

Summary
IgG antibodies are actively produced in response to antigenic challenge or passively administered as an effective form of immunotherapy to confer immunity against foreign antigens. Their protective activity is mediated through their bifunctional nature: a variable Fab domain mediates antigen binding specificity, whereas the constant Fc domain engages Fcγ receptors (FcγRs) expressed on the surface of leukocytes to mediate effector functions. While traditionally considered the invariant domain of an IgG molecule, the Fc domain displays remarkable structural heterogeneity determined primarily by differences in the amino acid sequence of the various IgG subclasses and by the composition of the complex, Fc-associated bi-antennary N-linked glycan. These structural determinants regulate the conformational flexibility of the IgG Fc domain and affect its capacity to interact with distinct types of FcγRs (type I or type II FcγRs). FcγR engagement activates diverse downstream immunomodulatory pathways with pleiotropic functional consequences including cytotoxicity and phagocytosis of IgG-coated targets, differentiation and activation of antigen presenting cells, modulation of T cell activation, plasma cell survival and regulation of antibody responses. These functions highlight the importance of FcγR-mediated pathways in the modulation of adaptive immune responses and suggest a central role for IgG-FcγR interactions during active and passive immunization.
Keywords: Fc receptors, Antibodies, Vaccination, Immunotherapies, Inflammation
Introduction
Antibodies are key components of our immune system that link the two main branches of immunity: innate and adaptive. These Y-shaped molecules are selected and produced by cells of the adaptive immune system (B cells), whereas their subsequent binding to innate leukocytes triggers a range of effector functions that provide effective host protection. Recognition and binding of antibodies to the surface of the leukocytes is mediated through their Fc domains interacting with specialized receptors, Fc receptors, expressed by several types of circulating and tissue-resident leukocytes. Contrary to the antigen-binding Fab domain that exhibits astonishing variability due to the almost unlimited diversification of its variable regions (VH and VL), the Fc domain is relatively constant, allowing thereby its recognition by innate receptors, like Fc receptors. Despite its invariant nature, the Fc domain exhibits some degree of heterogeneity, as a result of differences in the primary amino acid backbone sequence among the various IgG subclasses as well as in the structure and composition of the Fc-associated glycan (1, 2). Recent crystallographic studies revealed that the IgG Fc domain exhibits two main conformational states (‘open’ or ‘closed’) that are physiologically regulated by the Fc-associated glycan structure and consequently affect FcγR binding (3).
FcγRs are broadly categorized into type I or type II based on their capacity to interact with the two conformational states of the IgG Fc domain (4, 5). Type I FcγRs include the canonical FcγRs and can be engaged by the IgG Fc only in the ‘open’, but not in the ‘closed’ conformation. In contrast, type II FcγRs, which include C-type lectin receptors, like CD23 and DC-SIGN exhibit minimal affinity for the ‘open’ IgG Fc conformation, binding preferentially IgG Fcs at the ‘closed’ conformation. The dynamic flexibility of the IgG Fc domain and its capacity to interact with the different FcγR types greatly influences the in vivo effector functions of antibodies. Engagement of FcγRs expressed on the surface of effector leukocytes initiate a number of pro-inflammatory, anti-inflammatory and immunomodulatory functions with pleiotropic effects that contribute to the in vivo protective activity of antibodies, and modulate the host adaptive immune responses. In this review, we will focus on the mechanisms that regulate IgG binding to FcγRs and provide an overview of the activity and function of FcγRs. We will highlight the mechanisms by which FcγR engagement by passively-administered antibodies provide in vivo protection and discuss the evidence on the role of FcγR-mediated pathways in driving humoral and cellular immune responses upon active immunization.
IgG Fc domain structural and functional heterogeneity
IgG is the most abundant immunoglobulin class in serum, constituting over 75% of circulating immunoglobulin. It is the main immunoglobulin class that is produced during an immune response to provide efficient protection against foreign antigens. Common to the other immunoglobulin classes, IgG molecules are comprised of two identical heavy (H) and light (L) chain domains that are characterized by genetically variable (V) or constant (C) regions. Each domain is folded into a globular immunoglobulin motif (four for the heavy chain and two for the light chain), and the IgG molecule is divided into two main domains linked by a hinge region that ensures pairing of the two heavy chains through disulphide bonding. These domains include the Fab (fragment, antibody binding) domain, which includes the variable regions that mediate antigen binding), and the Fc (fragment, crystallizable) domain that primarily serves as the binding site for FcγRs, FcRn, complement and other receptors or proteins (e.g. TRIM, Streptococcal protein A or G)(6).
Contrary to the sequence variability of the Fab domain, the Fc domain is comprised of the constant domains (CH2 and CH3) of the two heavy chains and adopts a horseshoe-like conformation that is formed by the tight association of the two CH3 domains at the C-terminal proximal region of the IgG, while the CH2 domains remain further apart (7). This characteristic conformation is primarily achieved by the presence of a central N-linked glycan structure conjugated to the amino acid backbone of the CH2 domain at position Asn297 of both heavy chains (8, 9). This carbohydrate structure lies within the hydrophobic cleft of the Fc domain formed by the two CH2 domains and dynamically regulates the Fc domain conformational structure and thereby its capacity to interact with type I or type II FcγRs (3, 5, 10). The presence of the Fc-associated glycan is required to maintain the Fc domain in a structural conformation that would permit its interaction with FcγRs (10). Indeed, loss of this glycan either through enzymatic removal or mutation of the Asn297 residue greatly impacts the capacity of the Fc domain to interact with all classes of type I and type II FcγRs (10–13).
Like any other process of the immune system, IgG binding to the different FcγR types is tightly regulated and several layers of control exist that dynamically regulate this process to eliminate any potential for uncontrolled or inappropriate inflammation. Differences in the amino acid backbone between the various IgG subclasses (IgG1, IgG2, IgG3, IgG4 in humans) and the Gm allotypes readily affect the capacity of the hinge-proximal region of the Fc domain to interact with type I FcRs (14, 15). Likewise, the impact of specific mutations at the CH2 domain, particularly within the interface that participates in the interaction with type I FcγR, has been assessed in several studies and a number of protein sequence variants have been identified that specifically alter the affinity of the IgG Fc domain for particular classes of type I FcγRs (14, 16–18). These studies highlight the importance of the primary amino acid backbone sequence of the IgG Fc domain in regulating Fc-FcγR interactions and provided the basis for the generation and evaluation of Fc domain variants engineered for improved Fc effector activity through enhanced capacity for FcγR engagement (19–22).
Apart from the amino acid sequence of the IgG Fc domain, engagement of type I and type II FcγRs is also regulated by the structure and composition of the Fc-associated glycan (4). An ever-increasing body of evidence strongly suggests a crucial role for the Fc-associated glycan in the modulation of the IgG Fc effector activity (8, 10). The complex, biantennary Fc glycan consists of a core heptasaccharide structure attached to Asn297; a highly conserved N-linked glycosylation site among the different IgG subclasses and among many mammalian species (1). Analysis of the Fc glycan composition revealed substantial heterogeneity, due to the addition of fucose, galactose, N-acetylglucosamine and sialic acid residues to the core glycan structure. While the presence of the core glycan structure is necessary to preserve the CH2 domains of the two heavy chains at a conformation permissive for Fc-FcγR interactions (8, 11), it is the structure and composition of the glycan that finely tunes the affinity of the Fc domain for binding to the different classes of type I and type II FcγRs (3, 4, 10). For example, the presence of a branching fucose has been shown to reduce affinity specifically for the activating type I FcγR, FcγRIIIa, without any impact on the capacity of the Fc domain to interact with other classes of type I FcγRs (23, 24). Indeed, afucolylated IgG glycovariants exhibit improved in vivo Fc effector activity over their fucosylated counterparts and the approach of Fc glycoengineering has been successfully used to generate antibodies with improved in vivo efficacy (25–28). Likewise, the presence of terminal sialic acid residues abrogates binding to type I FcγRs and enables preferential engagement of type II FcγRs (29, 30). Molecular modeling and analysis of the crystal structure of sialylated IgG Fc revealed that sialylation induces a conformational change of the CH2 domains that affects type I and type II FcγR binding (3, 5). At the conformational state induced by the sialylated glycan, the type II FcγR binding site, located at the CH2-CH3 interface is exposed enabling the interaction with type II FcγRs, whereas the hinge-proximal region of the CH2 domain that mediates type I FcγR binding becomes inaccessible (3–5). It is therefore well-established that the Fc glycan structure plays a central role in regulating the dynamic flexibility of the IgG Fc domain and consequently its capacity to interact with type I or type II FcγRs, by adopting two mutually exclusive conformations: an ‘open’ that enables for type I, but not type II FcγR binding, and a ‘closed’ that is induced upon sialylation and preferentially engages type II, but not type I FcγRs (4).
Engagement of the different FcγR types induces divergent immunomodulatory responses that can greatly affect the outcome of IgG-mediated inflammation and immune responses. Given the significance of the effector responses induced upon FcγR engagement, it is anticipated that several regulatory mechanisms have evolved to control IgG Fc glycan composition. Although the precise mechanisms are still poorly characterized, there is substantial evidence suggestive of the existence of homeostatic processes that regulate Fc glycan composition, and more specifically the activity of ST6Gal1, the glycosyltransferase responsible for terminal Fc glycan sialylation. For example, Fc glycan analysis composition upon vaccination revealed specific modulation in the abundance of antigen-specific IgG with sialylated Fc domains, which potentially has immunomodulatory consequences for the subsequent immune response (31, 32). Likewise, analysis of the levels of sialylated IgG Fc in patients with autoimmune pathologies, like rheumatoid arthritis and Wegener’s granulomatosis revealed an association with clinical disease severity (33–36). In particular, higher levels of sialylated Fc in autoantibodies were observed during periods of clinical remission, whereas disease relapses were commonly associated with a significant decrease in IgG Fc sialylation (33, 34). These observations highlight the importance of the Fc glycosylation status in regulating IgG Fc effector activity and suggest the presence of homeostatic mechanisms that dynamically control IgG Fc glycan structure and composition.
FcγR function and activity
Type I FcγR Family
Based on their capacity to interact with the two main conformational states of the IgG Fc domain, FcγRs are broadly divided into type I and type II. Type I FcγRs is a group of immunoreceptors that are structurally and functionally related and belong to the immunoglobulin (Ig) superfamily (37, 38). Their extracellular, IgG-binding domain is comprised of two (or three for FcγRI) Ig domains that mediate interactions with the IgG through the hinge proximal region of the CH2 domain in a 1:1 complex (14, 15). They share highly conserved intracellular signaling components that mediate receptor signaling upon crosslinking by IgG-antigen complexes (39). In humans, eight different genes, each with multiple transcriptional isoforms, encode the various classes of type I FcγRs (FcγRI, FcγRIIa/b/c, and FcγRIIIa/b)(Figure 1). Most of these genes are mapped at a common FcγR locus (located at 1q23) that originated through sequential non-homologous recombination events of the ancestral FcγR locus that is highly conserved among mammalian species and can be traced back early in evolutionary history (40, 41). Despite the sequence similarity shared among the different classes of type I FcγRs, they present distinct structural and functional characteristics (37)(see also Hargraves et al. elsewhere in this volume for an analysis of the FcγR locus).
Figure 1: Structure and characteristics of the human type I FcγR family.
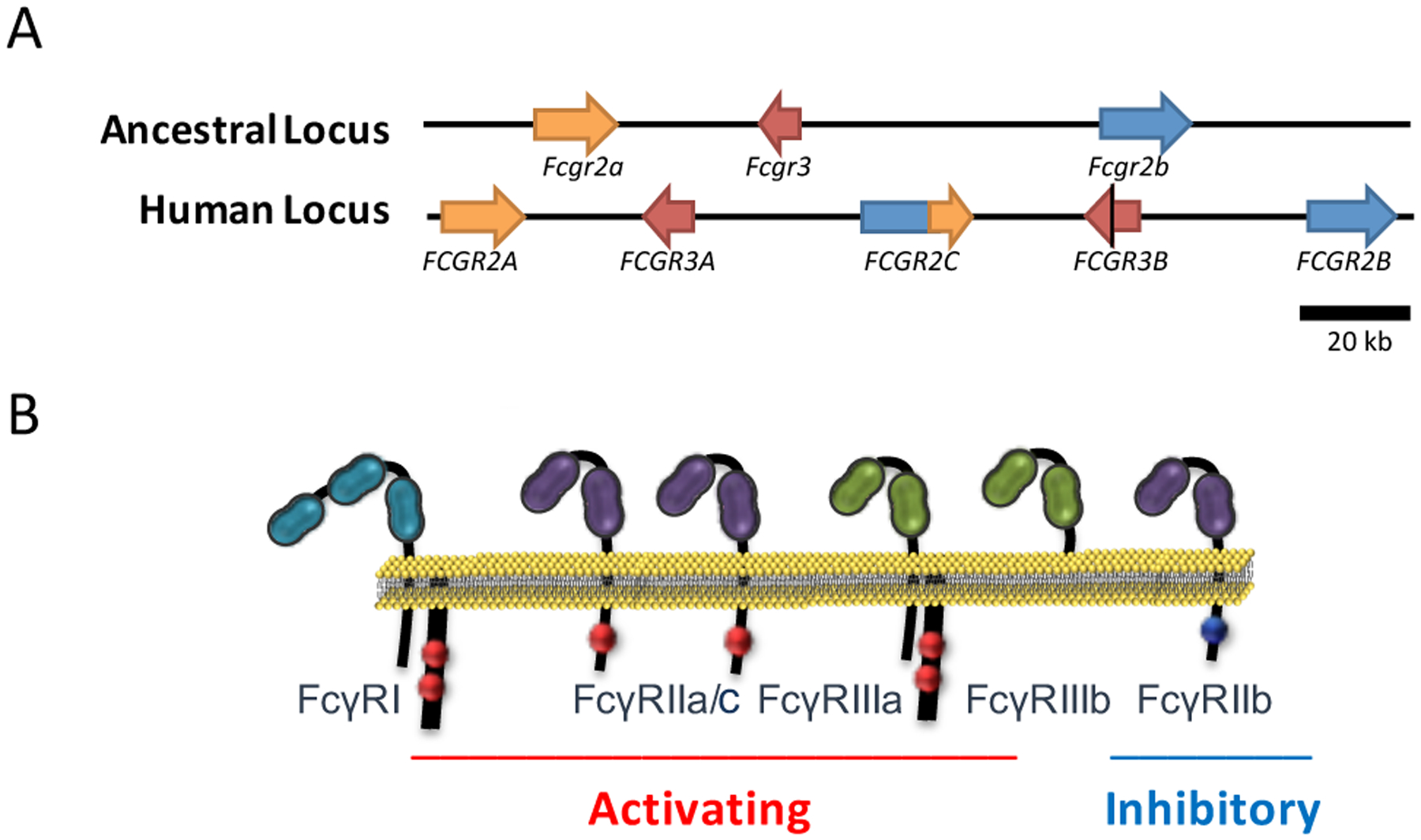
(A) The main human FcγR locus is located at 1q23 and comprises all the genes encoding the low affinity type I FcγRs. The unique genomic organization of the human FcγR locus is the result of a non-homologous recombination event of the ancestral FcγR locus, which is highly conserved among several mammalian species. This event gave rise to additional FcγR-encoding genes that are uniquely found in humans. (B) Human type I FcγRs are functionally categorized into activating or inhibitory based on the presence of an intracellular ITAM or ITIM motif. Engagement of activating or inhibitory FcγRs by IgG immune complexes induces immunostimulatory or immunosuppressive signals, respectively, influencing thereby the outcome of IgG-mediated inflammation and immunity.
FcγRI is a high affinity FcγR that is capable of binding monomeric IgG (KD 10−9-10−10 for human IgG1). Its increased affinity for IgG is due to its unique structure; the presence of a third (D3), extracellular Ig domain (compared to FcγRII or FcγRIII) acts as a spacer for the Fab domain, stabilizing thereby IgG-FcγRs interactions (42). FcγRI is constitutively expressed predominantly by monocytes and macrophages and can be induced in other myeloid leukocyte types, like neutrophils, primarily by interferon-γ and to a lesser extent by granulocyte-colony stimulating factor (G-CSF), interferon-α and interleukin-12 (43, 44). The ligand-binding, α chain of FcγRI associates with the FcR γ-chain homodimer; a process required for cell surface expression and signal transduction (45–47). Although three genes encoding FcγRI have been identified (FCGR1A, FCGR1B, FCGR1C), only FCGR1A expresses the functional FcγRI, whereas FCGR1B/C represent duplicated pseudogenes of FCGR1A that may express truncated or soluble forms of FcγRI due to the presence of a premature stop codon within the extracellular region of FcγRI (48, 49).
Contrary to FcγRI, the other type I FcγR classes, FcγRII and FcγRIII exhibit low affinity for IgG (KD for hIgG1: 10−5-10−7). While they are incapable of binding monomeric IgG, IgG-antigen complexes (immune complexes) can sufficiently engage and crosslink low affinity FcγRs through high avidity, multimeric interactions (50). There are three different FcγRII and two FcγRIII classes; each with unique structural and functional properties. All the different classes of FcγRII share a characteristic structure that includes a functional signaling motif within the cytoplasmic region of the receptor α chain and can therefore transduce intracellular signals following receptor crosslinking without the need for accessory signaling subunits, like the FcR γ-chain. FcγRIIa, FcγRIIb, and FcγRIIc are encoded by FCGR2A, FCGR2B, and FCGR2C, respectively. FcγRIIa encompasses an immunoreceptor tyrosine-based activation motif (ITAM) at its cytoplasmic tail and is widely expressed by several leukocyte types of myeloid origin, including granulocytes, monocytes, macrophages, dendritic cells, and platelets (37). By contrast, FcγRIIb is the sole inhibitory FcγR and its activity is mediated through an intracellular immunoreceptor tyrosine-based inhibitory motif (ITIM) (6). FcγRIIb is expressed as two splicing variants (FcγRIIb1 and FcγRIIb2) that differ in their capacity to become internalized following receptor crosslinking (51, 52). FcγRIIb1, the full-length transcript variant, includes a signal sequence that inhibits receptor internalization and is exclusively expressed by cells of the lymphoid lineage (B cells), whereas the expression of FcγRIIb2, the variant that is permissive for internalization, is restricted to myeloid cells, like monocytes, macrophages and dendritic cells (52). FcγRIIc represents essentially a chimeric receptor that is comprised of the extracellular domain of FcγRIIb and the intracellular of FcγRIIa. Indeed, FCGR2C, the gene coding for FcγRIIc is the result of a non-homologous recombination between the FCGR2A and FCGR2B genes and its expression is restricted to NK cells (37, 40, 41). However, FcγRIIc expression at protein level is limited to 10–25% of the population due to a common SNP (Q57X) at exon 3 that generates a stop codon (53, 54).
The other low affinity FcγR class, FcγRIII is encoded by the FCGR3A and FCGR3B genes. While both genes share very high levels of sequence homology, their products exhibit remarkable structural differences that influence their activity and function (41). FcγRIIIa is a transmembrane protein that associates with the FcR γ-chain for expression and signaling, similar to FcγRI (46, 55, 56). In contrast, FcγRIIIb is post-translationally processed as a glycophosphatidylinositol (GPI)-anchored protein and therefore lacks transmembrane and intracellular domains (56). It doesn’t require the FcR γ-chain for expression; however, it synergistically acts with the activating receptor FcγRIIa to mediate cellular responses and it has been shown to associate with the FcR γ-chain (57, 58). The structural differences between FcγRIIIa and FcγRIIIb are attributed to a point mutation (Phe203Ser) in the membrane proximal region of the extracellular domain of FcγRIIIb that created a GPI-anchor signal sequence. Additionally, FcγRIIIa and FcγRIIIb exhibit distinct expression patterns (58). While FcγRIIIb expression is restricted to neutrophils, FcγRIIIa is widely expressed by several leukocyte cell types, including macrophages, NK cells and monocyte subsets (inflammatory monocytes, CD14low)(4, 37).
From a functional point of view, type I FcγRs are classified into activating or inhibitory based on their capacity to transduce immunostimulatory or immunosuppressive signals following receptor crosslinking (12, 39). Both activating and inhibitory FcγRs are expressed by most leukocyte types. Notable exceptions include NK cells, which express only activating FcγRs (FcγRIIIa), and B cells, which only express the inhibitory FcγRIIb. Since most effector leukocytes co-express both activating and inhibitory FcγRs, the outcome of IgG-mediated inflammation and immunity is largely determined by balancing activating or inhibitory signals transduced by activating or inhibitory FcγRs, respectively (12). Activating FcγRs, which include FcγRI, FcγRIIa/c and FcγRIIIa are characterized by the presence of intracellular ITAMs either in the receptor α chain (in the case of FcγRIIa/c) or in the associated FcR γ- chain (FcγRI and FcγRIIIa)(4, 59, 60). Despite their structural differences, all ITAM-bearing Fc receptors follow an identical pattern of signal transduction following crosslinking by immune complexes (Figure 2). In particular, binding of multimeric IgG immune complexes to activating FcγRs causes receptor clustering and aggregation that in turn results in the phosphorylation of the ITAM domains (45, 58, 59, 61). Phosphorylation of the ITAM tyrosine residues induces the subsequent activation of a number of cytoplasmic tyrosine kinases, firstly those of the Src (Lyn, Lck, Hck and Fgr) and then of the Syk family (56, 59, 61–65). Furthermore, receptor aggregation and ITAM phosphorylation leads to the activation of ζ chain subunits, ZAP-70 and FAK that subsequently regulate actin polymerization, phagocytosis and receptor internalization (59, 61, 66). Src and Syk kinase family activation sequentially induces the activation of the PI3K-PKC pathway, leading to Ca2+ mobilization and cellular activation (67–70). In addition to these early signaling events, several other late signaling pathways become activated and include the MEK and MAP family kinases and the Ras pathway, resulting in the transcription of pro-inflammatory cytokines and chemokines with effects on cellular survival and differentiation (68, 71, 72). Although it is generally anticipated that any intracellular signals transduced upon receptor crosslinking ultimately lead to cellular activation and receptor internalization, the precise biological responses following activating FcγR engagement greatly vary between different leukocyte cell types. Such responses include antibody-dependent cellular cytotoxicity (ADCC) or, phagocytosis (ADCP), release of cytokines and chemokines, leukocyte differentiation and survival, as well as modulation of T and B cell responses (4, 39).
Figure 2: Overview of the downstream signaling events following crosslinking of activating type I FcγRs.
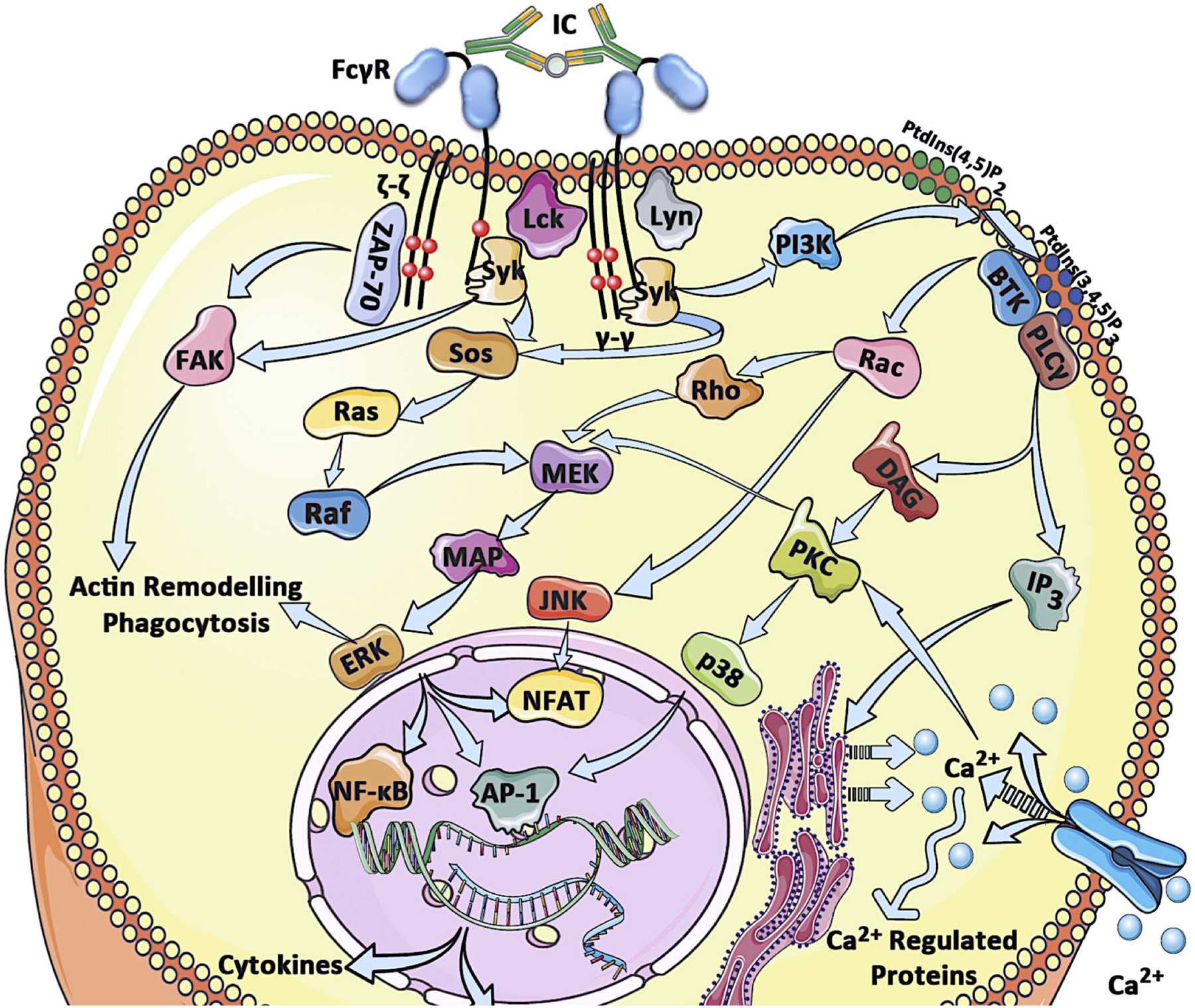
Engagement of activating type I FcγRs by IgG immune complexes triggers receptor crosslinking and phosphorylation of the ITAM domains by Src family kinases. This event subsequently initiates a cascade of signaling pathways that result in cellular activation, Ca2+ mobilization, activation of kinases involved in actin remodeling and at later stages transcription factor activation and expression of cytokines, chemokines and survival proteins that further augment inflammatory processes.
The signaling activity of the activating FcγRs is counterbalanced by the inhibitory type I FcγR, FcγRIIb. Its importance in regulating FcγR-mediated pathways is reflected by the association of several autoimmune and chronic inflammatory pathologies with functional polymorphisms within the promoter region or the coding sequence of the FCGR2B gene that affect receptor expression or activity, respectively (37). Several studies have elucidated the molecular mechanisms by which FcγRIIb mediate immunosuppressive signals. Critical for FcγRIIb activity is the recruitment of SHIP phosphates to its ITIM domain following receptor crosslinking and phosphorylation by Src family kinases (73–75). Recruited phosphatases, like SHIP and SHP2 promote the hydrolysis of phosphatidylinositol 3,4,5-triphosphate to phosphatidylinositol 4,5-biphosphate that in turn inhibit the recruitment and activation of PLCγ and BTK (73, 76, 77). These intracellular effects balance any signals mediated by activating FcγRs, effectively limiting cellular activation and effector responses. Alternatively, in the absence of activating FcγRs, as in the case of B cells that express only the inhibitory FcγRs, FcγRIIb engagement regulates cellular survival and antibody production (78).
Lastly, in contrast to activating or inhibitory type I FcγRs, FcγRIIIb lacks any intracellular signaling motifs and is therefore incapable of inducing or inhibiting cellular activation. Despite the absence of functional signaling domains, FcγRIIIb has been shown to transduce activation signals following receptor crosslinking, mainly by associating and acting synergistically with other receptors, such as FcγRIIa and complement receptors (57). In addition, due to its GPI-anchor, FcγRIIIb is preferentially localized in lipid raft microdomains that are rich in membrane-associated kinases, like Syk and might utilize such signal transduction machinery to initiate intrinsic signals following receptor engagement (57, 58, 79).
Type II FcγR Family
Type II FcγRs belong to the C-type lectin family of receptors and are capable of binding to the IgG Fc domain in the ‘closed’ conformation at the CH2-CH3 interface in a 2:1 complex (receptor:IgG)(3–5). At this conformational state, binding of type I FcγRs is inhibited, since the hinge proximal region of the CH2 domain, where type I FcγRs normally bind, is not accessible (3). So far, two receptors have been shown to mediate binding of the ‘closed’ conformation IgG Fc and exert in vivo activity: DC-SIGN and CD23 (3, 4). Other C-type lectins with similar binding capacity have been also suggested, including CD22 and DCIR; however, their exact role in vivo has not been clearly defined yet (80–82).
DC-SIGN, also referred as CD209, is a C-type lectin that is encoded by the CD209 gene and is expressed primarily by dendritic cells, macrophages, and monocytes (83, 84). Similar to other C-type lectins, DC-SIGN binds to a number of carbohydrate ligands, predominantly mannose-rich glycan structures that are commonly found on the surface of heavily-glycosylated proteins as well as bacterial and viral glycoproteins, like the HIV-1 envelope glycoprotein, gp160 (85). Given the diversity and the differential binding affinities of the molecules that have the capacity for DC-SIGN engagement, little is known about the exact signaling events initiated upon ligand binding. Downstream signaling cascades induced following receptor crosslinking appear to be highly dependent upon the type and the nature of the DC-SIGN – ligand interaction, as well as the effector leukocyte types involved. In the case of sialylated IgG Fc – DC-SIGN interaction, receptor engagement on regulatory macrophages induces the expression of interleukin 33 (IL-33) that limits T cell- and IgG-mediated inflammation (86, 87). Secreted IL-33, a potent Th2-polarizing cytokine known for its pleiotropic immunomodulatory effects, promotes Treg activation and expansion that effectively suppresses Th1 and Th17 responses (87). Additionally, IL-33 stimulates the release of IL-4 by basophils that in turn induces the upregulation of the inhibitory type I FcγR, FcγRIIb on effector inflammatory monocytes and macrophages at sites of inflammation to limit IgG-mediated inflammation, by balancing the activity of activating type I FcγRs (86).
CD23 is also a heavily glycosylated C-type lectin that is encoded by the FCER2 gene, which is located at the same locus with CD209 (19p13), probably reflecting the similarities in their function and activity between these two receptors. CD23 exists in two splicing isoforms: CD23a and CD23b, with no structural or functional differences. CD32a is constitutively expressed by mature B cells, whereas CD23b is only expressed upon induction by IL-4 by several leukocyte types, including T cells, monocytes, macrophages and granulocytes (88). CD23 has been initially identified as the low affinity receptor for IgE. Indeed, it has the capacity to bind IgE with low affinity (KD: 10−7); a property attributed to the intrinsic flexibility of the Cε3 domain of IgE (3, 89, 90). Molecular modelling revealed that sialylated IgG Fc also adopts increased flexibility that allows for binding to CD23 in analogy to the IgE:CD23 complex (3). Binding of IgE-containing immune complexes to CD23 on B cells has been shown to regulate IgE production by B cells, as well as to control antigen capture and presentation to dendritic cells (91, 92). CD23 – sialylated IgG interactions has been recently demonstrated to participate in the regulation of IgG affinity maturation and responses by inducing the expression of the inhibitory type I FcγR, FcγRIIb on B cells (32).
FcγR-IgG interactions in passive immunization
Passive immunization was the first effective therapeutic intervention against a number of infectious diseases, based on seminal work by Emil von Behring and Shibasaburo Kitasato during the late 19th century (93, 94). For over five decades, passive administration of anti-serum had been used with remarkable success to prevent or treat bacterial and viral infections. However, the systematic use of antibody therapy has been largely abandoned, mainly due to the lot-to-lot variability of the polyclonal antibody preparations, the relative difficulty of administration over conventional chemotherapeutic agents, and the reported side effects. Remarkable advances in hybridoma technologies over the past decades have revolutionized the approaches for the generation of highly specific monoclonal antibodies with exceptional in vivo activity. Currently, there are over 40 FDA-approved monoclonal antibodies in clinical use and more than a hundred in clinical trials. Indeed, antibody-based therapeutics are the first line of treatment for a number of chronic inflammatory and neoplastic disorders and have demonstrated unsurpassed efficacy and safety compared to conventional therapeutic approaches (95). Monoclonal antibody-based therapeutics have been developed primarily for the treatment of neoplastic diseases, including lymphoma and several types of solid tumors (96, 97). Additionally, a number of monoclonal antibodies exist that have been successfully used for the control of chronic inflammatory diseases, by directly targeting key inflammatory molecules, such as interleukins and TNF-α, as well as for the prophylaxis or therapy of a range of infectious diseases, including RSV, anthrax and Ebola (98–100).
Apart from the observed safety and efficacy of antibody-based therapeutics, their widespread clinical use has provided substantial evidence supporting a role for FcγR effector function in their in vivo activity. A number of genetic association studies indicated a strong correlation of allelic variants of type I FcγRs with clinical outcome following treatment with anti-tumor monoclonal antibodies. Indeed, patients carrying the allelic variants of FcγRIIa and FcγRIIIa that exhibit increased affinity for human IgG subclasses demonstrated greater responsiveness to anti-tumor antibody therapy in cases of B cell lymphomas, breast and colorectal cancers (101–106). Similarly, IgG Fc domain engineering for enhanced binding to the activating type I FcγR, FcγRIIIa resulted in a significant improvement in the therapeutic activity of anti-CD20 monoclonal antibodies (27). These clinical observations strongly suggest a key role for FcγR-mediated pathways in the in vivo activity of antibodies. A number of in vivo studies have dissected the exact mechanisms of Fc effector pathways and suggested novel approaches for optimizing the in vivo efficacy of passively administered antibodies in tumor and infectious disease models (12, 19, 21, 107, 108).
As described in the previous section, the outcome of IgG-mediated inflammation and immunity is the result of the balancing activity of activating and inhibitory type I FcγRs expressed by the various effector leukocyte types. This hypothesis has been initially tested in the mouse FcγR system in a model of lung metastasis of melanoma cells. Based on the differential capacity of mouse IgG subclasses to engage mouse activating type I FcγRs, IgG subclass variants of an antibody targeted against a glycoprotein (gp75) expressed by melanoma cells were generated and their in vivo activity was compared (12). The cytotoxic activity of the anti-gp75 antibodies perfectly correlated with their capacity for engaging activating type I FcγRs. In particular, mouse IgG2a subclass variants demonstrated augmented in vivo activity compared to mouse IgG2b, while the mouse IgG1 variant exhibited minimal activity. This hierarchy reflects the capacity of the mouse IgG subclasses to interact with the different classes of mouse FcγRs. Mouse IgG2a preferentially engages the activating FcγRIV (orthologue of human FcγRIIIa) with 100-fold greater affinity compared to the inhibitory FcγRIIb, whereas mouse IgG1 preferentially engages FcγRIIb, with minimal affinity for the activating FcγRIV (12, 109). These results have clearly demonstrated that the in vivo activity of antibodies is highly dependent on their capacity to engage activating FcγRs and suggested that manipulation of the FcγR-mediated pathways could greatly influence the in vivo protective activity.
Similar findings suggesting a role for FcγRs in the in vivo activity of monoclonal antibodies were observed for a number of different models ranging from tumors to bacterial (Bacillus anthracis (20, 107), Streptococcus pneumoniae (110), Staphylococcus aureus (111)), fungal (Cryprococcus neoformans (112, 113)) and viral (HIV-1 (19, 114, 115), influenza (108, 116), RSV (28)) infections. Common to all these models was the absolute requirement for Fc effector function for optimal antibody in vivo activity. Although the precise effector functions initiated upon activating type I FcγR engagement by IgG Fc differ between the various experimental models, it is anticipated that FcγR-mediated cellular activation of effector leukocytes, such as monocytes, macrophages, neutrophils, and NK cells, would result in antibody-dependent cellular cytotoxicity, phagocytosis, cytokine and chemokine release. Such effector functions would facilitate the cytotoxic lysis and clearance of tumor or infected cells, promote microbial opsonization and clearance, as well as enhance host immune responses, by altering the survival, mobilization and differentiation of leukocyte populations. Using examples from an infectious (HIV-1) and a neoplastic (CD20+ B cell lymphoma) disease, in the following sections we will discuss in detail the role of FcγR-mediated interactions in the in vivo protective activity of therapeutic antibodies.
HIV-1 passive immunization
The development of broadly neutralizing antibodies against HIV-1 and their clinical use for the prevention or treatment of the infection has been hampered by the unique immune evasion mechanisms of HIV-1. Indeed, HIV-1 displays remarkable diversity (117, 118), its main envelope glycoprotein is shielded in an elaborate glycan structure that masks all sites of vulnerability and it generally presents limited immunogenicity (119, 120). These structural and functional characteristics of the HIV-1 virus limit the host’s capacity to mount sustained antibody responses with broad neutralizing activity. However, a small fraction of patients develop potent neutralizing antibodies with broad activity against other viral clades that can efficiently suppress viremia for several years (121, 122). By B cell cloning of neutralizing antibodies from these patients, several broadly neutralizing anti-HIV-1 antibodies targeting virus’ envelope glycoprotein have been isolated and extensively characterized over the past 5 years (122). Passive administration of these antibodies confers protection against SHIV challenge in macaques (114, 123) and HIV-1 infection in humanized mice (124). Additionally, these broadly neutralizing antibodies have been shown to provide durable and sustained suppression of viremia in chronically SHIV-infected macaques (125, 126), and in humanized mice with established HIV-1 infection (19, 115, 127, 128). These encouraging findings suggest the potential clinical use of broadly neutralizing anti-HIV-1 antibodies for the prevention and treatment of HIV-1 infection in humans. Indeed, in a recent phase I/II trial of the broadly neutralizing anti-CD4bs antibody, 3BNC117, effective suppression of viremia in chronically infected HIV-1 patients was observed that lasted for several days post-antibody administration (129).
A few previous studies have suggested a role for FcγRs during HIV-1 infection. In a small patient cohort, the clinical progression of AIDS was found to be associated with genetic variants of FcγRIIa that affect IgG binding affinity (130). Similarly, the in vivo activity of the neutralizing antibody b12 has been shown to depend on FcγR, but not on complement interactions in a model of pre-exposure prophylaxis in SHIV-challenged Rhesus macaques (114). Given the recent isolation of anti-HIV-1 antibodies with potent and broad neutralizing activity, it was not until recently that the mechanisms of Fc effector function that contribute to their in vivo activity were investigated. Using models of HIV-1 entry (131), IgG subclass variants with preferential binding capacity for activating type I FcγRs exhibited improved in vivo activity and induced enhanced clearance of circulating virus particles (19). Similarly, in studies using HIV-1-infected humanized mice, the in vivo activity of broadly neutralizing antibodies has been shown to depend on Fc-FcγR interactions (19, 115). In particular, IgG Fc domain variants with diminished capacity to engage all classes of type I FcγRs failed to adequately suppress viremia in models of antibody-mediated post-exposure prophylaxis or therapy (19, 115). Likewise, Fc domain variants of anti-HIV-1 antibodies engineered for enhanced binding to activating type I FcγRs exhibited improved in vivo activity, as evidenced by prolonged and durable suppression of viremia in humanized mice with established HIV-1 infection (19). Collectively, these results highlight a key role for Fc effector function in the in vivo activity of anti-HIV-1 broadly neutralizing antibodies and suggest approaches for the development of improved antibody-based therapeutics with augmented in vivo efficacy through enhanced Fc-FcγR interactions.
Passive Immunotherapy of B-cell lymphoma
Several monoclonal antibodies are currently approved and used for the treatment of neoplastic diseases. The anti-tumor activity of these antibodies is achieved by directly targeting tumor cells through cytotoxic lysis, inducing cell death or blocking cell survival, antagonizing the activity of growth factors or their receptors, as well as altering the tumor microenvironment to stimulate host anti-tumor responses (96, 97). Anti-CD20 antibodies, like rituximab were among the first to be approved for cancer patients and have been used for several years to treat various classes of CD20+ leukemias and lymphomas as well as to control autoimmune syndromes.
A number of different mechanisms that account for the activity of anti-CD20 antibodies have been suggested, including induction of B cell apoptosis and complement-mediated cytolysis; however, in vivo evidence from animal and human studies strongly supports a dominant role for FcγR-mediated effector activity (103, 105, 132, 133). For example, allelic variants of the activating type I FcγRs, FcγRIIa and FcγRIIIa that exhibit altered affinity for human IgG have been shown to be associated with the efficacy of CD20+ cell depletion in lymphoma patients (103, 105). The requirement for activating type I FcγRs to mediate the in vivo activity of anti-CD20 antibodies has also been demonstrated in mouse models of B-cell lymphoma. Deletion of the FcR γ-chain that is required for the expression and signaling function of activating type I FcγR in mice, abrogated the in vivo activity of rituximab (134). Similarly, Fcgr2b-knockout mice that lack the inhibitory FcγRIIb exhibited improved therapeutic responses following anti-CD20 antibody administration (134). These findings have been recently extended in studies using mice humanized for type I FcγRs (21). These mice faithfully recapitulate the unique pattern of human FcγR expression and have been successfully used for the pre-clinical evaluation of cytotoxic antibodies in several models (18, 20, 108). A panel of Fc domain-engineered anti-CD20 antibodies were generated that exhibited selective binding to particular classes of human FcγRs and their activity was assessed in FcγR-humanized mice challenged with human CD20-expressing lymphoma cells (21). Fc domain variants with selectively enhanced binding either to all the classes of activating type I FcγRs or to the activating FcγRIIIa exhibited improved anti-tumor activity, achieving tumor cell clearance over a short time period. Investigation into the FcγRIIIa-mediated mechanisms that contributed to the antibody-dependent tumor clearance revealed that FcγRIIIa-expressing macrophages were the dominant effector cell population that mediated the anti-tumor effect, possibly through phagocytic processes (21).
Similarly, the importance of activating type I FcγRs in the in vivo activity of anti-CD20 antibodies is further highlighted by the recently approved anti-CD20 monoclonal antibody, obinutuzumab (27). This antibody exhibits the same antigen specificity as rituximab; however, it displays improved binding to the activating FcγRIIIa through engineering of the Fc domain-associated N-linked glycan (23, 26). Comparison of the therapeutic activity of this Fc-optimized antibody with the parental, unmodified one (rituximab) in chronic lymphocytic leukemia patients revealed that obinutuzumab extended progression-free survival for over 10 months compared to rituximab (27). These findings provide a great example on how the in vivo efficacy of a therapeutic antibody can be effectively improved through the study of the underlying mechanisms that govern Fc-FcγR interactions.
FcγR-mediated pathways in active immunization
As discussed in the previous sections, engagement of type I and type II FcγRs by IgG immune complexes can have diverse downstream consequences that initiate a number of immunomodulatory effector functions. Such functions not only provide immediate cytotoxicity and clearance of IgG-coated targets, but have also the capacity to modulate the immune responses through a range of effects on the various leukocyte types, including the differentiation and activation of antigen-presenting cells (APCs), cellular activation and release of chemokines and cytokines by innate effector leukocytes, enhanced antigen uptake and presentation by APCs, modulation of T cell activation by APCs, and regulation of their entry into the germinal centers, modulation of affinity maturation by FcγR-bound immune complexes retained on follicular dendritic cells, and finally plasma cell survival and regulation of antibody production (4).
These functions highlight the potential of FcγR-mediated pathways to modulate the adaptive immune response and have been exploited in immunization strategies, which employ immune complexes consisting of antigen-specific antibodies in combination with antigen to enhance the host immune responses against infections agents and tumor antigens. Indeed, the concept of IgG-mediated enhancement of T-cell and antibody responses can be traced back to as early as the end of the 19th century in immunization studies against diphtheria (135). Additionally, substantial evidence over the course of years clearly suggests that immunization with IgG immune complexes greatly enhances the immunogenicity of an antigen and improves host immune responses against a number of microbes, including Venezuela equine encephalitis virus, Newcastle disease virus, infectious bursal disease virus and HIV-1 (136–142). Indirect evidence supporting a role for Fc-FcγR interactions in modulating adaptive immune responses has come from passive administration of therapeutic antibodies to humans and non-human primates for the treatment of neoplastic and infectious diseases. For example, the treatment of non-Hodgkin lymphoma patients with anti-CD20 antibodies often results in the generation of a ‘vaccinal’ response such that relapsed patients who are re-treated with the same anti-tumor antibody show rapid and enhanced responses when compared to their response to initial therapy (143, 144). The basis for this vaccinal effect has been attributed to the development of robust anti-tumor CD8 responses in these patients, induced by the generation of immune complexes between the passively administered anti-CD20 antibody and the tumor cells (143). In another example, two independent studies using non-human primates demonstrated the contribution of passively administered anti-HIV-1 neutralizing antibody to the subsequent development of a host antiviral response with protective and sustained activity (145, 146). Administration of the neutralizing anti-HIV-1 antibody b12 in infected animals resulted in the modulation of their B cell responses and accelerated the production of neutralizing antibodies. Hyperimmune sera from these animals exhibited potent activity against SHIV and SIV infection when passively administered into naïve animals (145, 146). Similar augmentation in B cell responses was also observed following administration of anti-HIV-1 broadly neutralizing antibodies in chronically SHIV-infected rhesus monkeys (125).
Although these studies strongly suggest that active immunization with IgG immune complexes results in enhanced antigen immunogenicity and augmented humoral and cellular immune responses, the precise mechanisms of this phenomenon are poorly characterized. Several potential regulatory pathways have been suggested that could participate in the FcγR-mediated enhancement of adaptive immunity. Dendritic cells, follicular dendritic cells and macrophages are the dominant immune cell populations that could influence antibody responses through antigen presentation and activation of lymphoid cells. Activating type I FcγRs actively participate in the uptake of IgG immune complexes through endocytic and phagocytic processes (59, 147). Internalized immune complexes are efficiently processed and presented on MHC class I and class II molecules by dendritic cells, leading consequently to enhanced CD4+ and CD8+ T cell-mediated responses (4, 148)(Figure 3). Additionally, engagement of activating type I FcγRs expressed on the surface of dendritic cells induces cell maturation, upregulation of MHC and co-stimulatory molecules, and consequently enhanced antigen presentation (148–150). Likewise, the function of the activating type I FcγRs is balanced by the inhibitory FcγRIIb, as genetic deletion or blockade of FcγRIIb leads to spontaneous DC maturation and influences macrophage polarization (149, 151, 152). Studies using dendritic cells from Fcgr2b−/− mice, revealed a critical role for FcγRIIb in controlling IgG-mediated maturation of dendritic cells (149). Whereas IgG immune complexes are efficiently captured by dendritic cells, they fail to induce DC maturation without additional stimulatory signals (e.g. TLR signaling), as co-engagement of FcγRIIb sufficiently blocks signaling from the activating type I FcγRs (149). The potential of activating FcγR engagement on dendritic cells to induce T cell responses has been recently explored using FcγR-humanized mice in a model of human CD20+ lymphoma (21). Under homeostatic conditions, human dendritic cells (e.g. monocyte-derived dendritic cells) express only one class of activating type I FcγR, FcγRIIa, and its activity is controlled by the inhibitory FcγRIIb (18). In this study, investigation into the mechanisms underlying the induction of T cell responses following treatment with anti-CD20 antibodies revealed an absolute requirement for FcγRIIa expression by dendritic cells in the generation of T cell responses against CD20 (21). Indeed, Fc engineered variants of anti-CD20 antibody with enhanced affinity for FcγRIIa exhibited improved IgG-mediated T cell responses, by increasing the threshold for FcγRIIb inhibition (21). These studies strongly suggest that balancing activating and inhibitory type I FcγRs on antigen presenting cells, like dendritic cells is a key regulatory process that greatly influences subsequent immune responses (Figure 3).
Figure 3: The role of FcγR-mediated pathways in dendritic cell function.
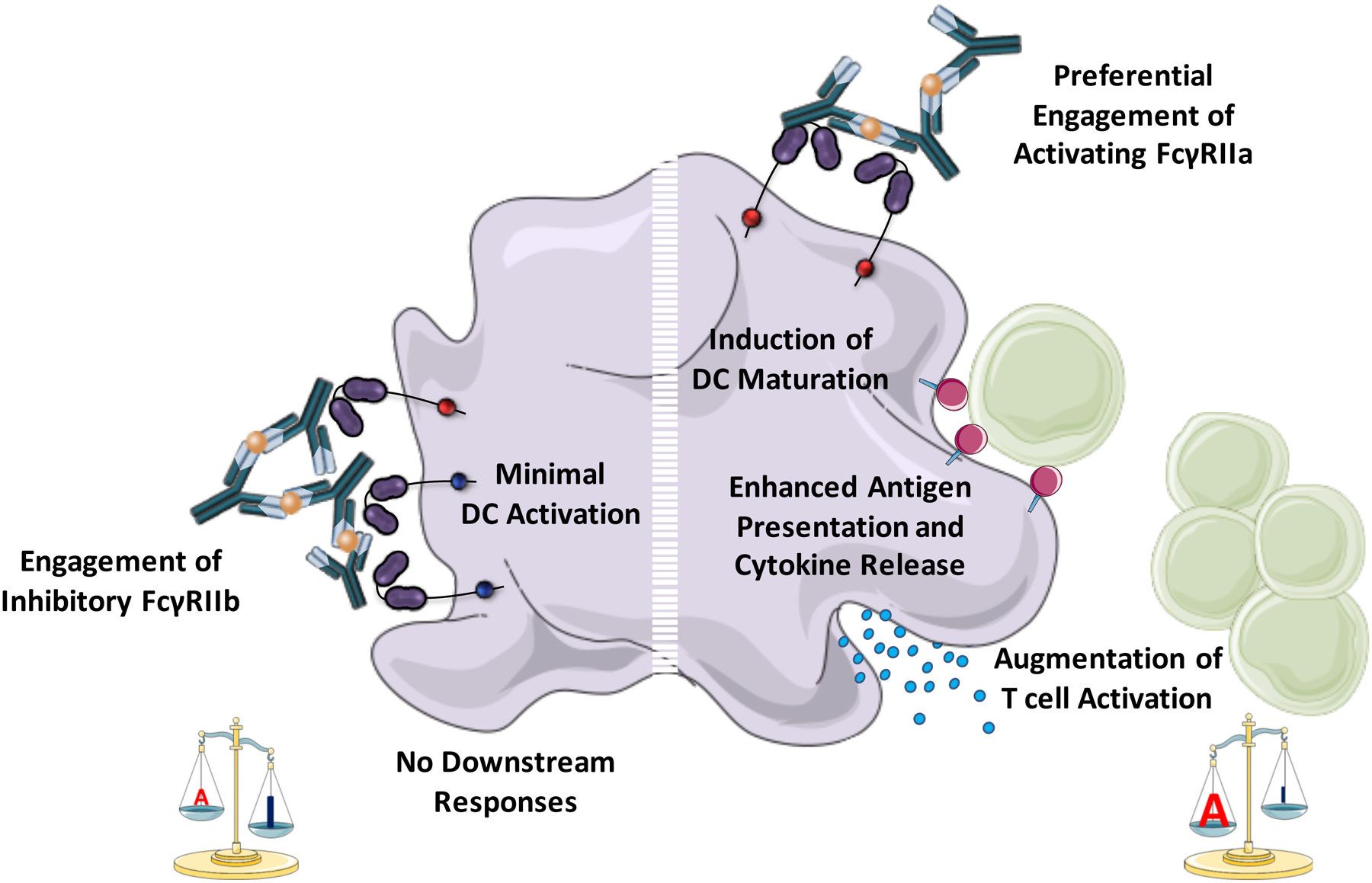
Human dendritic cells (DC) express two classes of type I FcγRs: the activating FcγRIIa, and the inhibitory FcγRIIb. Balancing the activity of these receptors is critical for DC function and for the regulation of T cell activation. FcγR engagement by IgG immune complexes fail to stimulate DC activation, as any pro-inflammatory signals initiated by FcγRIIa are counterbalanced by FcγRIIb. However, preferential engagement of the activating FcγRIIa (either through Fc domain engineering or FcγRIIb deletion or blockade) induces DC maturation and the upregulation of MHC and co-stimulatory molecules, leading to enhanced antigen presentation, and augmented T cell activation.
FcγR engagement on B cells by IgG immune complexes also induces a number of immunomodulatory processes with the potential to influence B cell selection and consequently the antibody affinity and production. B cells express only the inhibitory FcγRIIb throughout development, and not any other type I FcγRs. Despite the lack of activating type I FcγRs to counterbalance the activity of FcγRIIb, FcγRIIb engagement induces differential functional responses on B cells and several studies have highlighted its key regulatory role in shaping the antibody response and maintaining tolerance (78). Exclusive engagement of B cell FcγRIIb induces pro-apoptotic signals, whereas co-ligation of FcγRIIb with the B cell receptor attenuates these signals, leading to B cell survival (73, 77). This mechanism drives B cell selection, as B cells with higher affinity for the antigen are selected, whereas those with receptors exhibiting low or no affinity undergo apoptosis. Indeed, FcγRIIb-deficient mice fail to generate high affinity antibody responses, likely due to defects in B cell selection mechanisms (78). Loss of the B cell receptor expression in plasma cells leads to unopposed, pro-apoptotic signaling through FcγRIIb engagement, which represents a homeostatic regulatory mechanism to control IgG production by plasma cells (153). Engagement of FcγRIIb (human or mouse) on plasma cells by IgG immune complexes during an ongoing immune response acts as a negative feedback mechanism to prevent uncontrolled IgG production.
Apart from FcγRIIb, B cells also express the type II FcγR, CD23 at variable levels during B cell maturation, which is also part of a novel regulatory pathway that modulates B cell activation and affinity maturation (32)(Figure 4). Studies using IgE immune complexes have shown that CD23 engagement is required for the IgE-mediated enhancement of antibody and T cell responses (91). Likewise, CD23 contributes to the capture of IgE immune complexes by B cells and to their transfer to CD11c+ antigen presenting cells, leading to enhanced CD4+ T cell immunity (92). Given the capacity of CD23 to also interact with sialylated IgG Fc (3), a similar mechanism likely modulates immune responses through CD23 engagement by sialylated IgG immune complexes. Indeed, in a recent study, a key role for sialylated IgG Fc-CD23 interactions has been presented with great implications for the development of novel immunization strategies (32). Interaction of sialylated IgG immune complexes with CD23 upregulates FcγRIIb expression on B cells, which in turn leads to increased threshold of B cell selection. Using a model of influenza hemagglutinin (HA) immunization, vaccination with sialylated Fc anti-HA immune complexes induced higher affinity anti-HA IgG responses, with broadly neutralizing, in vivo protective activity (32)(Figure 4). By contrast, immunization of CD23−/− mice or with asialylated anti-HA immune complexes failed to generate high affinity anti-HA antibody responses, supporting thereby a key role for the sialylated IgG Fc-CD23-FcγRIIb pathway in modulating B cell selection and antibody affinity maturation (32).
Figure 4: Regulation of antibody responses through a CD23-FcγRIIb pathway.
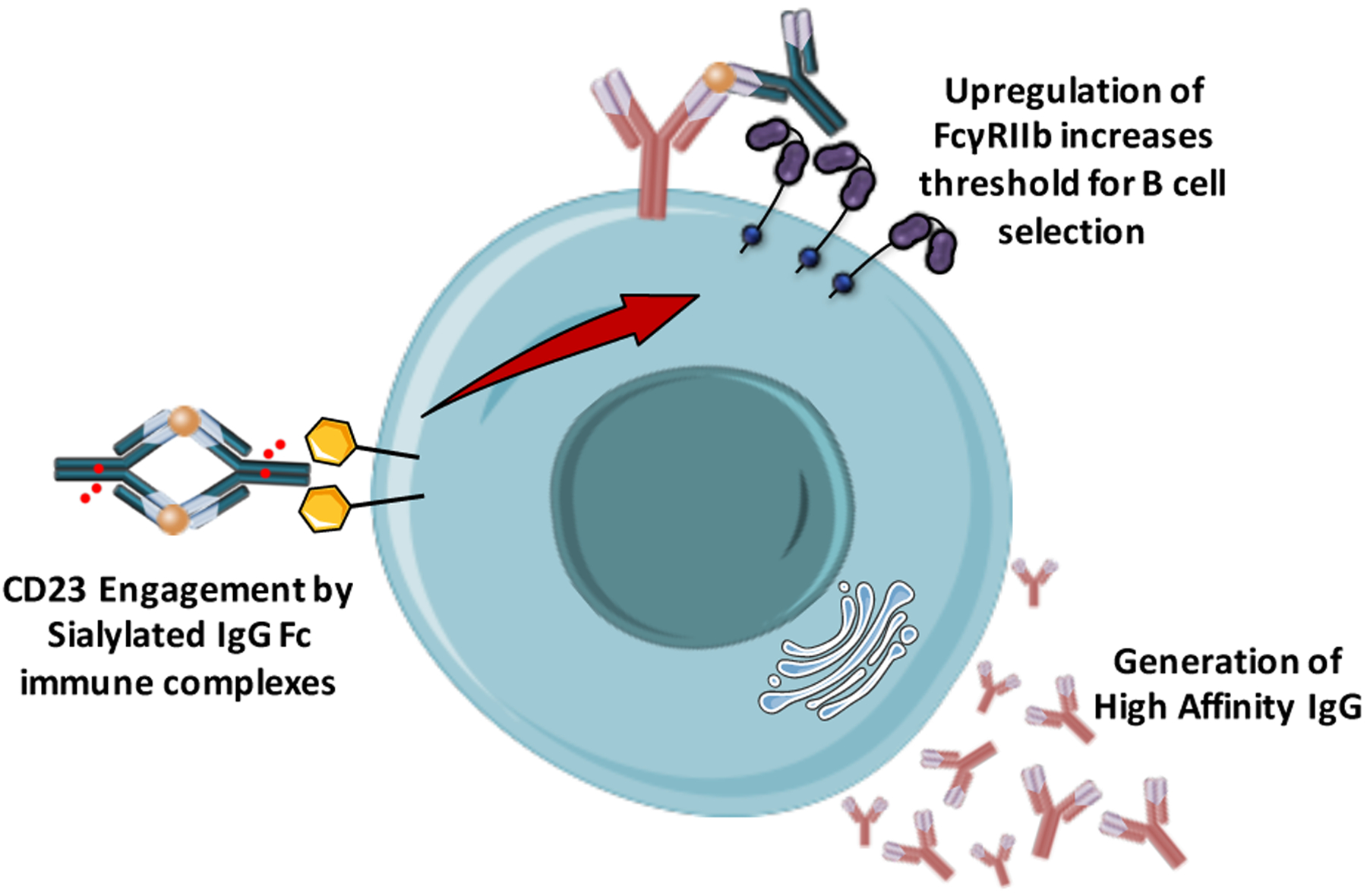
Sialylated IgG immune complexes interact with CD23 expressed by B cells, leading to the upregulation of FcγRIIb. In turn, increased FcγRIIb expression raises the threshold for B cell selection and B cells with higher affinity B cell receptor are selected over those with lower or no affinity. This process eventually leads to the generation of higher affinity antibody responses with potent in vivo activity.
Concluding Remarks
The immunomodulatory effects of the IgG Fc domain interactions with type I and type II FcγRs have the potential to dramatically influence the outcome of IgG-mediated inflammation and immunity. In a process primarily determined by the intrinsic flexibility of the IgG Fc domain that is tightly regulated by the Fc-associated glycan structure and composition, engagement of the different types of FcγRs modulates the activity of several distinct regulatory pathways that mediate Fc effector functions.
The development and clinical use of passive immunization strategies against several infectious and neoplastic diseases support a dominant role for FcγR-mediated pathways in the in vivo protective activity of antibody-based therapeutics. Manipulation of Fc-FcγR interactions through Fc domain engineering has already lead to the development of a new generation of monoclonal antibodies with improved in vivo efficacy through augmented Fc effector activity. Likewise, a number of studies strongly suggest a significant role for FcγR engagement by IgG immune complexes in the modulation of adaptive immune responses. Dissection of the precise mechanisms that drive IgG-mediated enhancement of cellular and humoral immunity would lead to the development of novel active immunization approaches that rely on the capacity of type I and type II FcγRs to regulate immune responses.
Acknowledgements
We wish to thank and acknowledge the Bill and Melinda Gates Foundation and NIH for supporting our research endeavors. SB is an amfAR Mathilde Krim Fellow in Basic Biomedical Research.
Footnotes
The authors have no conflicting financial interests.
References
- 1.Anthony RM, Wermeling F, Ravetch JV. Novel roles for the IgG Fc glycan. Ann N Y Acad Sci.2012;1253:170–180. [DOI] [PubMed] [Google Scholar]
- 2.Borrok MJ, Jung ST, Kang TH, Monzingo AF, Georgiou G. Revisiting the role of glycosylation in the structure of human IgG Fc. ACS Chem Biol.2012;7:1596–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sondermann P, Pincetic A, Maamary J, Lammens K, Ravetch JV. General mechanism for modulating immunoglobulin effector function. Proc Natl Acad Sci U S A.2013;110:9868–9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pincetic A, et al. Type I and type II Fc receptors regulate innate and adaptive immunity. Nat Immunol.2014;15:707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahmed AA, et al. Structural characterization of anti-inflammatory immunoglobulin G Fc proteins. J Mol Biol.2014;426:3166–3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ravetch JV, Bolland S. IgG Fc receptors. Annu Rev Immunol.2001;19:275–290. [DOI] [PubMed] [Google Scholar]
- 7.Narciso JE, et al. Analysis of the antibody structure based on high-resolution crystallographic studies. N Biotechnol.2011;28:435–447. [DOI] [PubMed] [Google Scholar]
- 8.Krapp S, Mimura Y, Jefferis R, Huber R, Sondermann P. Structural analysis of human IgG-Fc glycoforms reveals a correlation between glycosylation and structural integrity. J Mol Biol.2003;325:979–989. [DOI] [PubMed] [Google Scholar]
- 9.Teplyakov A, Zhao Y, Malia TJ, Obmolova G, Gilliland GL. IgG2 Fc structure and the dynamic features of the IgG CH2-CH3 interface. Mol Immunol.2013;56:131–139. [DOI] [PubMed] [Google Scholar]
- 10.Lux A, Nimmerjahn F. Impact of differential glycosylation on IgG activity. Adv Exp Med Biol.2011;780:113–124. [DOI] [PubMed] [Google Scholar]
- 11.Albert H, Collin M, Dudziak D, Ravetch JV, Nimmerjahn F. In vivo enzymatic modulation of IgG glycosylation inhibits autoimmune disease in an IgG subclass-dependent manner. Proc Natl Acad Sci U S A.2008;105:15005–15009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nimmerjahn F, Ravetch JV. Divergent immunoglobulin g subclass activity through selective Fc receptor binding. Science.2005;310:1510–1512. [DOI] [PubMed] [Google Scholar]
- 13.Nimmerjahn F, Anthony RM, Ravetch JV. Agalactosylated IgG antibodies depend on cellular Fc receptors for in vivo activity. Proc Natl Acad Sci U S A.2007;104:8433–8437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shields RL, et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J Biol Chem.2001;276:6591–6604. [DOI] [PubMed] [Google Scholar]
- 15.Sondermann P, Huber R, Oosthuizen V, Jacob U. The 3.2-A crystal structure of the human IgG1 Fc fragment-Fc gammaRIII complex. Nature.2000;406:267–273. [DOI] [PubMed] [Google Scholar]
- 16.Horton HM, et al. Fc-engineered anti-CD40 antibody enhances multiple effector functions and exhibits potent in vitro and in vivo antitumor activity against hematologic malignancies. Blood.2010;116:3004–3012. [DOI] [PubMed] [Google Scholar]
- 17.Richards JO, Karki S, Lazar GA, Chen H, Dang W, Desjarlais JR. Optimization of antibody binding to FcgammaRIIa enhances macrophage phagocytosis of tumor cells. Mol Cancer Ther.2008;7:2517–2527. [DOI] [PubMed] [Google Scholar]
- 18.Smith P, DiLillo DJ, Bournazos S, Li F, Ravetch JV. Mouse model recapitulating human Fcgamma receptor structural and functional diversity. Proc Natl Acad Sci U S A.2012;109:6181–6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bournazos S, Klein F, Pietzsch J, Seaman MS, Nussenzweig MC, Ravetch JV. Broadly Neutralizing Anti-HIV-1 Antibodies Require Fc Effector Functions for In Vivo Activity. Cell.2014;158:1243–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bournazos S, Chow SK, Abboud N, Casadevall A, Ravetch JV. Human IgG Fc domain engineering enhances antitoxin neutralizing antibody activity. J Clin Invest.2014;124:725–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DiLillo DJ, Ravetch JV. Differential Fc-Receptor Engagement Drives an Anti-tumor Vaccinal Effect. Cell.2015;161:1035–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li F, Ravetch JV. Inhibitory Fcgamma receptor engagement drives adjuvant and anti-tumor activities of agonistic CD40 antibodies. Science.2011;333:1030–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferrara C, et al. Unique carbohydrate-carbohydrate interactions are required for high affinity binding between FcgammaRIII and antibodies lacking core fucose. Proc Natl Acad Sci U S A.2011;108:12669–12674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shinkawa T, et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J Biol Chem.2003;278:3466–3473. [DOI] [PubMed] [Google Scholar]
- 25.Natsume A, Shimizu-Yokoyama Y, Satoh M, Shitara K, Niwa R. Engineered anti-CD20 antibodies with enhanced complement-activating capacity mediate potent anti-lymphoma activity. Cancer Sci.2009;100:2411–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shields RL, et al. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J Biol Chem.2002;277:26733–26740. [DOI] [PubMed] [Google Scholar]
- 27.Goede V, et al. Obinutuzumab plus chlorambucil in patients with CLL and coexisting conditions. N Engl J Med.2014;370:1101–1110. [DOI] [PubMed] [Google Scholar]
- 28.Hiatt A, et al. Glycan variants of a respiratory syncytial virus antibody with enhanced effector function and in vivo efficacy. Proc Natl Acad Sci U S A.2014;111:5992–5997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anthony RM, Nimmerjahn F, Ashline DJ, Reinhold VN, Paulson JC, Ravetch JV. Recapitulation of IVIG anti-inflammatory activity with a recombinant IgG Fc. Science.2008;320:373–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaneko Y, Nimmerjahn F, Ravetch JV. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science.2006;313:670–673. [DOI] [PubMed] [Google Scholar]
- 31.Selman MH, et al. Changes in antigen-specific IgG1 Fc N-glycosylation upon influenza and tetanus vaccination. Mol Cell Proteomics.2012;11:M111.014563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang T, et al. Anti-HA Fc glycoforms determine influenza vaccine efficacy through Type II Fc receptor signaling. Cell.2015;In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van de Geijn FE, et al. Immunoglobulin G galactosylation and sialylation are associated with pregnancy-induced improvement of rheumatoid arthritis and the postpartum flare: results from a large prospective cohort study. Arthritis Res Ther.2009;11:R193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Espy C, et al. Sialylation levels of anti-proteinase 3 antibodies are associated with the activity of granulomatosis with polyangiitis (Wegener’s). Arthritis Rheum.2011;63:2105–2115. [DOI] [PubMed] [Google Scholar]
- 35.Scherer HU, et al. Glycan profiling of anti-citrullinated protein antibodies isolated from human serum and synovial fluid. Arthritis Rheum.2010;62:1620–1629. [DOI] [PubMed] [Google Scholar]
- 36.Tomana M, Schrohenloher RE, Koopman WJ, Alarcón GS, Paul WA. Abnormal glycosylation of serum IgG from patients with chronic inflammatory diseases. Arthritis Rheum.1988;31:333–338. [DOI] [PubMed] [Google Scholar]
- 37.Bournazos S, Woof JM, Hart SP, Dransfield I. Functional and clinical consequences of Fc receptor polymorphic and copy number variants. Clin Exp Immunol.2009;157:244–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nimmerjahn F, Ravetch JV. Fcgamma receptors: old friends and new family members. Immunity.2006;24:19–28. [DOI] [PubMed] [Google Scholar]
- 39.Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol.2008;8:34–47. [DOI] [PubMed] [Google Scholar]
- 40.Qiu WQ, de Bruin D, Brownstein BH, Pearse R, Ravetch JV. Organization of the human and mouse low-affinity Fc gamma R genes: duplication and recombination. Science.1990;248:732–735. [DOI] [PubMed] [Google Scholar]
- 41.Su K, Wu J, Edberg JC, McKenzie SE, Kimberly RP. Genomic organization of classical human low-affinity Fcgamma receptor genes. Genes Immun.2002;3 Suppl 1:S51–56. [DOI] [PubMed] [Google Scholar]
- 42.Kiyoshi M, et al. Structural basis for binding of human IgG1 to its high-affinity human receptor FcγRI. Nat Commun.2015;6:6866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Y, et al. Increased expression of FcgammaRI/CD64 on circulating monocytes parallels ongoing inflammation and nephritis in lupus. Arthritis Res Ther.2009;11:R6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uciechowski P, Schwarz M, Gessner JE, Schmidt RE, Resch K, Radeke HH. IFN-gamma induces the high-affinity Fc receptor I for IgG (CD64) on human glomerular mesangial cells. Eur J Immunol.1998;28:2928–2935. [DOI] [PubMed] [Google Scholar]
- 45.Duchemin AM, Ernst LK, Anderson CL. Clustering of the high affinity Fc receptor for immunoglobulin G (Fc gamma RI) results in phosphorylation of its associated gamma-chain. J Biol Chem.1994;269:12111–12117. [PubMed] [Google Scholar]
- 46.Indik ZK, et al. The high affinity Fc gamma receptor (CD64) induces phagocytosis in the absence of its cytoplasmic domain: the gamma subunit of Fc gamma RIIIA imparts phagocytic function to Fc gamma RI. Exp Hematol.1994;22:599–606. [PubMed] [Google Scholar]
- 47.Ernst LK, Duchemin AM, Anderson CL. Association of the high-affinity receptor for IgG (Fc gamma RI) with the gamma subunit of the IgE receptor. Proc Natl Acad Sci U S A.1993;90:6023–6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ernst LK, Duchemin AM, Miller KL, Anderson CL. Molecular characterization of six variant Fcgamma receptor class I (CD64) transcripts. Mol Immunol.1998;35:943–954. [DOI] [PubMed] [Google Scholar]
- 49.Ernst LK, van de Winkel JG, Chiu IM, Anderson CL. Three genes for the human high affinity Fc receptor for IgG (Fc gamma RI) encode four distinct transcription products. J Biol Chem.1992;267:15692–15700. [PubMed] [Google Scholar]
- 50.Sondermann P, Kaiser J, Jacob U. Molecular basis for immune complex recognition: a comparison of Fc-receptor structures. J Mol Biol.2001;309:737–749. [DOI] [PubMed] [Google Scholar]
- 51.Lewis VA, Koch T, Plutner H, Mellman I. A complementary DNA clone for a macrophage-lymphocyte Fc receptor. Nature.1986;324:372–375. [DOI] [PubMed] [Google Scholar]
- 52.Brooks DG, Qiu WQ, Luster AD, Ravetch JV. Structure and expression of human IgG FcRII(CD32). Functional heterogeneity is encoded by the alternatively spliced products of multiple genes. J Exp Med.1989;170:1369–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ernst LK, Metes D, Herberman RB, Morel PA. Allelic polymorphisms in the FcgammaRIIC gene can influence its function on normal human natural killer cells. J Mol Med (Berl).2002;80:248–257. [DOI] [PubMed] [Google Scholar]
- 54.Metes D, Ernst LK, Chambers WH, Sulica A, Herberman RB, Morel PA. Expression of functional CD32 molecules on human NK cells is determined by an allelic polymorphism of the FcgammaRIIC gene. Blood.1998;91:2369–2380. [PubMed] [Google Scholar]
- 55.Hibbs ML, et al. Mechanisms for regulating expression of membrane isoforms of Fc gamma RIII (CD16). Science.1989;246:1608–1611. [DOI] [PubMed] [Google Scholar]
- 56.Selvaraj P, Carpén O, Hibbs ML, Springer TA. Natural killer cell and granulocyte Fc gamma receptor III (CD16) differ in membrane anchor and signal transduction. J Immunol.1989;143:3283–3288. [PubMed] [Google Scholar]
- 57.Zhou MJ, Brown EJ. CR3 (Mac-1, alpha M beta 2, CD11b/CD18) and Fc gamma RIII cooperate in generation of a neutrophil respiratory burst: requirement for Fc gamma RIII and tyrosine phosphorylation. J Cell Biol.1994;125:1407–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Unkeless JC, Shen Z, Lin CW, DeBeus E. Function of human Fc gamma RIIA and Fc gamma RIIIB. Semin Immunol.1995;7:37–44. [DOI] [PubMed] [Google Scholar]
- 59.Swanson JA, Hoppe AD. The coordination of signaling during Fc receptor-mediated phagocytosis. J Leukoc Biol.2004;76:1093–1103. [DOI] [PubMed] [Google Scholar]
- 60.Amigorena S, Salamero J, Davoust J, Fridman WH, Bonnerot C. Tyrosine-containing motif that transduces cell activation signals also determines internalization and antigen presentation via type III receptors for IgG. Nature.1992;358:337–341. [DOI] [PubMed] [Google Scholar]
- 61.Jouvin MH, Adamczewski M, Numerof R, Letourneur O, Vallé A, Kinet JP. Differential control of the tyrosine kinases Lyn and Syk by the two signaling chains of the high affinity immunoglobulin E receptor. J Biol Chem.1994;269:5918–5925. [PubMed] [Google Scholar]
- 62.Durden DL, Liu YB. Protein-tyrosine kinase p72syk in Fc gamma RI receptor signaling. Blood.1994;84:2102–2108. [PubMed] [Google Scholar]
- 63.Durden DL, Kim HM, Calore B, Liu Y. The Fc gamma RI receptor signals through the activation of hck and MAP kinase. J Immunol.1995;154:4039–4047. [PubMed] [Google Scholar]
- 64.Eiseman E, Bolen JB. Engagement of the high-affinity IgE receptor activates src protein-related tyrosine kinases. Nature.1992;355:78–80. [DOI] [PubMed] [Google Scholar]
- 65.Pignata C, Prasad KV, Robertson MJ, Levine H, Rudd CE, Ritz J. Fc gamma RIIIA-mediated signaling involves src-family lck in human natural killer cells. J Immunol.1993;151:6794–6800. [PubMed] [Google Scholar]
- 66.Hamawy MM, Minoguchi K, Swaim WD, Mergenhagen SE, Siraganian RP. A 77-kDa protein associates with pp125FAK in mast cells and becomes tyrosine-phosphorylated by high affinity IgE receptor aggregation. J Biol Chem.1995;270:12305–12309. [DOI] [PubMed] [Google Scholar]
- 67.García-García E, Sánchez-Mejorada G, Rosales C. Phosphatidylinositol 3-kinase and ERK are required for NF-kappaB activation but not for phagocytosis. J Leukoc Biol.2001;70:649–658. [PubMed] [Google Scholar]
- 68.Sánchez-Mejorada G, Rosales C. Fcgamma receptor-mediated mitogen-activated protein kinase activation in monocytes is independent of Ras. J Biol Chem.1998;273:27610–27619. [DOI] [PubMed] [Google Scholar]
- 69.Kanakaraj P, Duckworth B, Azzoni L, Kamoun M, Cantley LC, Perussia B. Phosphatidylinositol-3 kinase activation induced upon Fc gamma RIIIA-ligand interaction. J Exp Med.1994;179:551–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ninomiya N, et al. Involvement of phosphatidylinositol 3-kinase in Fc gamma receptor signaling. J Biol Chem.1994;269:22732–22737. [PubMed] [Google Scholar]
- 71.Bracke M, Coffer PJ, Lammers JW, Koenderman L. Analysis of signal transduction pathways regulating cytokine-mediated Fc receptor activation on human eosinophils. J Immunol.1998;161:6768–6774. [PubMed] [Google Scholar]
- 72.Aramburu J, Azzoni L, Rao A, Perussia B. Activation and expression of the nuclear factors of activated T cells, NFATp and NFATc, in human natural killer cells: regulation upon CD16 ligand binding. J Exp Med.1995;182:801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pearse RN, Kawabe T, Bolland S, Guinamard R, Kurosaki T, Ravetch JV. SHIP recruitment attenuates Fc gamma RIIB-induced B cell apoptosis. Immunity.1999;10:753–760. [DOI] [PubMed] [Google Scholar]
- 74.Amigorena S, et al. Cytoplasmic domain heterogeneity and functions of IgG Fc receptors in B lymphocytes. Science.1992;256:1808–1812. [DOI] [PubMed] [Google Scholar]
- 75.Muta T, Kurosaki T, Misulovin Z, Sanchez M, Nussenzweig MC, Ravetch JV. A 13-amino-acid motif in the cytoplasmic domain of Fc gamma RIIB modulates B-cell receptor signalling. Nature.1994;368:70–73. [DOI] [PubMed] [Google Scholar]
- 76.Ono M, Bolland S, Tempst P, Ravetch JV. Role of the inositol phosphatase SHIP in negative regulation of the immune system by the receptor Fc(gamma)RIIB. Nature.1996;383:263–266. [DOI] [PubMed] [Google Scholar]
- 77.Ono M, Okada H, Bolland S, Yanagi S, Kurosaki T, Ravetch JV. Deletion of SHIP or SHP-1 reveals two distinct pathways for inhibitory signaling. Cell.1997;90:293–301. [DOI] [PubMed] [Google Scholar]
- 78.Bolland S, Ravetch JV. Spontaneous autoimmune disease in Fc(gamma)RIIB-deficient mice results from strain-specific epistasis. Immunity.2000;13:277–285. [DOI] [PubMed] [Google Scholar]
- 79.Edberg JC, Kimberly RP. Modulation of Fc gamma and complement receptor function by the glycosyl-phosphatidylinositol-anchored form of Fc gamma RIII. J Immunol.1994;152:5826–5835. [PubMed] [Google Scholar]
- 80.Jellusova J, Nitschke L. Regulation of B cell functions by the sialic acid-binding receptors siglec-G and CD22. Front Immunol.2011;2:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schwab I, Seeling M, Biburger M, Aschermann S, Nitschke L, Nimmerjahn F. B cells and CD22 are dispensable for the immediate antiinflammatory activity of intravenous immunoglobulins in vivo. Eur J Immunol.2012;42:3302–3309. [DOI] [PubMed] [Google Scholar]
- 82.Böhm S, Kao D, Nimmerjahn F. Sweet and sour: the role of glycosylation for the anti-inflammatory activity of immunoglobulin G. Curr Top Microbiol Immunol.2014;382:393–417. [DOI] [PubMed] [Google Scholar]
- 83.Schwab I, Nimmerjahn F. Intravenous immunoglobulin therapy: how does IgG modulate the immune system? Nat Rev Immunol.2013;13:176–189. [DOI] [PubMed] [Google Scholar]
- 84.Anthony RM, Wermeling F, Karlsson MC, Ravetch JV. Identification of a receptor required for the anti-inflammatory activity of IVIG. Proc Natl Acad Sci U S A.2008;105:19571–19578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Soilleux EJ. DC-SIGN (dendritic cell-specific ICAM-grabbing non-integrin) and DC-SIGN-related (DC-SIGNR): friend or foe? Clin Sci (Lond).2003;104:437–446. [DOI] [PubMed] [Google Scholar]
- 86.Anthony RM, Kobayashi T, Wermeling F, Ravetch JV. Intravenous gammaglobulin suppresses inflammation through a novel T(H)2 pathway. Nature.2011;475:110–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fiebiger BM, Maamary J, Pincetic A, Ravetch JV. Protection in antibody- and T cell-mediated autoimmune diseases by antiinflammatory IgG Fcs requires type II FcRs. Proc Natl Acad Sci U S A.2015;112:E2385–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yokota A, et al. Two species of human Fc epsilon receptor II (Fc epsilon RII/CD23): tissue-specific and IL-4-specific regulation of gene expression. Cell.1988;55:611–618. [DOI] [PubMed] [Google Scholar]
- 89.Borthakur S, Andrejeva G, McDonnell JM. Basis of the intrinsic flexibility of the Cε3 domain of IgE. Biochemistry.2011;50:4608–4614. [DOI] [PubMed] [Google Scholar]
- 90.Dhaliwal B, et al. Crystal structure of IgE bound to its B-cell receptor CD23 reveals a mechanism of reciprocal allosteric inhibition with high affinity receptor FcεRI. Proc Natl Acad Sci U S A.2012;109:12686–12691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fujiwara H, et al. The absence of IgE antibody-mediated augmentation of immune responses in CD23-deficient mice. Proc Natl Acad Sci U S A.1994;91:6835–6839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Henningsson F, et al. IgE-mediated enhancement of CD4+ T cell responses in mice requires antigen presentation by CD11c+ cells and not by B cells. PLoS One.2011;6:e21760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.von Behring E, Kitasato S. Ueber das Zustandekommen der Diphtherie-Immunitat und der Tetanus-Immunitat bei Thieren. Dt Med Wochenschr.1890;16:1113–1114. [Google Scholar]
- 94.von Behring E Untersuchungen ueber das Zustandekommen der Diphtherie-Immunitat bei Thieren. Dt Med Wochenschr.1890;16:1145–1148. [Google Scholar]
- 95.Chan AC, Carter PJ. Therapeutic antibodies for autoimmunity and inflammation. Nat Rev Immunol.2010;10:301–316. [DOI] [PubMed] [Google Scholar]
- 96.Nimmerjahn F, Ravetch JV. Antibodies, Fc receptors and cancer. Curr Opin Immunol.2007;19:239–245. [DOI] [PubMed] [Google Scholar]
- 97.Page DB, Postow MA, Callahan MK, Allison JP, Wolchok JD. Immune modulation in cancer with antibodies. Annu Rev Med.2014;65:185–202. [DOI] [PubMed] [Google Scholar]
- 98.Yoshihara S, et al. Effect of palivizumab prophylaxis on subsequent recurrent wheezing in preterm infants. Pediatrics.2013;132:811–818. [DOI] [PubMed] [Google Scholar]
- 99.Qiu X, et al. Sustained protection against Ebola virus infection following treatment of infected nonhuman primates with ZMAb. Sci Rep.2013;3:3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Migone TS, et al. Raxibacumab for the treatment of inhalational anthrax. N Engl J Med.2009;361:135–144. [DOI] [PubMed] [Google Scholar]
- 101.Mellor JD, Brown MP, Irving HR, Zalcberg JR, Dobrovic A. A critical review of the role of Fc gamma receptor polymorphisms in the response to monoclonal antibodies in cancer. J Hematol Oncol.2013;6:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Musolino A, et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J Clin Oncol.2008;26:1789–1796. [DOI] [PubMed] [Google Scholar]
- 103.Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol.2003;21:3940–3947. [DOI] [PubMed] [Google Scholar]
- 104.Zhang W, et al. FCGR2A and FCGR3A polymorphisms associated with clinical outcome of epidermal growth factor receptor expressing metastatic colorectal cancer patients treated with single-agent cetuximab. J Clin Oncol.2007;25:3712–3718. [DOI] [PubMed] [Google Scholar]
- 105.Cartron G, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood.2002;99:754–758. [DOI] [PubMed] [Google Scholar]
- 106.Bibeau F, et al. Impact of Fc{gamma}RIIa-Fc{gamma}RIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J Clin Oncol.2009;27:1122–1129. [DOI] [PubMed] [Google Scholar]
- 107.Abboud N, et al. A requirement for FcgammaR in antibody-mediated bacterial toxin neutralization. J Exp Med.2010;207:2395–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.DiLillo DJ, Tan GS, Palese P, Ravetch JV. Broadly neutralizing hemagglutinin stalk-specific antibodies require FcγR interactions for protection against influenza virus in vivo. Nat Med.2014;20:143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nimmerjahn F, et al. FcgammaRIV deletion reveals its central role for IgG2a and IgG2b activity in vivo. Proc Natl Acad Sci U S A.2010;107:19396–19401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Weber S, Tian H, van Rooijen N, Pirofski LA. A serotype 3 pneumococcal capsular polysaccharide-specific monoclonal antibody requires Fcγ receptor III and macrophages to mediate protection against pneumococcal pneumonia in mice. Infect Immun.2012;80:1314–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Varshney AK, Wang X, Aguilar JL, Scharff MD, Fries BC. Isotype switching increases efficacy of antibody protection against staphylococcal enterotoxin B-induced lethal shock and Staphylococcus aureus sepsis in mice. MBio.2014;5:e01007–01014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sanford JE, Lupan DM, Schlageter AM, Kozel TR. Passive immunization against Cryptococcus neoformans with an isotype-switch family of monoclonal antibodies reactive with cryptococcal polysaccharide. Infect Immun.1990;58:1919–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schlageter AM, Kozel TR. Opsonization of Cryptococcus neoformans by a family of isotype-switch variant antibodies specific for the capsular polysaccharide. Infect Immun.1990;58:1914–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hessell AJ, et al. Fc receptor but not complement binding is important in antibody protection against HIV. Nature.2007;449:101–104. [DOI] [PubMed] [Google Scholar]
- 115.Halper-Stromberg A, et al. Broadly Neutralizing Antibodies and Viral Inducers Decrease Rebound from HIV-1 Latent Reservoirs in Humanized Mice. Cell.2014;158:989–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Corti D, et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science.2011;333:850–856. [DOI] [PubMed] [Google Scholar]
- 117.Gaschen B, et al. Diversity considerations in HIV-1 vaccine selection. Science.2002;296:2354–2360. [DOI] [PubMed] [Google Scholar]
- 118.Korber B, Gaschen B, Yusim K, Thakallapally R, Kesmir C, Detours V. Evolutionary and immunological implications of contemporary HIV-1 variation. Br Med Bull.2001;58:19–42. [DOI] [PubMed] [Google Scholar]
- 119.Burton DR, et al. A Blueprint for HIV Vaccine Discovery. Cell Host Microbe.2012;12:396–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Moore PL, et al. Evolution of an HIV glycan-dependent broadly neutralizing antibody epitope through immune escape. Nat Med.2012;18:1688–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Simek MD, et al. Human immunodeficiency virus type 1 elite neutralizers: individuals with broad and potent neutralizing activity identified by using a high-throughput neutralization assay together with an analytical selection algorithm. J Virol.2009;83:7337–7348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Klein F, Mouquet H, Dosenovic P, Scheid JF, Scharf L, Nussenzweig MC. Antibodies in HIV-1 vaccine development and therapy. Science.2013;341:1199–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mascola JR, et al. Protection of macaques against vaginal transmission of a pathogenic HIV-1/SIV chimeric virus by passive infusion of neutralizing antibodies. Nat Med.2000;6:207–210. [DOI] [PubMed] [Google Scholar]
- 124.Balazs AB, Chen J, Hong CM, Rao DS, Yang L, Baltimore D. Antibody-based protection against HIV infection by vectored immunoprophylaxis. Nature.2012;481:81–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Barouch DH, et al. Therapeutic efficacy of potent neutralizing HIV-1-specific monoclonal antibodies in SHIV-infected rhesus monkeys. Nature.2013;503:224–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Shingai M, et al. Antibody-mediated immunotherapy of macaques chronically infected with SHIV suppresses viraemia. Nature.2013;503:277–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Horwitz JA, et al. HIV-1 suppression and durable control by combining single broadly neutralizing antibodies and antiretroviral drugs in humanized mice. Proc Natl Acad Sci U S A.2013;110:16538–16543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Klein F, et al. HIV therapy by a combination of broadly neutralizing antibodies in humanized mice. Nature.2012;492:118–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Caskey M, et al. Viraemia suppressed in HIV-1-infected humans by broadly neutralizing antibody 3BNC117. Nature.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Forthal DN, Landucci G, Bream J, Jacobson LP, Phan TB, Montoya B. FcgammaRIIa genotype predicts progression of HIV infection. J Immunol.2007;179:7916–7923. [DOI] [PubMed] [Google Scholar]
- 131.Pietzsch J, et al. A mouse model for HIV-1 entry. Proc Natl Acad Sci U S A.2012;109:15859–15864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hamaguchi Y, Xiu Y, Komura K, Nimmerjahn F, Tedder TF. Antibody isotype-specific engagement of Fcgamma receptors regulates B lymphocyte depletion during CD20 immunotherapy. J Exp Med.2006;203:743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Uchida J, et al. The innate mononuclear phagocyte network depletes B lymphocytes through Fc receptor-dependent mechanisms during anti-CD20 antibody immunotherapy. J Exp Med.2004;199:1659–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med.2000;6:443–446. [DOI] [PubMed] [Google Scholar]
- 135.von Behring E, Wernicke E. Ueber Immunisierung und Heilung von Versuchstieren bei der Diphterie. Z Hyg Infektioskrankheit.1892;12:10–44. [Google Scholar]
- 136.Bouige P, Iscaki S, Pillot J. Immune complexes as immunizing agents to increase the number of monoclonal antibody producing hybrids and to deviate the response to poorly immunogenic epitopes. Hybridoma.1990;9:519–526. [DOI] [PubMed] [Google Scholar]
- 137.Haddad EE, et al. Efficacy of a novel infectious bursal disease virus immune complex vaccine in broiler chickens. Avian Dis.1997;41:882–889. [PubMed] [Google Scholar]
- 138.Hioe CE, et al. The use of immune complex vaccines to enhance antibody responses against neutralizing epitopes on HIV-1 envelope gp120. Vaccine.2009;28:352–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Houston WE, Kremer RJ, Crabbs CL, Spertzel RO. Inactivated Venezuelan equine encephalomyelitis virus vaccine complexed with specific antibody: enhanced primary immune response and altered pattern of antibody class elicited. J Infect Dis.1977;135:600–610. [DOI] [PubMed] [Google Scholar]
- 140.Kumar R, Tuen M, Li H, Tse DB, Hioe CE. Improving immunogenicity of HIV-1 envelope gp120 by glycan removal and immune complex formation. Vaccine.2011;29:9064–9074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kumar R, et al. Elicitation of broadly reactive antibodies against glycan-modulated neutralizing V3 epitopes of HIV-1 by immune complex vaccines. Vaccine.2013;31:5413–5421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Pokric B, Sladic D, Juros S, Cajavec S. Application of the immune complex for immune protection against viral disease. Vaccine.1993;11:655–659. [DOI] [PubMed] [Google Scholar]
- 143.Abes R, Gelize E, Fridman WH, Teillaud JL. Long-lasting antitumor protection by anti-CD20 antibody through cellular immune response. Blood.2010;116:926–934. [DOI] [PubMed] [Google Scholar]
- 144.Hilchey SP, et al. Rituximab immunotherapy results in the induction of a lymphoma idiotype-specific T-cell response in patients with follicular lymphoma: support for a “vaccinal effect” of rituximab. Blood.2009;113:3809–3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Haigwood NL, et al. Passive immunotherapy in simian immunodeficiency virus-infected macaques accelerates the development of neutralizing antibodies. J Virol.2004;78:5983–5995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ng CT, et al. Passive neutralizing antibody controls SHIV viremia and enhances B cell responses in infant macaques. Nat Med.2010;16:1117–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Guilliams M, Bruhns P, Saeys Y, Hammad H, Lambrecht BN. The function of Fcγ receptors in dendritic cells and macrophages. Nat Rev Immunol.2014;14:94–108. [DOI] [PubMed] [Google Scholar]
- 148.Regnault A, et al. Fcgamma receptor-mediated induction of dendritic cell maturation and major histocompatibility complex class I-restricted antigen presentation after immune complex internalization. J Exp Med.1999;189:371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kalergis AM, Ravetch JV. Inducing tumor immunity through the selective engagement of activating Fcgamma receptors on dendritic cells. J Exp Med.2002;195:1653–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Diaz de Ståhl T, Heyman B. IgG2a-mediated enhancement of antibody responses is dependent on FcRgamma+ bone marrow-derived cells. Scand J Immunol.2001;54:495–500. [DOI] [PubMed] [Google Scholar]
- 151.Dhodapkar KM, et al. Selective blockade of inhibitory Fcgamma receptor enables human dendritic cell maturation with IL-12p70 production and immunity to antibody-coated tumor cells. Proc Natl Acad Sci U S A.2005;102:2910–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Desai DD, et al. Fc gamma receptor IIB on dendritic cells enforces peripheral tolerance by inhibiting effector T cell responses. J Immunol.2007;178:6217–6226. [DOI] [PubMed] [Google Scholar]
- 153.Xiang Z, et al. FcgammaRIIb controls bone marrow plasma cell persistence and apoptosis. Nat Immunol.2007;8:419–429. [DOI] [PubMed] [Google Scholar]