

Abstract
Introduction:
Highly aggressive thoracic neoplasms characterized by SMARCA4 (BRG1) deficiency and undifferentiated round cell or rhabdoid morphology have been recently described and proposed to represent thoracic sarcomas. However, it remains unclear whether such tumors may instead represent sarcomatoid carcinomas, and how their clinicopathologic characteristics compare with those of nonsarcomatoid SMARCA4-deficient non–small cell lung carcinomas (SD-NSCC).
Methods:
We identified 22 SMARCA4-deficient thoracic sarcomatoid tumors (SD-TSTs) with round cell and/or rhabdoid morphology and 45 SD-NSCCs, and comprehensively analyzed their clinicopathologic, immunohistochemical, and genomic characteristics using 341–468 gene next-generation sequencing and other molecular platforms.
Results:
The relationship of SD-TSTs with NSCC was supported by (1) the presence of NSCC components juxtaposed with sarcomatoid areas in five cases, (2) focal expression of NSCC lineage markers TTF1 or p40 in four additional cases, (3) smoking history in all except one patient (mean = 51 pack-years), accompanied by genomic smoking signature, and (4) high tumor mutation burden (mean = 14.2 mutations per megabase) and mutations characteristic of NSCC in a subset. Compared with SD-NSCCs, SD-TSTs exhibited considerably larger primary tumor size (p < 0.0001), worse survival (p = 0.004), and more frequent presentation at younger age (30–50 years) despite heavier smoking history. Distinctive pathologic features of SD-TSTs included consistent lack of adhesion molecule claudin-4, SMARCA2 (BRM) codeficiency, and frequent expression of stem cell markers.
Conclusions:
SD-TSTs represent primarily smoking-associated undifferentiated/de-differentiated carcinomas rather than primary thoracic sarcomas. Despite their histogenetic relationship with NSCC, these tumors have unique clinicopathologic characteristics, supporting their recognition as a distinct entity. Further studies are warranted to determine therapeutic approaches to this novel class of exceptionally aggressive thoracic tumors.
Keywords: SMARCA4, BRG1, Thoracic, Lung, Rhabdoid, Sarcomatoid
Introduction
SMARCA4 (BRG1) is a central component of the Switch/Sucrose-Non-Fermentable (SWI/SNF) chromatin remodeling complex.1 Inactivating mutations and loss of expression in several components of this complex have been implicated in carcinogenesis.2,3 A prototypical example is inactivation of SMARCB1 (INI1) in pediatric malignant rhabdoid tumors (MRTs).4 The involvement of SMARCA4 in tumorigenesis has emerged only in the previous decade, after the initial description of SMARCA4 deficiency in a subset of MRTs.5,6 Subsequently, SMARCA4 deficiency was identified as a defining event in small cell carcinomas of the ovary, hypercalcemic type (SCCOHTs)7,8 — a tumor recently proposed to represent an ovarian MRT.9 In addition to MRTs and SCCOHTs— the tumors in which SMARCA4/B1 alterations are virtually pathognomonic events occurring in a distinctively simple genomic background10,11 — the loss of SMARCA4, SMARCB1, and other SWI/SNF components has been implicated in the process of de-differentiation in tumors of various sites, including carcinomas of endometrium, bladder, gastrointestinal tract, and other organs,11–13 and other tumor types such as melanoma.11,14 Among pulmonary tumors, SMARCA4 mutations and loss of expression occur in approximately 5% of non-small cell carcinomas (NSCC), predominantly adenocarcinomas, associated with aggressive clinical behavior.15–17
Recently, a novel entity designated “SMARCA4-deficient thoracic sarcoma” (SDTS) was described by Le Loarer et al.18 The initial landmark study comprised 19 patients with thoracic tumors characterized by undifferentiated round cell or rhabdoid morphology and SMARCA4 mutations with concomitant loss of expression. At the transcriptional level, these tumors were found to closely resemble MRTs and SCCOHTs, whereas their expression profiles were sharply distinct from those of conventional NSCCs, prompting a conclusion that they represented a type of thoracic sarcoma. In a subsequent series, Yoshida et al.19 and Sauter et al.20 described a set of analogous tumors (n = 12 each), confirming their distinctive clinicopatholologic features. Together with several additional reports,21–26 a total of 65 cases of SDTSs have been reported to date (Supplementary Table 1). Combined age range for the reported patients is broad (27–90 y; mean = 50), and most patients (85%) are smokers. Common to all studies is the dismal prognosis and typical presentation as large thoracic tumors, frequently involving both mediastinal and pulmonary structures.
Despite the phenotypic similarity of SDTS with MRT and SCCOHT, several features identified in prior studies were unusual for bona fide MRT-type sarcomas, including (1) history of smoking in most patients, (2) complex genomes with frequent TP53 mutations and presence of mutations typical of smoking-related NSCCs (KRAS, KEAP1, STK11) in a subset of tumors,18,19 and (3) focal expression of NSCC lineage markers TTF1 or p40 in several cases.18–20,23,26 In addition, none of the patients had evidence of germline SMARCA4 alterations, whereas familial transmission is a common feature of patients with MRT/SCCOHT-family tumors.12 The goal of our study was therefore to clarify the nosologic relationship of SDTS with sarcoma versus carcinoma, and to expand on clinicopathologic and genomic characteristics of this recently described entity. In particular, we aimed to clarify how these tumors differ from substantially more common SMARCA4-deficient NSCCs (SD-NSCCs) lacking undifferentiated/sarcomatoid features. We therefore identified thoracic tumors with SMARCA4 deficiency and undifferentiated round cell and/or rhabdoid morphology (n = 22), which given the aforementioned uncertainty regarding their histogenesis, we designated as SMARCA4-deficient thoracic sarcomatoid tumors (SD-TSTs). Here, we report on a comprehensive analysis of their clinicopathologic, immunohistochemical, and genomic features in comparison with a set of conventional SD-NSCCs (n = 45).
Materials and Methods
Study Design and Sample Selection
The study was performed with the approval of the institutional board of Memorial Sloan Kettering Cancer Center, New York. SD-TSTs were identified using three separate approaches. First, nine cases were identified prospectively during the period of 2016 to 2017, following the description SDTS.18 Second, eight cases were identified by means of a retrospective re-review of thoracic tumors harboring SMARCA4 truncating mutations or deletions in cBioPortal database of MSK-IMPACT next-generation sequencing (NGS) results.27,28 Third, five cases were identified by means of a retrospective search of pathology database for unclassified undifferentiated thoracic tumors in patients who are below 40 years or whose pathology reports contained search terms “rhabdoid,” “NUT,” or “SMARCB1/INI1/BAF47.” Original diagnoses for retrospectively identified cases are summarized in Supplementary Table 2. In addition, as a control group, 45 conventional lung NSCCs (comprising predominantly adenocarcinomas) with SMARCA4 truncating mutations and SMARCA4 loss by immunohistochemistry (IHC) (i.e., SD-NSCC) were selected from the retrospective search of cBioPortal.
The criteria for SD-TST in this study included (1) undifferentiated round cell and/or rhabdoid morphology (see Results), (2) SMARCA4 loss by IHC (see Supplementary Table 3), and (3) lack of epithelial adhesion molecule claudin-4 to confirm the loss of epithelial differentiation in specimens where full evaluation of morphology was limited by crush artifact and/or necrosis. Other markers (SMARCA2 loss and expression of 2–3 stem cell markers including SALL4, CD34, or SOX2) were recently proposed as additional criteria for SDTS.26 However, on the basis of review of published cases (Supplementary Table 1) and our exploratory analysis, expression of these markers is not fully sensitive or specific for SD-TSTs relative to SD-NSCC. In particular, SOX2 is known to be commonly expressed in squamous and neuroendocrine lung carcinomas29 and was reported to be positive in 33% of SD-NSCCs,19 which was in line with our exploratory findings; therefore, this marker was not utilized in the current series.
IHC
All 67 SMARCA4-deficient tumors (22 SD-TSTs and 45 SD-NSCCs) were analyzed for expression of SMARCA4, SMARCA2, claudin-4, SALL4, and CD34. In addition, SD-TSTs were analyzed for multiple additional IHC markers, including keratins, EMA, TTF1, and p40 among others, as summarized in Supplementary Table 3, which provides a detailed description of antibodies, IHC protocols, and scoring criteria.
Molecular and Cytogenetic Studies
A total of 16 SD-TSTs and 45 SD-NSCCs were analyzed using MSK-IMPACT (a Hybrid Capture-based NGS platform) for somatic mutations in 341 (v3), 410 (v4), or 468 (v5) cancer genes, as previously described.28 Two older, retrospectively identified SD-TSTs had been analyzed by Sequenom mass spectrometry genotyping for hotspot mutations in key lung NSCC oncogenes.30
Using NGS results, all silent and nonsilent mutations were used to identify different mutational signatures according to the distribution of the six substitution classes (C>A, C>G, C>T, T>A, T>C, T>G) and their trinucleotide context; for each sample, a weight corresponding to the percentage of mutations explained by each of 30 mutational signatures was calculated, as previously described.28,31 Fraction and Allele-Specific Copy Number Estimates from Tumor Sequencing analysis (FACETS) was performed to determine allele-specific copy number changes of SMARCA4 locus.32 Tumor mutation burden (TMB) was assessed as previously described.28
Fluorescence in situ hybridization (FISH) was performed on interphase nuclei using custom bacterial artificial chromosome probes flanking SMARCA4 (19p13) locus using a standard procedure.
Statistical Analysis
Clinicopathologic parameters were compared using Fisher’s exact or Student’s t test. Survival was estimated using the Kaplan-Meier method with time origin at the time of diagnosis and was compared using log-rank test. Statistical analysis was conducted using R 3.3.2 (https://www.R-project.org/).
Results
Clinicoradiologic Characteristics
As summarized in Table 1, patients with thoracic SD-TSTs (n = 22) were predominantly male (73%) with a mean age of 58 years (range = 30–80; six patients ≤ 50 y). All except one patient were smokers with mean packyear smoking history of 51 (range = 0–189). Most patients (n = 17) were heavy smokers (≥ 20 pack-years).
Table 1.
SMARCA4-Deficient Thoracic Sarcomatoid Tumors: Patient and Sample Characteristics
| Case ID | Age | Sex | Smoking Pack-Years | CT Findings in Non-Tumor Lung | Primary Tumor Locationa | Primary Tumor Size (cm) | SUV | Stage at Diagnosis | Specimen Siteb | Specimen Typeb |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 68 | M | 70 | Emphysema | Lung > Med | 4.9 | na | IV | Lymph node | Core bx |
| 2 | 59 | M | 98 | Emphysema | ? Lung vs. Med (2 masses) | 7.6 | 22.5 | IV | Lymph node | Excisional Bx |
| 3 | 64 | M | 5 | Emphysema | Lung > Med | 11.7 | 27.4 | IV | Lung | FNA |
| 4 | 53 | M | 48 | Emphysema, bullae | ? Lung vs. Med | 17.9 | 8.1 | IV | Mediastinum | Core bx |
| 5 | 33 | F | 0 | ILD | Lung > Med | 12.8 | 18 | IV | Lung | Core bx |
| 6 | 60 | M | 40 | Emphysema | ? Lung vs. Med | 10 | 17.7 | IV | Lung | Core bx |
| 7 | 45 | F | 12 | na | Lung | 6.5 | na | IV | Lung | Lobectomyc |
| 8 | 71 | M | 2 | Bullae, blebs | Lung > Med | 6.8 | 6.5 | IV | Lung | Core bx |
| 9 | 80 | M | 80 | Emphysema | Lung > Med | 13 | 13.2 | IV | Lung | Core bx |
| 10 | 58 | M | 126 | Emphysema, bullae | ? Lung vs. Med | 5.6 | 20.1 | IV | Lung | Bronchoscopic bx |
| 11 | 64 | M | 57 | Emphysema | ? Lung vs. Med | 12.7 | 16.3 | IV | Lymph node | FNA |
| 12 | 30 | M | 20 | Bullae | Lung | 4 | na | IV | Lung | Core bx |
| 13 | 74 | M | 60 | Emphysema | Lung > Med | 7.9 | 12.5 | IV | Lung | Bronchoscopic bx |
| 14 | 55 | F | 19 | Emphysema | Lung > Med | 18.3 | 11.6 | IV | Mediastinum | Core bx |
| 15 | 59 | F | 65 | Emphysema | Lung > Med | 4.4 | na | IV | Lung | Core bx |
| 16 | 78 | M | 189 | Emphysema | ? Lung vs. Med (2 masses) | 10 | na | IV | Mediastinum | Core bx |
| 17 | 37 | M | 23 | Emphysema, bullae | Lung | 15 | 24.6 | IV | Lung | Lobectomyc |
| 18 | 57 | F | 40 | Emphysema | ? Lung vs. Med | 7.5 | 10.2 | IV | Lung | Lobectomyc |
| 19 | 69 | M | 20 | Unremarkable | Lung | 10 | 11.4 | IV | Bone | Core bx |
| 20 | 67 | F | 53 | Emphysema | Lung | 2.2 | 12.3 | IB | Lung | Lobectomy |
| 21 | 46 | M | 60 | Unremarkable | Lung > Med | 6 | 11.2 | IV | Lung | Pneumonectomyc |
| 22 | 50 | M | 37 | Emphysema, bullae | Lung | 7.6 | 19.6 | IIIA | Lung | Core bx |
Lung > Med: involving both lung and mediastinum but with epicenter in the lung therefore favoring a lung origin; ? Lung vs. Med: extensively involving both sites with uncertain epicenter
For patients with >1 diagnostic sample, largest specimen site and type are shown.
Resections in patients with stage IV disease were performed for palliative purposes.
bx, biopsy; F, female; FNA, fine needle aspiration; ILD, interstitial lung disease; M, male; Med, mediastinum; na, not available; SUV, standardized uptake value.
Primary tumors were typically large (mean size = 9.2 cm; range = 2.2–18.3 cm) and highly PET-avid (mean standardized uptake value = 16; range = 7–27). Most patients had extensive involvement of thoracic structures, including infiltration of mediastinum, chest wall, or the entire hemithorax. On the basis of computed tomography scans, tumors were of definite pulmonary origin in six patients, whereas both lung and mediastinum were involved in 16 patients, of whom five had dominant epicenter in the lung and seven had unclear site of origin owing to either extensive infiltration of both structures (Fig. 1) or presence of separate large masses in both lung and mediastinum. Pulmonary origin in cases with unclear epicenter was subsequently supported by genomic studies (see section on Genomic Signature Analysis).
Figure 1.

Representative radiologic characteristics of SMARCA4-deficient thoracic sarcomatoid tumors. Axial (A) and coronal (B) Computed tomography images from “patient 11” illustrate large, central tumor extensively involving both pulmonary and mediastinal structures with unclear epicenter. Non-neoplastic lung shows marked emphysema in line with the patient’s history of heavy smoking. Subsequent molecular studies revealed presence of a smoking signature, supporting a lung origin.
Nearly all patients (91%) had stage IV disease at presentation. Common sites of metastases included lymph nodes (59%), bone (55%), and adrenal glands (27%) (Supplementary Table 4). Six patients had bulky (5–18 cm) abdominal tumors, usually involving peritoneum. In two patients, the size of metastatic tumors was similar to or exceeded that of the thoracic tumor, prompting initial consideration of an abdominal primary. No brain metastases were documented in any patients.
Notably, radiologic evidence of emphysema was evident in 76% of patients (16/21) with available computed tomography scans, including all patients with uncertain lung versus mediastinal origin (Fig. 1). Emphysema was also evident in a young (37-year-old) smoker. In addition, the non-tumor lung in the sole never-smoker (33-year-old woman) showed marked interstitial lung disease in the setting of a clinical diagnosis of scleroderma (Supplementary Fig. 1).
Morphological Features
As summarized in Supplementary Table 5, 17 of 22 SD-TSTs showed entirely sarcomatoid (undifferentiated round cells/rhabdoid) morphology, whereas five cases also contained areas of conventional NSCC (i.e., combined/composite SD-TSTs). The characteristics of sarcomatoid areas in all tumors are discussed jointly first, and the findings in combined tumors are discussed further below.
Morphologically, all sarcomatoid areas were characterized by round cell morphology with variable admixture of classic rhabdoid cells. Hallmarks of round cell morphology were discohesive round to oval cells with prominent nucleoli and overall monomorphism despite being overtly high grade (brisk mitotic activity and in most cases extensive necrosis) (Fig. 2A and B). Rhabdoid cells with distinctive hyaline cytoplasmic inclusions were present in 18 of 22 cases, in most cases focally (Figs. 2A–C). Despite the overall cytologic monomorphism, focally moderate pleomorphism, including scattered tumor giant cells, was seen in four cases (Fig. 2D). Notable morphologic variants included an alveolar pattern (focal in nine cases; diffuse in one case) and a reticular-myxoid pattern (three cases) (Supplementary Fig. 2), as noted in prior studies.19
Figure 2.

Histopathologic features of SMARCA4-deficient thoracic sarcomatoid tumors. A and B illustrate hallmark morphology: undifferentiated round to plasmacytoid cells with prominent nucleoli, discohesion, and overall monomorphism. Classic rhabdoid cells with hyaline cytoplasmic inclusions indenting the nuclei were present focally in most cases (A, B; black circles); in few cases they were a predominant feature (C; inset). Several cases showed increased pleomorphism, entering in close differential diagnosis with NSCC (D). Distinctive cells with compressed crescent-shaped peripheral nuclei were noted in most cases (blue circles). Typical immunohistochemical features included the loss of SMARCA4 (E) and SMARCA2 (F) with retained expression in normal inflammatory and stromal cells, lack of claudin-4 (G) and weak (or in other cases entirely negative) keratins (H).
Immunohistochemical Features
Key immunohistochemical features in sarcomatoid areas of 22 SD-TSTs are summarized in Supplementary Table 5 and illustrated in Figure 2. On average, two keratin stains were performed per case (range = 1–6), revealing either negative or focal/weak labeling in 17 cases (77%), whereas four cases had focally moderate labeling, and one case had diffuse dot-like reactivity, as previously noted in MRTs.12 All tested cases showed patchy to diffuse labeling for EMA. Epithelial adhesion molecule claudin-4 was consistently negative in all cases but showed focal (≤ 5%) weak to moderate membranous labeling in six cases. Vimentin was strongly positive in six of seven tested cases. Weak/focal expression of p40 and TTF1 was seen in one and three cases, respectively. Stem cell markers SALL4 and CD34 were expressed in 32% and 18% of cases, respectively. In addition, a subset of cases was tested for CD99, revealing membranous labeling in three of 10 cases (one diffuse, two focal). All tested cases exhibited retained expression of SMARCB1 (n = 7) and were negative for NUT (n = 9).
In line with the inclusion criteria, all 22 cases were deficient for SMARCA4 (BRG1) expression, manifesting either as either complete loss (n = 18) or “severe global reduction” (n = 4; see Discussion). SMARCA2 (BRM) was lost in 18 of 22 tumors. Notably, tumors with retained SMARCA2 exhibited identical sarcomatoid morphology and lacked claudin-4 as other SD-TSTs.
Synaptophysin labeling was seen in 16 cases (71%) and was diffuse in most cases. Ki67 proliferation rate was consistently high (mean = 79%, range = 40%–100%). The combination of strong synaptophysin expression, high Ki67, and frequent crush artifact with geographic necrosis closely resembled the appearance of neuroendocrine carcinomas, resulting in an initial diagnosis of large cell or small cell neuroendocrine carcinomas in five cases (23%) (Supplementary Table 2).
Combined SD-TSTs: Morphological and Immunohistochemical Features
Five SD-TSTs contained areas of conventional NSCC juxtaposed with sarcomatoid areas (case ID 18–22; Supplementary Table 5). Areas of NSCC were focal in four cases and predominant in one case. As illustrated in Figures 3 and 4, the morphology of sarcomatoid areas was identical to that of pure tumors, featuring discohesive round/rhabdoid cells. Transition from NSCC to sarcomatoid areas was abrupt in most areas, but some areas showed gradual transition through intermixed/indeterminate histology in between NSCC and sarcomatoid. This transition was strikingly highlighted by a sharp drop in epithelial markers claudin-4 and keratins, although a spectrum of staining intensities was seen in the areas of gradual transition (Supplementary Fig. 3).
Figure 3.

Histopathologic features of SMARCA4-deficient composite tumors (case #20). (A, B) Hematoxylin and eosin section showing an abrupt transition from cohesive NSCC (right) to discohesive undifferentiated round cell/rhabdoid histology (left; Undif). Cell-to-cell cohesion is evident in NSCC component (right inset) compared with discohesion of round cell/rhabdoid component (left inset). The transition is accompanied by a sharp loss of SMARCA2 (D), keratins (E) and claudin-4 (F), superimposed on SMARCA4 deficiency in both components (C). SMARCA4 and SMARCA2 positive cells within undifferentiated component in C and D represent lymphocytes and stromal cells. Undif, undifferentiated.
Figure 4.

Histopathologic features of SMARCA4-deficient composite tumors (case #22). Hematoxylin and eosin section (A, B) illustrates a tumor with predominantly undifferentiated round cell morphology with focal myxoid features containing scattered islands of NSCC with squamoid morphology. Transition between carcinomatous and undifferentiated areas is highlighted by the sharp loss of SMARCA2 (D), keratins (E), claudin-4 (F) and p40 (not shown) superimposed on homogeneous SMARCA4-deficiency in both components (C).
In all combined cases, SMARCA4 staining was negative in both NSCC and sarcomatoid areas. Remarkably, SMARCA2 showed a striking dichotomous expression in two cases, exhibiting a sharp loss of expression in transition from NSCC to sarcomatoid areas (Figs. 3D and 4D). In other composite cases, SMARCA2 expression was either retained (n = 2) or absent (n = 1) throughout.
Comparison of Clinicopathologic Characteristics of SD-TSTs Versus SD-NSCCs
We next compared clinicopathologic features of SD-TSTs (n = 22) to those of SMARCA4-deficient conventional NSCCs lacking undifferentiated round cell/rhabdoid features (SD-NSCC; n = 45) (Figs. 5A–B). As summarized in Figure 5C, compared with SD-NSCC, SD-TSTs were over-represented in males and were associated with heavier smoking history despite more frequent occurrence in younger patients. The most striking difference was the substantially larger primary tumor size of SD-TSTs compared with SD-NSCCs (mean = 9.2 cm versus 3.2 cm, respectively; p < 0.0001). By IHC, in stark contrast with SD-TSTs, all SD-NSCCs exhibited diffuse or near-diffuse membranous labeling for claudin-4 (p = 0.0001), and none of the cases exhibited complete loss of SMARCA2 (p = 0.0001), whereas expression of stem-cell markers was either absent (SALL4) or rare (CD34; 7% of cases).
Figure 5.
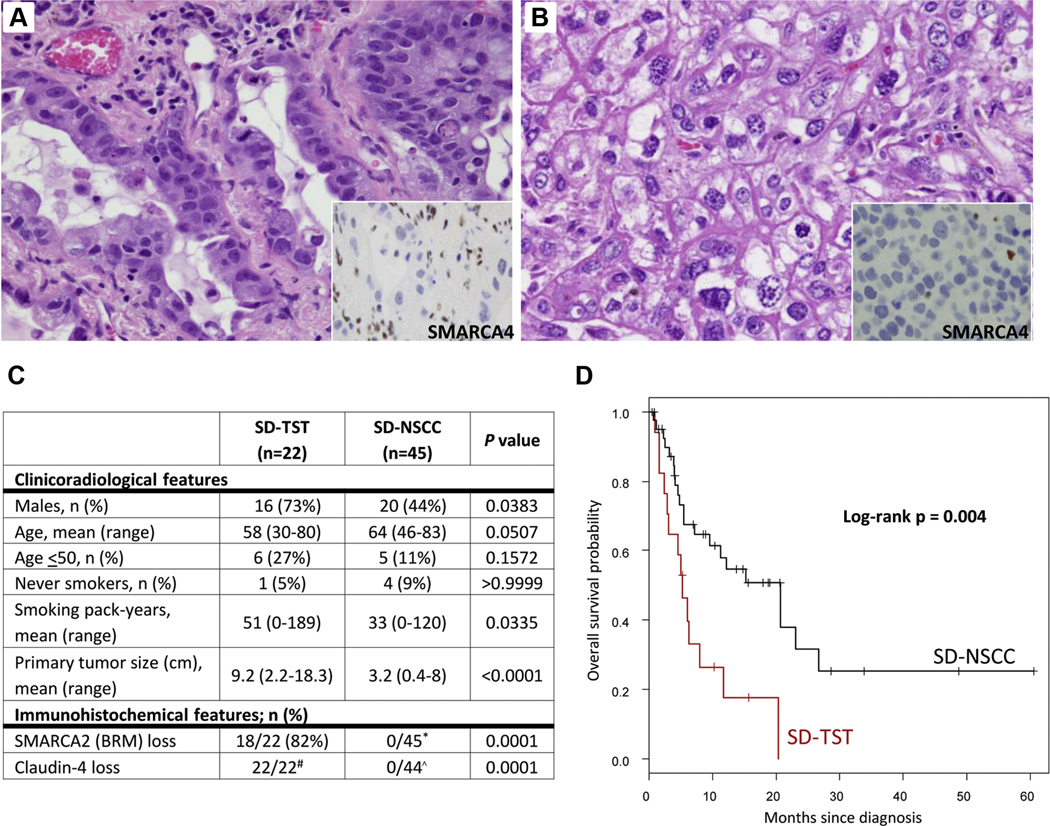
Comparison of SMARCA4-deficient thoracic sarcomatoid tumors (SD-TST) versus SMARCA4-deficient NSCC (SD-NSCC). (A, B) Histologic features of SD-NSCC illustrating conventional acinar (A) or solid (B) morphology lacking undifferentiated round cell/rhabdoid features. Hallmarks include cellular cohesion and lack of monomorphism. Insets show the loss of SMARCA4 expression in tumor cells compared with entrapped benign cells. (C) Comparison of clinicopathologic features in SD-TSTs and SD-NSCCs. *Mosaic pattern of SMARCA2 expression (positive cells intermixed with low/negative cells) was seen in some SD-NSCCs but was not regarded as a loss. #Claudin-4 loss in SD-TSTs was diffuse in 16 cases, and near-diffuse (retained expression in < 5% of tumor cells) in six cases (see Supplementary Tables 3 and 5). ^All SD-NSCCs had diffuse or near-diffuse expression of claudin-4. (D) Comparison of overall survival for stage IV SD-TSTs (n = 19) versus SD-NSCCs (n = 40).
Survival analysis was performed by log-rank test for stage-matched (stage IV) patients with the median time to death or last follow-up from the time of diagnosis of 7 months (range = 1–20). This revealed that overall survival of patients with SD-TSTs was considerably worse than that for patients with SD-NSCC (median survival = 5.2 mo versus 20.7 mo, respectively; p = 0.004) (Fig. 5D).
Molecular and Cytogenetic Features of SD-TSTs
A total of 16 SD-TSTs were analyzed by NGS and two additional cases by Sequenom genotypic assay (Fig. 6, Supplementary Table 6). Tumors analyzed by NGS harbored on average 16 nonsynonymous mutations per case (range = 4–34). TP53 was the most frequently mutated gene (88%). Overall, 50% of cases with molecular analysis (9/18) harbored alterations typical of smoking-associated NSCC,33 including STK11 (n = 6), KEAP1 (n = 4), or KRAS (n = 5); three cases harbored concurrent KRAS/STK11/KEAP1 mutations. KRAS mutations comprised 2 G12C, 2 G12V, and Q61H alterations. In addition, three cases harbored NF1 mutations — another alteration seen in NSCC with predominance in smokers.34
Figure 6.

Genomic profiles of SMARCA4-deficient thoracic sarcomatoid tumors. (A) OncoPrint illustrating recurrent genomic alterations in SD-TSTs. Key characteristics of patients and SMARCA4 locus analysis by FISH and FACETS are annotated. CN-LOH: copy-neutral LOH, HOM: homozygous deletion. For FISH, ++ indicates both alleles intact, ↓+ heterozygous loss/deletion, and ↓↓ homozygous loss/deletion. CT:? lung versus mediast denotes tumors with uncertain site of origin on initial imaging studies. (B) Comparison of genomic alterations in SD-TSTs and SD-NSCC. None of the parameters showed statistically significant differences. (C) Analysis of genomic smoking signature in 16 SD-TSTs versus 45 ST-NSCC versus 44 sarcomas, including soft tissue (n = 38) and thoracic (n = 6) sarcomas.
For comparison, review of published NGS data for 933 soft tissue sarcomas in cBioPortal revealed that mutations in KRAS, STK11, or KEAP1 are either absent or extremely rare in those tumors (Supplementary Fig. 4).
SMARCA4 gene alterations were identified in 14 of 16 SD-TSTs; two cases had no detectable mutations in SMARCA4 by NGS despite complete loss of SMARCA4 expression by IHC. SMARCA4 alterations comprised nonsense mutations (n = 7), splice site mutations (n = 4), and deletions (n = 3) (Supplementary Fig. 4). By FACETS, SMARCA4 mutations were accompanied by loss of heterozygosity (LOH) in all cases, which was primarily copy-neutral (i.e., deletion of wild-type allele accompanied by duplication of mutant allele). LOH was detected in both cases that lacked detectable SMARCA4 mutations.
FISH for SMARCA4 locus was performed on 20 SD-TSTs (14 with NGS and six without NGS), revealing detectable alterations in only seven cases (35%) (Supplementary Table 7). As expected, most cases with copy-neutral LOH by FACETS were detected as diploid by FISH.
All SMARCA4 alterations detected by NGS were somatic, given that the MSK-IMPACT bioinformatic pipeline filters out germline variants found in the patient’s paired normal DNA. In addition, the 33-year-old never-smoker consented to dedicated germline testing, which revealed no evidence of germline SMARCA4 alterations.
Comparison of mutation profiles in SD-TST and SD-NSCC (Fig. 6B) revealed overall comparable distribution of major genomic alterations. However, the percentage of cases lacking major alterations characteristic of NSCC (KRAS, STK11, KEAP1, or any other drivers) was enriched among SD-TSTs (56%) compared with SD-NSCC (27%).
TMB in SD-TSTs was comparable with that of SD-NSCC (mean = 14.2 versus 15.8 mutations per mega-base, respectively).
Genomic Signature Analysis
Analysis of NGS data revealed a dominant smoking/tobacco signature (reflecting the predominance of G>T transversion mutations) in both SD-TSTs (14/16 cases, 88%) and SD-NSCC (64%) (Fig. 6C, Supplementary Table 8). Conversely, genomic signature analysis using the same method on 44 conventional sarcomas (including six thoracic sarcomas, of which three patients were smokers) revealed no smoking/tobacco signature in any cases (Fig. 6C, Supplementary Table 9).
Notably, six SD-TSTs with available genomic analysis were from patients with unclear site of origin as lung versus mediastinum on the basis of radiologic features; all of these tumors exhibited smoking/tobacco signature and/or NSCC-type mutations (Fig. 6A).
Clinicopathologic and Molecular Comparison of Pure Versus Combined SD-TSTs
As summarized in Supplementary Table 10, SD-TSTs with and without epithelial elements had overall comparable clinicopathologic characteristics. However, although not statistically significant because of low case number, combined SD-TSTs were more frequently encountered in resected tumors, and were enriched in cases with retained SMARCA2, negative stem cell markers, and presence of NSCC-type alterations.
Discussion
This is the largest series to date on SMARCA4-deficient thoracic tumors (n = 67), encompassing both tumors with sarcomatoid round cell/rhabdoid features, which in the literature to date have been referred to as “SMARCA4-deficient thoracic sarcomas,” and SD-NSCCs lacking sarcomatoid morphology. We confirm and expand on the prior findings that SD-TSTs have a highly distinctive clinicopathologic characteristics compared with SD-NSCC. However, our findings provide several lines of evidence that in most cases SD-TSTs are histogenetically related to carcinomas/epithelial progenitors rather than primary thoracic sarcomas.
Nosologic Relationship to Sarcoma Versus Carcinoma
In this series, the relationship of SD-TST with NSCC was supported by the following observations: (1) documentation of a NSCC component in five of 22 SD-TSTs; (2) focal expression of NSCC lineage markers, TTF1 or p40, in four additional cases; (3) smoking history in all except one patient, accompanied by radiologic evidence of emphysema, and (4) genomic alterations characteristic of smoking-related NSCC, including a dominant smoking signature, high TMB (average = 14.2 mutations per megabase) and smoking-associated NSCC-type mutations (KRAS, STK11, KEAP1) in a subset of cases. Furthermore, the pattern of metastatic spread for SD-TSTs was more typical of carcinoma than sarcoma, involving the lymph nodes, bones, and adrenal glands. Although several aforementioned features were noted in prior studies and hinted at the epithelial derivation of SD-TSTs, the current study included the first detailed description of epithelial components in some SD-TSTs. Furthermore, our study included comprehensive NGS with genomic signature analysis on the largest set of SD-TSTs to date, which in aggregate with other observations provide compelling arguments linking SD-TSTs to NSCC.
On the basis of the above considerations, we postulate that pulmonary SD-TSTs are conceptually analogous to sarcomatoid carcinomas — tumors that in various organs are understood fundamentally as carcinomas that have undergone epithelial-mesenchymal transition. A similar concept of “epithelial-rhabdoid transition” or “rhabdoid/round cell de-differentiation” is a well-recognized phenomenon in tumors of various sites,14 which has been recently linked to inactivation of SWI/SNF components superimposed on the genomic alterations of the parent neoplasm.11,13 Our findings for SD-TSTs are thus in line with the general concept of the rhabdoid/round cell phenotype in tumors arising in visceral organs of adults representing the final common pathway of de-differentiation mediated by SWI/SWF complex abnormalities. Although the lineage of rhabdoid cells has been long debated, recent data linking these cells to primitive mesenchymal cells supports the concept of SD-TSTs as a special type of sarcomatoid carcinoma.35 Although epithelial differentiation is evident histologically in only a minority of SD-TSTs, sarcomatoid carcinomas are known to frequently consist entirely of sarcomatoid elements.36
SMARCA4-Deficient Sarcomatoid Tumors Versus SMARCA4-Deficient NSCC: Distinctive Histopathologic Features and Marker Expression
Beyond the issue of nosologic status of SD-TSTs as sarcomatoid carcinoma versus sarcoma, the other major question investigated in this study was a comparison of clinicopathologic features of SD-TSTs and the substantially more prevalent SMARCA4-deficient NSCCs lacking sarcomatoid features (SD-NSCC). Prior studies included a small number of SD-NSCCs for direct comparison with SDTS,18,19 whereas here we expanded this analysis to a cohort of 45 such cases.
We confirm that at histologic level, SD-NSCC represent conventional carcinomas, whereas SD-TSTs have a distinct and recognizable undifferentiated round cell or fully rhabdoid morphology that is closely analogous to that of pediatric MRTs. Predominant round cell morphology also occurs in MRTs and can be thought of as a spectrum of rhabdoid phenotype.37 Marker expression in SD-TSTs is also distinctive, comprising negative or low keratin expression, consistent lack of epithelial adhesion molecule claudin-4, codeficiency with the second major catalytic subunit of SWI/SNF complex SMARCA2 (BRM) in most cases, and frequent but variable expression of stem cell markers. In particular, the lack of claudin-4 and SMARCA2 represents important parallels between SD-TSTs and MRT/SCCOHT, which consistently lack claudin-4 even in the presence of some keratin expression,38 and in which SMARCA4 deficiency is consistently accompanied by SMARCA2 co-inactivation.39 We also note striking parallels in morphology and marker expression between SD-TSTs and the recently described de-differentiated/undifferentiated endometrial carcinomas.40
Notably, in our series, the loss of both claudin-4 and SMARCA2 was entirely specific to SD-TSTs compared with SD-NSCCs, although variable results have been previously reported for SMARCA2 codeficiency in SD-NSCC,19,21,41 suggesting that the dual SMARCA4/SMARCA2 loss may uncommonly occur in NSCC. We note that SMARCA4 deficiency in NSCC is known to be associated with poor differentiation41 and features of epithelial-mesenchymal transition16; however, rhabdoid-like phenotype has distinctive aforementioned characteristics representing a special and distinct type of sarcomatoid carcinomas.
Thus, in the thorax, SMARCA4 deficiency occurs in two different phenotypic settings: conventional carcinomas (SD-NSCC—claudin-4-positive, SMARCA2proficient) and sarcomatoid round cell/rhabdoid tumors (SD-TST—claudin-4-negative, most SMARCA2deficient). Overall, we found that in virtually all cases, these two groups of SMARCA4-deficient tumors were readily separable morphologically and immunohistochemically. However, in a minority of cases, atypical IHC profiles, including low level of epithelial marker expression or retained SMARCA2 expression, combined with focal areas of increased pleomorphism/cohesion, blurred the distinction between sarcomatoid and carcinomatous differentiation. In fact, the possibility that a minority of SMARCA4-deficient tumors exist in a biological continuum between these phenotypic states — as exemplified by transitional areas in combined tumors — cannot be excluded, and further studies will be required to clarify the diagnostic approach to such cases.
Composite SD-TSTs and Potential Model of Pathogenesis
To our knowledge, only one SD-TST containing mature epithelial elements has been reported to date.21 Here we describe in detail five such cases, which provide direct evidence for the link between SD-TSTs and NSCC.
A remarkable and novel observation in several composite cases in our series was that of a sharp loss of SMARCA2 in transition from carcinomatous to sarcomatoid areas, in line with the suggested importance of SMARCA2 co-inactivation in the pathogenesis of rhabdoid-like phenotype,39 notwithstanding few SD-TSTs with retained SMARCA2 in the current and prior series.19 Apparent enrichment of retained SMARCA2 in combined SD-TSTs may suggest that de-differentiation in tumors with more pronounced epithelial differentiation may be more likely to be mediated by SMARCA2-independent mechanisms.
A potential model of pathogenesis of thoracic SD-TSTs is depicted in Figure 7. It illustrates stepwise inactivation of SMARCA2 from preexisting SMARCA4-deficient carcinomatous elements, as directly reported by several composite cases in our series. However, the fact that most SD-TSTs lack evidence of carcinomatous components may suggest that SMARCA4/A2 co-inactivation tends to occur as an early event in the pathogenesis of SD-TSTs, in line with prior suggestion that the timing of SWI/SNF inactivation may determine the extent of residual carcinomatous components.42 Apparent enrichment of NSCC-type mutations and lower stem cell marker expression in combined cases may also suggest that the timing of rhabdoid divergence could be associated with a spectrum of phenotypic and genomic features in SD-TSTs. Shared clinicopathologic characteristics of fully undifferentiated tumors and those with residual NSCC components supports their understanding as a single family of tumors.
Figure 7.

A model for SMARCA4-mediated pathogenesis of thoracic tumors, depicting solitary SMARCA4 deficiency in conventional NSCC, and dual SMARCA4/A2 deficiency in sarcomatoid/undifferentiated tumors. The timing of SMARCA4/A2 may determine the presence and extent of carcinomatous components and likelihood of NSCC-type alterations (see Discussion). *denotes that SMARCA2-independent mechanisms may cooperate with SMARCA4 inactivation in a minority of cases.
SD-TST Versus Other Entities: Diagnostic Considerations
Beyond NSCC, a variety of other entities come up in the differential diagnosis with SD-TSTs. Given the lack of familiarity with histologic features of this recently described entity combined with peculiar marker expression, SD-TSTs may present a major diagnostic challenge, as reflected by the variety of original diagnostic consideration in tumors identified retrospectively or submitted to us for consultation (Supplementary Table 2). In particular, the combination of overtly high-grade features (high proliferation rate, necrosis) with overall monomorphic cytomorphology evokes the differential diagnosis of NUT carcinoma, round cell sarcomas, and neuroendocrine carcinomas (particularly given frequent synaptophysin expression). The lack of keratin expression in combination with stem cell marker expression may raise a consideration of germ cell tumors (SALL4+), epithelioid angiosarcoma/epithelioid sarcoma/leukemic infiltrate (CD34+), and Ewing sarcoma (CD99+). Awareness of SD-TSTs morphology and marker expression should lead to their increasingly accurate diagnosis and distinction from various mimics in practice.
Genomic Findings
Our study offers several novel insights related to SMARCA4 genomic alterations and expression. First, our series supports prior observation that although most SD-TSTs exhibit complete loss of SMARCA4 expression, some tumors retain protein expression at a low level, previously termed “severe global reduction.”19,22 In such cases, truncated SMARCA4 could be partially expressed and detected by the SMARCA4 antibody that recognizes the N-terminal epitope.
Second, our findings confirm prior observations that genomic mechanisms that mediate SMARCA4 inactivation are variable, and comprise primarily truncating mutations combined with LOH, whereas large chromosomal losses/deletions account for only a minority of cases. Notably, we found that in most cases, LOH is copy-neutral (i.e., accompanied by duplication of the mutated allele), and is therefore not detected by FISH. Thus, in contrast to SMARCB1-deficient neoplasms, FISH has only a limited role in the evaluation of SD-TSTs.
A potentially surprising finding was that in two SD-TSTs featuring complete loss of SMARCA4 expression by IHC, SMARCA4 mutations could not be identified by NGS despite full exon coverage by our NGS panel. This phenomenon has also been documented in several SMARCA4-deficient adenocarcinomas.21 It is possible that the loss of SMARCA4 in such cases is mediated by structural variants (such as translocations) involving the poorly covered intronic regions. Overall, these data highlight the importance of IHC for establishing SMARCA4 deficiency.
Although our NGS panel did not encompass the SMARCA2 gene, prior studies in SCCOHT found a consistent lack of mutations, suggesting epigenetic or post-translational regulation.39
Our findings regarding overall mutation profiles of SD-TSTs in smokers are notable for the parallels with SD-NSCC, including the high TMB (defined as > 13.8 mutations per megabase28) and the presence of classic smoking-associated NSCC mutations involving KRAS, SKT11, and KEAP1 in a subset of cases.33 Apparent enrichment in NF1 mutations may be of interest given their previously reported association with undifferentiated histology in NSCC.19 We did note, however, that SD-TSTs were enriched in cases lacking any established or putative NSCC drivers compared with SD-NSCC, suggesting that SMARCA4/A2 codeficiency may itself serve as a major mitogenic driver in some cases or may potentiate concurrent drivers in other cases.
Regarding comparison with sarcomas, we demonstrate the presence of a dominant smoking/tobacco signature in virtually all SD-TSTs in smokers, whereas such signature was entirely absent in soft tissue and thoracic sarcomas analyzed on the same platform. Furthermore, no smoking signature has been detected in soft tissue sarcomas in prior studies.43 Similarly, high TMB in SD-TSTs contrasts sharply with characteristically low TMB in soft tissue sarcomas (mean = 1.06 per Mb),43 MRTs,44 and SCCOHTs.45 Finally, NSCC-type mutations found in SD-TSTs are distinctly uncommon in sarcomas.
Distinctive Clinical Features: Exceptionally Aggressive, Large Tumor Size, Unusual Pattern of Metastases, Broad Age Range With Predilection for Young Smokers
Our study substantially expands on the distinctive clinicoradiologic features of SD-TSTs and highlights the differences from SD-NSCC.
First, we confirm that SD-TSTs are associated with exceptionally poor prognosis (median survival = 5.2 mo) and show that the prognosis is substantially worse than that for SD-NSCCs. Furthermore, most SD-TSTs are extremely large tumors, some filling the entire hemithorax, which on average are considerably larger than SD-NSCCs. Massive size of carcinomas with undifferentiated/rhabdoid features and their explosive clinical behavior is well documented in various organs.13,42
Second, these tumors have a distinctive pattern of metastasis. In particular, we noted the lack of brain metastases for SD-TSTs. We also confirm the observation by Yoshida et al.19 that these tumors have a predilection for bulky peritoneal metastases, which may raise a consideration of an abdominal primary.19 Thus, the pattern of spread of SD-TST both overlaps with NSCC (involving nodes, adrenal glands, and bones) but also has several distinctive features (lack of brain metastases, large peritoneal masses).
Finally, we confirm that SD-TST is predominantly a disease of smokers with predilection for men and a peculiar broad age distribution, comprising patients with the typical age for NSCC (> 50–80 years old), but also patients who are unusually young for NSCC (30–50 years old; 27% of patients). The typical molecular alterations for lung carcinomas in patients below 50 years include EGFR, ALK, and ROS1, all of which are associated with never-smoker status.46 Remarkably, this appears to be the first instance of a genomically defined pulmonary tumor to be associated with young smokers.
SD-TST in Never-Smokers and Young Patients
A particularly interesting minor subgroup of patients with SD-TST is that of young never-smokers. In this study, only one patient (33-year-old woman) was a never-smoker. In prior studies, 15% of patients were never-smokers, all of whom were young (31–38 years old) (Supplementary Table 1). A potentially remarkable feature of our patient was background interstitial lung disease in the setting of scleroderma. Intriguingly, in a study by Yoshida et al.,19 both young never-smokers with SD-TSTs had “emphysema/bullae,”19 raising the possibility of an etiologic link between interstitial lung injury and increased risk of SD-TSTs in young patients. In fact, precocious onset of emphysema in a young (37-year-old) smoker in our study and several similar patients in prior series19 may also suggest exaggerated sensitivity to smoking-induced cell injury as a potential etiologic factor in young smokers with SD-TST.
The question that is not entirely resolved by our study is whether SD-TSTs arising in young never-smokers could represent true thoracic sarcomas, unlike the phenotypically identical tumors that occur in smokers. This especially concerns rare tumors reported to arise in soft tissue of the chest wall.19 Continued study of this rare subset of SMARCA4-deficient thoracic tumors is needed to clarify their histogenesis.
Site of Origin
Another notable clarification provided by the current study relates to the site of origin of SD-TSTs. In prior studies, most of such tumors were regarded as being of mediastinal origin.18,20 Indeed, mediastinal involvement was seen in most patients in our series given the tumors’ central location, large size, and highly infiltrative nature; and for a subset of our patients, the site of origin was initially uncertain or was considered to be potentially mediastinal. However, consistent history of smoking, radiologic evidence of emphysema, and—most importantly—presence of genomic smoking signature and NSCC-type alterations in such tumors support their pulmonary origin. These findings do not argue against the possibility that some SD-TSTs may arise in the mediastinum, because a tumor with undifferentiated/rhabdoid features can occur in any organ including the thymus.47 However, our data suggest that most thoracic SD-TSTs are of pulmonary origin, despite extensive involvement of mediastinal structures.
SD-TST and Pulmonary Tumors With Rhabdoid Features
Although the description of SMARCA4 deficiency in undifferentiated round cell/rhabdoid thoracic tumors is a recent finding, we note that the concept of rhabdoid pulmonary tumors (both pure and composite) is not novel in thoracic pathology literature.48–51 Notably, association of such tumors with dismal prognosis, stem cell marker expression, and presentation at a young age has been recognized in those earlier studies, which undoubtedly included some tumors now recognized as SD-TSTs. In fact, SMARCA4 may not be the exclusive molecular event underlying round cell/rhabdoid de-differentiation in lung tumors, because SMARCA4 loss was found in most but not all thoracic tumors with such phenotype in our series (data not shown) and in the study by Sauter et al.20
Potential Treatment Implications
Therapeutic approaches to tumors with SMARCA4 and other SWI/SNF complex alterations is an area of active investigation.52 In particular, inhibitors of Enhancer of Zeste Homolog 2 histone methyltransferase have emerged as promising therapeutic agents for MRTs and SCCOHTs, with activity linked to SMARCB1 or dual SMARCA4/A2 inactivation.53 However, whether therapeutic vulnerabilities of SWI/SNF-inactivated tumors are shared across tumor types or may be influenced by cell lineage and overall mutational context (which are highly distinct for MRT/SCCOHT and SD-TSTs) remains an open question. Similarly, the effectiveness of NSCC-type therapies for SD-TSTs awaits clinical investigation. We note that the finding of high TMB nominates immune checkpoint inhibitors as a potentially attractive consideration for SD-TSTs,54 which could synergize with the increased immunotherapy responsiveness of tumors with SWI/SNF alterations,55 as supported by a recent case report.24 Overall, the determination of treatment approaches for SD-TSTs should be facilitated by the more precise pathologic and molecular understanding of these tumors.
Conclusion
In this study we provide clinicopathologic and genomic evidence that thoracic SMARCA4-deficient sarcomatoid tumors represent primarily smoking-related undifferentiated/de-differentiated carcinomas rather than primary thoracic sarcomas, although histogenesis of a minor subset of tumors arising in never-smokers requires further clarification. We also confirm and markedly expand on the distinctive clinicopathologic characteristics of these tumors, which should aid in their wider recognition in clinical practice. Studies are in progress at our institution to determine optimal clinical management of these exceptionally aggressive tumors.
Supplementary Material
Acknowledgments
This work was supported in part by NIH P01 CA129243 (NR, ML), and was made possible by the infrastructural support from Marie-José and Henry R. Kravis Center for Molecular Oncology, and the National Cancer Institute Cancer Center Core Grant P30-CA008748. We thank Cristina Antonescu and Lei Zhang for performing fluorescence in situ hybridization (FISH) assays, and Marina Asher and Denise Frosina for performing immunohistochemical studies.
Footnotes
Disclosure: The authors have no relevant conflicts of interest pertaining to this article.
Supplementary Data
Note: To access the supplementary material accompanying this article, visit the online version of the Journal of Thoracic Oncology at www.jto.org and at https://doi.org/10.1016/j.jtho.2019.10.023.
References
- 1.Roberts CW, Orkin SH. The SWI/SNF complex–chromatin and cancer. Nat Rev Cancer. 2004;4:133–142. [DOI] [PubMed] [Google Scholar]
- 2.Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. 2011;11:481–492. [DOI] [PubMed] [Google Scholar]
- 3.Arnaud O, Le Loarer F, Tirode F. BAFfling pathologies: alterations of BAF complexes in cancer. Cancer Lett. 2018;419:266–279. [DOI] [PubMed] [Google Scholar]
- 4.Hollmann TJ, Hornick JL. INI1-deficient tumors: diagnostic features and molecular genetics. Am J Surg Pathol. 2011;35:e47–e63. [DOI] [PubMed] [Google Scholar]
- 5.Schneppenheim R, Fruhwald MC, Gesk S, et al. Germline nonsense mutation and somatic inactivation of SMARCA4/BRG1 in a family with rhabdoid tumor predis-position syndrome. Am J Hum Genet. 2010;86:279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hasselblatt M, Gesk S, Oyen F, et al. Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am J Surg Pathol. 2011;35:933–935. [DOI] [PubMed] [Google Scholar]
- 7.Witkowski L, Carrot-Zhang J, Albrecht S, et al. Germline and somatic SMARCA4 mutations characterize small cell carcinoma of the ovary, hypercalcemic type. Nat Genet. 2014;46:438–443. [DOI] [PubMed] [Google Scholar]
- 8.Ramos P, Karnezis AN, Craig DW, et al. Small cell carcinoma of the ovary, hypercalcemic type, displays frequent inactivating germline and somatic mutations in SMARCA4. Nat Genet. 2014;46:427–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Foulkes WD, Clarke BA, Hasselblatt M, Majewski J, Albrecht S, McCluggage WG. No small surprise - small cell carcinoma of the ovary, hypercalcaemic type, is a malignant rhabdoid tumour. J Pathol. 2014;233:209–214. [DOI] [PubMed] [Google Scholar]
- 10.Fahiminiya S, Witkowski L, Nadaf J, et al. Molecular analyses reveal close similarities between small cell carcinoma of the ovary, hypercalcemic type and atypical teratoid/rhabdoid tumor. Oncotarget. 2016;7:1732–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fuller CE. All things rhabdoid and SMARC: an enigmatic exploration with Dr. Louis P. Dehner. Semin Diagn Pathol. 2016;33:427–440. [DOI] [PubMed] [Google Scholar]
- 12.Agaimy A, Foulkes WD. Hereditary SWI/SNF complex deficiency syndromes. Semin Diagn Pathol. 2018;35:193–198. [DOI] [PubMed] [Google Scholar]
- 13.Agaimy A. The expanding family of SMARCB1(INI1)deficient neoplasia: implications of phenotypic, biological, and molecular heterogeneity. Adv Anat Pathol. 2014;21:394–410. [DOI] [PubMed] [Google Scholar]
- 14.Wick MR, Ritter JH, Dehner LP. Malignant rhabdoid tumors: a clinicopathologic review and conceptual discussion. Semin Diagn Pathol. 1995;12:233–248. [PubMed] [Google Scholar]
- 15.Bell EH, Chakraborty AR, Mo X, et al. SMARCA4/BRG1 is a novel prognostic biomarker predictive of cisplatin-based chemotherapy outcomes in resected non-small cell lung cancer. Clin Cancer Res. 2016;22:2396–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsubara D, Kishaba Y, Ishikawa S, et al. Lung cancer with loss of BRG1/BRM, shows epithelial mesenchymal transition phenotype and distinct histologic and genetic features. Cancer Sci. 2013;104:266–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reisman DN, Sciarrotta J, Wang W, Funkhouser WK, Weissman BE. Loss of BRG1/BRM in human lung cancer cell lines and primary lung cancers: correlation with poor prognosis. Cancer Res. 2003;63:560–566. [PubMed] [Google Scholar]
- 18.Le Loarer F, Watson S, Pierron G, et al. SMARCA4 inactivation defines a group of undifferentiated thoracic malignancies transcriptionally related to BAF-deficient sarcomas. Nat Genet. 2015;47:1200–1205. [DOI] [PubMed] [Google Scholar]
- 19.Yoshida A, Kobayashi E, Kubo T, et al. Clinicopathological and molecular characterization of SMARCA4-deficient thoracic sarcomas with comparison to potentially related entities. Mod Pathol. 2017;30:797–809. [DOI] [PubMed] [Google Scholar]
- 20.Sauter JL, Graham RP, Larsen BT, Jenkins SM, Roden AC, Boland JM. SMARCA4-deficient thoracic sarcoma: a distinctive clinicopathological entity with undifferentiated rhabdoid morphology and aggressive behavior. Mod Pathol. 2017;30:1422–1432. [DOI] [PubMed] [Google Scholar]
- 21.Agaimy A, Fuchs F, Moskalev EA, Sirbu H, Hartmann A, Haller F. SMARCA4-deficient pulmonary adenocarcinoma: clinicopathological, immunohistochemical, and molecular characteristics of a novel aggressive neoplasm with a consistent TTF1(neg)/CK7(pos)/HepPar-1(pos) immuno-phenotype. Virchows Arch. 2017;471:599–609. [DOI] [PubMed] [Google Scholar]
- 22.Kuwamoto S, Matsushita M, Takeda K, et al. SMARCA4-deficient thoracic sarcoma: report of a case and insights into how to reach the diagnosis using limited samples and resources. Hum Pathol. 2017;70:92–97. [DOI] [PubMed] [Google Scholar]
- 23.Kunimasa K, Nakamura H, Sakai K, et al. Patients with SMARCA4-deficient thoracic sarcoma and severe skeletal-related events. Lung Cancer. 2019;132:59–64. [DOI] [PubMed] [Google Scholar]
- 24.Henon C, Blay JY, Massard C, et al. Long lasting major response to pembrolizumab in a thoracic malignant rhabdoid-like SMARCA4-deficient tumor [e-pub ahead of print]. Ann Oncol. 10.1093/annonc/mdz160, accessed December 6, 2019. [DOI] [PubMed] [Google Scholar]
- 25.Stewart BD, Kaye F, Machuca T, et al. SMARCA4-deficient thoracic sarcoma: A case report and review of literature [e-pub ahead of print]. Int J Surg Pathol. 10.1177/1066896919865944. accessed December 6, 2019. [DOI] [PubMed] [Google Scholar]
- 26.Perret R, Chalabreysse L, Watson S, et al. SMARCA4-deficient Thoracic Sarcomas: clinicopathologic Study of 30 Cases with an Emphasis on their Nosology and Differential Diagnoses. Am J Surg Pathol. 2019;43:455–465. [DOI] [PubMed] [Google Scholar]
- 27.Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multi-dimensional cancer genomics data. Cancer Discov. 2012;2:401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Silva EG, Deavers MT, Bodurka DC, Malpica A. Association of low-grade endometrioid carcinoma of the uterus and ovary with undifferentiated carcinoma: a new type of dedifferentiated carcinoma? Int J Gynecol Pathol. 2006;25:52–58. [DOI] [PubMed] [Google Scholar]
- 30.Rekhtman N, Tafe LJ, Chaft JE, et al. Distinct profile of driver mutations and clinical features in immunomarker-defined subsets of pulmonary large-cell carcinoma. Mod Pathol. 2013;26:511–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44:e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jordan EJ, Kim HR, Arcila ME, et al. Prospective comprehensive molecular characterization of lung adenocarcinomas for efficient patient matching to approved and emerging therapies. Cancer Discov. 2017;7:596–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Redig AJ, Capelletti M, Dahlberg SE, et al. Clinical and molecular characteristics of NF1-mutant lung cancer. Clinical Cancer Res. 2016;22:3148–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Torchia J, Picard D, Lafay-Cousin L, et al. Molecular subgroups of atypical teratoid rhabdoid tumours in children: an integrated genomic and clinicopathological analysis. Lancet Oncol. 2015;16:569–582. [DOI] [PubMed] [Google Scholar]
- 36.Kerr KM, Pelosi G, Austin JHM, et al. Pleomorphic, spindle cell and giant cell carcinoma In: Travis WD, Brambilla E, Burke AP, et al., eds. WHO Classification of Tumours of the Lung, Pleura, Thymus and Heart. Lyon: IARC Press; 2015:88–90. [Google Scholar]
- 37.Weeks DA, Beckwith JB, Mierau GW, Luckey DW. Rhabdoid tumor of kidney. A report of 111 cases from the National Wilms’ tumor Study Pathology Center. Am J Surg Pathol. 1989;13:439–458. [PubMed] [Google Scholar]
- 38.Schaefer IM, Agaimy A, Fletcher CD, Hornick JL. Claudin-4 expression distinguishes SWI/SNF complex-deficient undifferentiated carcinomas from sarcomas. Mod Pathol. 2017;30:539–548. [DOI] [PubMed] [Google Scholar]
- 39.Karnezis AN, Wang Y, Ramos P, et al. Dual loss of the SWI/SNF complex ATPases SMARCA4/BRG1 and SMARCA2/BRM is highly sensitive and specific for small cell carcinoma of the ovary, hypercalcaemic type. J Pathol. 2016;238:389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tessier-Cloutier B, Soslow RA, Stewart CJR, Köbel M, Lee CH. Frequent loss of claudin-4 expression in dedifferentiated and undifferentiated endometrial carcinomas. Histopathology. 2018;73:299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yoshimoto T, Matsubara D, Nakano T, et al. Frequent loss of the expression of multiple subunits of the SWI/SNF complex in large cell carcinoma and pleomorphic carcinoma of the lung. Pathol Int. 2015;65:595–602. [DOI] [PubMed] [Google Scholar]
- 42.Agaimy A, Rau TT, Hartmann A, Stoehr R. SMARCB1 (INI1)-negative rhabdoid carcinomas of the gastrointestinal tract: clinicopathologic and molecular study of a highly aggressive variant with literature review. Am J Surg Pathol. 2014;38:910–920. [DOI] [PubMed] [Google Scholar]
- 43.The Cancer Genome Atlas Network. Comprehensive and integrated genomic characterization of adult soft tissue sarcomas. Cell. 2017;171:950–965.e928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abro B, Kaushal M, Chen L, et al. Tumor mutation burden, DNA mismatch repair status and checkpoint immunotherapy markers in primary and relapsed malignant rhabdoid tumors. Pathol Res Pract. 2019;215: 152395. [DOI] [PubMed] [Google Scholar]
- 45.Lin DI, Chudnovsky Y, Duggan B, et al. Comprehensive genomic profiling reveals inactivating SMARCA4 mutations and low tumor mutational burden in small cell carcinoma of the ovary, hypercalcemic-type. Gynecol Oncol. 2017;147:626–633. [DOI] [PubMed] [Google Scholar]
- 46.Tanaka K, Hida T, Oya Y, et al. Unique prevalence of oncogenic genetic alterations in young patients with lung adenocarcinoma. Cancer. 2017;123:1731–1740. [DOI] [PubMed] [Google Scholar]
- 47.Toprani TH, Tamboli P, Amin MB, Ordoñez NG, Ayala AG, Ro JY. Thymic carcinoma with rhabdoid features. Ann Diagn Pathol. 2003;7:106–111. [DOI] [PubMed] [Google Scholar]
- 48.Tamboli P, Toprani TH, Amin MB, et al. Carcinoma of lung with rhabdoid features. Hum Pathol. 2004;35:8–13. [DOI] [PubMed] [Google Scholar]
- 49.Cavazza A, Colby TV, Tsokos M, Rush W, Travis WD. Lung tumors with a rhabdoid phenotype. Am J Clin Pathol. 1996;105:182–188. [DOI] [PubMed] [Google Scholar]
- 50.Shimazaki H, Aida S, Sato M, Deguchi H, Ozeki Y, Tamai S. Lung carcinoma with rhabdoid cells: a clinicopathological study and survival analysis of 14 cases. Histopathology. 2001;38:425–434. [DOI] [PubMed] [Google Scholar]
- 51.Falconieri G, Moran CA, Pizzolitto S, Zidar A, Angione V, Wakely PE. Intrathoracic rhabdoid carcinoma: a clinicopathological, immunohistochemical, and ultrastructural study of 6 cases. Ann Diagn Pathol. 2005;9:279–283. [DOI] [PubMed] [Google Scholar]
- 52.Orlando KA, Nguyen V, Raab JR, Walhart T, Weissman BE. Remodeling the cancer epigenome: mutations in the SWI/SNF complex offer new therapeutic opportunities. Expert Rev Anticancer Ther. 2019;19:375–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Januario T, Ye X, Bainer R, et al. PRC2-mediated repression of SMARCA2 predicts EZH2 inhibitor activity in SWI/SNF mutant tumors. Proc Natl Acad Sci U S A. 2017;114:12249–12254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rizvi NA, Hellmann MD, Snyder A, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abou Alaiwi S, Nassar A, El Bakouny Z, et al. Association of polybromo-associated BAF (PBAF) complex mutations with overall survival (OS) in cancer patients (pts) treated with checkpoint inhibitors (ICIs). J Clin Oncol. 2019;37: 103–103. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.