeffer I. et al. Biochemical characterization of New Delhi metallo-β-lactamase variants reveals differences in protein stability. J Antimicrob Chemother 2015; 70: 463–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Edwards IA, Elliott AG, Kavanagh AM. et al. Contribution of amphipathicity and hydrophobicity to the antimicrobial activity and cytotoxicity of β-hairpin peptides. ACS Infect Dis 2016; 2: 442–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Trent MS, Ribeiro AA, Lin S. et al. An inner membrane enzyme in Salmonella and Escherichia coli that transfers 4-amino-4-deoxy-L-arabinose to lipid A: induction on polymyxin-resistant mutants and role of a novel lipid-linked donor. J Biol Chem 2001; 276: 43122–31. [DOI] [PubMed] [Google Scholar]
- 61. Raetz CRH, Reynolds CM, Trent MS. et al. Lipid A modification systems in gram-negative bacteria. Annu Rev Biochem 2007; 76: 295–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Peterson AA, Fesik SW, McGroarty EJ.. Decreased binding of antibiotics to lipopolysaccharides from polymyxin-resistant strains of Escherichia coli and Salmonella typhimurium. Antimicrob Agents Chemother 1987; 31: 230–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lehrer RI, Barton A, Daher KA. et al. Interaction of human defensins with Escherichia coli. Mechanism of bactericidal activity. J Clin Invest 1989; 84: 553–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Klingler F-M, Wichelhaus TA, Frank D. et al. Approved drugs containing thiols as inhibitors of metallo-β-lactamases: strategy to combat multidrug-resistant bacteria. J Med Chem 2015; 58: 3626–30. [DOI] [PubMed] [Google Scholar]
- 65. Yusof Y, Tan DTC, Arjomandi OK. et al. Captopril analogues as metallo-β-lactamase inhibitors. Bioorg Med Chem Lett 2016; 26: 1589–93. [DOI] [PubMed] [Google Scholar]
- 66. Arjomandi OK, Hussein WM, Vella P. et al. Design, synthesis, and in vitro and biological evaluation of potent amino acid-derived thiol inhibitors of the metallo-β-lactamase IMP-1. Eur J Med Chem 2016; 114: 318–27. [DOI] [PubMed] [Google Scholar]
- 67. Brem J, van Berkel SS, Aik W. et al. Rhodanine hydrolysis leads to potent thioenolate mediated metallo-β-lactamase inhibition. Nat Chem 2014; 6: 1084–90. [DOI] [PubMed] [Google Scholar]
- 68. Brem J, Cain R, Cahill S. et al. Structural basis of metallo-β-lactamase, serine-β-lactamase and penicillin-binding protein inhibition by cyclic boronates. Nat Commun 2016; 7: 12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yang S-K, Kang JS, Oelschlaeger P. et al. Azolylthioacetamide: a highly promising scaffold for the development of metallo-β-lactamase inhibitors. ACS Med Chem Lett 2015; 6: 455–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.