

 In this review we provide a brief historic overview of covalent inhibitors and summarize recent advances focusing on developments in the last decade. Applications in challenging targets and future perspectives are also discussed.
In this review we provide a brief historic overview of covalent inhibitors and summarize recent advances focusing on developments in the last decade. Applications in challenging targets and future perspectives are also discussed.
Abstract
Covalent inhibitors are recognized as an important component in drug discovery and therapeutics. Since the first appearance of covalent inhibitors in the late 18th century, the field has advanced significantly and currently about 30% of the marketed drugs are covalent inhibitors. The numerous advantages of covalent inhibitors are counteracting the initial concerns regarding potential off-target toxicity. Thus, continuous research, especially for cancer targets is reported. The aim of this review is to provide a short historic overview and focus on recently developed covalent inhibitors (2011–2019), including structural aspects and examples on challenging targets.
1. Introduction
Traditionally, drug design of small molecules is based on their ability to interact with their biological targets under equilibrium binding states. These binding states occur in a fast and reversible process, which affects the duration of the therapeutic response. Thus, prolongation of therapeutic response can be extended by increasing the duration of interaction between a drug and its target. This type of prolonged interaction can be achieved by covalent inhibitors. Covalent inhibitors bind to their target in two steps, starting with equilibrium bond formation, and ending with covalent bond formation. The final state is considered to be irreversible, resulting in a drug–protein complex that is different from that formed with a normal equilibrium bond (Fig. 1).
Fig. 1. General mechanism of covalent interaction between a small molecule and its target.

Covalent inhibitors in general are compounds that by design are intended to form a covalent bond with a specific molecular target. The covalent bond can be either reversible or irreversible, depending on the chosen warhead. A number of different warheads have been exploited to target specific amino acid residues, including among others cysteine, serine, threonine, tyrosine and lysine. A detailed overview of covalent warheads, as well as the amino acids that they target was recently published.1
Despite the initial skepticism of pharmaceutical industry to develop covalent inhibitors, in the last 50 years, the development of covalent inhibitors has shown a significant increase (Fig. 2).2,3 Numerous drug candidates are progressing through clinical trials or being approved by the FDA. Today, there are at least 50 FDA-approved drugs that act as covalent inhibitors.
Fig. 2. Number of publications per decade obtained from a search of the term “covalent drug” in SciFinder®.

Due to the growing interest in covalent inhibitors as drugs, it is worthwhile to examine the design of these drugs in detail. In this review, after a short historic overview, the advantages and disadvantages of covalent inhibitors are discussed. Then, we focus on covalent inhibitors that reached the market or are in advanced clinical trials (2011–2019). In the last part, we review recent applications of covalent inhibitors.
2. Historic background of covalent inhibitors
The earliest covalent inhibitor to be introduced was acetylsalicylic acid (aspirin), which has been marketed since 1899 (Fig. 3). The acetyl warhead of aspirin acetylates Ser-530 of prostaglandin endoperoxide (PGH) synthase-1, inactivating the cyclooxygenase activity of the enzyme.4 Penicillin, which was discovered in 1928, also acts as a covalent inhibitor, as the β-lactam binds covalently to the active site serine (Ser-36) of the bacterial enzyme dd-transpeptidase to form penicilloyl-enzyme which is inactive, thus making it incapable of synthesizing the cell-wall.5
Fig. 3. A historic overview of covalent inhibitors and their approval dates. The warheads are highlighted in red.

In 1975, timoprazole showed inhibition towards gastric acid secretion.6 However, due to its toxicity, the development of timoprazole was stopped. Its derivative, omeprazole, was discovered in 1979, as the first class of drug which is now known as proton pump inhibitor (PPI).7,8 Omeprazole forms a disulfide bond with the sulfhydryl group of the hydrogen–potassium ATPase. The second of the PPI drugs to reach the market was lansoprazole, which was patented in 1984, launched in early 1990s, and approved by FDA in 1995.
In the 1990s, two examples of the most successful covalent drugs were fosfomycin and clopidogrel. Fosfomycin was found in 1969,9 however, the approval and medical usage started in 1996. The drug inhibits bacterial cell wall biogenesis through inactivation of the enzyme MurA.10 It bears an epoxide warhead, which is targeting the active site residue Cys-115 of MurA. Clopidogrel was patented in 1982, and approved for medical use in 1997. Clopidogrel is a prodrug, which is used as antiplatelet medication. The prodrug undergoes a two-step metabolic activation; the thiophene ring is first oxidized towards the inactive in vitro 2-oxo-clopidogrel and then by ring opening, the active thiol–carboxylic acid metabolite is released. The latter binds irreversibly via the –SH group to the P2Y21 purinergic receptor, thus preventing binding of ADP to the P2Y21 receptor. Bortezomib, one of the few covalent inhibitors with boronic acid warhead, is an anticancer medication. It was approved and marketed in 2003. In 2009, a drug containing nitrile warhead saxagliptin was developed. Saxagliptin is an antidiabetic drug which was used to treat type 2 diabetes mellitus, succeeding vildagliptin which was withdrawn in 2008. Today, there are at least 50 approved covalent inhibitors used to treat ailments ranging from obesity to cancer.3 Zanubrutinib (approved November 2019, brand name Brukinsa®) and dacomitinib (approved September 2018, marketed under the name Vizimpro®) are two of the most recently approved covalent inhibitors of BTK and EGFR, respectively.
3. Advantages and disadvantages of covalent inhibitors
Toxicity and efficacy are the main reasons for the attrition of drug candidates during clinical study.11 In these regards, covalent inhibitors have several advantages: (1) improving efficiency, (2) lowering the dose, (3) increasing compliance due to less-frequent dosing, (4) reducing the possibility of drug resistance, and (5) targeting shallow binding sites. Each of these advantages is interconnected. For example, since they are highly potent, with low IC50 values, and long binding duration, the necessary dose and intake frequency are lower compared to normal drugs. Indeed, it has been shown that the compliance of patients increases significantly with lower dosing frequency, especially with once-daily dosing,12 which can be achieved with a covalent inhibitor. It has also been reported that covalent inhibitors have potential advantages against resistance–prone targets. In 2013, Walter et al.13 showed that a novel covalent inhibitor, CO-1686, inhibits a drug-resistance mutant of T790M EGFR. In this case, the warhead is forming a covalent bond to Cys-797.
On the other hand, covalent inhibitors are often considered to have poor selectivity due to their high reactivity. However, several studies have shown that covalent inhibitors are amazingly selective. In a study on quinazoline inhibitors, Zeng et al.14 found that a targeted small covalent inhibitor suppresses KRAS G12C, and the selectivity was higher than that of allosteric compounds. Similarly, a study by Kempson et al.15 on Janus kinases (JAKs) showed high selectivity against JAK3 due to the addition of a covalent warhead in the compound. The same study also showed that the compound has lower activity towards other JAK family kinases, thus reducing the chance of nonspecific binding. Another example in 2010, Hagel et al.16 designed and synthesized small covalent binders towards protease that showed inhibition towards HCV NS3/4A viral protease (HCVP), while having weak reactivity towards other thiols, including glutathione.
Meanwhile, covalent inhibitors have their disadvantages. For example, they (1) may cause unexpected toxicity or hypersensitivity, (2) may cause drug-induced toxicity, (3) may not be suitable for targets that are rapidly turned over/degraded by enzymes, and (4) may cause problems in choosing the correct warhead. The recently published article regarding covalent warheads may become a guideline, problem, and limitation in developing a covalent warhead, especially when it comes to the actual design, synthesis, and in vivo studies.1 However, the increasing amount of knowledge regarding the mechanism of action and the reactivity of many types of warheads may facilitate the future design of covalent inhibitors and assist in tuning their properties to minimize their disadvantages.
4. Covalent drugs on the market and advanced clinical trials
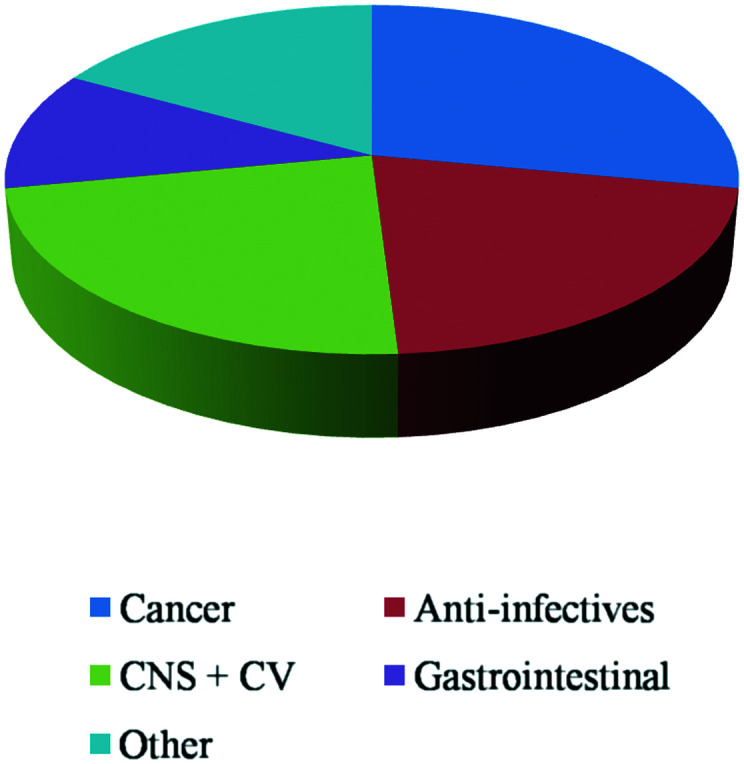
Among the covalent inhibitors on the market, about 28% are used in oncology related targets, 23% are used in CNS and cardiovascular disorders, 21% are anti-infectives (mostly the β-lactam class of antibiotics), and about 11% are used in gastrointestinal diseases (Fig. 4).
Fig. 4. Approved covalent drugs by therapeutic indication.
In the last 10 years, there are 14 newly approved covalent drugs (Table 1). However, two of them, telaprevir and boceprevir, were discontinued in 2014 and 2015 respectively due to their low demand compared to newer generation of anti HCV agents. Among these newly approved covalent drugs, there are 9 drugs with α,β-unsaturated carbonyl as their warhead. Here, a structural analysis of the binding mode for the recently approved covalent inhibitors is provided.
Table 1. FDA-approved covalent drugs (2011–2019).
| No | Name | Therapeutic area | Warhead | Year |
| 1. | Telaprevir | Anti-HCV | α-Ketoamide | 2011 |
| 2. | Boceprevir | Anti-HCV | α-Ketoamide | 2011 |
| 3. | Abiraterone | Anticancer | — | 2011 |
| 4. | Afatinib | Anticancer | α,β-Unsaturated carbonyl | 2013 |
| 5. | Dimethyl fumarate | Multiple sclerosis | α,β-Unsaturated carbonyl | 2013 |
| 6. | Ibrutinib | Anticancer | α,β-Unsaturated carbonyl | 2014 |
| 7. | Osimertinib | Anticancer | α,β-Unsaturated carbonyl | 2015 |
| 8. | Olmutinib | Anticancer | α,β-Unsaturated carbonyl | 2015 |
| 9. | Narlaprevir | Anti-HCV | α-Ketoamide | 2016 |
| 10. | Acalabrutinib | Anticancer | α,β-Unsaturated propargylamide | 2017 |
| 11. | Neratinib | Anticancer | α,β-Unsaturated carbonyl | 2017 |
| 12. | Dacomitinib | Anticancer | α,β-Unsaturated carbonyl | 2018 |
| 13. | Selinexor | Anticancer | α,β-Unsaturated carbonyl | 2019 |
| 14. | Zanubrutinib | Anticancer | α,β-Unsaturated carbonyl | 2019 |
Abiraterone is a drug to treat prostate cancer. Unlike most covalent inhibitors, there is no warhead present in it. Instead, the nitrogen atom from pyridine ring in abiraterone binds covalently to HEM-600 of the enzyme CYP17A1 (Fig. 5).17
Fig. 5. Abiraterone complex with human cytochrome P450 CYP17A1 (PDB ID 3RUK). Abiraterone is shown as cyan sticks and HEM-600 is shown as yellow sticks.

Telaprevir, narlaprevir and boceprevir are serine protease inhibitors targeting, in particular, the hepatitis C virus NS3/4A protease. All of them contain the α-ketoamide warhead. Telaprevir binds to Ser-1139, whereas narlaprevir and boceprevir bind to Ser-139 (Fig. 6).
Fig. 6. Chemical structures of covalent inhibitors and their crystal structures with the NS3/4A protease complex (a) telaprevir binding to Ser-1139 (PDB ID 3SV6), (b) narlaprevir (PDB ID ; 3LON), and (c) boceprevir (PDB ID ; 3LOX) binding to Ser-139. Inhibitors are shown as cyan sticks and Ser-1139, Ser-139 are shown as yellow sticks.

Dimethyl fumarate, dacomitinib, and ibrutinib and zanubrutinib are examples of covalent inhibitors containing the α,β-unsaturated carbonyl warhead (Fig. 7). Dimethyl fumarate is used for the treatment of relapse-remitting multiple sclerosis. It binds to Cys-599 of p90 Ribosomal S6 Kinase. Dacomitinib, ibrutinib, and zanubrutinib are anticancer drugs. Dacomitinib is an EGFR kinase inhibitor to treat non-small cell lung cancer; it binds to Cys-797 of T790M EGFR kinase. Ibrutinib and zanubrutinib are BTK inhibitors used widely to treat mantle cell lymphoma. Both inhibitors bind to Cys-481 of BTK.
Fig. 7. Chemical structures and binding modes with the target protein: (a) dimethyl fumarate binding to Cys-599 (PDB ID 5O1S), (b) EGFR kinase inhibitor dacomitinib, binding Cys-797 (PDB ID ; 4I24), (c) ibrutinib (PDB ID ; 5P9J), and (d) zanubrutinib (PDB ID ; 6J6M) binding to Cys-481 of BTK. Inhibitors are shown as cyan sticks and Cys-599, Cys-797, Cys-481 are shown as yellow sticks.

Apart from the marketed covalent drugs, there are many more inhibitors currently undergoing clinical trials in phase II or higher. These inhibitors are aiming at cysteine residues of the target proteins (Table 2).
Table 2. Examples of covalent inhibitors in clinical trials.
| No. | Structure and name | Target (therapeutic) | Warhead | Clinical stage |
| 1 |
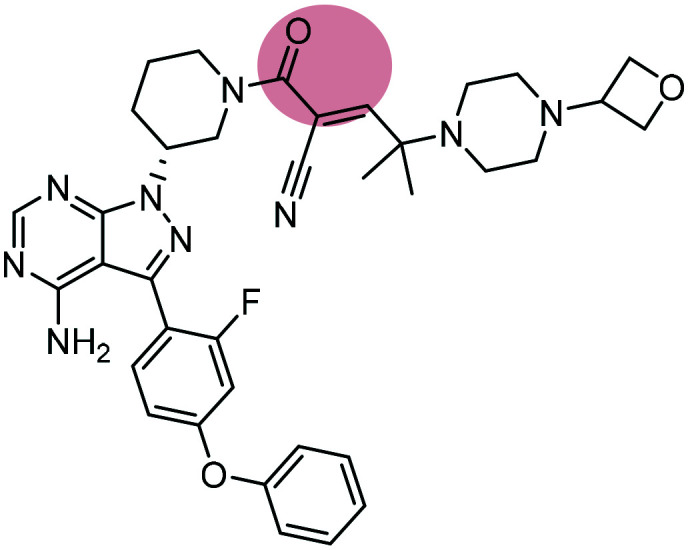 PRN1008 PRN1008 |
BTK inhibitor | α,β-Unsaturated carbonyl | III |
| 2 |
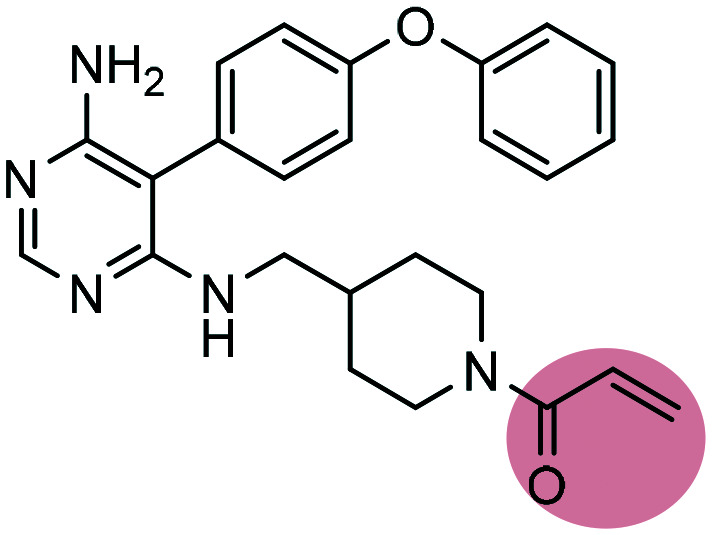 Evobrutinib Evobrutinib |
BTK inhibitor | α,β-Unsaturated carbonyl | III |
| 3 |
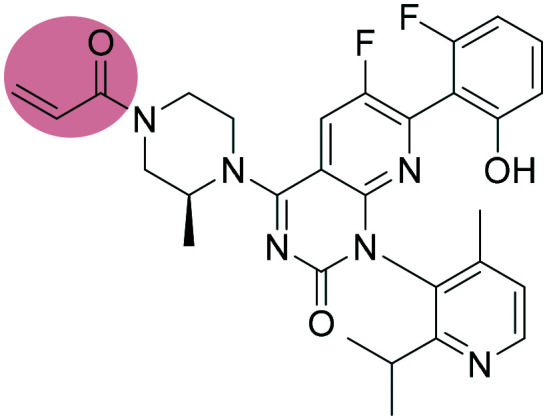 AMG510 AMG510 |
KRAS inhibitor | α,β-Unsaturated carbonyl | II |
| 4 |
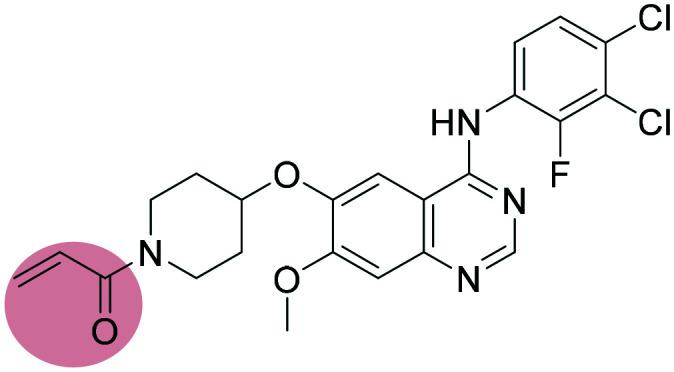 Poziotinib Poziotinib |
EGFR inhibitor | α,β-Unsaturated carbonyl | II |
| 5 |
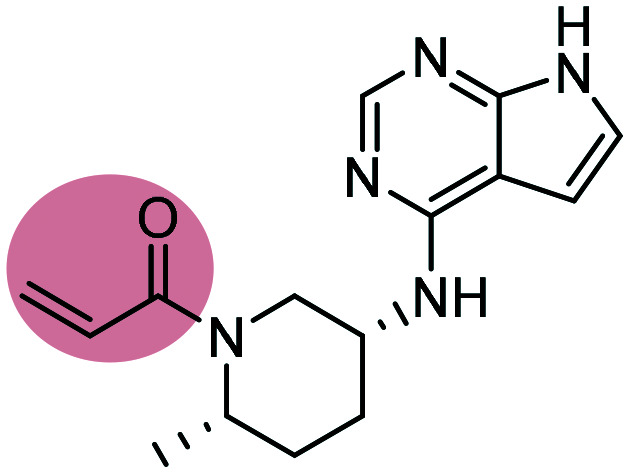 PF-06651600 PF-06651600 |
JAK3 inhibitor | α,β-Unsaturated carbonyl | II |
| 6 |
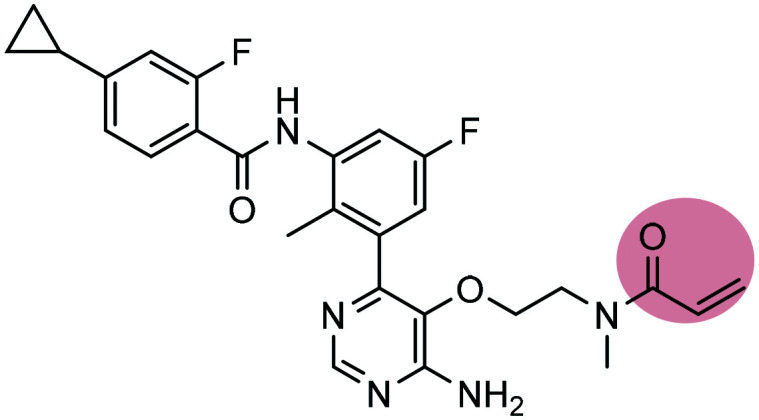 Remibrutinib Remibrutinib |
BTK inhibitor | α,β-Unsaturated carbonyl | II |
| 7 |
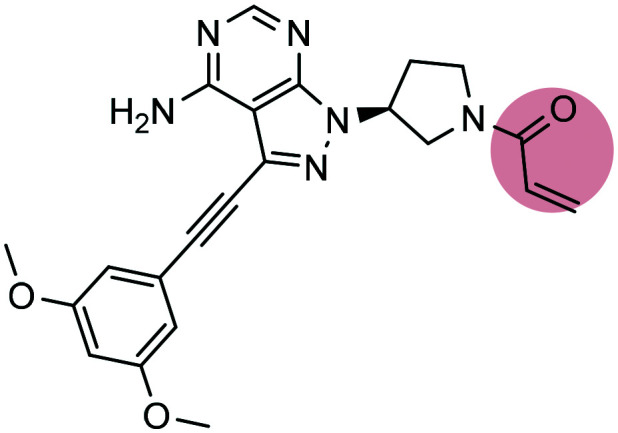 Futibatinib Futibatinib |
FGFR inhibitor | α,β-Unsaturated carbonyl | II |
| 8 |
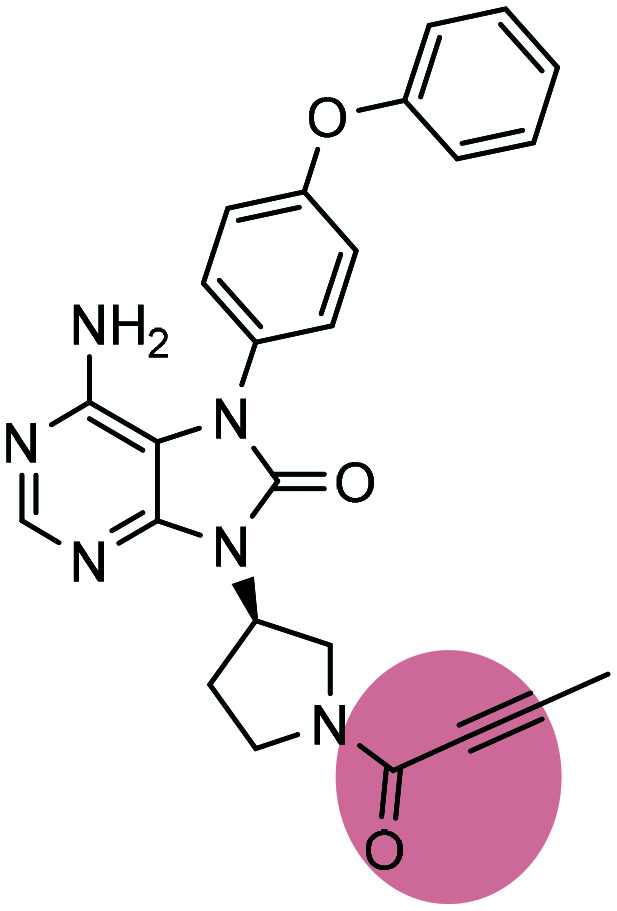 Tirabrutinib Tirabrutinib |
BTK inhibitor | α,β-Unsaturated propargylamide | II |
| 9 |
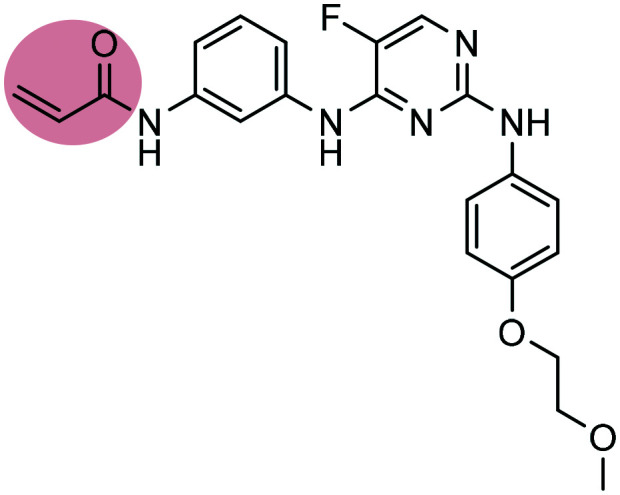 Spebrutinib Spebrutinib |
BTK inhibitor | α,β-Unsaturated carbonyl | II |
5. Recent development of covalent inhibitors
As mentioned before, there is increasing interest in developing covalent inhibitors. Here, we highlight a few applications of covalent inhibitors.
5.1. RAS
One of the most notorious oncology targets is still the RAS protein. RAS is a GTPase, transmitting signals for cell growth and division.18 For decades RAS was considered an undruggable target.19,20 One of the first successful attempts to target the KRAS mutant G12C was described by Shokat et al. involving covalent inhibitors bearing the acrylamide moiety.21 After many different attempts to tackle KRAS, in 2018 Amgen developed AMG510, a covalent inhibitor targeting KRASG12C. Notably, AMG510 was the first compound to undergo clinical trials after three decades of research. MRTX-849 (developed by Mirati Therapeutics) is also currently under clinical investigation (Fig. 8).22–24 Recently, KRAS was also explored in the field of proteolysis targeting chimeras (PROTACs). PROTACs are an exciting new modality of heterobifunctional molecules aiming to degrade the protein of interest instead of inhibiting it.25,26 In 2019, Gray et al.27 reported the development of a cereblon-based degrader molecule library targeting KRASG12C and bearing acrylamide warheads. Although the lead PROTAC (Fig. 8c) of the series showed cereblon engagement in cells and bound to KRASG12C in vitro, it was unable to effectively polyubiquitinate endogenous KRAS and thus degradation was not observed in pancreatic and lung cancer cells. Further studies will be necessary to fully understand the challenges of targeting KRAS using PROTACs.
Fig. 8. Chemical structures and binding modes with the target protein: (a) AMG510 (PDB ID 6OIM) and (b) MRTX-849 (PDB ID ; 6UT0) binding to Cys-12 of KRAS (G12C). Inhibitors are shown as cyan sticks and Cys-12 is shown as yellow sticks, (c) structure of CRBN–KRASG12C lead PROTAC described by Gray et al.27.

5.2. BTK and PROTACs
In the last decade, several covalent inhibitors targeting Bruton's tyrosine kinase (BTK) were discovered, including ibrutinib and zanubrutinib. These compounds contain an acrylamide warhead, targeting Cys-481 in the ATP binding domain of BTK. Recently, these covalent kinase inhibitors were further developed as PROTACs. In 2019, Xue et al.28 developed a series of PROTACs bearing covalent warheads. Ibrutinib and PLS-123 as covalent inhibitors were chosen as the backbone of the modified compounds 1 and 2 (Fig. 9).
Fig. 9. Structure of ibrutinib analogue 1, PLS-123, and PLS-123 analogue 2.

The analogues 1 and 2 were then combined with ligands targeting E3 ligases, either pomalidomide or VH032, through a series of linkers and their activity towards BTK was examined. The active group of acrylamide was also replaced with propanamide as comparison towards covalent vs. non-covalent inhibition. The combination between analogue 1 (ibrutinib analogue) and VH032 (PROTAC a, Fig. 10) showed excellent results regarding BTK degradation compared to other combinations and non-covalent PROTACs,27 highlighting a new application of covalent warheads in protein degradation.
Fig. 10. Structure of PROTAC a targeting BTK.

Conclusions
The rational development of covalent inhibitors has been steadily increasing in the past decades. Numerous warheads have been developed, expanding the covalent warhead toolbox and allowing for selective targeting of specific amino acid residues. More than 50 covalent compounds are on the market or in advanced clinical trials. As more data emerge regarding safety and efficacy of covalent drugs, the structure-guided optimization and rational development will be facilitated in the future. Thus, covalent inhibitors remain an attractive alternative for difficult targets, including protein–protein interactions, where selectivity may remain elusive with non-covalent inhibitors. Moreover, the recent examples of PROTACs bearing covalent warheads show their potential in new modalities, whereas the development of diverse covalent warheads allows for careful tuning of their reactivity and selectivity.
Conflicts of interest
There are no conflicts to declare.
Acknowledgments
This research has been supported (to AD) through ITN “Accelerated Early stage drug dIScovery” (AEGIS, grant agreement No. 675555), the National Institute of Health (NIH) (2R01GM097082-05), the European Lead Factory (IMI) (grant agreement number 115489), the Qatar National Research Foundation (NPRP6-065-3-012) and COFUNDs ALERT (grant agreement No. 665250), Prominent (grant agreement No. 754425) and KWF Kankerbestrijding grant (grant agreement No. 10504). Fandi Sutanto acknowledges Indonesian Endowment Fund for Education (Lembaga Pengelola Dana Pendidikan) for financial support.
Biographies

Fandi Sutanto
Fandi Sutanto obtained his Pharmacist Degree (2011) and his M. Sc. in Pharmacochemistry (2013) from the Bandung Institute of Technology, Indonesia. He then joined an analytical department in a pharmaceutical industry as a scientist. Later on, he started his PhD journey in 2016 under the guidance of Prof. A. Dömling in the Drug Design Group in the University of Groningen, The Netherlands. His PhD research focuses on the development of covalent inhibitors utilizing the multicomponent reactions.

Markella Konstantinidou
Markella Konstantinidou obtained her Degree in Pharmacy (2012) and her M.Sc. in Medicinal Chemistry (2014) from the Aristotle University of Thessaloniki, Greece. Afterwards, she joined the National Hellenic Research Foundation in Athens, as a research assistant. In 2016, she joined as a PhD candidate in the Drug Design Group in the University of Groningen in The Netherlands under the guidance of Prof. A. Dömling. She received her PhD in February 2020. Her PhD research included multicomponent reactions, medicinal chemistry applications and new modalities.

Alexander Dömling
Prof. Alexander Dömling has held the chair for Drug Design at the University of Groningen since 2011. He studied chemistry and biology at the Technische Universität München and obtained his Ph.D. under the guidance of Ivar Ugi. After a postdoc under a Humboldt Fellowship in the group of the Nobel Laureate Barry Sharpless, he founded the biotech company Morphochem and later Carmolex Inc. After his habilitation he worked as full professor at the University of Pittsburgh in the School of Pharmacy. His interests are centered around multicomponent reaction (MCR) chemistry and its application to problems in drug design. His special focus is on MCR-centered pharmacophore methods, structure-based drug design and MCR-centered fragment-based drug design. A creto of his lab is automation + miniaturization = acceleration. He is the author of more than 270 scientific articles, reviews and book contributions. He has applied for more than 50 patents. His long-term vision is to bring a novel drug to patients in an indicated area of unmet medical need.
References
- Gehringer M., Laufer S. A. J. Med. Chem. 2018;62:5673–5724. doi: 10.1021/acs.jmedchem.8b01153. [DOI] [PubMed] [Google Scholar]
- Singh J., Petter R. C., Baillie T. A., Whitty A. Nat. Rev. Drug Discovery. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- Bauer R. A. Drug Discovery Today. 2015;20:1061–1073. doi: 10.1016/j.drudis.2015.05.005. [DOI] [PubMed] [Google Scholar]
- Shimokawa T., Smith W. J. Biol. Chem. 1992;267:12387–12392. [PubMed] [Google Scholar]
- Waxman D. J., Strominger J. L. Annu. Rev. Biochem. 1983;52:825–869. doi: 10.1146/annurev.bi.52.070183.004141. [DOI] [PubMed] [Google Scholar]
- Shin J. M., Munson K., Vagin O., Sachs G. Pflugers Arch. 2009;457:609–622. doi: 10.1007/s00424-008-0495-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munson K., Garcia R., Sachs G. Biochemistry. 2005;44:5267–5284. doi: 10.1021/bi047761p. [DOI] [PubMed] [Google Scholar]
- Fellenius E., Berglindh T., Sachs G., Olbe L., Elander B., Sjöstrand S.-E., Wallmark B. Nature. 1981;290:159–161. doi: 10.1038/290159a0. [DOI] [PubMed] [Google Scholar]
- Michalopoulos A. S., Livaditis I. G., Gougoutas V. Int. J. Infect. Dis. 2011;15:e732–e739. doi: 10.1016/j.ijid.2011.07.007. [DOI] [PubMed] [Google Scholar]
- Brown E. D., Vivas E. I., Walsh C. T., Kolter R. J. Bacteriol. 1995;177:4194–4197. doi: 10.1128/jb.177.14.4194-4197.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kola I., Landis J. Nat. Rev. Drug Discovery. 2004;3:711–716. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- Coleman C. I., Limone B., Sobieraj D. M., Lee S., Roberts M. S., Kaur R., Alam T. J. Manage. Care Pharm. 2012;18:527–539. doi: 10.18553/jmcp.2012.18.7.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter A. O., Sjin R. T. T., Haringsma H. J., Ohashi K., Sun J., Lee K., Dubrovskiy A., Labenski M., Zhu Z., Wang Z. Cancer Discovery. 2013;3:1404–1415. doi: 10.1158/2159-8290.CD-13-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng M., Lu J., Li L., Feru F., Quan C., Gero T. W., Ficarro S. B., Xiong Y., Ambrogio C., Paranal R. M. Cell Chem. Biol. 2017;24:1005–1016. e1003. doi: 10.1016/j.chembiol.2017.06.017. [DOI] [PubMed] [Google Scholar]
- Kempson J., Ovalle D., Guo J., Wrobleski S. T., Lin S., Spergel S. H., Duan J. J.-W., Jiang B., Lu Z., Das J. Bioorg. Med. Chem. Lett. 2017;27:4622–4625. doi: 10.1016/j.bmcl.2017.09.023. [DOI] [PubMed] [Google Scholar]
- Hagel M., Niu D., Martin T. St, Sheets M. P., Qiao L., Bernard H., Karp R. M., Zhu Z., Labenski M. T., Chaturvedi P. Nat. Chem. Biol. 2011;7:22–24. doi: 10.1038/nchembio.492. [DOI] [PubMed] [Google Scholar]
- Fernández-Cancio M., Camats N., Flück C. E., Zalewski A., Dick B., Frey B. M., Monné R., Torán N., Audí L., Pandey A. V. Pharmaceuticals. 2018;11:37. doi: 10.3390/ph11020037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodsell D. S. Stem Cells. 1999;17:235–236. doi: 10.1002/stem.170235. [DOI] [PubMed] [Google Scholar]
- Cox A. D., Fesik S. W., Kimmelman A. C., Luo J., Der C. J. Nat. Rev. Drug Discovery. 2014;13:828–851. doi: 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downward J. Nat. Rev. Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- Ostrem J. M., Peters U., Sos M. L., Wells J. A., Shokat K. M. Nature. 2013;503:548–551. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallin J., Engstrom L. D., Hargis L., Calinisan A., Aranda R., Briere D. M., Sudhakar N., Bowcut V., Baer B. R., Ballard J. A. Cancer Discovery. 2020;10:54–71. doi: 10.1158/2159-8290.CD-19-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ClinicalTrials.gov Identifier: NCT03600883. A Phase 1/2, Study Evaluating the Safety, Tolerability, PK, and Efficacy of AMG 510 in Subjects With Solid Tumors With a Specific KRAS Mutation (CodeBreak 100), 2018, Available from: https://clinicaltrials.gov/ct2/show/NCT03600883.
- Canon J., Rex K., Saiki A. Y., Mohr C., Cooke K., Bagal D., Gaida K., Holt T., Knutson C. G., Koppada N. Nature. 2019;575:217–223. doi: 10.1038/s41586-019-1694-1. [DOI] [PubMed] [Google Scholar]
- Konstantinidou M., Li J., Zhang B., Wang Z., Shaabani S., Ter Brake F., Essa K., Dömling A. Expert Opin. Drug Discovery. 2019;14:1255–1268. doi: 10.1080/17460441.2019.1659242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scudellari M. Nature. 2019;567:298. doi: 10.1038/d41586-019-00879-3. [DOI] [PubMed] [Google Scholar]
- Zeng M., Xiong Y., Safaee N., Nowak R. P., Donovan K. A., Yuan C. J., Nabet B., Gero T. W., Feru F., Li L., Gondi S., Ombelets L. J., Quan C., Jänne P. A., Kostic M., Scott D. A., Westover K. D., Fischer E. S., Gray N. S. Cell Chem. Biol. 2020;27:19–31. e16. doi: 10.1016/j.chembiol.2019.12.006. [DOI] [PubMed] [Google Scholar]
- Xue G., Chen J., Liu L., Zhou D., Zuo Y., Fu T., Pan Z. Chem. Commun. 2020;56:1521–1524. doi: 10.1039/c9cc08238g. [DOI] [PubMed] [Google Scholar]