

cGAS controls replication dynamics and suppresses replication-associated DNA damage.
Abstract
The cyclic GMP-AMP synthase (cGAS), a sensor of cytosolic DNA, is critical for the innate immune response. Here, we show that loss of cGAS in untransformed and cancer cells results in uncontrolled DNA replication, hyperproliferation, and genomic instability. While the majority of cGAS is cytoplasmic, a fraction of cGAS associates with chromatin. cGAS interacts with replication fork proteins in a DNA binding–dependent manner, suggesting that cGAS encounters replication forks in DNA. Independent of cGAMP and STING, cGAS slows replication forks by binding to DNA in the nucleus. In the absence of cGAS, replication forks are accelerated, but fork stability is compromised. Consequently, cGAS-deficient cells are exposed to replication stress and become increasingly sensitive to radiation and chemotherapy. Thus, by acting as a decelerator of DNA replication forks, cGAS controls replication dynamics and suppresses replication-associated DNA damage, suggesting that cGAS is an attractive target for exploiting the genomic instability of cancer cells.
INTRODUCTION
Cellular DNA is normally confined within the nucleus and mitochondria in eukaryotic cells. However, under conditions such as viral infection and DNA damage, double-stranded DNA (dsDNA) accumulates in the cytosol (1). Cyclic guanosine monophosphate (GMP)–adenosine monophosphate (AMP) synthase (cGAS) catalyzes the formation of cyclic GMP-AMP (cGAMP) in response to cytosolic dsDNA and triggers an immune defense against multiple types of pathogens (2–6). It is known that cGAS stimulates the type I interferon response via the cGAMP-STING (stimulator of interferon genes) pathway (7–9). cGAMP binds and activates STING on the endoplasmic reticulum, triggering the phosphorylation of IRF3 and its nuclear entry. STING also activates nuclear factor κB (NF-κB), which functions with IRF3 to activate the transcription program that promotes innate immunity. In STING-deficient cells, such as U2OS and human embryonic kidney (HEK) 293T cells, cGAS is unable to activate type I interferon expression (10). In addition to sensing foreign DNA in the cytosol, cGAS also contributes to the surveillance of DNA damage in the nuclear genome (11). As cells progress through mitosis, chromosome fragments generated by DNA double-strand breaks may give rise to micronuclei, and rupture of micronuclei has been shown to recruit cGAS. Furthermore, the aberrant nucleolytic processing of stalled replication forks in the nucleus could also lead to an increase in cytosolic DNA, activating the cGAS-STING pathway (12). The effects of the cGAS-STING pathway on tumors are complex. On one hand, activation of the cGAS-STING pathway promotes antitumor immunity, although some cancers escape this immunity by deactivating the cGAS-STING pathway (13). Conversely, in certain contexts, the cGAS-STING pathway may promote tumorigenesis or metastasis by promoting inflammation (14). Dysregulation of the cGAS-STING pathway has also been implicated in autoimmune disorders.
Although the role of cGAS in immunity is well established, whether and how cGAS functions in other cellular processes remain elusive. While STING is often repressed in cancer, cGAS is rarely lost, raising the possibility that cGAS has a STING-independent function that may be indispensable in cancer cells. In this study, we identified a STING-independent function of cGAS in regulating DNA replication. We found that loss of cGAS accelerated cell proliferation, whereas cGAS overexpression slowed the process. cGAS deficiency endows a hyper-replicative cell state associated with genomic instability. Ablation of cGAS in a number of noncancerous and cancer cell lines increased cellular sensitivity to a variety of DNA-damaging agents, whereas cGAS overexpression conferred resistance. The ability of cGAS to bind DNA in the nucleus and interact with replication proteins is important for slowing replication forks and suppressing DNA damage sensitivity, suggesting that cGAS protects the genome by acting as a “decelerator” of DNA replication. Thus, in addition to its role as a sensor of the cytosolic DNA arising from DNA damage, cGAS is an important regulator of replication dynamics and a suppressor of genomic instability, suggesting that inhibition of cGAS in STING-deficient tumors may exploit the replication stress in cancer cells, enhancing cancer therapy.
RESULTS
cGAS slows cell proliferation through DNA binding in the nucleus
In CRISPR-Cas9–generated cGAS−/− BJ fibroblast cells in which the first exon containing ATG is targeted, we found that cGAS−/− cells displayed shorter doubling time and faster proliferation rates compared to wild-type (WT) cells (Fig. 1A and fig. S1A). Similar observations were made in cGAS−/− and WT mouse embryonic fibroblast (MEF) cells (Fig. 1B and fig. S1B). The proliferation rate of cGAS−/− U2OS cells, which do not express STING, was also increased compared to WT U2OS cells (Fig. 1C). Stable overexpression of WT cGAS in WT U2OS cells (cGAS-OE) reduced the proliferation rate (Fig. 1C), reversing the effect of cGAS loss on cell proliferation. These results demonstrate that a loss of cGAS results in accelerated cell proliferation.
Fig. 1. Nuclear localization and DNA binding activity of cGAS are required to suppress cell proliferation, independently of STING and cGAMP.
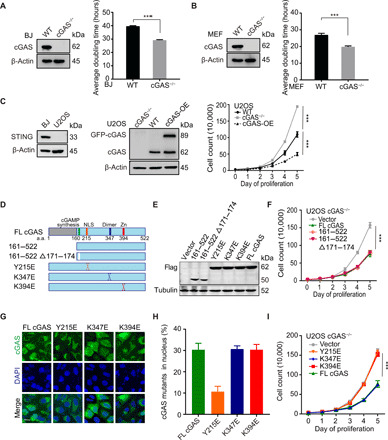
(A and B) Western blot (WB) of cGAS in WT and cGAS−/− BJ and MEF cells. Thirty-thousand cells were seeded into each well of a six-well plate. The number of BJ WT/cGAS−/− (A) and MEF WT/cGAS−/− (B) cells was counted on the indicated day to determine the average population doubling time. (C) WB of STING in BJ and U2OS WT cells and cGAS in WT, cGAS−/−, and green fluorescent protein (GFP)–cGAS stably overexpressed WT U2OS cells (cGAS-OE). Proliferation rates of WT, cGAS−/−, and cGAS-OE U2OS cells were determined as described in (A) and (B). (D and E) Schematic structure of cGAS truncated proteins. WB of flag-tagged cGAS deletions and mutants in U2OS cGAS−/− cells is shown. (F) Thirty-thousand cells were seeded into each well of a six-well plate. The number of U2OS cGAS−/− cells transfected with flag-tagged deletion and flag-vector plasmids was counted every day until reaching high density. (G) U2OS cGAS−/− cells were transfected with flag-tagged cGAS point mutant plasmids and tested by immunostaining (scale bar, 10 μm). (H) Quantification of percentage of cells with nuclear cGAS (n = 50). Three independent experiments were done. (I) Thirty-thousand cells were seeded into each well of a six-well plate. The number of U2OS cGAS−/− cells transfected with flag-tagged full length (FL), Y215E, K347E, K394E, and flag-vector plasmids was counted every day until reaching high density. Note that the same vector control is used in (F) and (I). ***P < 0.001, Mann-Whitney test.
Human cGAS is composed of an unstructured, poorly conserved N terminus and a highly conserved C terminus (Fig. 1, D and E). When activated by cytosolic DNA, cGAS generates cGAMP, which binds to and activates STING (15). The cGAS fragment 161–522 a.a. retains the cGAMP synthesis activity but cannot undergo phase separation (16), whereas the fragment 161–522 a.a. Δ 171–174 a.a. loses the cGAMP synthesis activity (2). Both 161–522 a.a. and 161–522 a.a. Δ 171–174 a.a. rescued fast proliferation of cGAS−/− cells as efficiently as full-length (FL) cGAS did (Fig. 1F), indicating that cGAS slows cell proliferation in a cGAMP-independent manner. We also examined several other cGAS mutants for their abilities to slow cell proliferation (Fig. 1, D and E): The Y215E mutation of cGAS abolishes its nuclear translocation (17); the K347E mutation disrupts the dimerization of cGAS, but does not abolish DNA binding (18, 19); and the K394E mutation abolishes the DNA binding of cGAS (18–21). Both the K394E and K347E mutants were localized to the cytoplasm and the nucleus as FL cGAS, whereas the Y215E mutant was only detected in the cytoplasm (Fig. 1, G and H). When expressed in cGAS−/− U2OS cells, the K347E mutant reversed the accelerated proliferation as did FL cGAS, whereas the Y215E and K394E mutants did not reverse accelerated proliferation (Fig. 1I). Thus, cGAS entering the nucleus and binding DNA, but not its ability to dimerize, is required for slowing cell proliferation.
cGAS deficiency endows a hyper-replicative cell state
To understand how loss of cGAS accelerates cell proliferation, we examined the cell cycle profiles of WT and cGAS−/− cells by flow cytometry, after labeling nascent DNA with 5-ethynyl-2′-deoxyuridine (EdU). The fractions of S-phase cells (EdU+) were higher in asynchronously growing cGAS−/− BJ, MEF, and U2OS cell populations than in the corresponding WT cell populations (Fig. 2, A and B, and fig. S1C). Notably, the S-phase cells undergoing robust DNA synthesis (EdUhigh) were substantially increased in cGAS−/− cells compared to WT cells. In contrast to that in cGAS−/− cells, cGAS-OE in WT U2OS cells reduced the fraction of S-phase cells (fig. S1C). These results suggest that cGAS deficiency promotes DNA replication.
Fig. 2. cGAS deficiency endows a hyper-replicative cell state associated with replication defects.
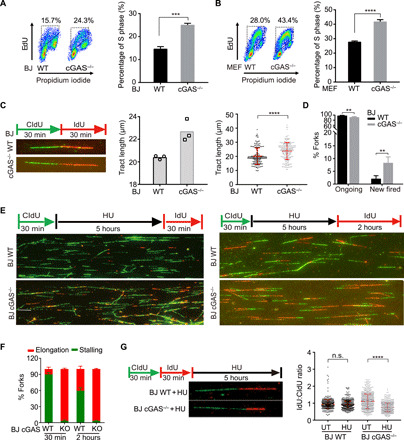
(A and B) The staining of EdU and propidium iodide (PI) in BJ WT (A), MEF WT (B), and corresponding cGAS−/− cells was analyzed by flow cytometry. The percentage of S phase from flow cytometry analysis is shown. (C) Representative images of nascent DNA tract length in BJ WT and cGAS−/− cells (left), quantification of DNA tract length in either BJ WT or cGAS−/− cells from three independent experiments (middle), and nascent DNA tract length of one representative experiment (right) (n = 200, mean ± SD in each experiment). Statistical analysis was done with the two-sided Mann-Whitney test; ****P < 0.0001. (D) DNA fiber analysis of ongoing and new fired forks (n = 600) in BJ WT and cGAS−/− cells. Mean ± SD is shown. (E) The replication fork status of BJ WT and cGAS−/− cells treated with 4 mM HU for 5 hours and released for 30 min or 2 hours. Representative images of fiber assay are shown (scale bar, 10 μm). (F) DNA fiber analysis of replication fork stalling and elongation (n = 1800) in BJ WT and cGAS−/− cells treated with HU. Means and SD of three independent experiments are shown. (G) Ratios of IdU and CldU length (each group, n > 660) are shown in BJ WT and cGAS−/− cells treated with or without [untreated (UT)] 4 mM HU for 5 hours. Median ratio is indicated in red. **P < 0.01, ***P < 0.001, Mann-Whitney test. n.s., not significant.
Next, we used DNA fiber assays to directly examine the impact of cGAS on DNA replication forks. Nascent DNA was sequentially labeled with thymidine analogs 5-chloro-2′-deoxyuridine (CldU) and 5-iodo-2′-deoxyuridine (IdU). CldU- and IdU-labeled replication tracts were longer in cGAS−/− BJ cells than in WT cells (Fig. 2C). Moreover, increased new fired forks (IdU alone) were observed in cGAS−/− BJ cells compared to WT cells (Fig. 2D). These results indicate that cGAS deficiency accelerates the progression of replication forks.
The acceleration of replication forks in cGAS−/− cells prompted us to ask whether cGAS alters the restart of stalled replication forks, which occurs throughout the genome in response to intrinsic replication stress. To follow fork restart in WT and cGAS−/− cells, we stalled replication forks with hydroxyurea (HU) and then released them in the presence of IdU. Fewer than 10% of WT cells incorporated IdU at 30 min after the release, and ~40% of WT cells became IdU+ at 2 hours (Fig. 2, E and F), showing that stalled replication forks restarted gradually in WT cells. In stark contrast, even 30 min after the release, >90% of cGAS−/− cells already incorporated IdU (Fig. 2, E and F), showing that cGAS deficiency alters the kinetics of fork restart.
In addition to replication fork speed, the stability of stalled replication forks is another key aspect of replication dynamics. To analyze the effects of cGAS on the stability of stalled forks, we treated WT and cGAS−/− cells with HU after sequential CldU and IdU labeling. When HU-stalled forks are not properly protected, the IdU-labeled nascent DNA at replication forks is degraded by nucleases, leading to a reduced IdU:CldU ratio. The IdU:CldU ratio of replication tracts was not altered by HU in WT cells, but was reduced by HU in cGAS−/− cells (Fig. 2G), suggesting that cGAS deficiency reduces the stability of stalled replication forks.
Collectively, our results suggest that replication forks progress faster and restart prematurely after transient stalling in the absence of cGAS. Furthermore, the stability of replication forks is compromised in cGAS-deficient cells, suggesting that cGAS deficiency endows a hyper-replicative cell state that is associated with replication defects.
cGAS deficiency induces replication stress
To further characterize the impacts of cGAS loss, we performed RNA sequencing (RNA-seq) on WT and cGAS−/− BJ cells. Consistent with the hyper-proliferative and replicative state of cGAS−/− cells, the genes involved in DNA replication and the cell cycle are significantly up-regulated by the loss of cGAS (Fig. 3A). For example, DNA replication genes such as MCM3, MCM5, RFC2, RFC5, and RPA3 are up-regulated in cGAS−/− cells (Fig. 3B). In addition, a group of cell cycle genes, such as E2F, CDK2, CDC25, CDK1, Cyclin B2, and PLK1, is also increasingly expressed in cGAS−/− cells (Fig. 3B). RRM2 expression was increased by about twofold in cGAS knockout (KO) cells. To test whether this increase of RRM2 affects fork speed, we used small interfering RNA (siRNA) to reduce RRM2 in cGAS KO cells by about twofold (fig. S1D). Fork speed was not affected in the cGAS KO with reduced RRM2, suggesting that the increase of RRM2 is not responsible for increased fork speed (fig. S1D). Another notable change of gene expression in cGAS−/− cells is the up-regulation of DNA repair genes, including the genes involved in homologous recombination, mismatch repair, and base excision repair. The up-regulation of DNA repair genes in cGAS−/− cells raised the possibility that genomic instability is increased in the absence of cGAS. To test whether genomic instability is induced in cGAS−/− cells, we analyzed the activation of ATR (Ataxia Telangiectasia and Rad3 Related) gene kinase and Chk1, the master sensor of DNA replication stress. Both the autophosphorylation of ATR (p-ATR) and the phosphorylation of Chk1 (p-Chk1), a substrate and effector of ATR, were increased in cGAS−/− cells compared with WT cells (Fig. 3C), suggesting that cGAS deficiency elicits an ATR response. Thus, the hyper-replicative state of cGAS−/− cells induces replication stress and activates ATR; however, ATR activity in cGAS−/− cells is not sufficient to slow cell proliferation, leaving cGAS−/− cells in a vulnerable state.
Fig. 3. Fast replication renders cGAS-deficient cells sensitive to radiation and chemo-drugs.
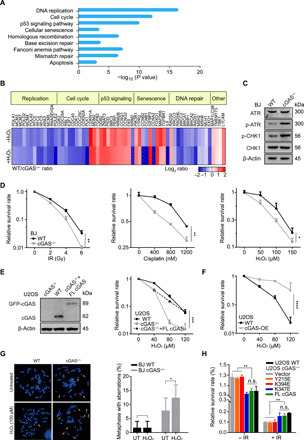
(A) RNA-seq expression data of BJ WT and cGAS−/− cell were analyzed. The enrichment KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway comparison between gene expression from RNA-seq in BJ WT and cGAS−/− cells is shown. (B) Heat map of RNA expression in BJ WT and cGAS−/− cells. A gene list was generated using genes showing a 1.5-fold change cutoff, 10 reads or more, and P value of <0.05. (C) Level of phospho-ATR and CHK1 in BJ WT and cGAS−/− cells by WB. (D) Colony formation assay for BJ WT and cGAS−/− cells with treatment of the indicated dose of ionizing radiation (IR), cisplatin, or H2O2. (E) Colony formation assay for U2OS WT, cGAS−/−, and GFP-cGAS stably expressed cGAS−/− (cGAS−/− + FL cGAS) cells with the indicated dose of H2O2. (F) Colony formation assay for U2OS WT and cGAS-OE with treatment of indicated dose of H2O2. (G) Representative metaphases of BJ WT and cGAS−/− cells treated with 100 μM H2O2 for 24 hours. Quantification of incidence of total chromosomal aberrations (n > 500 in one experiment). Three independent experiments were done. (H) Colony formation assay for U2OS WT and cGAS−/− cells transfected with mutant plasmids treated without and with 4-Gy IR. **P < 0.01, ****P < 0.0001. Mann-Whitney test. n.s., not significant.
cGAS deficiency increases cellular sensitivity to radiation and chemotherapy
Ionizing radiation (IR) and genotoxic chemotherapeutics kill cancer cells by inflicting DNA damage. Given that cGAS−/− cells are hyper-proliferative, we aimed to determine whether cGAS deficiency alters the sensitivity of cells to IR and chemotherapy. We treated WT and cGAS−/− cells with IR and genotoxic agents, including H2O2, etoposide (ETO), cisplatin, HU, and ultraviolet C (UVC), and measured cell viability with a colony formation assay. cGAS−/− BJ cells were more sensitive to all of these genotoxic insults compared to WT BJ cells (Fig. 3D and fig. S2A). Similarly, cGAS−/− U2OS cells were more sensitive to H2O2, ETO, cisplatin, and IR than were WT U2OS cells (Fig. 3E and fig. S2B). cGAS expression in cGAS−/− U2OS cells restored H2O2 resistance back to the WT level (Fig. 3E, dotted line). Knockdown of cGAS in U2OS cells with siRNA recapitulated the effects of cGAS KO on cell proliferation and cell survival after DNA damage (fig. S2C). In contrast, cGAS-OE in WT U2OS cells rendered these cells resistant to H2O2 (Fig. 3F), ETO, and UVC (fig. S2D). These results demonstrate that cGAS deficiency increases cellular sensitivity to radiation and chemotherapy.
Given that STING is not detectable in U2OS cells (Fig. 1C) (22), the increased DNA damage sensitivity of cGAS−/− U2OS cells is likely independent of STING. Consistent with this idea, STING knockdown in cGAS−/− and WT BJ cells did not alter DNA damage sensitivity (fig. S3, A and B). Furthermore, the H2O2 sensitivity of WT and cGAS−/− U2OS cells was not affected by increasing concentrations of cGAMP (fig. S3C). Thus, cGAS promotes cell survival after DNA damage in a cGAMP- and STING-independent manner.
In metaphase spreads, we observed significantly higher levels of chromosomal aberrations in cGAS−/− BJ cells than in WT BJ cells, especially after DNA damage (Fig. 3G). The cGAS mutants defective for phase separation, cGMP synthesis, and dimerization all reversed the hyper-proliferation and H2O2 sensitivity of cGAS−/− cells, as did WT cGAS (fig. S3D). In contrast, the cGAS Y215E and K394E mutants, which are defective for nuclear entry and DNA binding, respectively, did not reverse the IR or H2O2 sensitivities (Fig. 3H and fig. S3D). Together with the data in Fig. 1 (D to I), our results show that cGAS suppresses both proliferation and DNA damage sensitivity through its DNA binding in the nucleus, suggesting that the hyper-replicative state of cGAS-deficient cells underlies the increased sensitivity to DNA damage.
cGAS associates with chromatin and encounters replication forks in DNA
Next, we investigated how cGAS slows replication by binding DNA in the nucleus. A fraction of cGAS was detected in chromatin in U2OS cells (Fig. 4A), consistent with a recent report (23, 24). To understand how cGAS interacts with other proteins on chromatin, we pulled down cGAS and analyzed its interactome with mass spectrometry. A group of replication fork components, including MCM2, MCM3, MCM7, replication factor C 1 (RFC1), and proliferation cell nuclear antigen (PCNA), was identified as cGAS-interacting proteins (Fig. 4B and fig. S4A). Furthermore, in a proximity ligation assay (PLA), cGAS colocalized with PCNA in the nucleus after extraction by detergent (Fig. 4, C and D), suggesting that replication forks may encounter cGAS on chromatin. Our results also raised the possibility that cGAS may directly bind to the dsDNA at replication forks and compromise the replisome. To test this possibility, we immunoprecipitated PCNA from cGAS WT and KO cells and tested the interactions of PCNA with replication fork proteins MCM2, MCM7, and FEN1 (fig. S4B). None of these interactions was affected by the loss of cGAS, suggesting that cGAS probably slows replication forks as a “roadblock” on dsDNA instead of a destabilizer of the replisome.
Fig. 4. DNA binding of cGAS is required for fork stalling after damage and drug resistance.
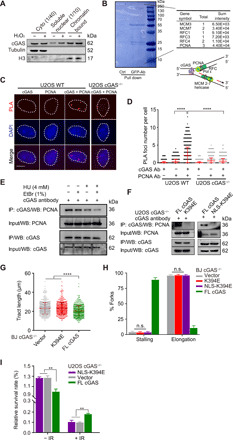
(A) Cytosolic, soluble nuclear, and chromatin fractions from U2OS cells were immunoblotted for cGAS and indicated proteins. (B) U2OS cells expressing GFP-cGAS were pulled down for interactome analysis. The results of mass spectrometry identified several major replication fork components. The schematic structure of cGAS pulled down with a replication fork complex via DNA or based on the proximity at replication forks. (C) Representative PLA of U2OS WT and cGAS−/− cells (scale bar, 10 μm). (D) Quantification of colocalization between cGAS and PCNA. Three independent experiments were done, mean ± SD is shown, and statistical analysis was done with the two-sided Mann-Whitney test; ****P < 0.001 and **** P < 0.0001. (E) Interaction of PCNA and cGAS detected with coimmunoprecipitation (Co-IP). U2OS cells were treated with or without 4 mM HU or 1% EtBr before Co-IP analysis with anti-cGAS antibody. (F) Interaction of cGAS mutants and PCNA detected with Co-IP. U2OS cGAS−/− cells were transfected with cGAS flag-tagged FL, K394E mutant, or NLS-K394E mutant for 36 hours before Co-IP analysis with anti-cGAS antibody. (G) Analysis of nascent DNA tract length (n = 600) in BJ cGAS−/− cells transfected with K394E mutant and corresponding control plasmids. For all experiments in this study, three independent experiments were done, mean ± SD is shown, and statistical analysis was done with the two-sided Mann-Whitney test; **** P < 0.0001. (H) DNA fiber analysis of replication fork stalling and elongation (n = 1800) in BJ cGAS−/− cells transfected with DNA binding defective mutants. Means and SD of three independent experiments are shown. (I) Colony formation assay for U2OS cGAS−/− cells expressing NLS-K394E mutants without and with 4-Gy IR treatment.
If replication forks encounter cGAS on DNA, the interaction between replication proteins and cGAS should be dependent on the DNA binding of cGAS. The cGAS K394E mutant, which is defective for DNA binding (18–21), did not interact with PCNA (Fig. 4, E and F). While analyzing the localization of cGAS during the DNA damage response, we noticed that endogenous cGAS was increasingly localized in the nucleus after DNA damage (fig. S4, C to E). To ensure that the defect of the cGAS K394E mutant in PCNA binding is not a result of compromised nuclear localization, we fused K394E with a nuclear localization signal (NLS). NLS-K394E localized to the nucleus efficiently, but did not interact with PCNA (Fig. 4F and fig. S4F), confirming that replication forks interact with DNA-bound cGAS.
DNA-bound cGAS slows replication forks and suppresses DNA damage sensitivity
To directly test the effects of DNA-bound cGAS on replication forks, we measured the length of CldU/IdU double-positive replication tracts in cGAS−/− BJ cells expressing WT cGAS or the K394E cGAS mutant. Expression of WT cGAS, but not K394E, in cGAS−/− BJ cells reduced replication tract length (Fig. 4G). Furthermore, after HU release, only WT cGAS—not K394E or NLS-K394E—slowed the restart of stalled forks (Fig. 4H and fig. S4G). These results suggest that replication forks, including restarting replication forks, are slowed down when they encounter cGAS on DNA.
If the slowing of replication forks by DNA-bound cGAS is important for preventing replication-associated DNA damage after radiation, one would expect that the DNA binding–defective cGAS mutant cannot suppress the IR sensitivity of cGAS KO cells. Unlike WT cGAS, the cGAS NLS-K394E mutant did not reverse the hyper-proliferation and IR sensitivity of cGAS KO U2OS cells (Fig. 4I and fig. S4H), establishing a strong link between the cGAS-mediated slowing of DNA replication and cellular resistance to IR.
While the function of cGAS in sensing cytosolic DNA is well established, its function in the nucleus is poorly understood. Recent studies suggest that cGAS promotes tumorigenesis by suppressing DNA repair (23). However, using multiple untransformed and cancer cell lines, we demonstrated that cells lacking cGAS are more sensitive to radiation and a variety of DNA-damaging agents, suggesting that cGAS protects cells from DNA damage. We have shown that the ability of cGAS to suppress DNA damage sensitivity is dependent on the binding of cGAS to DNA in the nucleus. Furthermore, cGAS interacts with replication forks in DNA and reduces fork speed, suggesting that DNA-bound cGAS is a decelerator of DNA replication (fig. S5). It should be noted that although cGAS may slow replication forks as a roadblock, it does not affect the integrity of the replisome and induce replication stress. The unique effect of cGAS on replication fork is distinct from those of the DNA lesions interfering with DNA replication forks and compromising replisomes. In the absence of cGAS, replication forks progress at an excessive speed, leading to genomic instability. In addition to its effects on replication forks, cGAS may also suppress replication origin firing as a dsDNA-binding protein. The increase of origin firing in cGAS KO cells may also contribute to the enhanced DNA replication in these cells and their increased sensitivity to DNA damage.
Together with previous investigations, this study suggests that cGAS may integrate multiple functions to counter genomic instability. cGAS senses the cytosolic DNA arising from the genomic instability of cancer cells and triggers antitumor immunity. cGAS is also essential for DNA damage–induced senescence and promotes autophagy in response to cytosolic DNA (17). cGAS not only promotes senescence through STING but also suppresses genomic instability in a STING-independent manner, which may reduce the signals that trigger senescence. The ability of cGAS to regulate DNA replication enables it to restrict genomic instability during S phase. A recent study showed that ISG15, a protein induced by the STING-mediated interferon response, accelerates replication fork progression and induces chromosomal breakage (25), suggesting that cGAS may affect replication forks through STING-dependent and STING-independent mechanisms in different contexts. Notably, STING is repressed in a significant fraction of cancers, rendering these cancers refractory to antitumor immunity. Thus, the STING-independent function of cGAS in suppressing genomic instability contributes to cell viability after radiation and chemotherapy, and it may be particularly important for the survival of cancer cells that are intrinsically unstable, positioning cGAS as a potential therapeutic target in STING-deficient tumors.
MATERIALS AND METHODS
Cell lines and CRISPR-Cas9 KO
U2OS-SCE, BJ, and MEF cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Lonza, catalog no. 12-604F) with 10% (v/v) fetal bovine serum (FBS) at 37°C, 5% CO2. BJ, MEF cells, and their KO lines were described previously (26). All cells included in the study were treated with BM cyclin (10799050001, Sigma-Aldrich) and were mycoplasma free [tested using the PCR Detection Kit (MP0035, Sigma-Aldrich)].
For cGAS KO, oligonucleotides that delete the first exon containing ATG sites and target the following sequence: upstream 5′-GGCCAGCCTCTTCGCGGCAT-3′ and downstream 5′-GGCCCCCATTCTCGTACGGA-3′ (Invitrogen), were synthesized by Integrated DNA Technologies. They were inserted into PX330 plasmids and transfected into U2OS-TRE cells. Primers designed for genotyping polymerase chain reaction (PCR) were as follows: forward, 5′-TGTTTTGTGATGGACTCTTTTTC-3′; reverse, 5′-CGTGCTCATAGTAGCTCCCGGTG-3′ (Invitrogen). After 48 hours, single cells were spread in a 96-well plate to obtain monoclonal colonies.
For U2OS-TRE green fluorescent protein (GFP)–cGAS stable expression, complementary DNAs of WT and mutant cGAS were subcloned into the pTY-IRES-Hygro vector with an N-terminal flag tag. The vectors were cotransfected with packaging plasmids into 293FT cells for virus packaging. The culture medium was changed 8 hours after transfection. Forty-eight hours later, the medium was collected and filtered with a 0.45-μm filter (Millex-HA, SLHAM33SS). The U2OS-TRE cells were cultured in a medium mixed with normal DMEM (10% FBS) at a 1:1 ratio. Polybrene (10 μg/ml) was added to the culture system to promote efficiency. Forty-eight hours later, the cells were cultured in DMEM (10% FBS) with hygromycin (1 μg/ml), and the medium was changed once every 2 days.
Plasmids, siRNAs, and chemicals
Plasmids and siRNAs were transfected with Lipofectamine 2000 (Thermo Fisher Scientific, 11668019) and DharmaFECT 1 (Dharmacon, T-2001-03), respectively, using standard protocols. MB21D1 (cGAS) siRNA was purchased from Dharmacon (M-015607-01-0005) and SMARTpool; siGENOME TMEM173 siRNA was purchased from Dharmacon (M-024333-00-0005). siRNA targeting RRM2 (sense, 5′-GCGAUUUAGCCAAGAAGUUCA-3′; antisense, 5′-UGAACUUCUUGGCUAAAUCGC-3′) was purchased from Dharmacon. Flag-cGAS truncation and the mutant plasmid transfection protocol were also described in a previous study (19). Hydrogen peroxide solution (216763, Sigma-Aldrich), HU (H8627, Sigma-Aldrich), ETO (E1383, Sigma-Aldrich), and cisplatin (232120, Sigma-Aldrich) were purchased from the company.
Proliferation assay and cell cycle profiling via flow cytometry
For determining proliferation rate, cells (3 × 104 cells/ml) were seeded in coverslipped six-well plates and incubated at 37°C for 24 hours to adhere. The number of BJ cGAS−/−, U2OS cGAS−/− or overexpressed GFP-cGAS U2OS, and MEF cGAS−/− cells compared with their controls was counted every day until arriving at high density.
For cell cycle profiling, the cells were treated with 10 μM EdU for 1 hour and then trypsinized and washed once with phosphate-buffered saline (PBS). Then, cells were fixed in cold 70% ethanol at 4°C overnight, which can be stored for up to 1 week. The Click-iT EdU Alexa Fluor 488 Flow Cytometry Assay Kit (Invitrogen) was used. The cells were washed once with 2% bovine serum albumin (BSA) in PBS and incubated in PBS with 2% BSA, propidium iodide (PI) (50 μg/ml), and RNase A (100 μg/ml) in the dark for 30 min at 37°C. Cells were selected by SSC (side scatter) and FSC (forward scatter) through flow cytometry. DNA stained with PI reflected the cell cycle stage.
DNA fiber assay
Cells were pulse-labeled with 25 μM CldU (Sigma-Aldrich, C6891mg) and 250 μM IdU (Sigma-Aldrich, I7125-5g) at indicated times, with or without treatment, as reported in the experimental schemes. DNA fibers were prepared as previously reported (27). For the immunodetection of labeled tracks, the following primary antibodies were used: anti-CldU [rat monoclonal anti–5-bromo-2′-deoxyuridine (BrdU)/CldU; BU1/75 ICR1, Novus, 1:100] and anti-IdU (mouse monoclonal anti-BrdU/IdU; clone B44, Becton Dickinson, 1:50). The secondary antibodies were goat anti-mouse Alexa Fluor 594 (Abcam, ab150116, 1:200) or goat anti-rat Alexa Fluor 488 (Abcam, ab150157, 1:200). The incubation with antibodies was accomplished in a humidified chamber for 1 hour at room temperature. Images were acquired randomly from fields with untangled fibers using a Nikon Eclipse 80i fluorescence microscope, equipped with a video confocal system. The length of labeled tracks was measured using Fiji software, and values were converted into kilobases using the conversion factor of 1 μm = 2.59 kb. A minimum of 100 individual fibers was analyzed for each experiment, and the mean of at least three independent experiments is presented. Statistical analysis was performed using GraphPad Prism Software.
Immunofluorescence staining, microscopy, and immunoblots
Cells in a 35-mm dish were rinsed with PBS and fixed in 4% paraformaldehyde (PFA; Affymetrix, 19943 1 LT) for 15 min at room temperature. They were washed three times with PBS, permeabilized with 0.2% Triton X-100 in PBS for 10 min, and then washed again three times using PBS. Next, they were blocked by 5% BSA (Sigma-Aldrich, A-7030) in 0.1% PBS-Tween (PBST) for 30 min at room temperature. Primary antibodies were diluted in blocking buffer and incubated with cells overnight at 4°C. Then, the cells were washed three times with 0.05% PBST and incubated with secondary antibodies for 1 hour at room temperature, including Alexa Fluor 405/488/594 goat anti-mouse/rabbit immunoglobulin G conjugate (1:5000). Last, cells were washed three times with 0.05% PBST and stained with DAPI (4′,6-diamidino-2-phenylindole; 1:1000 in PBS) for 5 min at room temperature. The primary antibodies for immunoassays were anti-flag M2 mouse monoclonal antibody (Sigma-Aldrich, catalog no. F1804) and anti-cGAS rabbit monoclonal antibody (Novus Biologicals, catalog no. NBP1-86761). For Western blot analysis, samples were heated at 95°C for 5 to 8 min in SDS loading buffer. Then, they were subjected to electrophoresis in 10 to 12% SDS–polyacrylamide gels and transferred to a polyvinylidene difluoride (PVDF) membrane. The membranes were prestained with Ponceau S Red solution (Sigma-Aldrich, catalog no. P3504-10G). For block and antibody dilution, 5% nonfat milk in PBS was used. After primary antibody incubation at 4°C overnight and secondary antibody incubation at room temperature for 1 hour, the membranes were washed three times in 0.1% PBST. The following primary antibodies and corresponding secondary antibodies (1:5000) were applied (Supplementary table 1). Chemiluminescent horseradish peroxidase substrate was purchased from Millipore (catalog no. WBKLS0500). Images were acquired using Bio-Rad Universal Hood II with ImageJ software.
Metaphase chromosome spreading assay
To analyze BJ cells and chromosomal aberrations, cells were grown until they reached 60% of their cell cycle before they were treated with H2O2 (100 μM) for 2 hours. After the treatment, the cells were incubated in the dark for 12 hours at 37°C. One hundred microliters of colcemid (10 μg/ml; Gibco BRL) was added to the cells to reach a final concentration of 100 μg/ml and was incubated at 37°C for 3 hours. The cells were trypsinized, and the floating cells were collected and incubated in 8 ml of 75 mM KCl at 37°C for 30 min. The cells were fixed three times with freshly prepared 3:1 solution of methanol:glacial acetic acid, and three or four drops of cell suspension were placed onto the microscope slide at a 45° vertical angle. The microscope slides were then washed with fixative, allowed to dry, and mounted with DABCO (1,4-diazabicyclo[2.2.2]octane) and DAPI (Sigma-Aldrich).
Colony formation assay
All cells subjected to a survival assay were trypsinized thoroughly to ensure that they were completely separated from each other. The cells were centrifuged at 500 rpm for 5 min and resuspended in DMEM with 10% FBS. The number of cells was counted manually in a cell meter glass. Approximately 400 single cells were seeded in a 6-cm dish with 3 ml of 10% FBS in DMEM. The cells were treated immediately with the corresponding chemicals or UVC for 6 hours after seeding. After 7 to 10 days, colonies were fixed and stained with 0.3% crystal violet in methanol for 5 min at room temperature, and the numbers of colonies were counted manually.
RNA sequencing
After 24 hours of 100 μM H2O2 treatment, cells were harvested by trypsinization, centrifuged, and rinsed once with ice-cold PBS. Total RNA was extracted using the PureLink RNA Mini Kit (Invitrogen), according to the manufacturer’s instructions. RNAs isolated from all fibroblast lines (1 μg per sample) were reverse-transcribed to generate sequencing libraries using the TruSeq Stranded Total RNA Library Prep Kit (Illumina) and sequenced by HiSeq2000 (Illumina) (Novogene Corporation, 8801 Folsom Blvd., Suite 290, Sacramento, CA). Approximately 27 million to 39 million sequencing reads were generated for each fibroblast mRNA preparation, and 81 to 94% of fragments were mapped by both ends to the human genome (hg19) using TopHat (version 2.0.7) and bowtie2 (version 2.1.0).
Differentially expressed gene identification, KEGG, and pathway analysis
The normalized expression of all samples was estimated in fragments per kilobase of exon per million fragments mapped (FPKM) using Cuffdiff v2.2.1. Genes with significant differences were those with less than the adjusted P value of 0.05, and the fold change cutoff was set from 1.5 to −1.5. All genes with a log2 fold change >1.5 were considered up-regulated, whereas all genes with a log2 fold change <−1.5 were regarded as down-regulated to generate the heat map representation for differentially expressed genes.
Coimmunoprecipitation
The lysates of U2OS, U2OS-TRE GFP-cGAS stable cells, and cGAS−/− U2OS transfected with flag-tagged mutant using Lipofectamine 2000 (Invitrogen) were prepared with lysis buffer. Lysates were cleared by centrifugation at 13,000 rpm for 15 min and incubated with G-Sepharose protein beads (GE Healthcare Bio-Sciences) attached with and without corresponding antibody overnight at 4°C on a rocking platform. Beads were then collected by centrifugation at 8200 rpm for 5 s at 4°C, extensively washed in lysis buffer, and resuspended in SDS gel loading buffer. The proteins were separated on a 10% SDS–polyacrylamide gel, transferred to a PVDF membrane, and analyzed by immunoblotting with the corresponding antibodies.
Proximity ligation assay
Cells were washed once with 1× PBS before being treated with CSK extraction buffer [0.2% Triton X-100, 20 mM Hepes-KOH (pH 7.9), 100 mM NaCl, 3 mM MgCl2, 300 mM sucrose, 1 mM EGTA] on ice for 3 min. Cells were fixed with 4% PFA on ice for 5 min, followed by ice-cold methanol treatment at 20°C for 20 min. Subsequently, cells were permeabilized with 1× PBS containing 0.5% Triton X-100 for 5 min and then blocked with 3% BSA in PBST at room temperature for 1 hour. Afterward, cells were incubated with the indicated primary antibodies PCNA (PC10) (Santa Cruz Biotechnology, sc-56, 1:500) and cGAS (D1D3G) [CST (Cell Signaling Technology), 15102, 1:500] at 4°C overnight. After three washes with 1× PBST, cells were incubated with anti-mouse minus and anti-rabbit plus PLA probes (PLA kit from Sigma-Aldrich) at 37°C for 1 hour. Following the manufacturer’s instructions, the PLA reaction was performed with the Duolink In Situ Detection Reagents (PLA kit). Last, cells were washed three times with buffer B, stained with DAPI during the second wash, mounted on slides with Prolong Gold, and sealed with nail polish. Images were captured with a Nikon 90i microscope and quantified using ImageJ.
Supplementary Material
Acknowledgments
Funding: This work was supported, in part, by grants from the NIH to L.L. (GM118833) and L.Z. (GM076388 and CA197779). H.C. (second author) was a visiting international research scholar at the University of Pittsburgh Medical School from September 2016 to August 2018 from Tsinghua University School of Medicine, and is funded by the Tsinghua Educational Foundation of North America (TEFNA), with the cost of living covered by the China Scholarship Council (CSC) through University of Pittsburgh Medical School. Author contributions: H.C. (first author) conducted the major experiments. H.C. (second author), J.Z., Y.W., A.S., and H.Y. participated in the experiments and data analysis. A.S.L., L.Z., and Z.C. participated in experimental design and data analysis. L.L. provided overall experimental guidance. All authors described their specific contributions and reviewed and edited the manuscript. Competing interest: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/42/eabb8941/DC1
REFERENCES AND NOTES
- 1.Jackson S. P., Bartek J., The DNA-damage response in human biology and disease. Nature 461, 1071–1078 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sun L., Wu J., Du F., Chen X., Chen Z. J., Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li T., Chen Z. J., The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J. Exp. Med. 215, 1287–1299 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang H., Hu S., Chen X., Shi H., Chen C., Sun L., Chen Z. J., cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc. Natl. Acad. Sci. U.S.A. 114, 1637–1642 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pépin G., Gantier M. P., cGAS-STING activation in the tumor microenvironment and its role in cancer immunity. Adv. Exp. Med. Biol. 1024, 175–194 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Li X.-D., Wu J., Gao D., Wang H., Sun L., Chen Z. J., Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 341, 1390–1394 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xia P., Wang S., Gao P., Gao G., Fan Z., DNA sensor cGAS-mediated immune recognition. Protein Cell 7, 777–791 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schoggins J. W., MacDuff D. A., Imanaka N., Gainey M. D., Shrestha B., Eitson J. L., Mar K. B., Richardson R. B., Ratushny A. V., Litvak V., Dabelic R., Manicassamy B., Aitchison J. D., Aderem A., Elliott R. M., García-Sastre A., Racaniello V., Snijder E. J., Yokoyama W. M., Diamond M. S., Virgin H. W., Rice C. M., Corrigendum: Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 525, 144 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schoggins J. W., MacDuff D. A., Imanaka N., Gainey M. D., Shrestha B., Eitson J. L., Mar K. B., Richardson R. B., Ratushny A. V., Litvak V., Dabelic R., Manicassamy B., Aitchison J. D., Aderem A., Elliott R. M., García-Sastre A., Racaniello V., Snijder E. J., Yokoyama W. M., Diamond M. S., Virgin H. W., Rice C. M., Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 505, 691–695 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma F., Li B., Liu S.-y., Iyer S. S., Yu Y., Wu A., Cheng G., Positive feedback regulation of type I IFN production by the IFN-inducible DNA sensor cGAS. J. Immunol. 194, 1545–1554 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Oliveira Mann C. C., Kranzusch P. J., cGAS conducts micronuclei DNA surveillance. Trends Cell Biol. 27, 697–698 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Coquel F., Silva M.-J., Técher H., Zadorozhny K., Sharma S., Nieminuszczy J., Mettling C., Dardillac E., Barthe A., Schmitz A.-L., Promonet A., Cribier A., Sarrazin A., Niedzwiedz W., Lopez B., Costanzo V., Krejci L., Chabes A., Benkirane M., Lin Y.-L., Pasero P., SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 557, 57–61 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Chen Y.-A., Shen Y.-L., Hsia H.-Y., Tiang Y.-P., Sung T.-L., Chen L.-Y., Extrachromosomal telomere repeat DNA is linked to ALT development via cGAS-STING DNA sensing pathway. Nat. Struct. Mol. Biol. 24, 1124–1131 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Kwon J., Bakhoum S. F., The cytosolic DNA-sensing cGAS-STING pathway in cancer. Cancer Discov. 10, 26–39 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ablasser A., Chen Z. J., cGAS in action: Expanding roles in immunity and inflammation. Science 363, eaat8657 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Du M., Chen Z. J., DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 361, 704–709 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu H., Zhang H., Wu X., Ma D., Wu J., Wang L., Jiang Y., Fei Y., Zhu C., Tan R., Jungblut P., Pei G., Dorhoi A., Yan Q., Zhang F., Zheng R., Liu S., Liang H., Liu Z., Yang H., Chen J., Wang P., Tang T., Peng W., Hu Z., Xu Z., Huang X., Wang J., Li H., Zhou Y., Liu F., Yan D., Kaufmann S. H. E., Chen C., Mao Z., Ge B., Nuclear cGAS suppresses DNA repair and promotes tumorigenesis. Nature 563, 131–136 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Dai J., Huang Y.-J., He X., Zhao M., Wang X., Liu Z.-S., Xue W., Cai H., Zhan X.-Y., Huang S.-Y., He K., Wang H., Wang N., Sang Z., Li T., Han Q.-Y., Mao J., Diao X., Song N., Chen Y., Li W.-H., Man J.-H., Li A.-L., Zhou T., Liu Z.-G., Zhang X.-M., Li T., Acetylation blocks cGAS activity and inhibits self-DNA-induced autoimmunity. Cell 176, 1447–1460.e14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang X., Wu J., Du F., Xu H., Sun L., Chen Z., Brautigam C. A., Zhang X., Chen Z. J., The cytosolic DNA sensor cGAS forms an oligomeric complex with DNA and undergoes switch-like conformational changes in the activation loop. Cell Rep. 6, 421–430 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gui X., Yang H., Li T., Tan X., Shi P., Li M., Du F., Chen Z. J., Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 567, 262–266 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barnett K. C., Coronas-Serna J. M., Zhou W., Ernandes M. J., Cao A., Kranzusch P. J., Kagan J. C., Phosphoinositide interactions position cGAS at the plasma membrane to ensure efficient distinction between self- and viral DNA. Cell 176, 1432–1446.e11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deschamps T., Kalamvoki M., Impaired STING pathway in human osteosarcoma U2OS cells contributes to the growth of ICP0-null mutant herpes simplex virus. J. Virol. 91, e00006-17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang H., Xue X., Panda S., Kawale A., Hooy R. M., Liang F., Sohn J., Sung P., Gekara N. O., Chromatin-bound cGAS is an inhibitor of DNA repair and hence accelerates genome destabilization and cell death. EMBO J. 38, e102718 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Volkman H. E., Cambier S., Gray E. E., Stetson D. B., Tight nuclear tethering of cGAS is essential for preventing autoreactivity. eLife 8, e47491 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raso M. C., Djoric N., Walser F., Hess S., Schmid F. M., Burger S., Knobeloch K.-P., Penengo L., Interferon-stimulated gene 15 accelerates replication fork progression inducing chromosomal breakage. J. Cell Biol. 219, e202002175 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang H., Wang H., Ren J., Chen Q., Chen Z. J., cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. U.S.A. 114, E4612–E4620 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buisson R., Boisvert J. L., Benes C. H., Zou L., Distinct but concerted roles of ATR, DNA-PK, and Chk1 in countering replication stress during S phase. Mol. Cell 59, 1011–1024 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/42/eabb8941/DC1