

Abstract

For respiratory conditions, targeted drug delivery to the lungs could produce higher local concentrations with reduced risk of adverse events compared to systemic administration. Despite the increasing interest in pulmonary delivery, the pharmacokinetics (PK) of drugs following pulmonary administration remains to be elucidated. In this context, the application of modeling and simulation methodologies to characterize PK properties of compounds following pulmonary administration remains a scarcity. Pseudomonas aeruginosa (PA) lung infections are resistant to many of the current antibiotic therapies. Targeted treatments for pulmonary delivery could be particularly beneficial for these local conditions. In this study, we report the application of biopharmaceutical pharmacometrics (BPMX) for the analysis of PK data from three investigational antimicrobial agents following pulmonary administration of a suspension formulation. The observed drug concentration–time profiles in lungs and plasma of the compound series were combined for simultaneous analysis and modeling. The developed model describes the PK data, taking into account formulation properties, and provides a mechanism to predict dissolved drug concentrations in the lungs available for activity. The model was then used to evaluate formulation effects and the impact of variability on total and dissolved drug concentrations in lungs and plasma. The predictions suggest that these therapies for lung delivery should ideally be delivered in a sustained release formulation with high solubility for maximum local exposure in lungs for efficacy, with rapid systemic clearance in plasma for reduced risk of unwanted systemic adverse effects. This work shows the potential benefits of BPMX and the role it can play to support drug discovery and development in pulmonary delivery.
1. Introduction
In the treatment of respiratory conditions, targeted drug delivery to its required site of action could provide unique benefits compared to systemic administration. Local delivery to the lungs could achieve a higher local concentration at the target site with reduced systemic exposure and risk of adverse events.1 For instance, pulmonary delivery of antimicrobial therapies could be beneficial in the treatment of respiratory infections. For these local treatments, the pharmacokinetics (PK) at the site of infection in the lungs is a crucial determinant for drug efficacy. However, despite increasing interest in pulmonary drug delivery over the past few decades, the PK of drug molecules following pulmonary administration remains to be elucidated. In this context, the application of modeling and simulation to characterize the impact of formulation behavior on local and systemic PK following pulmonary administration remains in its infancy.
Antibiotic resistance is a growing challenge and a major public health threat worldwide. In particular, Pseudomonas aeruginosa (PA), the causative bacterial species in a wide range of pulmonary conditions, has recently developed resistance to many of the current antibiotic therapies available.2,3 Previous reports have indicated that about 80% of patients with cystic fibrosis present with chronic PA lung infections.4,5 New treatment options for these infections are therefore of much importance. There have been ongoing efforts to explore new targets on PA infections. To this end, the inhibition of the quorum-sensing (QS) signaling pathway, which regulates the production of multiple virulence factors including traits required for PA biofilm formation and their resistance to antibiotics, has been suggested as a promising target.3,6−9 We therefore aim to develop a novel class of anti-virulence agents, quorum-sensing inhibitors (QSIs), for the inhibition of biofilm formation to sensitize PA to antibiotic treatments and attenuate its virulence.
Preclinical studies are invaluable in the lead optimization process in order to evaluate the PK of compounds. For pulmonary drug delivery, formulations are dosed into the lungs of the animals. While sufficiently soluble compounds can be administered into the lungs as solutions, poorly soluble compounds are commonly administered as a suspension. However, due to the dissolution of solid drug particles, the absorption of drugs from lung to plasma depends on the properties of the formulation, which affects both local and systemic drug concentrations. Consequently, it could be misleading to interpret the PK profiles of drug compounds without taking into account the properties of the formulation used for administration. During lead optimization and candidate selection, PK studies are conducted to identify compounds with the best potential for development into a clinical therapy. It is often impractical to invest heavily in formulation development for a specific compound in this early phase of drug discovery when a wide range of compounds are still being considered. For pulmonary suspensions, solubility and dissolution of drug particles in the formulation vehicle are the major determinants of local retention and systemic absorption. While slow dissolution of drug particles can provide sustained drug concentrations, only dissolved drugs are available for disposition and activity. However, it is practically difficult to separate dissolved drugs in lung samples following administration of a suspension. For instance, both dissolved and undissolved drugs will appear in an assay for homogenized lung samples. Hence, a methodology that could support efficient PK evaluation of drug compounds following pulmonary drug delivery taking into account formulation properties and prediction of concentrations of dissolved drugs in lungs would be beneficial.
In this study, we propose and report the application of biopharmaceutical pharmacometrics (BPMX) – i.e., pharmacometric modeling incorporating biopharmaceutical principles – to support the PK evaluation of three discovery drug compounds following pulmonary administration of a suspension. SEN023, SEN066, and SEN089 are three novel drug molecules that are structural analogues of a prototype QSI, which has shown promising activity against P. aeruginosa.10 The compounds were characterized in vitro and in vivo for their therapeutic potential and suitability as drug candidates. The lung and plasma concentration data of the compound series following pulmonary administration of a suspension in rats were collected and compiled for simultaneous analysis and modeling. The developed model was then applied to predict concentrations of dissolved drugs in the lung available for activity, to investigate the potential impact of solubility and dissolution on lung and plasma PK. The sensitivity of drug exposures to interindividual variability on systemic clearance and formulation properties was also evaluated.
2. Materials and Methods
2.1. Compounds
SEN023, SEN066, and SEN089 are structural QSI analogues designed, synthesized, and supplied by the University of Nottingham (Nottingham, UK). The structures of the compounds are not disclosed for intellectual property protection since the compounds are potentially patent pending. They are, however, structural analogues of SEN001 as shown previously.11 Polyethylene 400 (PEG400) and polysorbate 80 (PS80) were purchased from Sigma-Aldrich (Stockholm, Sweden).
2.2. Physicochemical Properties
The physicochemical properties of the compounds were predicted using ADMET Predictor version 7.2 (Simulations Plus, Inc., Lancaster, CA, USA). Two-dimensional SDF files of SEN023, SEN066, and SEN089 were introduced into the software, and the physiochemical and biopharmaceutical properties were calculated at pH 7.4. The physicochemical properties of the compound series are summarized in Table 1.
Table 1. In Silico Predicted ADMET Properties of SEN023, SEN066, and SEN089a.
| CPD | MW | acidic pKa | basic pKa | c log P | human jejunal permeability (cm/s, ×104) |
|---|---|---|---|---|---|
| SEN023 | 368.8 | none | 7.1; 1.3 | 2.4 | 2.0 |
| SEN066 | 383.9 | 10.7 | 1.6 | 3.1 | 4.1 |
| SEN089 | 398.9 | none | 5.4 | 3.7 | 2.8 |
Abbreviations: CPD = compound; MW = molecular weight.
2.3. Solubility Determination
The kinetic solubility was determined by adding 5 μL of 10 mM DMSO stocks of the compounds into 495 μL of phosphate buffer at pH 7.4. Samples were incubated at 37 °C for >20 h, and the supernatant was separated from the excess solid by centrifugation at 2465g (Eppendorf Centrifuge 5810 R) for 30 min at 37 °C. The drug concentrations in the supernatant were then determined by liquid chromatography–tandem mass spectrometry (LC–MS/MS). Depending on the materials available, the solubility measurements were performed in 1–3 replicates. The fraction of compound dissolved in the suspension formulation was determined at Saretius (Nottingham, UK). Briefly, 1 mg of drug was added to 2 mL of the formulation vehicle containing PS80 and PEG400 at 2% w/v each in water, and the mixture was stirred overnight at room temperature. The formulation was then filtered, and the filtrate was sent to XenoGesis (Nottingham, UK) for analysis to determine compound concentrations.
2.4. Metabolic Stability
Briefly, metabolic stability at 37 °C was determined in 0.5 mg/mL human and rat liver microsomes at a compound concentration of 1 μM in 100 mM KPO4 buffer at pH 7.4 and a total incubation volume of 500 μL. The reaction was initiated by addition of 1 mM NADPH. Samples were withdrawn after 0, 5, 10, 20, 40, and 60 min of incubation, and the reaction was terminated by addition of cold acetonitrile. The amount of parent compound remaining was analyzed by liquid chromatography–tandem mass spectrometry (LC–MS/MS). The measured clearance in the microsomes (CLm) was then used to calculate the predicted intrinsic clearance (CLint), hepatic clearance (CLH), and hepatic extraction efficiency (EE) using the scaling parameters of the respective species, as shown in eqs 1–3.
| 1 |
| 2 |
| 3 |
where MPPGL is milligram microsomal protein per gram liver (46 mg/g for rat and 39.8 mg/g for human), GLPBW is gram liver per body weight (40 g/kg for rat and 21 g/kg for human), BW is body weight (0.25 kg for rat and 70 kg for human), fu is fraction unbound (assumed to be 1), and LBF is liver blood flow (13.8 mL/min for rat and 1500 mL/min for human).
2.5. Pharmacokinetics Studies
The PK study of the compound series was performed at XenoGesis (Nottingham, UK) and its animal research facility Saretius (Nottingham, UK). The study protocols were reviewed and approved by the animal ethics committee at Saretius (UK Home Office licence 70/8420, 19b 2). Male Sprague–Dawley (CD) rats (Charles River UK, 0.273 ± 0.048 kg (mean ± s.d.) on initiation of dosing) were housed in groups of up to 5 prior to the study in the Saretius animal facility. Animals were maintained under a 12 h light/dark cycle (lights on at 07:00 h), where temperature (19–22 °C) and humidity (45–65%) were controlled according to UK Home Office regulations with free access to food (laboratory chow) and water. Animals were allowed to acclimatize for at least 2 days prior to initiation of the study.
Intratracheal dosing was performed under general anesthetic using gaseous isoflurane (Merial Animal Health Ltd., Harlow, UK, 5% isoflurane/95% oxygen). Briefly, rats were suspended under anesthesia by their incisors resting on a board angled at approximately 45°, and the tongue was gently moved aside using forceps. A laryngoscope was then inserted to provide a clear view of the top of the trachea, and any mucous was removed with a cotton bub prior to insertion of the dosing needle, which is angled to enable a clear view of the trachea during insertion. Correct insertion of the needle was confirmed by detection of the cartilage rings within the trachea using the needle tip before administration of the suspension. The suspension vehicle was composed of 2% PEG400 and 2% PS80 in water, and the compounds were added to a concentration of 0.5 mg/mL. The compounds were administered to the animals at a dose of 0.5 mg/kg body weight in a dose volume of 1 mL/kg. Plasma and lung homogenate samples were collected at 0.17, 0.5, 1, 2, 3, 6, 12, and 24 h after dosing.
2.6. Analytical Methods
The amounts of drugs in the formulation and PK samples were quantified using LC–MS/MS. Briefly, quantitative analysis was performed using a Thermo Scientific TSQ Quantiva triple quadrupole mass spectrometer (Thermo Scientific, San Jose, California, USA) in positive electrospray MRM mode coupled to a Thermo Scientific Vanquish Binary UHPLC system (Thermo Scientific, Germering, Germany). MS ion transitions were SEN023 370 > 132 and 370 > 352, SEN066 384 > 229 and 384 > 257, and SEN089 399 > 206 and 399 > 357. The analytes were separated using an Accucore, Phenyl-X 2.6 μm, 100 × 2.1 mm (Thermo Scientific, Runcorn, UK) over 2.10 min using a gradient method at a column temperature of 60 °C. The mobile phase was composed of Milli-Q water with formic acid (0.1%) and acetonitrile with formic acid (0.1%).
2.7. Pharmacokinetics Modeling
Initially, models were independently developed for SEN023, SEN066, and SEN089 to obtain parameter values for each compound. The observed concentrations were then combined to simultaneously analyze the lung and plasma pharmacokinetic profiles of the three drugs. Briefly, the model consisted of one lung compartment (L) where doses were given and lung samples were taken, one central compartment (C) where plasma samples were taken, one peripheral tissue compartment (P) to describe the disposition of drugs following systemic absorption, one solution compartment (Dd) to describe the amount dissolved drugs, and one suspension compartment (Ds) to describe the solid drug particles in the dosing suspension. The total amount of drugs in the lung compartment was the sum of drugs in the solution and suspension compartment (Dd + Ds). Only dissolved drugs in solution can be distributed into the systemic circulation. In addition to the suspension compartment for dosing of the formulation, one-, two-, three-, and four-compartment drug disposition models were evaluated. The transfers of drug between these compartments over time (t) are shown in eqs 4–7.
| 4 |
| 5 |
| 6 |
| 7 |
where V, CL, and Q are the volume of distribution, clearance, and intercompartmental clearance parameters, respectively, between the corresponding compartments and kd is the dissolution rate constant of the solid particles in the suspension. The structure of the final pharmacokinetic model is illustrated in Figure 1. Clearance (CL and Q) and volume of distribution parameters were allometrically scaled and parametrized during the estimation as shown in eqs 8 and 9:
| 8 |
| 9 |
where CLi and Vi are the individual animal parameters, WT (kg) is the animal body weight, and CLTV and VTV are the typical clearance and volume of distribution, respectively, of an animal with a typical body weight of 0.25 kg.
Figure 1.
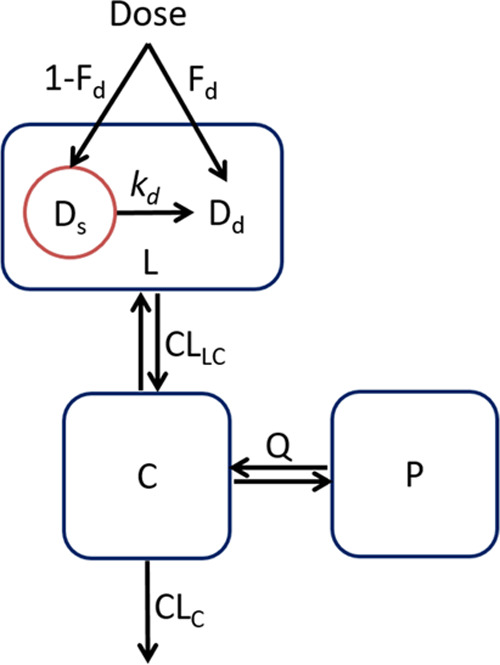
Schematic diagram showing the structure of the final pharmacokinetic model. Abbreviations: CLC = central clearance; CLLC = distribution clearance from lung to central compartment; Q = intercompartmental clearance; C = central compartment; L = lung compartment; P = peripheral tissue compartment; Dd = solution compartment for dissolved drugs; Ds = suspension compartment for solid drug particles in the suspension; Fd = fraction dissolved; kd = dissolution rate constant, where kd = kd0·(e–kdt1·t + e–kdt2·t), and kd0 = dissolution rate constant at time 0, kdt1 = first decline rate constant of kd, and kdt2 = second decline rate constant of kd.
For the dissolution of the suspension particles, a first-order dissolution and a time-varying dissolution were evaluated. For time-varying dissolution, two functions where the dissolution rate constant declined over time in a monoexponential and a biexponential manner were evaluated as shown in eqs 10 and 11
| 10 |
| 11 |
where kd0 is the dissolution rate constant at time 0, representing the baseline dissolution rate of the formulation and kdt1 and kdt2 are the first and second decline rate constants of kd. The fraction of the dose dissolved (Fd) in the suspension, based on solubility measurements of the formulation, determined the initial amount of dissolved drug in the lung compartment.
In the model, the observed plasma concentrations were predicted from the concentrations in the central compartment. To reflect the gradual dissolution of the suspension in the lungs, the observed lung concentrations were predicted from the total amount of drugs in the lung compartment, as a sum of drug in both the solution compartment and the suspension compartment.
2.8. Simulation Study
The final PK model was used to perform simulation studies to investigate the potential impact of formulation properties on the concentration–time profiles of total drugs in lungs, dissolved drugs in lungs, and drugs in plasma following pulmonary administration of a suspension formulation. For the simulations, key formulation parameters were evaluated while the drug disposition parameters were fixed. Lung and plasma PK profiles were simulated for a range of values of initial fraction dissolved (Fd = 0.1 to 1.0) and baseline dissolution rate constant (kd0 = 0.2 to 2.0 h–1) parameters. The evaluated values of Fd represent formulations containing 10 to 100% of the compounds initially dissolved and reflect the solubility of the drugs in the formulation. The evaluated values of kd0 represent half-lives of 0.35 to 3.47 h based on approximation from first-order kinetics. Summary PK indices over a 24 h period following a single dose including the cumulative area-under-the-curve (AUC) of the PK profiles up to 24 h, maximum concentration (Cmax), and the percentage of time over a 24 h period where the PK profiles exceeded the target concentration (% T > target) were computed from the simulations. Since the effective concentrations of these discovery compounds have not been determined, for the purpose of comparing the compounds and formulation effect in this study, 1 mg/L (i.e., 2.5 to 2.7 μM) was chosen as the target concentration to demonstrate the application of the model.
To investigate the sensitivity of lung and plasma concentrations to interindividual variability (IIV) of systemic clearance and formulation properties, simulations were performed with variability on the parameters of interest. The predictions were generated from a virtual population composed of 1000 simulated data sets with variability on the systemic clearance (CLC), initial fraction dissolved (Fd), and baseline dissolution rate constant (kd0) parameters. Since physiological and formulation characteristics are positively constrained and typically log-normally distributed, IIV was applied to the parameters using an exponential model as shown in eq 12
| 12 |
where θi is the individual value of the parameter for each subject, θTV is the typical value of the parameter in the population, and η is normally distributed with a coefficient of variance (CV) of 50% and mean 0.
2.9. Data Analysis and Software
Model development was performed in the nonlinear mixed-effects modeling software program, NONMEM (version 7.4; ICON Development Solutions),12 using the Laplacian estimation with interaction algorithm. Perl-speaks-NONMEM (PsN) was used for model execution and generation of visual predictive checks (VPCs).13 R (version 3.6; R Foundation for Statistical Computing, Vienna, Austria) was used for data management, and the Xpose4 package14 was used to support graphical evaluation of results for model diagnostics. The run records and numerical comparison of models were maintained and facilitated by Pirana.15
The likelihood ratio test (LRT) was used to evaluate statistical significance for inclusion of additional parameters in nested models, where the objective function value (OFV) was assumed to be χ2 distributed. A decrease in OFV of 3.84 between nested models with one parameter difference was considered to be a statistically significant difference at the 5% significance level. Model development was guided by the scientific plausibility of the parameter estimates, the change in objective function value (ΔOFV), parameter precision, and evaluation of goodness-of-fit and residual diagnostic plots including the simulation-based VPCs.16
Since the majority of samples were terminal samples and the primary interest of central tendency in the preclinical study, IIV of the parameters was not estimated. For VPCs, 1000 data sets were simulated from the final parameter estimates using the original data set as a template. The predicted medians and the 2.5th and 97.5th percentiles and their corresponding 95% confidence intervals were computed from the simulated data and overlaid with the observed values. Data below the lower limit of quantification (LLOQ) were handled using the likelihood-based M3 method.17 The observed data were natural log-transformed for model development (i.e., the transform-both-side approach), and a proportional error model, as approximated by a natural log-transformed additive error model, was used to describe the residual unexplained variability.
For the simulation study, prediction and visualization of results were performed in R. The mrgsolve package (version 0.9.0; Metrum Research Group, USA)18 was used to perform the simulations. The shiny package (version 1.2.0; RStudio, USA)19 was used for application development to support interactive evaluation and visualization of the simulation output.
3. Results
3.1. Solubility Determination
The kinetic solubility measurements of SEN023, SEN066, and SEN089 in the formulation used were 16.4, 14.9, and 27.3 μg/mL, respectively. The results were consistent with the rank order observed in the suspension formulation, in which the fractions of SEN023, SEN066, and SEN089 dissolved were 10, 5, and 21%, respectively. These solubility values were included in the PK model.
3.2. Metabolic Stability
The metabolic stability of the compounds in human and rat liver microsomes are summarized in Tables 2 and 3. The results showed that the compounds were all likely to be rapidly metabolized in rats after systemic absorption with high hepatic extraction efficiency (EE) ranging from 0.72 to 0.86. However, the compounds were more stable in human than in rat liver microsomes, with their EE ranging from 0.34 to 0.72, suggesting that these compounds might be less extensively metabolized in humans.
Table 2. Metabolic Stabilities of SEN023, SEN066, and SEN089 in Rat Liver Microsomesa.
| CPD | half-life (min) | CLint (mL/min) | CLH (mL/min) | EE |
|---|---|---|---|---|
| SEN023 | 18.14 | 35.15 | 9.91 | 0.72 |
| SEN066 | 11.26 | 56.63 | 11.10 | 0.80 |
| SEN089 | 7.50 | 85.07 | 11.87 | 0.86 |
Abbreviations: CPD = compound; CLint = intrinsic clearance; CLH = hepatic clearance; EE = extraction efficiency.
Table 3. Metabolic Stabilities of SEN023, SEN066, and SEN089 in Human Liver Microsomesa.
| CPD | half-life (min) | CLint (mL/min) | CLH (mL/min) | EE |
|---|---|---|---|---|
| SEN023 | 67.51 | 1201.17 | 667.03 | 0.44 |
| SEN066 | 106.73 | 759.72 | 504.30 | 0.34 |
| SEN089 | 21.25 | 3816.07 | 1076.76 | 0.72 |
Abbreviations: CPD = compound; CLint = intrinsic clearance; CLH = hepatic clearance; EE = extraction efficiency.
3.3. Plasma Pharmacokinetics Following Pulmonary Administration
Following pulmonary administration of the suspension, the compounds were rapidly distributed into the systemic circulation as shown by the rapid appearance of the drugs in plasma and the lack of a distinct absorption phase in the plasma concentration–time profiles (Figure 2). Among the three compounds, the plasma concentration of SEN089 declined most rapidly, with a steep distribution phase that lasted throughout the sampling period. In contrast, following the initial distribution phase, the decline of SEN066 was the slowest as characterized by a relatively flat plasma concentration–time profile after 6 h.
Figure 2.

Visual predictive check of the final pharmacokinetic model showing the observed lung and plasma concentrations of SEN023, SEN066, and SEN089 with the observed median and the 2.5th and 97.5th percentiles following intratracheal administration of the suspension formulation and their corresponding 95% confidence intervals computed from the simulated data (shaded).
3.4. Lung Pharmacokinetics Following Pulmonary Administration
The lung concentrations of the compounds declined slowly following pulmonary administration of the suspension and remained in the lungs at high concentrations over the sampling period (Figure 2). The lung concentrations of SEN089 declined most rapidly, while the ones of SEN066 lasted the longest in the lungs. It is worth noting that the measured lung concentrations of the compounds were more than 2 orders of magnitude higher than their plasma concentrations. The lung concentration–time profiles also declined slower than their plasma profiles, indicating that the measured concentrations in lung and plasma were not at equilibrium.
3.5. Pharmacokinetics Modeling
The observed concentrations of the compounds were described by the final model with acceptable fit and predictive performance as demonstrated in the VPC (Figure 2). While a peripheral tissue compartment was needed to describe the observed concentrations for SEN089, it did not improve the model fit for SEN023. Although an additional peripheral tissue compartment improved the model fit for SEN066, parameter estimates became physiologically implausible. Further investigation revealed that the difference in model fit was primarily contributed by the limited plasma samples at 24 h, and there was no improvement when these samples were excluded. The peripheral tissue compartment was therefore not included in the final model for SEN066. Thereafter, the addition of a deep lung compartment to the model before systemic absorption did not further improve model fit for any of the compounds.
For the formulation components of the model, the fractions of the nominal dose in solution were fixed to the values determined in the solubility study. Dissolution of solid particles was described by the release of drugs from the suspension compartment to the solution compartment in the lung, in which the dissolved drugs in solution were available for systemic absorption and distribution. For the dissolution process, compared to a typical first-order kinetics, the time-varying dissolution rate constant in the final model improved model fit for all of the compounds significantly (p < 0.001, ΔOFV for SEN023 = −95.2, SEN066 = −78.1, SEN089 = −81.9). Compared to a monoexponential decline, a time-varying dissolution rate constant with a biexponential decline further improved model fit significantly (p < 0.01–0.001, ΔOFV for SEN023 = −15.5, SEN066 = −22.7, SEN089 = −17.5). The parameter estimates of the final model and their relative standard errors are summarized in Table 4.
Table 4. Parameter Estimates and Relative Standard Errors (RSE %) of the Final Modela.
| parameter | unit | description | SEN023 | RSE % | SEN066 | RSE % | SEN089 | RSE % |
|---|---|---|---|---|---|---|---|---|
| CLLC | mL/min | clearance – lung to central | 1.19 | 47.2 | 1.16 | 9.1 | 2.06 | 45.7 |
| VL | mL | volume of distribution – lung | 3.22 | 11.6 | 3.29 | 12.2 | 5.97 | 14.5 |
| CLC | mL/min | clearance – central | 6.33 | 7.2 | 11.9 | 11.2 | 2.51 | 8.4 |
| VC | mL | volume of distribution – central | 47.9 | 30.7 | 34.2 | 12.1 | 12.7 | 18.9 |
| Q | mL/min | intercompartmental clearance | -- | -- | -- | -- | 0.0483 | 46.6 |
| VP | mL | volume of distribution – peripheral | -- | -- | -- | -- | 17.7 | 45.0 |
| Fd | -- | initial fraction dissolved | 0.10* | -- | 0.05* | -- | 0.21* | -- |
| kd0 | h–1 | baseline dissolution rate constant | 0.304 | 9.6 | 0.235 | 11.4 | 0.806 | 4.4 |
| kdt1 | h–1 | first decline rate constant of kd | 0.687 | 35.4 | 1.32 | 14.2 | 1.16 | 7.4 |
| kdt2 | h–1 | second decline rate constant of kd | 0.0582 | 14.6 | 0.12 | 8.0 | 0.138 | 10.6 |
| ERRPL | % | proportional error – plasma | 45.9 | 9.2 | 77.1 | 9.3 | 43.9 | 6.5 |
| ERRLG | % | proportional error – lung | 39.4 | 17.7 | 34.2 | 25.3 | 55.0 | 15.8 |
3.6. Formulation Effects on Drug Exposures in Lungs and Plasma
The predicted lung and plasma PK profiles resulting from suspension formulations with different initial fractions of drugs dissolved and varying dissolution rates are shown in Figure 3. The simulation results showed that increasing the initial amount of dissolved drugs in the suspension would increase the amount of drugs available for rapid distribution from lung to plasma immediately after administration, leading to higher initial plasma concentrations. For a given drug, the concentrations in lungs and plasma then declined at the same rate for all suspension formulations after the rapid initial distribution phase. When the formulation was a complete solution (i.e., Fd = 1), the initial concentrations in both lungs and plasma declined much more rapidly than any suspension formulations, reaching the terminal elimination phase in less than 2 h after administration. The results also showed that increasing dissolution rate would lead to a more rapid decline of drug concentrations in both lungs and plasma due to faster distribution of the compounds to the systemic circulation, after which they were rapidly metabolized by the liver. However, peak concentrations in plasma increased with dissolution rate before the more rapid decline.
Figure 3.

Predicted concentration–time profiles of SEN023, SEN066, and SEN089 in lungs (total and dissolved) and plasma resulting from formulations with different values of (a) initial fraction dissolved (Fd) and (b) baseline dissolution rate (kd0).
Drug exposures, as demonstrated by the PK indices, resulting from suspension formulations with different initial amounts of dissolved drugs and dissolution rates, are shown in Figures 4–6. The results showed that after 24 h following a single dose, AUCs of the total lung and the plasma concentration–time profiles would both increase with decreased initial fraction of dissolved drugs in the suspension. However, AUC of the dissolved drugs in lungs would increase with solubility. In contrast, slower dissolution would lead to higher AUC for total drugs in lungs but lower AUCs for dissolved drugs in lungs and drugs in plasma. The AUC of total drugs in lungs was most affected by the range of solubility and dissolution rates evaluated, with a 39-, 57-, and 47-fold change for SEN023, SEN066, and SEN089, respectively, across the formulation design space. In contrast, the changes of AUCs for dissolved drugs in lungs and drugs in plasma ranged from 2.0 to 2.4 and from 1.1 to 1.4, respectively.
Figure 4.

The predicted (a) area-under-the-curve at 24 h (AUC), (b) maximum concentration (Cmax), and (c) fraction of time above the target concentration (% T > target) over a 24 h period of SEN023 in lungs and plasma resulting from a range of initial fractions dissolved (Fd) and baseline dissolution rate constants (kd0). The shade of the color represents the values in the formulation design space explored where dark purple is highest and white is lowest.
Figure 6.

The predicted (a) area-under-the-curve at 24 h (AUC), (b) maximum concentration (Cmax), and (c) fraction of time above the target concentration (% T > target) over a 24 h period of SEN089 in lungs and plasma resulting from a range of initial fractions dissolved (Fd) and baseline dissolution rate constants (kd0). The shade of the color represents the values in the formulation design space explored where dark purple is highest and white is lowest.
Figure 5.

The predicted (a) area-under-the-curve at 24 h (AUC), (b) maximum concentration (Cmax), and (c) fraction of time above the target concentration (% T > target) over a 24 h period of SEN066 in lungs and plasma resulting from a range of initial fractions dissolved (Fd) and baseline dissolution rate constants (kd0). The shade of the color represents the values in the formulation design space explored where dark purple is highest and white is lowest.
In the predictions, the peak concentrations of dissolved drugs in lungs and drugs in plasma increased with increasing solubility and dissolution rate. Within the study design space, the Cmax of the compounds in plasma changed by about 7-fold, increasing with initial fraction dissolved. The peak concentrations of total drugs in lungs occurred immediately after dosing and were independent of formulation properties, while peak concentrations of dissolved drugs in lungs were dependent only on solubility. Low solubility and slow dissolution were also predicted to increase % T > target in lungs. However, the range of solubility and dissolution rates influenced the % T > target of dissolved drugs in lungs and drugs in plasma differently for the three compounds. The % T > target value in plasma increased with increasing solubility and dissolution rate for SEN023 and SEN066. For SEN089, however, low solubility and fast dissolution led to increased T > target in plasma. For dissolved drugs in lungs, while the overall % T > target values were low in all scenarios, low solubility with a moderate dissolution rate resulted in the highest % T > target for all of the compounds. The PK indices of the compounds are summarized in Table 5.
Table 5. Summary of PK Indices across the Study Design Space for SEN023, SEN066, and SEN089a.
| CPD | AUCLT | AUCLD | AUCPL | CMXLT | CMXLD | CMXPL | TATLT | TATLD | TATPL | |
|---|---|---|---|---|---|---|---|---|---|---|
| (μg/L·h) | (μg/L) | (%) | ||||||||
| SEN023 | max | 222131.3 | 5720.5 | 395.1 | 46583.9 | 46583.9 | 1613.1 | 100.0 | 3.4 | 0.7 |
| min | 5720.5 | 2696.3 | 347.3 | 46583.9 | 4658.4 | 209.0 | 1.1 | 0.7 | 0.0 | |
| ratio | 38.8 | 2.1 | 1.1 | 1.0 | 10.0 | 7.7 | 90.9 | 4.9 | NA | |
| SEN066 | max | 314073.5 | 5545.2 | 205.3 | 45592.7 | 45592.7 | 1101.1 | 100.0 | 3.0 | 0.4 |
| min | 5545.2 | 2289.3 | 148.1 | 45592.7 | 4559.3 | 153.5 | 0.9 | 0.6 | 0.0 | |
| ratio | 56.6 | 2.4 | 1.4 | 1.0 | 10.0 | 7.2 | 111.1 | 5.0 | NA | |
| SEN089 | max | 182530.9 | 3881.0 | 985.4 | 25125.6 | 25125.6 | 4002.7 | 100.0 | 3.2 | 1.7 |
| min | 3881.0 | 1965.3 | 796.1 | 25125.6 | 2512.6 | 531.6 | 1.5 | 0.6 | 0.0 | |
| ratio | 47.0 | 2.0 | 1.2 | 1.0 | 10.0 | 7.5 | 66.7 | 5.3 | NA | |
Abbreviations: CPD = compound; AUC = area-under-the-curve at 24 h; CMX = maximum concentration; TAT = %T > target; LT = total lung drug concentrations; LD = dissolved lung drug concentrations; PL = plasma drug concentrations; NA = not applicable.
3.7. Impact of Interindividual Variability on Drug Exposures in Lungs and Plasma
The predicted lung and plasma PK profiles taking into account IIV on systemic clearance, solubility, and dissolution rate of the formulation are shown in Figure 7. The results showed that only plasma drug exposure, and dissolved drugs in lungs to a small extent, but not the exposure of total drugs in lungs, was sensitive to variability in systemic clearance. Both plasma and lung exposures were insensitive to IIV of solubility of the drug in the formulation at the level of variability evaluated. Drug exposures were most sensitive to IIV of dissolution rate in the formulation, which was predicted to impact both lung and plasma drug exposures. Among the compound series, SEN089 was the most sensitive to the IIV of dissolution rate while SEN066 was the least sensitive to it.
Figure 7.

Predicted lung and plasma concentration–time profiles of SEN023, SEN066, and SEN089 and their corresponding 95% prediction intervals (shaded) with interindividual variability on (a) systemic clearance, (b) solubility, and (c) baseline dissolution rate constant.
4. Discussion
This study set out to explore the application of BPMX in preclinical drug evaluation given the typical constraints in a drug discovery setting, based on our investigation of the PK of three prototype QSIs following pulmonary administration. Due to the limited solubility, the compounds were administered into the lungs as a suspension formulation. The suspension allowed administration of doses higher than the aqueous solubility and provided a sustained release of compounds in lungs. Drug absorption and clearance from lungs are typically rapid, leading to a short residence time in lungs with low local drug exposure. If high drug concentration in lungs is desirable for a local effect, sustained release formulations as exemplified here by a suspension could be beneficial. The relatively simple formulation vehicle used in this study was established previously to improve solubility of the compound series with excipients that have been shown to be suitable for lung administration.11,20−22
The local and systemic PK profiles of drugs were dependent on the solubility of the compound in the formulation and the dissolution of particles in the suspension. Despite the influential roles of these formulation properties, extensive investment in formulation development for each compound during this early phase of drug discovery is often not feasible due to time constraints and limited availability of compounds, typically in batch sizes of milligrams in a laboratory. The compound is then prioritized for different assessments such as in vitro absorption, distribution, metabolism, and excretion (ADME) assays, formulation, and PK studies. Another limiting factor when working with formulations intended for pulmonary administration is that it is often difficult to determine dissolved drug concentrations in lungs available for activity. Hence, a methodology that can support efficient evaluation of local and systemic PK following pulmonary administration, with predictive capacity of dissolved drug concentrations in lungs, taking into account the biopharmaceutical effects of formulation properties could be beneficial.
The three compounds in the present study were all poorly water soluble and lipophilic, with their c log P values ranging from 2.4 to 3.7. It was expected that the excipients in the formulation vehicle would increase the solubility of the compound series as previously shown with the analogues.11 However, despite the use of the formulation medium, only 5 to 21% of the compounds were dissolved when prepared at a concentration of 0.5 mg/mL. The compounds were therefore given as a suspension formulation into the lungs of the animals. Following pulmonary administration, the compounds were rapidly absorbed into the systemic circulation as seen by the immediate appearance of the drugs in plasma and the lack of a distinct absorption phase. Previous analogues of the compounds have been shown to be rapidly absorbed when given as a solution formulation.11 The results in the present study showed that these compound analogues were easily absorbed even when given as a suspension formulation. Such rapid absorption has also been reported with other antimicrobial agents.23−27 This is consistent with the high absorptive capacity of lungs given the large surface area and the thin alveolar epithelium linings.28−30
From the PK profiles, it was evident that the drug concentrations in lungs and plasma declined at different rates. This lack of parallelism indicated that the observed drug concentrations in lungs and plasma were not at distribution equilibrium. Given the rapid distribution between lung and plasma, the sustained and slow decline of the observed lung concentrations reflected the gradual dissolution of drugs in the suspension. Drug dissolution in suspension formulations has been shown to be the rate-limiting step in absorption using an isolated perfused rat lung model.31 The present study provided further insight into this phenomenon in vivo by investigating the lung and plasma PK profiles following pulmonary administration of a suspension formulation. However, since whole lung samples were collected, both dissolved and undissolved drugs in the suspension would have appeared in the sample assays. The dissolution of drug in lungs is a dynamic process. It is not straightforward to determine concentrations of dissolved drugs in lungs available for activity and disposition. It would have required special sample handling and separation of any remaining drug particles in the lung samples with specific assays to capture and quantify the undissolved and dissolved drugs at each time point. Hence, we developed a BPMX model to describe the PK profiles and predict dissolved drug concentrations in lungs as a function of formulation properties.
Ideally, comprehensive comparison of the compounds would have required substantial investment to develop multiple formulations for each compound across the vast chemical and formulation design space with subsequent in vivo studies to thoroughly evaluate the PK of these compounds and formulations. In the present study, the modeling approach helped to maximize the information obtained from the study by supporting the analysis and interpretation of the data, taking into account the properties of the formulation used in the study. The model allowed prediction of the potential PK of the compounds across the formulation design space in a semimechanistic manner for inference and compound comparison when exhaustive testing is not practically feasible. Although the comparison of sampling methods is beyond the scope of this study, it is worth noting that the implications resulting from the use of tissue homogenates have been discussed.32 Without endorsing a specific sampling method, the present study focused on investigating the potential application of semimechanistic pharmacometric models to support compound selection in a drug discovery setting.
The developed model described both the central tendency and the variability of the observed drug concentrations in lungs and plasma for the three compounds with good predictive performance as shown by the VPC. Since whole lung samples were collected in the study, clearance mechanisms within the lung, e.g., mucociliary clearance and macrophages, were not considered in the model separately. The study was neither sufficiently powered with spatial resolution to distinctively identify these mechanisms nor was this the objective of the study. Therefore, the clearance described in the model reflects the net clearance of the drug as observed in the study. From the model parameters, SEN089 seemed to be the most widely distributed into peripheral tissue, as reflected by the additional distribution compartment. During model development, the solubility of the compounds in the formulation, as reflected by the fraction of drugs initially dissolved in the suspension, were included in the model. This information helped maintain model identifiability and allowed the estimation of the in vivo dissolution parameters. SEN089, being the most soluble compound in the formulation, had the highest baseline dissolution rate constant in the model. In contrast, the solubility of SEN023 and SEN066 was lower than that of SEN089 in the formulation with a lower baseline dissolution rate constant in the model. Importantly, the model was not able to describe the data when first-order kinetics was used to describe the dissolution process. Instead, a time-varying rate constant, which declined over time, was needed to describe the observed concentrations. In particular, a dissolution rate constant with a biexponential decline significantly improved the model fit. This suggests that the in vivo dissolution process observed was a complex phenomenon with potentially subpopulations of particles dissolving at different rates. This could be caused by the range of particle sizes as they dissolved at different rates and/or the regional distribution of drugs in the airways leading to different dissolution and clearance of the drugs. It should be noted that the suspension used in this study was not a controlled suspension with a narrow particle size distribution, but a rather coarse suspension of the material as received thoroughly dispersed and stirred overnight prior to administration. As a coarse approximation from visual examination, we estimated the particle size of the visible materials to be around 50–100 μm. The small quantities of the QSIs restricted the development of controlled suspensions through ball milling or ultrasonication.33,34 For such formulations, typically larger volumes need to be prepared than the ones prepared herein, and much material is lost during processing.
The developed PK model was used to evaluate the potential impact of formulation properties on local and systemic PK of the drugs. From the predicted PK profiles, the role of suspension formulations in providing sustained concentrations in lungs was clearly demonstrated. When the solubility parameter (Fd) of the model increased from 0.1 to 0.9, the absorption of drugs from lung to plasma became increasingly rapid, as shown by the sharper peak concentrations in plasma. However, following the initial distribution, drugs in plasma were also more rapidly cleared leading to lower concentrations in both lung and plasma with higher solubility in the formulation. After initial absorption of the dissolved drugs, drug concentrations declined at the same rate regardless of solubility. When the drug was completely dissolved in the formulation (i.e., Fd = 1), the concentrations in both lung and plasma declined much more rapidly than the ones resulting from suspension formulations and decreased by more than 3 orders of magnitude within 2 h. This was consistent with the rapid absorption and clearance observed when the analogues of the compound series were administered as solution formulations to the lungs.11 Without the sustained release properties resulting from the slow dissolution of drug particles, drugs in solution were rapidly absorbed and cleared systemically. The high metabolism, as suggested by the results from the rat liver microsome study, likely also contributed to the rapid systemic clearance. It should be noted that, in contrast to systems pharmacology-based models that are developed to quantitatively describe a biological or disease process with less emphasis on describing specific observations, if they are available at all, pharmacometric models as applied in the present study are data-driven, relying on robust statistical models and algorithms derived to describe the available data, and models are rigorously assessed for their ability to reproduce the observations.35 The measured metabolic stability values were therefore not specifically included as a parameter in this model. The results of the metabolic stability study, however, provided insight into the potential clearance mechanisms of these compounds in vivo.
It was apparent from the predictions that lung concentrations declined at a faster rate with increased dissolution rate, causing a more rapid absorption and an increase in initial plasma concentrations. However, this was also followed by a faster decline of plasma concentrations after absorption. In contrast, slower dissolution led to more sustained concentration–time profiles in both lung and plasma, despite the slower systemic absorption and lower peak concentrations in plasma. This suggests that a sustained-release lung formulation is expected to provide sustained plasma concentrations, which could impact systemic exposure for therapeutic effect or toxicity, depending on the target site of treatment. SEN023 and SEN066, being the less widely distributed compounds in peripheral tissues compared to SEN089, were more susceptible to changes in the baseline dissolution rate. Their plasma concentrations declined more rapidly with increasing baseline dissolution rate than SEN089. This rapid decline could be further contributed by the increased availability of drugs for systemic clearance due to the relatively low tissue distribution. The decline of dissolved drug concentrations in lungs mirrored the concentrations in plasma for the compound series, consistent with the rapid distribution between lung and plasma.
To evaluate formulation effects on overall drug exposures, we computed the AUC, Cmax, and % T > target over a 24 h period as a function of both solubility and dissolution rate of the compounds in the formulation. Similar metrics have long been used for the calculation of PK/PD indices to relate drug concentrations and efficacy of antimicrobial drugs.36−39 From the predictions, it was evident that low solubility and slow dissolution would lead to maximum total drug exposure in lungs as shown by the local AUC and % T > target. However, high solubility was needed for maximum dissolved drug exposure in lungs for activity. In contrast, for systemic drug delivery, low solubility with fast dissolution was expected to lead to maximum drug exposure after 24 h as demonstrated by the plasma AUC. The AUC of total drugs in lungs increases with decreasing initial fraction dissolved since undissolved drugs stay in the lung longer than dissolved drugs. The net outcome is slower but more sustained absorption from the lung into the systemic circulation, resulting in a lower peak plasma concentration but a more sustained plasma concentration–time profile and hence a higher AUC. Conversely, the AUC of dissolved drugs in lungs is directly related to solubility and the initial fraction of dissolved drugs. Higher solubility leads to higher concentrations of dissolved drugs and therefore higher AUC. Hence, treatments for local conditions such as these QSIs may be engineered to minimize unwanted systemic drug exposure and adverse effects. The results suggest that, depending on the target site of action, systemic and local drug exposures may be optimized for maximal efficacy and safety. Hence, if the compounds are intended for local conditions in lungs, as in the case of chronic lung infections, a sustained-release formulation with high solubility and slow dissolution could be preferable. In contrast, for systemic treatment, a formulation with low solubility and fast dissolution would be beneficial. While this is the ideal scenario, it is worth acknowledging the formulation challenge since highly soluble compounds tend to have fast dissolution rates. Therefore, sustained-release formulations with careful design would be needed to implement such a drug delivery strategy. For example, dry powder inhaler formulations with sophisticated particle engineering and design using spray-drying could play a valuable role here.40,41
It should be noted that the AUC at 24 h in plasma was only moderately affected by formulation properties with a maximum of 1.4-fold difference across the formulation design space in the study. In contrast, the AUC of total drug concentrations in lungs was much more susceptible to changes in formulation properties, differed by 39- to 57-fold for the scenarios evaluated. Interestingly, peak concentrations in lungs and plasma were affected differently by these formulation properties. Cmax in plasma differed by about 7-fold for all of the compounds across the study design space. It increased with both solubility and dissolution rate and was most affected by solubility. In contrast, Cmax of total drug concentrations in lungs was independent of formulation properties and was only dictated by the dose. The effect of formulation on % T > target of total drugs in lungs was most dramatic, differed by >111-fold for SEN066 and ranging from <1 to 100% for the three compounds. In contrast, % T > target in plasma was barely affected, ranging from 0 to 1.7% for the three compounds. Overall, despite their effect on Cmax in plasma, formulation properties were much more influential on drug exposure in lungs, both total and dissolved, than in plasma, as indicated by the much wider range of AUC and % T > target values for total drug concentrations in lungs. It should be noted that the objective of the present study was to demonstrate an application of BPMX in a drug discovery setting and hence focused on only the PK metrics at 24 h following a single dose for comparison. Other metrics such as exposures after repeated dosing at a later time-point may be more appropriate for other therapies. Further studies should be considered to more thoroughly evaluate these drug delivery strategies for other treatments and chronic therapies.
From the metabolic stability results, it was apparent that the metabolism of the compounds was variable and species-dependent as noted by the different metabolism in human and rat liver microsomes. In addition, formulation properties including solubility and dissolution measurements are known to be variable depending on experimental conditions. The developed model was therefore used to evaluate the sensitivity of lung and plasma exposures to IIVs on systemic clearance and formulation properties. It was clear that IIV on systemic clearance only impacted plasma concentrations with a negligible effect on lung concentrations. This is consistent with previous results reported with an earlier series of QSI analogues.11 For formulation properties, IIV on solubility had a limited influence on drug concentrations in both lungs and plasma, likely due to the low solubility of the compounds. SEN089, the most soluble compound in the series, was most affected by IIV in solubility. In contrast, IIV on the dissolution rate parameter was much more influential than on solubility and impacted drug concentrations in both lungs and plasma. In fact, IIV of the dissolution rate parameter was the major contributor resulting in variability of drug concentrations in all scenarios. This reflects the importance of dissolution as a rate-limiting step in drug absorption since the compounds were administered at a concentration higher than their solubility in the suspension. The finding highlights the potential impact of variability in dissolution studies and how this uncertainty should be considered when interpreting measurements. It also indicates the importance of careful particle engineering for consistent dissolution rates of formulations designed for pulmonary delivery. The results resonate with previous work in the literature that suggests that dissolution is the rate-limiting step governing the systemic absorption rate for lipophilic drugs with low solubility following pulmonary administration.42 This supports that the modeling approach adopted in the present study is appropriate and has the potential to be more broadly applicable to inform the development of other treatments.
5. Conclusions
This study has demonstrated an application of BPMX and how it can be a valuable tool to inform drug discovery and development in pulmonary delivery. The modeling approach supported quantitative evaluation of the PK properties of the investigational compounds following pulmonary administration of a suspension formulation, given the available data and constraints typically encountered in such a preclinical setting. The developed model adequately described the PK data in lungs and plasma, taking into account formulation properties of the suspension, and thus allowed the prediction of dissolved drug exposure in lungs as a function of formulation properties. Depending on the target site of drug action and PK driver for efficacy, formulations can be engineered for optimal drug exposure accordingly for maximal efficacy and safety. The results suggest that these QSI treatments designed for pulmonary delivery should ideally be given in a sustained-release formulation with high solubility and slow dissolution for extended local residence time and exposure and rapid clearance after absorption into the systemic circulation. Similar formulation strategies may be applicable for other local treatments. This work shows that BPMX has the potential to play an increasingly valuable role in modern drug discovery and development of new therapies.
Acknowledgments
This work was supported by the Joint Programming Initiative on Antimicrobial Resistance (JPIAMR) grant SENBIOTAR (Swedish Research Council grant reference 2014-7513; UK Medical Research Council grant reference MR/N501852/1). The authors would like to acknowledge Uppsala University Drug Optimisation and Pharmaceutical Profiling Platform (UDOPP) for their assistance in the measurements of kinetic solubility and metabolic stability, XenoGesis (Nottingham, UK) for their assistance in the in vivo studies, and the National Biofilms Innovation Centre (NBIC), which is an Innovation and Knowledge Centre funded by the Biotechnology and Biological Sciences Research Council, Innovate UK, and the Hartree Centre [Award Number BB/R012415/1], for the support of chemical synthesis of the compounds. We are also grateful to Simulations Plus (Lancaster, CA) for providing us a reference site license for the software ADMET Predictor.
The authors declare no competing financial interest.
References
- Falagas M. E.; Michalopoulos A.; Metaxas E. I. Pulmonary drug delivery systems for antimicrobial agents: facts and myths. Int. J. Antimicrob. Agents 2010, 35, 101–106. 10.1016/j.ijantimicag.2009.10.001. [DOI] [PubMed] [Google Scholar]
- Chatterjee M.; Anju C. P.; Biswas L.; Anil Kumar V.; Gopi Mohan C.; Biswas R. Antibiotic resistance in Pseudomonas aeruginosa and alternative therapeutic options. Int. J. Med. Microbiol. 2016, 306, 48–58. 10.1016/j.ijmm.2015.11.004. [DOI] [PubMed] [Google Scholar]
- Ciofu O.; Tolker-Nielsen T.; Jensen P. Ø.; Wang H.; Høiby N. Antimicrobial resistance, respiratory tract infections and role of biofilms in lung infections in cystic fibrosis patients. Adv. Drug Delivery Rev. 2015, 85, 7–23. 10.1016/j.addr.2014.11.017. [DOI] [PubMed] [Google Scholar]
- Crull M. R.; Ramos K. J.; Caldwell E.; Mayer-Hamblett N.; Aitken M. L.; Goss C. H. Change in Pseudomonas aeruginosa prevalence in cystic fibrosis adults over time. BMC Pulm. Med. 2016, 16, 176. 10.1186/s12890-016-0333-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Høiby N. Recent advances in the treatment of Pseudomonas aeruginosa infections in cystic fibrosis. BMC Med. 2011, 9, 32. 10.1186/1741-7015-9-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brackman G.; Cos P.; Maes L.; Nelis H. J.; Coenye T. Quorum Sensing Inhibitors Increase the Susceptibility of Bacterial Biofilms to Antibiotics In Vitro and In Vivo. Antimicrob. Agents Chemother. 2011, 55, 2655–2661. 10.1128/AAC.00045-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui N.; Fan J.; Cen K. Effect of particle-particle collision in decaying homogeneous and isotropic turbulence. Phys. Rev. E 2008, 78, 046307 10.1103/PhysRevE.78.046307. [DOI] [PubMed] [Google Scholar]
- Brackman G.; Coenye T. Quorum Sensing Inhibitors as Anti-Biofilm Agents. Curr. Pharm. Des. 2015, 21, 5–11. 10.2174/1381612820666140905114627. [DOI] [PubMed] [Google Scholar]
- Hurley M. N.; Cámara M.; Smyth A. R. Novel approaches to the treatment of Pseudomonas aeruginosa infections in cystic fibrosis. Eur. Respir. J. 2012, 40, 1014–1023. 10.1183/09031936.00042012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilangovan A.; Fletcher M.; Rampioni G.; Pustelny C.; Rumbaugh K.; Heeb S.; et al. Structural basis for native agonist and synthetic inhibitor recognition by the Pseudomonas aeruginosa quorum sensing regulator PqsR (MvfR). PLoS Pathog. 2013, 9, e1003508 10.1371/journal.ppat.1003508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sou T.; Kukavica-Ibrulj I.; Soukarieh F.; Halliday N.; Levesque R. C.; Williams P.; et al. Model-Based Drug Development in Pulmonary Delivery: Pharmacokinetic Analysis of Novel Drug Candidates for Treatment of Pseudomonas aeruginosa Lung Infection. J. Pharm. Sci. 2019, 108, 630–640. 10.1016/j.xphs.2018.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal S., Sheiner L. B., Boeckmann A., Bauer R. J.. NONMEM User’s Guides (1989–2009); Icon Development Solutions: Ellicott City, MD, USA. 2009. [Google Scholar]
- Lindbom L.; Ribbing J.; Jonsson E. N. Perl-speaks-NONMEM (PsN)—a Perl module for NONMEM related programming. Comput. Methods Programs Biomed. 2004, 75, 85–94. 10.1016/j.cmpb.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Jonsson E. N.; Karlsson M. O. Xpose—an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput. Methods Programs Biomed. 1998, 58, 51–64. 10.1016/S0169-2607(98)00067-4. [DOI] [PubMed] [Google Scholar]
- Keizer R. J.; Karlsson M. O.; Hooker A. Modeling and Simulation Workbench for NONMEM: Tutorial on Pirana, PsN, and Xpose. CPT: Pharmacometrics Syst. Pharmacol. 2013, 2, e50 10.1038/psp.2013.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T. H. T.; Mouksassi M. S.; Holford N.; Al-Huniti N.; Freedman I.; Hooker A. C.; et al. Model Evaluation of Continuous Data Pharmacometric Models: Metrics and Graphics. CPT: Pharmacometrics Syst. Pharmacol. 2017, 6, 87–109. 10.1002/psp4.12161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal S. L. Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn. 2001, 28, 481–504. 10.1023/A:1012299115260. [DOI] [PubMed] [Google Scholar]
- Baron K. T.. mrgsolve: Simulate from ODE-Based Population PK/PD and Systems Pharmacology Models 2018 [Available from: https://CRAN.R-project.org/package=mrgsolve.
- Chang W., Cheng J., Allaire J., Xie Y., McPherson J.. shiny: Web Application Framework for R. R package. version 1.2.0. ed2018.
- Pilcer G.; Amighi K. Formulation strategy and use of excipients in pulmonary drug delivery. Int. J. Pharm. 2010, 392, 1–19. 10.1016/j.ijpharm.2010.03.017. [DOI] [PubMed] [Google Scholar]
- Traini D.; Young P. M.; Rogueda P.; Price R. Investigation into the influence of polymeric stabilizing excipients on inter-particulate forces in pressurised metered dose inhalers. Int. J. Pharm. 2006, 320, 58–63. 10.1016/j.ijpharm.2006.04.016. [DOI] [PubMed] [Google Scholar]
- Respaud R.; Marchand D.; Parent C.; Pelat T.; Thullier P.; Tournamille J. F.; et al. Effect of formulation on the stability and aerosol performance of a nebulized antibody. mAbs 2014, 6, 1347–1355. 10.4161/mabs.29938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yapa S. W. S.; Li J.; CJH P.; Nation R. L.; Patel K.; MP M. I. Population Pharmacokinetics of Colistin Methanesulfonate in Rats: Achieving Sustained Lung Concentrations of Colistin for Targeting Respiratory Infections. Antimicrob. Agents Chemother. 2013, 57, 5087–5095. 10.1128/AAC.01127-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.-W.; Zhou Q. T.; Cheah S.-E.; Zhao J.; Chen K.; Wang J.; et al. Pharmacokinetics/Pharmacodynamics of Pulmonary Delivery of Colistin against Pseudomonas aeruginosa in a Mouse Lung Infection Model. Antimicrob. Agents Chemother. 2017, 61, e02025–e02016. 10.1128/AAC.02025-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stass H.; Delesen H.; Nagelschmitz J.; Staab D. Safety and Pharmacokinetics of Ciprofloxacin Dry Powder for Inhalation in Cystic Fibrosis: A Phase I, Randomized, Single-Dose, Dose-Escalation Study. J. Aerosol Med. Pulm. Drug Delivery 2015, 28, 106–115. 10.1089/jamp.2013.1056. [DOI] [PubMed] [Google Scholar]
- Stass H.; Weimann B.; Nagelschmitz J.; Rolinck-Werninghaus C.; Staab D. Tolerability and Pharmacokinetic Properties of Ciprofloxacin Dry Powder for Inhalation in Patients With Cystic Fibrosis: A Phase I, Randomized, Dose-Escalation Study. Clinical Therapeutics. 2013, 35, 1571–1581. 10.1016/j.clinthera.2013.08.003. [DOI] [PubMed] [Google Scholar]
- Marchand S.; Grégoire N.; Brillault J.; Lamarche I.; Gobin P.; Couet W. Biopharmaceutical Characterization of Nebulized Antimicrobial Agents in Rats: 3. Tobramycin. Antimicrob. Agents Chemother. 2015, 59, 6646–6647. 10.1128/AAC.01647-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labiris N. R.; Dolovich M. B. Pulmonary drug delivery. Part I: Physiological factors affecting therapeutic effectiveness of aerosolized medications. Br. J. Clin. Pharmacol. 2003, 56, 588–599. 10.1046/j.1365-2125.2003.01892.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton J. S.; Fishburn C. S.; Weers J. G. The lungs as a portal of entry for systemic drug delivery. Proc. Am. Thorac Soc. 2004, 1, 338–344. 10.1513/pats.200409-049TA. [DOI] [PubMed] [Google Scholar]
- Uchenna Agu R.; Ikechukwu Ugwoke M.; Armand M.; Kinget R.; Verbeke N. The lung as a route for systemic delivery of therapeutic proteins and peptides. Respir. Res. 2001, 2, 198–209. 10.1186/rr58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson J.; Thörn H.; Sjögren E.; Holmstén L.; Rubin K.; Lennernäs H. Pulmonary Dissolution of Poorly Soluble Compounds Studied in an ex Vivo Rat Lung Model. Mol. Pharmaceutics 2019, 16, 3053–3064. 10.1021/acs.molpharmaceut.9b00289. [DOI] [PubMed] [Google Scholar]
- Mouton J. W.; Theuretzbacher U.; Craig W. A.; Tulkens P. M.; Derendorf H.; Cars O. Tissue concentrations: do we ever learn?. J. Antimicrob. Chemother. 2007, 61, 235–237. 10.1093/jac/dkm476. [DOI] [PubMed] [Google Scholar]
- Andersson S. B. E.; Alvebratt C.; Bergström C. A. S. Controlled Suspensions Enable Rapid Determinations of Intrinsic Dissolution Rate and Apparent Solubility of Poorly Water-Soluble Compounds. Pharm. Res. 2017, 34, 1805–1816. 10.1007/s11095-017-2188-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergström C. A. S.; Box K.; Holm R.; Matthews W.; McAllister M.; Müllertz A.; et al. Biorelevant intrinsic dissolution profiling in early drug development: Fundamental, methodological, and industrial aspects. Eur. J. Pharm. Biopharm. 2019, 139, 101–114. 10.1016/j.ejpb.2019.03.011. [DOI] [PubMed] [Google Scholar]
- Jamei M. Where Do PBPK Models Stand in Pharmacometrics and Systems Pharmacology?. CPT: Pharmacometrics Syst. Pharmacol. 2020, 9, 75–76. 10.1002/psp4.12493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig W. A. Pharmacokinetic/Pharmacodynamic Parameters: Rationale for Antibacterial Dosing of Mice and Men. Clin. Infect. Dis. 1998, 26, 1–10. 10.1086/516284. [DOI] [PubMed] [Google Scholar]
- Mouton J. W.; Dudley M. N.; Cars O.; Derendorf H.; Drusano G. L. Standardization of pharmacokinetic/pharmacodynamic (PK/PD) terminology for anti-infective drugs: an update. J. Antimicrob. Chemother. 2005, 55, 601–607. 10.1093/jac/dki079. [DOI] [PubMed] [Google Scholar]
- Nielsen E. I.; Cars O.; Friberg L. E. Pharmacokinetic/Pharmacodynamic (PK/PD) Indices of Antibiotics Predicted by a Semimechanistic PKPD Model: a Step toward Model-Based Dose Optimization. Antimicrob. Agents Chemother. 2011, 55, 4619–4630. 10.1128/AAC.00182-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelman B.; Leggett J.; Turnidge J.; Ebert S.; Gudmundsson S.; Craig W. A. Correlation of Antimicrobial Pharmacokinetic Parameters with Therapeutic Efficacy in an Animal Model. J. Infect. Dis. 1988, 158, 831–847. 10.1093/infdis/158.4.831. [DOI] [PubMed] [Google Scholar]
- Weers J. G.; Miller D. P. Formulation Design of Dry Powders for Inhalation. J. Pharm. Sci. 2015, 104, 3259–3288. 10.1002/jps.24574. [DOI] [PubMed] [Google Scholar]
- Hoppentocht M.; Hagedoorn P.; Frijlink H. W.; de Boer A. H. Technological and practical challenges of dry powder inhalers and formulations. Adv. Drug Delivery Rev. 2014, 75, 18–31. 10.1016/j.addr.2014.04.004. [DOI] [PubMed] [Google Scholar]
- Hastedt J. E.; Bäckman P.; Clark A. R.; Doub W.; Hickey A.; Hochhaus G.; et al. Scope and relevance of a pulmonary biopharmaceutical classification system AAPS/FDA/USP Workshop March 16-17th, 2015 in Baltimore. MD. AAPS Open 2016, 2, 1–20. 10.1186/s41120-015-0002-x. [DOI] [Google Scholar]