

Abstract

The technological advances in mass spectrometry allow us to collect more comprehensive data with higher quality and increasing speed. With the rapidly increasing amount of data generated, the need for streamlining analyses becomes more apparent. Proteomics data is known to be often affected by systemic bias from unknown sources, and failing to adequately normalize the data can lead to erroneous conclusions. To allow researchers to easily evaluate and compare different normalization methods via a user-friendly interface, we have developed “proteiNorm”. The current implementation of proteiNorm accommodates preliminary filters on peptide and sample levels followed by an evaluation of several popular normalization methods and visualization of the missing value. The user then selects an adequate normalization method and one of the several imputation methods used for the subsequent comparison of different differential expression methods and estimation of statistical power. The application of proteiNorm and interpretation of its results are demonstrated on two tandem mass tag multiplex (TMT6plex and TMT10plex) and one label-free spike-in mass spectrometry example data set. The three data sets reveal how the normalization methods perform differently on different experimental designs and the need for evaluation of normalization methods for each mass spectrometry experiment. With proteiNorm, we provide a user-friendly tool to identify an adequate normalization method and to select an appropriate method for differential expression analysis.
1. Introduction
With the latest technological advances in the development of mass spectrometers, the detection accuracy of the instruments and the speed with which data is being produced have drastically increased. The amount of data generated by these high-throughput devices increases rapidly, making the need for tools that allow users to streamline analyses more and more apparent.
In a single mass spectrometry run, peptide levels of thousands of proteins can be measured for several patients. However, mass spectrometry data is often affected by systemic bias, variations caused by non-biological sources, which can lead to erroneous conclusions.1 These sources of variation can include sample preparation and handling, device calibration, changes in temperature, and other unknown sources.2 Unlike the effect of batching, these sources of variation are either not measured or simply cannot be measured and therefore cannot be controlled for during the statistical analysis. Normalization is a technique that aims to account for these systematic biases and make samples more comparable while preserving the signal. Many normalization methods have been proposed, with most of them being adopted from DNA microarray technology.3 Välikangas et al.2 systemically evaluated normalization methods used in quantitative label-free proteomics. While there are certain normalization methods that consistently rank among the top performing methods (i.e., variance stabilizing normalization (VSN), linear regression normalization, and local regression normalization), it is crucial to select a suitable normalization method for a specific data set.2,4 The need for tools that identify proper methods for normalization was recognized and addressed by Webb-Robertson et al.4 and Chawade et al.,1 with their tools SPAN and Normalyzer, respectively. SPAN combines eight peptide selection methods to select peptides subsequently used during the normalization and five methods for normalization.4 Normalyzer includes popular normalization methods such as linear regression, local regression, total intensity, average intensity, median intensity, VSN, and quantile normalization. The performance of each normalization method is individually evaluated by comparing its pooled coefficient of variance (PCV), pooled median absolute deviation (PMAD), and pooled estimate of variance (PEV).1 However, there remains an outstanding need for a user-friendly and publicly available tool that provides a systematic evaluation of normalization methods and also facilitates imputation of missing data and compares different methods to assess differential abundance of proteins and/or peptides.
Here, we present proteiNorm, a user-friendly tool for a systematic evaluation of normalization methods for mass spectrometric data sets that do not have a protein standard or spike-in proteins for reference, imputation of missing values, and comparisons of different differential abundance methods. Our tool provides a point-and-click R-Shiny interface for researchers that are less comfortable or unfamiliar with the R programming environment. However, the functions are run in R and can be easily modified to include additional normalization methods.
After loading the raw peptide data from the database search results into the peptide input box in the “data” tab in the Shiny interface, the user can choose to filter the peptide data using the “Top3” method,5 where the three most intense peptides of a protein are averaged to calculate a stable protein intensity and create a filtered protein file. Next, either the filtered protein file or the original/raw protein file from the database search results is uploaded in the protein input box in the “data” tab. The tool is designed to normalize the data using the protein relative abundance directly from MaxQuant, which is generated by summing the peptide intensities. The Top3 method provided in the “data” upload for peptide data is used when outlier peptides are present that then skew the relative protein abundance values provided by MaxQuant. The ultimate goal is to perform downstream differential abundance analysis to identify significantly differentiating proteins between sample conditions. However, if someone is interested in performing an analysis at the peptide level, then a matrix of peptides as rows and peptide intensities for each sample as columns can be loaded into the protein input box in the “data” tab in the Shiny interface.
The meta-data (i.e., custom sample names, group labels, and batch labels) can be specified in the user-interface after loading protein data. The custom sample names will be used for labeling all subsequent plots in the tool. On the next “filter” tab in the Shiny interface, any unwanted samples such as blanks, samples with poor quality, or pooled samples can be removed from subsequent steps. The remaining data is then subjected to different normalization methods, which are compared among each other in the “Normalization” tab. Once a suitable normalization method is identified, missing values can be imputed (optional) and the normalized protein data can be saved from the “DAtest” tab in the Shiny interface. In addition, different methods for differential abundance analysis can be evaluated for a selected normalization method (“DAtest” tab), and the statistical power of a given test of differential abundance can be assessed (“DApower” tab).
2. Results
Our tool “proteiNorm” is implemented in R statistical language (http://r-project.org, version 3.6) using the package “shiny”6 to provide a user-friendly user interface. It is publicly available at https://sbyrum.shinyapps.io/proteiNorm/ and can also be downloaded from GitHub (https://github.com/ByrumLab/proteiNorm). Figure 1 shows its workflow and illustrates an overview involving the following steps: upload peptide-level data, filter peptides to create new filtered protein-level data (optional), upload protein-level data (original or filtered), provide meta-data (e.g., batch and treatment groups), normalize data with different normalization methods and evaluate their performances, select appropriate normalization methods, select an appropriate imputation method (optional), and export normalized (and imputed) protein-level data. If the user is uncertain about the choice of method for the differential abundance analysis, proteiNorm provides the option to compare different methods for differential abundance analysis provided by the R package “DAtest”.7
Figure 1.
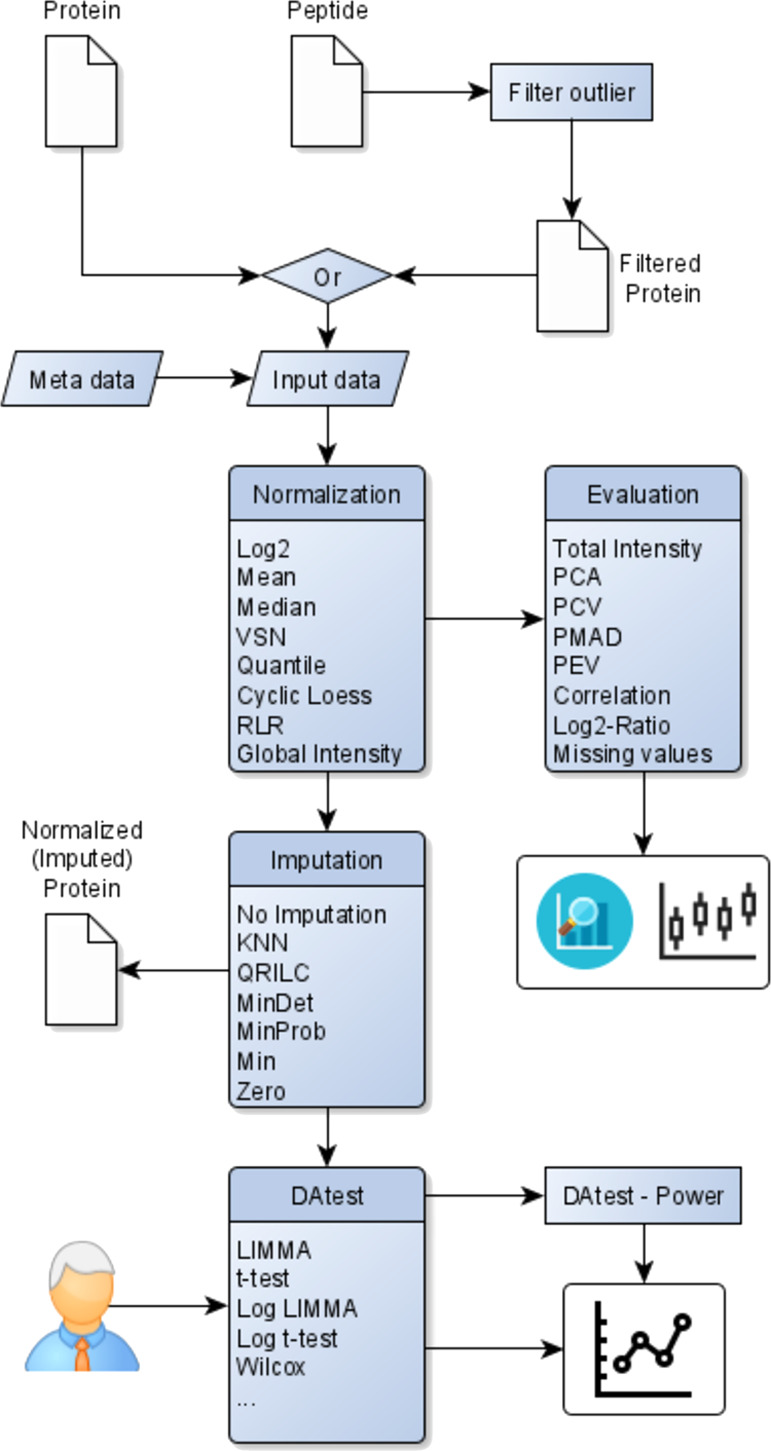
Overview of proteiNorm’s workflow, inputs, and outputs.
To demonstrate the application of proteiNorm and the interpretation of results in different scenarios, we analyzed the following proteomics data sets: mouse data set with a weak signal, breast cancer cell lines with a strong signal, and spike-in data set with weak to strong known signals (described in Sections 2.1–2.3).
2.1. Mouse Data Set
First, the raw peptide file (“peptides.txt”) produced by MaxQuant is uploaded and filtered using the “Top3” method5 and then exported as the filtered proteinGroups file (e.g., “proteinGroups_filtered.txt”). Next, the newly created and filtered proteinGroups file is uploaded (alternatively, the unfiltered raw “proteinGroups.txt” file generated by MaxQuant can be uploaded), and the corresponding meta-data is specified, such as treatment group, batch number, and an optional custom name (as shown in Table 1). In this example, we focus on the striatum samples (“S_T_1” – “S_T_3” and “S_1” – “S_3”) and therefore unselected all nucleus accumbens samples (“NA_T_1” – “NA_T_3” and “NA_1” – “NA_3”) within the “Filter” tab. No additional samples were excluded as no sample was identified as an outlier sample or a sample with poor quality based on the intensity distributions and the PCA plot (see Figures S1–S2). Next, the performance of each normalization method is evaluated at the protein level. Therefore, the following figures are inspected (Figure 2): (A) total intensity, (B) PCA, (D) PCV, (E) PEV, (F) PMAD, (G) intragroup correlation, (H) correlation heatmap, and (I) LogRatio density. Panels (A), (B), and (H) visualize the total intensity, PCA, and correlation heatmap of VSN, respectively (panels (A), (B), and (H) of the remaining normalization are shown in Figures S3–S5, respectively).
Table 1. Meta-Data for the Mouse Data Seta.
| Protein.Sample.Names | Custom.Sample.Names | group | batch |
|---|---|---|---|
| Reporter.intensity.corrected.1.TMT1 | NA_T_1 | NA_T | 1 |
| Reporter.intensity.corrected.2.TMT1 | NA_T_2 | NA_T | 1 |
| Reporter.intensity.corrected.3.TMT1 | NA_T_3 | NA_T | 1 |
| Reporter.intensity.corrected.4.TMT1 | NA_1 | NA | 1 |
| Reporter.intensity.corrected.5.TMT1 | NA_2 | NA | 1 |
| Reporter.intensity.corrected.6.TMT1 | NA_3 | NA | 1 |
| Reporter.intensity.corrected.1.TMT2 | S_T_1 | S_T | 2 |
| Reporter.intensity.corrected.2.TMT2 | S_T_2 | S_T | 2 |
| Reporter.intensity.corrected.3.TMT2 | S_T_3 | S_T | 2 |
| Reporter.intensity.corrected.4.TMT2 | S_1 | S | 2 |
| Reporter.intensity.corrected.5.TMT2 | S_2 | S | 2 |
| Reporter.intensity.corrected.6.TMT2 | S_3 | S | 2 |
“Protein.Sample.Names” care automatically generated from the proteinGroups file. “Custom.Sample.Names” is optional and replaces protein sample names when provided (needs to be a unique name). “Group” specifies individual treatment groups. “Batch” is optional and indicates the batch for each sample.
Figure 2.

Evaluation of normalization and missing values. (A) Sum of normalized intensities using VSN by sample. (B) Principal component analysis plot based on data normalized by VSN. (C) Sample-clustered heatmap of missing values. (D) Pooled intragroup coefficient of variance comparing different normalization methods. (E) Pooled intragroup estimate of variance comparing different normalization methods. (F) Pooled intragroup median absolute deviation comparing different normalization methods. (G) Pairwise sample correlations within a group for different normalization methods. (H) Correlation heatmap (all pairwise samples). (I) Distribution of log2-ratios (all two-group combinations) for different normalization methods.
With regard to the total intensity, the samples are nearly indistinguishable after applying VSN (Figure 2A) and the remaining normalization methods produced very similar results (Figure S3). Unless the treatment between groups, such as in a knock out experiment, is expected to have a major impact on a subset of the most abundant proteins, it can be expected that the sums of the intensities by sample are similar within a batch (small changes or changes in low abundant proteins will be dominated by highly abundant proteins; differences between total intensities comparing multiple batches are not unusual). Also, in cases where a sample did not sequence well on the instrument, the total intensity for that sample will be lower than the rest and can indicate a bad sequencing run, especially in the case of label-free designs. Panel (B) of Figure 2 shows the PCA figure of VSN (see Figure S2 for the PCA plot before normalization and Figure S4 for remaining normalization methods) and demonstrates an unclear separation among the sample groups. In Figure 2C, a clustered heatmap of missing values for each sample is shown. Only proteins with at least one measured value and one missing value are shown (proteins with complete measurement or only missing values are not shown). The purpose of this heatmap is to assist researchers to identify patterns of missing values (i.e., MAR or MNAR) and guide the choice of imputation method, if desired. Here, the missing values seem to not demonstrate an obvious pattern. Figure 2D–F compares different measurements of intragroup variations (PCV, PEV, and PMAD, respectively) for different normalization methods. Small values are desirable here as they indicate limited variation of sample replicates. It can be observed that VSN seems to perform the best in reducing the intragroup variation throughout all three metrics. Figure 2G compares the pairwise correlation within the treatment groups of the normalization methods. High intragroup correlation values indicate high correlation of replicates within a treatment group and are desirable. All pairwise correlations using VSN are presented in a clustered correlation heatmap (Figure 2H; clustered correlation heatmaps of the remaining normalization methods in Figure S5). It can be observed that the untreated and treated samples from the same specimen cluster together. This could indicate that the differences between sample specimens outweigh the treatment effect. In panel (I) of Figure 2, the density distributions of log2-ratios of all pairwise treatment combinations are compared for each normalization method. Unless an unbalanced regulation (primarily up- or downregulation) in the data is to be expected, the densities should be centered around zero. The density distributions of most normalization are more or less off center and could potentially indicate a bias in the normalized data. Only the density distributions of “VSN” and “RLR” normalization are centered around zero, with “VSN” demonstrating the highest peak.
Based on the evaluation and comparison of normalization methods and the heatmap of missing values, the user can choose an adequate normalization method and has the choice to select an imputation method. Here, we have selected VSN for the mouse data set. Because some statistical methods do not require complete data (e.g., limma8), imputation is not always necessary, and we have decided not to impute our example data set. Following the selection of normalization and imputation method (“no imputation” is an imputation choice), the normalized protein data was exported from the “DAtest” tab in the Shiny interface.
2.2. Breast Cancer Cell Lines
Analogously, the raw peptide file (“peptides.txt”) of the breast cancer cell lines is first uploaded, filtered using the “Top3” method,5 and then exported as the filtered proteinGroups file. Next, the newly created and filtered “proteinGroups.txt” file is uploaded, and the corresponding meta-data is specified (as shown in Table 2). Next, pooled samples are unselected and excluded from the subsequent evaluation. The pool sample helps to evaluate TMT batch effects in the PCA plot. The pool also serves as a mixture of all proteins present in all samples so that mass spectrometry has the potential to switch on those proteins in both TMT batches and limit the number of missing values across two TMT runs. However, the pool is not part of the differential abundance analysis and should be removed from the final normalized protein data that will be utilized for differential abundance analysis.
Table 2. Meta-Data for the Breast Cancer Data Seta.
| Protein.Sample.Names | Custom.Sample.Names | group | batch |
|---|---|---|---|
| Reporter.intensity.corrected.1.TMT3 | ER/PR+ MCF7_UT_19 | ERPR | 3 |
| Reporter.intensity.corrected.2.TMT3 | ER/PR+ MCF7_UT_20 | ERPR | 3 |
| Reporter.intensity.corrected.3.TMT3 | ER/PR+ MCF7_UT_21 | ERPR | 3 |
| Reporter.intensity.corrected.4.TMT3 | MCF 10A_UT_22 | control | 3 |
| Reporter.intensity.corrected.5.TMT3 | MCF 10A_UT_23 | control | 3 |
| Reporter.intensity.corrected.6.TMT3 | MCF 10A_UT_24 | control | 3 |
| Reporter.intensity.corrected.7.TMT3 | HER2+ HCC 1954_UT_25 | HER2 | 3 |
| Reporter.intensity.corrected.8.TMT3 | HER2+ HCC 1954_UT_26 | HER2 | 3 |
| Reporter.intensity.corrected.9.TMT3 | HER2+ HCC 1954_UT_27 | HER2 | 3 |
| Reporter.intensity.corrected.10.TMT3 | pool_3 | pool | 3 |
| Reporter.intensity.corrected.1.TMT4 | MCF 10A_TR_28 | ERPR_TR | 4 |
| Reporter.intensity.corrected.2.TMT4 | MCF 10A_TR_29 | ERPR_TR | 4 |
| Reporter.intensity.corrected.3.TMT4 | MCF 10A_TR_30 | ERPR_TR | 4 |
| Reporter.intensity.corrected.4.TMT4 | ER/PR+ MCF7_TR_31 | control_TR | 4 |
| Reporter.intensity.corrected.5.TMT4 | ER/PR+ MCF7_TR_32 | control_TR | 4 |
| Reporter.intensity.corrected.6.TMT4 | ER/PR+ MCF7_TR_33 | control_TR | 4 |
| Reporter.intensity.corrected.7.TMT4 | HER2+ HCC 1954_TR_34 | HER2_TR | 4 |
| Reporter.intensity.corrected.8.TMT4 | HER2+ HCC 1954_TR_35 | HER2_TR | 4 |
| Reporter.intensity.corrected.9.TMT4 | HER2+ HCC 1954_TR_36 | HER2_TR | 4 |
| Reporter.intensity.corrected.10.TMT4 | pool_4 | pool | 4 |
“Protein.Sample.Names” are automatically generated from the proteinGroups file. “Custom.Sample.Names” is optional and replaces protein sample names when provided (needs to be a unique name). “Group” specifies individual treatment groups. “Batch” is optional and indicates the batch for each sample.
No additional samples were excluded as no sample was identified as an outlier sample or a sample with poor quality based on the intensity distributions and the PCA plot (see Figures S6–S7). It can be observed that the batch effect is more prevalent in the raw peptide data than in the filtered protein data (Figure S7). Next, the performance of each normalization method is evaluated by inspecting the following figures (Figure S8): (A) total intensity, (B) PCA, (D) PCV, (E) PEV, (F) PMAD, (G) intragroup correlation, (H) correlation heatmap, and (I) LogRatio density. We selected the cyclic loess normalization to normalize this data set. Panels (A), (B), and (H) visualize the total intensity, PCA, and correlation heatmap of the cyclic loess normalization, respectively (panels (A), (B), and (H) of the remaining normalization methods produced similar results; Figures S9–S11).
With regard to total intensity, the samples are nearly indistinguishable after applying cyclic loess normalization (Figure S8A) and the remaining normalization methods produced very similar results (Figure S9). Panel (B) of Figure S8 shows the PCA figure after cyclic loess normalization (see Figure S10 for remaining normalization methods) and demonstrates a stronger separation of cell lines after normalization, while the cell-line-treatment combinations cluster tighter than before normalizing. In Figure S8C, a clustered heatmap of missing values for each sample is shown. It can be observed that samples cluster primarily by batch and secondarily by cell-line-treatment combination. It is apparent that measurements are not missing at random, but rather the missingness of values is influenced by batch and cell-line-treatment combination. Figure S8D–F compares different measurements of intragroup variations (PCV, PEV, and PMAD, respectively) for different normalization methods. It can be observed that all evaluated normalization methods produced similar results, with cyclic loess normalization ranking among the better performing methods. Figure S8G compares the pairwise correlation within cell-line-treatment combinations of the normalization methods. All pairwise correlations using cyclic normalization are presented in a clustered heatmap (Figure S8H; correlation heatmaps of remaining normalization methods in Figure S11). The highest correlations are observed among samples from the same cell-line with either no treatment or treatment. The next strongest correlations are demonstrated between treated and untreated samples from the same cell-line. The second weakest correlations are produced by samples from different cell-lines with the same treatment, which are also samples within the same batch. Samples originating from different cell-lines with different treatments (in different batches) exhibited the weakest correlation. In panel (H) of Figure S8, the density distributions of log2-ratios of all treatment combinations are compared for each normalization method. No differences are apparent among the different normalization methods.
2.3. Spike-in
For the spiked-in data, the raw peptide files (“peptides.txt”) from MaxQuant are uploaded and filtered using the “Top3” method5 before exporting the filtered “proteinGroups_filtered.txt” file. Next, the filtered “proteinGroups_filtered.txt” file is uploaded, the corresponding meta-data is specified (as shown in Table 3), and no samples are excluded. Analogously using Figure S14 to evaluate the different normalization methods, it can be observed that the methods perform similarly. For the spike-in data set, “VSN” was selected because of the slightly lower intragroup variations and slightly higher intragroup correlation. The advantage of this spike-in data set is that the true signal is known and the performance of the normalization can be evaluated by comparing the intensity ratios of the spiked-in proteins.
Table 3. Meta-Data for the Spiked-in Data Seta.
| Protein.Sample.Names | Custom.Sample.Names | group | batch |
|---|---|---|---|
| Intensity.12500amol_R1 | 12500amol_R1 | 12500 | 1 |
| Intensity.12500amol_R2 | 12500amol_R2 | 12500 | 1 |
| Intensity.12500amol_R3 | 12500amol_R3 | 12500 | 1 |
| Intensity.125amol_R1 | 125amol_R1 | 125 | 1 |
| Intensity.125amol_R2 | 125amol_R2 | 125 | 1 |
| Intensity.125amol_R3 | 125amol_R3 | 125 | 1 |
| Intensity.25000amol_R1 | 25000amol_R1 | 25000 | 1 |
| Intensity.25000amol_R2 | 25000amol_R2 | 25000 | 1 |
| Intensity.25000amol_R3 | 25000amol_R3 | 25000 | 1 |
| Intensity.2500amol_R1 | 2500amol_R1 | 2500 | 1 |
| Intensity.2500amol_R2 | 2500amol_R2 | 2500 | 1 |
| Intensity.2500amol_R3 | 2500amol_R3 | 2500 | 1 |
| Intensity.250amol_R1 | 250amol_R1 | 250 | 1 |
| Intensity.250amol_R2 | 250amol_R2 | 250 | 1 |
| Intensity.250amol_R3 | 250amol_R3 | 250 | 1 |
| Intensity.50000amol_R1 | 50000amol_R1 | 50000 | 1 |
| Intensity.50000amol_R2 | 50000amol_R2 | 50000 | 1 |
| Intensity.50000amol_R3 | 50000amol_R3 | 50000 | 1 |
| Intensity.5000amol_R1 | 5000amol_R1 | 5000 | 1 |
| Intensity.5000amol_R2 | 5000amol_R2 | 5000 | 1 |
| Intensity.5000amol_R3 | 5000amol_R3 | 5000 | 1 |
| Intensity.500amol_R1 | 500amol_R1 | 500 | 1 |
| Intensity.500amol_R2 | 500amol_R2 | 500 | 1 |
| Intensity.500amol_R3 | 500amol_R3 | 500 | 1 |
| Intensity.50amol_R1 | 50amol_R1 | 50 | 1 |
| Intensity.50amol_R2 | 50amol_R2 | 50 | 1 |
| Intensity.50amol_R3 | 50amol_R3 | 50 | 1 |
“Protein.Sample.Names” are automatically generated from the proteinGroups file. “Custom.Sample.Names” is optional and replaces protein sample names when provided (needs to be a unique name). “Group” specifies individual treatment groups. “Batch” is optional and indicates the batch for each sample.
Figure 3 shows the ratios between different concentrations of the spiked-in protein averages across replicates comparing the normalized protein data to the raw protein data. It can be observed that normalized data is performing better in the estimation of the true concentration ratio (gray dashed line), especially when the concentration and consequently the intensities increase.
Figure 3.

Distribution ratios of spiked-in proteins (averages across replicates) of different spike-in concentrations for raw and normalized protein intensities. Intensities of spiked-in proteins were averaged across concentration replicates to calculate the ratio of averaged proteins between different spike-in concentrations. The distributions of ratios of the measured proteins are compared between raw and normalized protein data. Spiked-in concentrations of 50–500amol were excluded due to the absence of most measurements.
3. Discussion
In our example data sets, we evaluated different popular normalization methods. While most methods performed very similarly, we decided to use the VSN or cyclic loess normalization for our data sets based on their performance. In addition, both normalization methods rank consistently among the top performing methods.2 While it is important to identify an adequate normalization method for the data set at hand, Välikangas et al.2 have demonstrated that most normalization methods result in better performance than a simple log2 transformation.
In experimental designs where there are expected to be thousands of significantly differentiating proteins, such as the case for the breast cancer cell lines, the choice of normalization method does not have a big impact on the final results. However, in the case of the mouse data where it is expected to have small protein changes, then the normalization method can make an impact on the final significant protein lists as shown in the log2 ratio plots. Each data set should be evaluated based on its own metrics.
The current implementation of proteiNorm is limited to the evaluation of normalization methods utilizing intragroup variance and correlation. A more suitable form of evaluations for normalization methods is the assessment of true spike-in signals and/or proteomics standards. However, this requires additional steps during the sample preparation and might not always be possible. In future versions of proteiNorm, we intend to include the option of evaluating normalization methods by assessing spike-in signals and/or proteomic standards. Further extensions include a broader spectrum of the input file, additional peptide filter methods, and additional normalization and imputation methods.
4. Conclusions
When analyzing proteomics data measured using mass spectrometers, normalization of the data is an integral data processing step to adjust unwanted systematic biases. Failing to acknowledge such issues can lead to erroneous results and inferences, which limits reproducibility and scientific progress and increases the cost of science.
With proteiNorm, we present a user-friendly tool with the goal of assisting researchers in the evaluation of popular normalization methods and of identifying an adequate normalization method suited for their data sets. Researchers also have the option to impute missing values and/or compare different differential abundance methods and their power for a range of effect sizes. Once the data has been evaluated in proteiNorm, the normalized output is now ready for differential abundance analysis using other available tools such as limma.8
5. Methods
5.1. Data Requirements
As input, proteiNorm expects tab-separated peptide (optional) and protein data (not on logarithmic scale) as produced by software such as MaxQuant,9 where each row represents a peptide or protein and the column names of the measured intensities (samples) beginning with “Reporter intensity corrected” followed by an integer and an optional label (e.g., “Reporter intensity corrected 5 TMT2”) for TMT experiments. The column names for samples in label-free experiments should begin with “Intensity” followed by an integer (“Intensity 01”). Data from both mass spectrometry quantitation methods, tandem mass tag (TMT) and label-free mass spectrometry, are supported. The TMT reporter intensities should be corrected using the error correction factors provided by Thermo Fisher for the specific TMT lot that was used during the sample preparation. These correction factors are imported into MaxQuant when setting up the database search parameters. The MaxQuant “proteinGroups.txt” output file will then contain column names for each TMT reporter ion labeled as “Reporter intensity corrected” and “Reporter intensity”. In the following examples, we are using the “Reporter intensity corrected” columns from MaxQuant since these intensities apply the TMT correction factors. The reporter ion isotopic distributions (−2, −1, +1, +2) are primarily for carbon isotopes with reporter ion interference for each mass tag. ProteiNorm automatically detects these column names and determines if the data is TMT or label-free data type. ProteiNorm also removes proteins flagged as common contaminants and reverse sequences in the MaxQuant output using the column names “Potential contaminant” and “Reverse” provided in the proteinGroups.txt MaxQuant output file. If you prefer to load in a different matrix of data, then the column labels for the samples will need to be modified to match the naming structure provided here. An example of the data input files is hosted on Github for both TMT and label-free data sets.
Due to the detection limits of mass spectrometry instruments, many measurements of peptides or proteins result in intensity levels of zero. These values will be considered as missing values (denoted as NA) and can be imputed with precaution. A modified heatmap of missing values (Figure 2C) from the DEP Bioconductor package10 helps to determine if data is missing at random (MAR) or missing not at random (MNAR), and the MSnbase vignette describes different imputation methods used for different types of missing data.11
5.2. Normalization Methods
The current implementation of proteiNorm includes several popular normalization methods, including log2 (Log2) (a data transformation method), median normalization (Median), mean normalization (Mean), variance stabilizing normalization (VSN),12 quantile normalization (Quantile),13 cyclic loess normalization (Cyclic Loess),8 global robust linear regression normalization (RLR),1 and global intensity normalization (Global Intensity).1 The individual performance of each method can be evaluated by comparing the following metrics: total intensity (Figure 2A), principal component analysis (PCA; Figure 2B), pooled intragroup coefficient of variation (PCV; Figure 2D), pooled intragroup estimate of variance (PEV; Figure 2E), pooled intragroup median absolute deviation (PMAD; Figure 2F); intragroup correlation (Figure 2G), correlation heatmap (Pearson; Figure 2H), and log2-ratio distributions (Figure 2I). These plots are automatically generated and can be viewed on the “Normalization” tab. When samples are unselected on the “Filters” tab, the normalization plots automatically update to include only the selected samples. The overall goal is to select the normalization method with the lowest variance and the highest correlation within the replicates within a group. The plots included in proteiNorm display a boxplot of each sample group where each dot represents a group so the variance effects of the normalization method can easily be evaluated by examining the metrics on the y-axis. The correlation plots include multiple dots per group for all the pairwise intragroup correlations. The log2 ratio plot helps to evaluate if the normalization method introduces skewing of the fold change values when comparing the sample group conditions. The log2-ratios are calculated from all two-group combinations. The log2 ratio should be centered on zero and not introduce bias toward up- or downregulation of proteins. All of the plots in proteiNorm can be saved by clicking on the “Save Figures” button in the “Normalization” tab in the interface.
5.3. Differential Abundance Analysis
If the user is undecided about the statistical method for the differential abundance analysis, methods can be compared using the incorporated functions of the R package DAtest7 using the “DAtest” tab in the Shiny interface. As the DAtest package requires complete data (no missing values), the user can select an appropriate method for normalization and imputation for the differential abundance analysis. This choice should be guided by the comparison of the normalization methods and the heatmap visualizing patterns of missing values (Figure 2C). The current implementation of proteiNorm provides the following imputation methods: k-nearest neighbors (KNN),14 Quantile Regression Imputation of Left-Censored (QRILC),15 deterministic minimal value imputation (MinDet),15 stochastic minimal value imputation (MinProb),15 minimal value imputation (Min), and zero imputation (Zero).
In addition, proteiNorm provides the opportunity to adjust the DAtest-specific parameters (i.e., number of times to run the tests, effect size for the spike-ins (on a natural intensity scale), and number of cores to use for parallel computing) and to exclude certain statistical tests. The latter option can be beneficial as the exclusion of tests with high computation demand (such as the permutation-based test) will drastically reduce the run-time.
DAtest automatically selects appropriate statistical tests, depending on the data type (count versus intensities) from the pool of tests and repeatedly evaluates their performance. To do so, the data is shuffled and spiked-in by multiplying the values in one group by the selected effect size in the “spike-in” parameter box on the “DAtest” Shiny tab for randomly chosen features within the normalized protein data set. For instance, if you are interested in a differential analysis method that can detect a 2-fold change, then the spike-in parameter can be set to 2. DAtest then assesses whether the spiked-in features are correctly identified by each test and if the false discovery rate is controlled. The results are summarized in a table and figures comparing and ranking the performance of each individual test. Additionally, proteiNorm provides the opportunity to investigate the performance of each individual test by estimating statistical power (probability of correctly detecting the true signal) over a range of effect sizes (centered around the previously specified effect size for the spike-ins), utilizing the “powerDA” function from the “DAtest” package. This DA power analysis is run on the “DAtest Power” tab in the Shiny interface. Once the differential analysis method is determined, then the saved normalized protein data file will need to be uploaded into a downstream tool or R to perform the differential analysis. ProteiNorm is used to evaluate normalization methods and save the normalized matrix using the “Save file” button after selecting the normalization method on the “DAtest” data tab.
5.4. Data
Our examples consist of three data sets: (1) a TMT6plex mouse data set, where proteins were extracted from the striatum of the brain after treating the mice with methamphetamine or saline; (2) three breast cancer cell lines (MCF10A non-tumorigenic epithelial cell line, MCF7 ER/PR+ cells, and HCC1954 HER2+ cells) with and without hydroxyurea treatment; (3) a publicly available yeast lysate with known spiked-in UPS1 concentrations (ProteomeXchange ID: PXD001819).16 The sample preparation and mass spectrometry methods for each data set are included in the Supporting Information Methods section.
Acknowledgments
The authors would like to acknowledge the University of Arkansas for Medical Sciences Proteomics Core Facility.
Glossary
Abbreviations
- VSN
variance stabilizing normalization
- PCV
pooled coefficient of variance
- PMAD
pooled median absolute deviation
- PEV
pooled estimate of variance
- TMT
tandem mass tag
- MAR
missing at random
- MNAR
missing not at random
- RLR
robust linear regression
- PCA
principal component analysis
- KNN
k-nearest neighbors
- QRILC
Quantile Regression Imputation of Left-Censored
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c02564.
Author Contributions
S.G., J.T., and S.D.B. performed tool development, manuscript writing, and editing. M.K.Z. and A.K.B. performed the sample preparation for the breast cancer data set and editing. E.C.P. and C.B. performed the sample preparation for the mouse data set and editing.
This study was supported by the Arkansas Children’s Research Institute, the Arkansas Biosciences Institute, the Little Rock Envoys Seeds of Science, and the Center for Translational Pediatric Research funded under the National Institutes of Health National Institute of General Medical Sciences (NIH/NIGMS) grant P20GM121293, the UAMS Barton Pilot Study Grant, and National Institutes of Health National Institute of Drug Abuse (NIH/NIDA) R21 DA041822.
The authors declare no competing financial interest.
Notes
All of the mass spectrometry data is available via ProteomeXchange with identifier PXD018152. ProteiNorm and example data is freely available at https://github.com/ByrumLab/ProteiNorm.
Supplementary Material
References
- Chawade A.; Alexandersson E.; Levander F. Normalyzer: a tool for rapid evaluation of normalization methods for omics data sets. J. Proteome Res. 2014, 13, 3114–3120. 10.1021/pr401264n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Välikangas T.; Suomi T.; Elo L. L. A systematic evaluation of normalization methods in quantitative label-free proteomics. Briefings Bioinf. 2018, 19, 1–11. 10.1093/bib/bbw095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpievitch Y. V.; Dabney A. R.; Smith R. D. Normalization and missing value imputation for label-free LC-MS analysis. BMC Bioinf. 2012, 13, S5. 10.1186/1471-2105-13-S16-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb-Robertson B.-J. M.; Matzke M. M.; Jacobs J. M.; Pounds J. G.; Waters K. M. A statistical selection strategy for normalization procedures in LC-MS proteomics experiments through dataset-dependent ranking of normalization scaling factors. Proteomics 2011, 11, 4736–4741. 10.1002/pmic.201100078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva J. C.; Gorenstein M. V.; Li G.-Z.; Vissers J. P. C.; Geromanos S. J. Absolute quantification of proteins by LCMSE - A virtue of parallel MS acquisition. Mol. Cell. Proteomics 2006, 5, 144–156. 10.1074/mcp.M500230-MCP200. [DOI] [PubMed] [Google Scholar]
- Easy web applications in R; RStudio, Inc.:2013 [Google Scholar]
- Russel J.; Thorsen J.; Brejnrod A. D.; Bisgaard H.; Sørensen S. J.; Burmølle M. DAtest: a framework for choosing differential abundance or expression method. bioRxiv 2018, 241802. 10.1101/241802. [DOI] [Google Scholar]
- Ritchie M. E.; Phipson B.; Wu D.; Hu Y.; Law C. W.; Shi W.; Smyth G. K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S.; Temu T.; Cox J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Smits A. H.; van Tilburg G. B. A.; Ovaa H.; Huber W.; Vermeulen M. Proteome-wide identification of ubiquitin interactions using UbIA-MS. Nat. Protoc. 2018, 13, 530–550. 10.1038/nprot.2017.147. [DOI] [PubMed] [Google Scholar]
- Gatto L.; Lilley K. S. MSnbase-an R/Bioconductor package for isobaric tagged mass spectrometry data visualization, processing and quantitation. Bioinformatics 2012, 28, 288–289. 10.1093/bioinformatics/btr645. [DOI] [PubMed] [Google Scholar]
- Huber W.; von Heydebreck A.; Sültmann H.; Poustka A.; Vingron M. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 2002, S96–S104. 10.1093/bioinformatics/18.suppl_1.S96. [DOI] [PubMed] [Google Scholar]
- Bolstad B.preprocessCore: A collection of pre-processing functions; 2019.
- Hastie T.; Tibshirani R.; Narasimhan B.; Chu G.. impute: impute: Imputation for microarray data; 2018.
- Lazar C.imputeLCMD: A collection of methods for left-censored missing data imputation; 2015.
- Ramus C.; Hovasse A.; Marcellin M.; Hesse A.-M.; Mouton-Barbosa E.; Bouyssié D.; Vaca S.; Carapito C.; Chaoui K.; Bruley C.; Garin J.; Cianférani S.; Ferro M.; Van Dorssaeler A.; Burlet-Schiltz O.; Schaeffer C.; Couté Y.; de Peredo A. G. Spiked proteomic standard dataset for testing label-free quantitative software and statistical methods. Data Brief 2016, 6, 286–294. 10.1016/j.dib.2015.11.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.