

Abstract
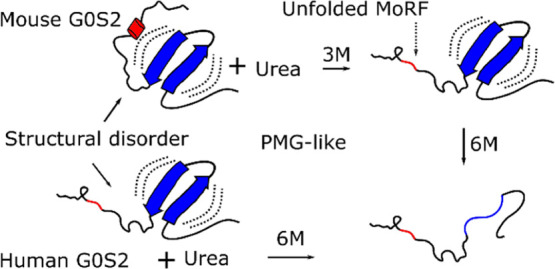
G0S2 is a small protein of 103 residues in length that is involved in multiple cellular processes. To date, several reports have shown that G0S2 functions by making direct protein–protein interactions with key proteins. In lipolysis, G0S2 specifically interacts with adipose triglyceride lipase, inhibiting its activity and resulting in lipolysis being downregulated. In a similar way, G0S2 also participates in the regulation of apoptosis, cell proliferation, and oxidative phosphorylation; however, information regarding G0S2 structural and biophysical properties is limited. In this work, we conducted a comparative structural analysis of human and mouse G0S2 proteins. Bioinformatics suggests the presence of a disordered C-terminal region in human G0S2. Experimental characterization by size-exclusion chromatography and dynamic light scattering showed that human and mouse G0S2 have different hydrodynamic properties. In comparison to the mouse G0S2, which behaves similar to a globular protein, the human G0S2 shows an elongated conformation, most likely by displaying a disordered C-terminal region. Further analysis of hydrodynamic properties under denaturing conditions suggests the presence of a structural element in the mouse protein that undergoes an order to disorder transition at low urea concentration. Structural analysis by circular dichroism revealed that in native conditions, both proteins are mainly unstructured, showing the presence of beta sheet structures. Further analysis of CD data suggests that both proteins belong to the premolten globule family of intrinsically disordered proteins. We suggest that the intrinsic disorder observed in the G0S2 protein may facilitate its interaction with multiple partners in the regulation of cellular metabolism.
Introduction
The G0/G1 switch gene 2 (G0S2) was first identified in blood mononuclear cells. The gene was differentially expressed during the drug-induced cell cycle transition from the G0 to G1 phase.1 The G0S2 gene encodes a small protein of 103 residues in length that has only been identified in vertebrates; orthologs in organisms including nematodes or flies have not been reported.2 G0S2 is conserved among species, human and mouse proteins share 77.7% of global identity with major differences located at the C-terminal region.3 The best-known function of G0S2 is in lipolysis. The protein inhibits the hydrolase activity of adipose triglyceride lipase (ATGL) by interacting with the patatin domain of the enzyme. Inhibition is observed even in the presence of CGI-58, the natural co-activator of ATGL.2 The participation of G0S2 in direct protein–protein interactions with key proteins has been described in multiple cellular processes. In mitochondria, G0S2 specifically interacts with Bcl-2, preventing its interaction with Bax. In this way, G0S2 inhibits the formation of the antiapoptotic heterodimeric complex Bcl-2/Bax, favoring apoptosis.4 In hematopoietic stem cells, G0S2 interacts with a domain rich in RGG repeats of nucleolin. By doing this, G0S2 retains nucleolin in the cytosol and arrests cell proliferation.5 Furthermore, the interaction of G0S2 with F0F1-ATP synthase in cardiomyocytes stimulates ATP production during hypoxia.6
Recent studies have shown that G0S2 displays a lysophosphatidic acid acyltransferase activity. As a result, G0S2 promotes triacylglycerol (TG) accumulation in the liver in addition to its function as an inhibitor of TG hydrolysis by interacting with ATGL. Moreover, the G0S2 gene is upregulated by the lipogenic transcription factor liver X receptor α.7 In addition, the G0S2 gene has been found to be hypermethylated in certain types of cancer, indicating a possible role in cancer development.4,8
To date, no experimental three-dimensional G0S2 structure has been published and predictions of its structure by homology modeling fail because of the lack of adequate structural homologues. Moreover, the progress made in elucidating its structure–function has focused on identifying minimal domains of interaction with other proteins, which it is proposed to involve the hydrophobic domain (HD), though there are currently no reports of further structural studies. In this work, we describe the results of a bioinformatic analysis and biochemical and biophysical characterization of human and mouse G0S2 proteins. Our results show that both proteins behave as intrinsically disordered proteins (IDPs) of the premolten globule like (PMG).
Results and Discussion
In Silico Analysis Suggests the Disordered Character of the Human G0S2 C-Terminal Region
In silico analysis of human and mouse G0S2 sequences included the prediction of disordered regions using the GeneSilico MetaDisorder server.9 The analysis of the sequences indicates similar but not identical disorder trends for both proteins. Human and mouse G0S2 proteins share the same HD, which is predicted to be the most structured region of the protein (residues 27–42, Figure 1A,B), whereas the N- and C-terminal regions tend toward disorder. It is remarkable that in comparison with the C-terminal region of mouse G0S2, which shows some tendency to disorder, the C-terminal region of human G0S2 is predicted to be fully disordered (Figure 1B).
Figure 1.

In silico analysis of human and mouse G0S2. (A) Schematic representation of G0S2; the HD is shown in orange and the C-terminal region (G0S2C) in yellow. A graphical representation showing protein identity between human and mouse G0S2 is shown below. (B) Disorder prediction of human (red) and mouse (blue) G0S2 calculated by the GeneSilico MetaDisorder server with the panel showing the MetaDisorder analysis; values above 0.5 indicate a high disorder probability. (C) Amino acid distribution of human (red) and mouse (blue) G0S2C relative to the SWISS-PROT database; data are shown combined with the amino acid distribution found in IDPs (gray); composition enrichment (>0) or depletion (<0) is shown, generated with Composition Profiler. (D) Charge-hydropathy plot. The absolute, mean net charge versus mean hydropathy values for a set of disordered (open circles) and ordered (gray circles) proteins were plotted together with human and mouse G0S2C, red and blue circles, respectively.
A recent computational proteomic analysis has shown an overall disorder content of 20.5% in eukaryotes. This work considered segments of more than 30 residues in length as they might be implicated in protein recognition.10 Based on these observations, we performed additional in silico analyses using the C-terminal segments of both proteins, which correspond to the last 35 residues (G0S2C, Figure 1A). The abundance or depletion of amino acid content for both G0S2C sequences relative to proteins found in nature was calculated by taking the amino acid composition of proteins deposited in the SWISS-PROT 51 database as a reference.11,12 The resulting distribution was combined and plotted together with the amino acid distribution of IDPs deposited in the DisProt 3.4 database13 (Figure 1C). In comparison to the SWISS-PROT database, mouse G0S2C is enriched in the disorder-promoting residues glutamine, alanine and serine, but it also contains leucine and cysteine, an α-helix former residues, as well as histidine, which is classified as disorder-neutral14,15 (Figure 1C). In contrast, human G0S2C is substantially depleted of order-promoting amino acids, showing few leucine residues and is significantly enriched in glutamine and other disorder-promoting residues commonly found in IDPs (K, R, G, and A).16 This analysis shows that mouse G0S2C has a more balanced composition of order-promoting and disorder-promoting residues than human G0S2C, which is more similar to proteins deposited in the DisProt database, suggesting that the C-terminal region of human G0S2 may display properties of an intrinsic disordered region. To gain additional insights into the possible conformation of the C-terminal region of G0S2, we analyzed the mean net charge and the mean hydrophobicity properties of G0S2C sequences as these properties have been used to distinguish IDPs from ordered proteins.16Figure 1D shows that human G0S2C is located in a well-defined position that corresponds to IDPs; however, mouse G0S2C is located in the corresponding region for ordered proteins. These results suggest the presence of a disordered C-terminal region in the human G0S2 protein.
G0S2 Expression and Purification
To experimentally evaluate the disordered nature of G0S2, we cloned the cDNAs encoding the full-length human and mouse G0S2 proteins into the pET28 expression vector. Initial attempts to express the N-terminal 6XHis-tagged proteins were unsuccessful, despite modifying expression conditions such as temperature incubation, expression time, IPTG concentration, and BL21 strains (results not shown). Therefore, we decided to express the recombinant proteins as fusions with the SUMO protein from S. cerevisiae as previously reported.17 The SUMO-G0S2 proteins were successfully expressed and purified using nickel-nitrilotriacetic acid (Ni-NTA) affinity chromatography. The complete cleavage of the SUMO-G0S2 fusion proteins was achieved in reducing conditions [1–2 mM dithiothreitol (DTT)] with the HRV 3C protease. Soluble G0S2 proteins were further purified by ionic exchange and size-exclusion chromatography. The inset of Figure 2A shows the electrophoretic profile of both proteins after purification. The purity of both proteins, calculated with ImageJ software, was ≥95%.
Figure 2.
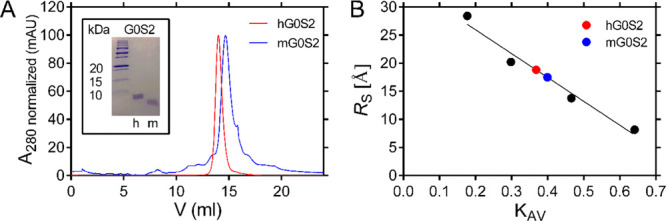
Determination of Stokes radii for human and mouse G0S2 by gel filtration. (A) Analytical SEC chromatogram; the red and blue curves represent the elution profile of human and mouse G0S2, respectively. The inset shows a sodium dodecyl sulfate–polyacrylamide gel electrophoresis of the purified human and mouse G0S2 proteins used in SEC analysis. (B) Calibration curve prepared by plotting the KAV values of standard proteins (black dots) versus their corresponding RS value. The red and blue circles correspond to human and mouse G0S2, respectively.
Hydrodynamic Analyses of Human and Mouse G0S2 Proteins
Size exclusion chromatography (SEC) and dynamic light scattering (DLS) experiments were performed to obtain information on the hydrodynamic parameters related to the shape and size of both proteins.18,19 In analytical SEC experiments, human G0S2 eluted as a single peak (Figure 2A) with a retention volume corresponding to a Stokes radius (RS) of 18.8 Å. This value is 1.1 times larger than the theoretical value of 17.5 Å that can be calculated assuming that human G0S2 has a globular structure and a molecular weight of 11.3 kDa (Figure 2B).20 This difference resulted in being significant by means of a t-test of one sample (P = 0.0003). The experimental volume was 28.0 Å3, which was higher than the theoretical volume (22.4 Å3), while the experimental density of 0.4 kDa/Å3 was lower than the theoretical value (0.5 kDa/Å3, Table 1). The SEC elution profile for mouse G0S2 showed a single peak (Figure 2A) with an RS of 17.5 Å having no significant difference (P = 0.0625) to the theoretical value of 17.4 Å, which was calculated with a molecular weight of 11.1 kDa (Figure 2B).20 The experimental values for volume (22.5 Å3) and density (0.5 kDa/Å3) were very similar to the theoretical values of 22.0 Å3 and 0.5 kDa/Å3, respectively (Table 1). These results suggest that the human G0S2 protein has a slightly extended structure compared to the more compact conformation observed in the mouse G0S2 protein. The extended conformation in human G0S2 could be explained in at least two possible ways. It may be due to a partially swelled tertiary structure, which would generate a globular conformation with larger dimensions, as in the case of a molten globule (MG). In this regard, the observed RS value (18.8 Å) is smaller compared to the expected RS value for an MG (19.9 Å).20 Alternatively, the extended conformation could also be attained by the presence of a disordered and extended region exposed to the solvent; in this case, the protein would have a globular domain with an extended segment that would generate an elongated conformation with the observed Rs.
Table 1. Characterization of Human and Mouse G0S2 by SEC.
|
Rs [Å] |
Vs × 103 [Å3] |
p × 103 [kDa/Å3] |
||||
|---|---|---|---|---|---|---|
| MM [kDa] | theor | exp | theora | expb | theora | expb |
| 11.3h | 17.5 | 18.8 ± 0.03 | 22.4 | 28.0 | 0.5 | 0.4 |
| 11.1m | 17.4 | 17.5 ± 0.05 | 22.0 | 22.5 | 0.5 | 0.5 |
Calculated using the theoretical Rs.
Calculated using the experimental Rs. h Human. m Mouse.
To gain further insights into the molecular shape of human and mouse G0S2 proteins, DLS measurements were subsequently carried out to calculate the hydrodynamic radius (Rh) for each protein. The size distribution of protein samples showed the presence of a single species with an apparent Rh of 1.4 ± 0.03 and 1.7 ± 0.02 nm, human and mouse G0S2, respectively (Figure 3A). The hydrodynamic radius calculated for mouse G0S2 confirms that it has a compact and globular conformation as the Rh is in agreement with the RS, which is also 1.7 nm (Table 1). Interestingly, the Rh obtained for the human G0S2 protein was smaller than the calculated RS value of 1.9 nm determined by SEC, indicating that this protein diffuses like a smaller protein in solution with an apparent size of 8 kDa. The apparent discrepancy could be explained as follows. DLS detects the translational diffusion motion in solution of macromolecules and estimates the corresponding translational diffusion coefficient (D), which is used to calculate the particle size using the Stokes–Einstein equation. The hydrodynamic dimensions are calculated by assuming that the diffusion particle is a sphere that diffuses at the same speed in all directions.19 Nonspherical particles (i.e., elongated proteins) can be represented by two axes of different lengths. These particles diffuse faster along one axis (the shorter) than the other (the longer) and as a result the size of the sphere calculated by DLS has a diameter that is shorter than the longer axis of the particle. Thus, an elongated protein will show an apparent Rh value that corresponds to a smaller size. In SEC, proteins are separated by their frictional coefficient (f) if the appropriate pore volume of the column has been selected. The calculated Rs is defined as the radius of a sphere that would have the actual f of the protein. The size of the sphere determined by the Rs is a good approximation to the size of a globular protein but elongated proteins usually show larger Rs values.21 Thus, an elongated protein will show an apparent Rs that corresponds to a larger size. Our SEC and DLS results suggest that the human G0S2 protein has an elongated shape with a globular domain of approximately 8 kDa. Similar results have been observed in the MG of the CaLB protein, whose radius calculated by DLS is smaller than that calculated by SEC.22 The elongated conformation of the human G0S2 could be explained by the presence of a disordered C-terminal region, as suggested by the bioinformatic analysis. Indeed, a theoretical molecular weight of 7.5 kDa can be calculated for human G0S2 by omitting the C-terminal region, which is in accordance with the observed Rh value.
Figure 3.
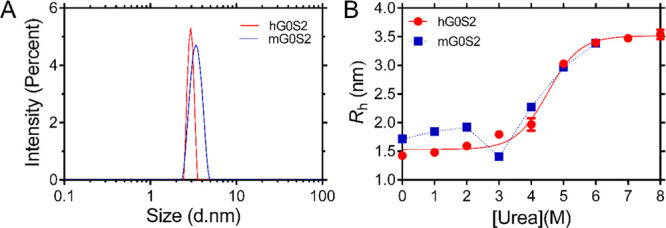
Hydrodynamic radii of human and mouse G0S2 in native and unfolded state. (A) Sample size distributions of human (red) and mouse (blue) G0S2 obtained by DLS under native conditions. (B) Urea denaturation curve of human (red) and mouse (blue) G0S2 followed by DLS. The continuous line is the best fit using a multistate equilibrium display model.
Given these Rh differences in the native states of both proteins, we investigated their behavior under denaturing conditions; as both proteins are 103 residues in length and share an overall identity of 77.7%, we anticipated that unfolded proteins should have a similar Rh. Denaturation curves of human and mouse G0S2 were obtained in the presence of increasing urea concentrations, and at each point Rh was estimated by DLS (Figure 3B). The denaturation curve for human G0S2 follows sigmoidal behavior with an increase in the experimental Rh; a single transition is observed from 2 to 6 M urea with midpoint transition CM of 4.4 M. The protein loses its structure and fully unfolds at 6 M urea, reaching an Rh of 3.5 ± 0.06 nm; this value is in agreement with the expected Rh value of 2.96 nm that can be calculated for an unfolded protein of 103 residues.23 The denaturation process of mouse G0S2 initiates with an Rh of 1.7 ± 0.02 nm. A swelling process in which the side chains of the amino acids stop interacting with each other and start interacting with urea is observed from 0 to 2 M urea.24 Interestingly, when the protein is in 3 M urea its Rh collapses to a value of 1.4 ± 0.01 nm. At higher concentrations, mouse G0S2 follows the same pattern observed for human G0S2 until it reaches a value of 3.4 ± 0.01 nm (Figure 3B). This result was interpreted as the loss of a structural element in the mouse protein, suggesting that at low urea concentration a structured region unfolds before the rest of the protein. The close similarity of mouse G0S2 Rh value in 3 M urea (1.4 ± 0.01 nm) compared to the Rh value observed for human G0S2 in native conditions (1.4 ± 0.03 nm) suggests that the labile structural element found in the mouse protein must be very similar in size and properties to that described for the human protein, therefore pointing to the C-terminal region of mouse G0S2 as the region that may contain the labile structural element.
Previous studies have shown that IDPs usually have regions that undergo disorder to order transitions that facilitate interaction with multiple partners.25 In order to identify a potential structural element in the C-terminal region of G0S2, we searched for molecular recognition regions (MoRFs) using the MoRFpred server.26 The prediction identified eight residues from 95 to 102 (ALSLRQHA) in the mouse G0S2 sequence as a possible MoRF. Additionally, the Jpred4 server27 predicted an α-helix structure for the same region (seven underlined residues). In the case of the human G0S2 protein, the region from residues 95 to 102 (ALSNRQHA) was also predicted as an MoRF but with a lower probability (six bolded residues out of eight) and only three residues were predicted to be in α-helix conformation. Additionally, a hydrophobic cluster analysis (HCA) was performed. HCA provides a two-dimensional helical representation of protein sequences in which hydrophobic clusters are plotted along the sequence.28 This approach has proven useful in identifying coiled coil regions, regions with biased composition, and regions with potential for induced folding via disorder–order transitions.28,29 HCA analyses of human and mouse G0S2 distinguish three segments that overlap to the N-terminal region, the HD and the C-terminal region. The N-terminal region displays short hydrophobic clusters; the main hydrophobic cluster is found within the HD, while the C-terminal region (residues 50 to 103) shows isolated hydrophobic residues with a couple of short hydrophobic clusters of two or three residues in length (Figure 4A,B). This is in agreement with the fact that long regions devoid of clusters correspond to disordered regions and small clusters within disordered regions correspond to putative MoRFs.30 The most striking difference is in residues 95 to 102 of mouse G0S2, which show “mosaic clusters” of leucine residues. Such clusters have been associated with secondary structure elements on the surface of proteins, thus suggesting the presence of a structural element in the C-terminal region of mouse G0S2 (Figure 4B).28 As a result of these analyses, the prediction of an MoRF region of eight residues, of which seven are predicted to be in an α-helix conformation, as well as the presence of “mosaic clusters” of leucine residues, allow us to suggest that residues 95 to 102 of mouse G0S2 are part of the structural element that unfolds at low urea concentrations. In the case of human G0S2, the lower probability of MoRF, the prediction of a shorter α-helix, and the absence of “mosaic clusters” support the disordered character of its C-terminal region.
Figure 4.
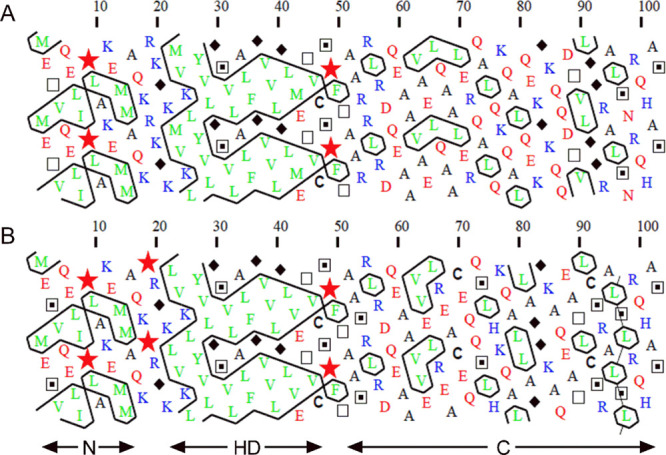
HCA analysis of (A) human and (B) mouse G0S2. The shape of the clusters is mainly associated to each type of secondary structure. Hence, horizontal and vertical clusters often correspond to α-helices and β-strands, respectively. Hydrophobic amino acids are shown in green. Basic and acidic residues are shown in blue and red, respectively. Symbols are used to designate specific amino acids: star for proline, diamond for glycine, square for threonine, and dotted square for serine.
Secondary Structure Content Analysis Reveals That G0S2 Is Largely Unstructured
To gain more insight into the structural content of G0S2, we used circular dichroism (CD) spectroscopy in the far UV region.31 The human and mouse G0S2 CD spectra exhibit properties previously observed for IDPs. They show a strong minimum signal close to 200 nm (−11.67 × 10–3 and −12.05 × 10–3 deg·cm2·dmol–1, human and mouse G0S2, respectively) and a small negative contribution close to 222 nm (−2.55 × 10–3 and −1.48 × 10–3 deg·cm2·dmol–1, human and mouse G0S2, respectively) (Figure 5A). These characteristics have been associated with the presence of residual structures in the order of 10–20%.32 The analysis of CD data using the BestSel algorithm33 indicates that human and mouse G0S2 proteins are mainly unstructured, showing content of ∼55% of coil, ∼25% of β-strands, ∼19% of β-turns, and virtually no contribution of α-helices (Table 2). The high content of coils confirms the tendency toward disorder suggested by the bioinformatic analysis (Figure 1B). The content of β-structures is in agreement with the secondary structure prediction of G0S2. Previous reports have suggested the presence of two alpha-helices separated by a hydrophobic sequence with the potential to assume β-sheet conformation.1 A detailed examination using the JPred4 and the fast estimator of latent local structure (FELLS) prediction showed that the proposed β-sheet should comprise residues 22 to 28. Additionally, there are two other segments with the potential to generate β-sheet conformation, these being located at residues 44 to 48 and 87 to 97.27,34 Furthermore, G0S2 sequences contains ∼55% of low complexity regions, which is in agreement with the observed content of coils.35
Figure 5.
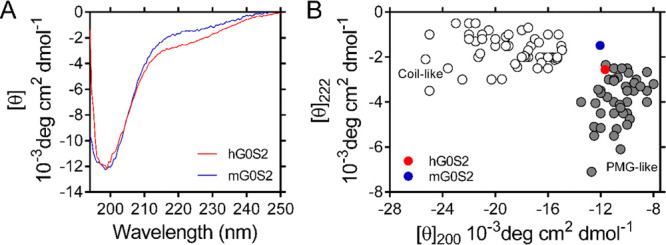
Far-UV CD spectra of human and mouse G0S2. (A) CD spectra of human (red) and mouse (blue) G0S2 were recorded at 22 μM protein concentration. (B) Double-wavelength plot showing ellipticity [θ] at 200 and 222 nm of a set of coil-like (white circles) and PMG-like (gray circles) proteins. The positions of human and mouse G0S2 are indicated by red and blue circles, respectively.
Table 2. Summary of Human and Mouse G0S2 Secondary Structure Content As Estimated by the BeStSel Algorithm.
| type of structure | hG0S2 | mG0S2 |
|---|---|---|
| Helix | 0.4 | 0 |
| Antiparallel | 24.8 | 25.8 |
| Parallel | 0 | 0 |
| Turn | 19.1 | 19.6 |
| Other (coil) | 55.7 | 54.6 |
| Total | 100 | 100 |
The content of secondary structures has been used to classify IDPs in MG, PMG, and random-coil (RC). MG proteins have collapsed conformations with a significant content of secondary structures; PMG proteins show partially collapsed hydrophobic regions with residual secondary structures, while RC proteins have large hydrodynamic dimensions that lack secondary structures.29 We analyzed CD data using a double-wavelength plot of the ellipticity values at 200 and 222 nm, as this approach has been used to differentiate coil-like from PMG IDPs.36Figure 5B shows that human and mouse G0S2 localizes in the corresponding position of PMG IDPs. Altogether, our results show that human and mouse G0S2 behaves like collapsed IDPs in solution, with the difference that human G0S2 shows a more elongated conformation, which is most likely due to the presence of an unstructured conformation of its C-terminal domain.
It is well known that IDPs’ backbone is not rigid. Instead, it is highly dynamic and flexible and it usually contains residual structural elements required for proper function.29 In many cases, this flexibility allows IDPs to interact with multiple partners by molecular direct interaction. When IDPs bind a target protein, DNA, or RNA and inhibit or activate the functions of the target, it is called an effector.29 In line with this, our results are in agreement with previous works that have described the direct binding of G0S2 to different partners. G0S2 binds and inhibits ATGL in lipolysis,2 promotes apoptosis by interacting with Bcl-2,4 arrests cell proliferation by binding and retaining nucleolin in the cytosol,5 and stimulates ATP production by direct binding with the F0F1-ATP synthase.6 Therefore, we suggest that the unstructured character of G0S2 allows it to interact as an effector with multiple partners with regulatory functions. Furthermore, the exposure and flexibility of residues in IDPs is a key factor that facilitates post-translational modifications (PTMs) such as phosphorylation.29 For example, the activity of Ets1, a transcription factor involved in cancer and autoimmunity diseases, is regulated by phosphorylation of its disordered N- and C-terminus.37 In the case of p53, phosphorylation and acetylation have a profound effect on the conformational preferences of its intrinsically disordered negative regulatory domain, thus regulating the activity of this important protein.38 In line with this, bioinformatic prediction of PTMs shows two phosphorylation sites (S97 and T89) for the unstructured C-terminal region of human G0S2, while only one site is predicted (S97) for the labile C-terminal region of mouse G0S2C.
Materials and Methods
In Silico Analysis
The following servers were used online with the default parameters. Protein disorder prediction was carried out using the GeneSilico MetaDisorder server9 (http://genesilico.pl/metadisorder/). Composition Profiler11 (http://www.cprofiler.org) was used to analyze the amino acid distribution. Charge-hydropathy plot analyses16 were generated using PONDR (http://www.pondr.com). HCA28 was performed at (http://mobyle.rpbs.univ-paris-diderot.fr/cgi-bin/portal.py?form=HCA#forms::HCA). Protein secondary structure prediction was done by Jpred4 (https://www.compbio.dundee.ac.uk/jpred/) and FELLS (http://old.protein.bio.unipd.it/fells/) servers.27,34
Protein Expression and Purification
The coding DNA for human (NP_056529.1) and mouse (NP_032085.1) G0S2 and S. cerevisiae SUMO protein residues 1–97 (6xHis-SUMO-HRV3C) was acquired optimized for bacterial expression from Integrated DNA Technologies. The SUMO fragment was cloned into a pET28a expression vector, followed by the insertion of the G0S2 fragment to obtain the pET28SUMO-hG0S2 and pET28SUMO-mG0S2 constructs. Escherichia coli BL21 (DE3) competent cells were transformed with the recombinant vector and cultured for 4 h at 37 °C. Protein expression was induced with 1 mM IPTG at 28 °C overnight. The cells were harvested and sonicated at 4 °C in lysis buffer [50 mM Tris (pH 8.0), 300 mM NaCl, and 10 mM imidazole]. The supernatant was loaded onto a Ni-NTA column equilibrated with the lysis buffer. Fusion proteins were eluted in one step with the lysis buffer supplemented with 500 mM imidazole. 6xHis-SUMO fusion was removed with HRV 3C protease overnight at 4 °C. Human G0S2 was then loaded into a HiTrap-Q HP column (GE Healthcare) in buffer A [50 mM Tris buffer (pH 8.0), 0.5 mM ethylenediaminetetraacetate (EDTA), and 1 mM DTT], and mouse G0S2 was loaded into a HiTrap-S HP column in buffer B [50 mM HEPES (pH 8.2), 0.5 mM EDTA, and 1 mM DTT]. Eluted proteins were concentrated (Amicon Ultra-15, Merck) and further purified through a Superdex-75 10/300 GL column (GE Healthcare) with buffer C [20 mM Tris (pH 8.0), 150 mM NaCl, 0.5 mM EDTA, and 1.0 mM DTT]. Protein concentration was estimated at 205 nm;39 extinction coefficient values (E2050.1%) of 29.70 and 30.23 were estimated directly40 for human and mouse G0S2, respectively.
Analytical Size-Exclusion Chromatography
Protein samples of 100 μL at 1 mg/mL were injected independently on a Superdex 75 10/300 GL column (GE Healthcare), equilibrated with SEC buffer [20 mM Tris–HCl pH 8.0 and 150 mM NaCl] at a flow rate of 0.5 mL/min. The column was calibrated with protein markers: ovalbumin (44 kDa), myoglobin (17 kDa), insulin (5.8 kDa), and vitamin B12 (1.35 kDa). Stokes radii (RS) were calculated by means of eq 1.20
| 1 |
The total volume of the column (VT) was 24 mL and the void volume (VO) of 8.2 mL was calculated using thyroglobulin. The gel phase distribution coefficients (KAV) for the standard proteins were determined using eq 2,41 where VE is the elution volume of the protein.
| 2 |
Dynamic Light Scattering
DLS experiments were performed at 25 °C using an APS2000 system (Malvern Instruments). All hydrodynamic radii (Rh) measurements were made at 1 mg/mL protein concentration in buffer D [20 mM Tris (pH8.0) and 1 mM beta-mercaptoethanol]. For denaturation curves, proteins were incubated with increasing concentrations of urea overnight at room temperature prior to DLS measurements being taken. All values are the mean of three independent measurements.
CD Spectroscopy
CD measurements were made on a JASCO J-815 spectropolarimeter (Jasco Inc., Easton, MD) equipped with a PFD-425S Peltier temperature controller cell. CD spectra were recorded from 194 to 250 nm using a 0.1 cm path length cell. Protein samples were prepared in buffer E [20 mM potassium phosphate (pH 8.0) and 20 mM NaCl] at 0.25 mg/mL. Ellipticity is reported as mean ellipticity per residue [θ]. Secondary structure content was estimated from the CD data (range 200–250 nm) using BestSel (http://bestsel.elte.hu/).33
Acknowledgments
This work was supported by grants from CONACyT [CB 168710 to SLG, and INFRA 204373]. EDPP would like to thank CONACyT for providing fellowship [453649].
Glossary
Nomenclature
- G0S2
G0/G1 switch gene 2
- IDP
intrinsic disorder protein
- PMG
premolten globule
- SEC
size-exclusion chromatography
- DLS
dynamic light scattering
- CD
circular dichroism
- MoRFs
molecular recognition regions
- HCA
hydrophobic cluster analysis
The authors declare no competing financial interest.
References
- Russell L.; Forsdyke D. R. A human putative lymphocyte G0/G1 switch gene containing a CpG-rich island encodes a small basic protein with the potential to be phosphorylated. DNA Cell Biol. 1991, 10, 581–591. 10.1089/dna.1991.10.581. [DOI] [PubMed] [Google Scholar]
- Yang X.; Lu X.; Lombès M.; Rha G. B.; Chi Y.-I.; Guerin T. M.; Smart E. J.; Liu J. The G0/G1 switch gene 2 regulates adipose lipolysis through association with adipose triglyceride lipase. Cell Metab. 2010, 11, 194–205. 10.1016/j.cmet.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerk I. K.; Wechselberger L.; Oberer M. Adipose triglyceride lipase regulation: an overview. Curr. Protein Pept. Sci. 2018, 19, 221–233. 10.2174/1389203718666170918160110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch C.; Santra M. K.; El-Assaad W.; Zhu X.; Huber W. E.; Keys R. A.; Teodoro J. G.; Green M. R. Identification of a protein, G0S2, that lacks Bcl-2 homology domains and interacts with and antagonizes Bcl-2. Cancer Res. 2009, 69, 6782–6789. 10.1158/0008-5472.can-09-0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T.; Park C. S.; Shen Y.; Rabin K. R.; Lacorazza H. D. G0S2 inhibits the proliferation of K562 cells by interacting with nucleolin in the cytosol. Leuk. Res. 2014, 38, 210–217. 10.1016/j.leukres.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kioka H.; Kato H.; Fujikawa M.; Tsukamoto O.; Suzuki T.; Imamura H.; Nakano A.; Higo S.; Yamazaki S.; Matsuzaki T.; Takafuji K.; Asanuma H.; Asakura M.; Minamino T.; Shintani Y.; Yoshida M.; Noji H.; Kitakaze M.; Komuro I.; Asano Y.; Takashima S. Evaluation of intramitochondrial ATP levels identifies G0/G1 switch gene 2 as a positive regulator of oxidative phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 273–278. 10.1073/pnas.1318547111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Xie X.; Heckmann B. L.; Saarinen A. M.; Gu H.; Zechner R.; Liu J. Identification of an intrinsic lysophosphatidic acid acyltransferase activity in the lipolytic inhibitor G0/G1 switch gene 2 (G0S2). FASEB J. 2019, 33, 6655–6666. 10.1096/fj.201802502r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusakabe M.; Watanabe K.; Emoto N.; Aki N.; Kage H.; Nagase T.; Nakajima J.; Yatomi Y.; Ohishi N.; Takai D. Impact of DNA demethylation of the G0S2 gene on the transcription of G0S2 in squamous lung cancer cell lines with or without nuclear receptor agonists. Biochem. Biophys. Res. Commun. 2009, 390, 1283–1287. 10.1016/j.bbrc.2009.10.137. [DOI] [PubMed] [Google Scholar]
- Kozlowski L. P.; Bujnicki J. M. MetaDisorder: a meta-server for the prediction of intrinsic disorder in proteins. BMC Bioinf. 2012, 13, 111. 10.1186/1471-2105-13-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Z.; Yan J.; Fan X.; Mizianty M. J.; Xue B.; Wang K.; Hu G.; Uversky V. N.; Kurgan L. Exceptionally abundant exceptions: comprehensive characterization of intrinsic disorder in all domains of life. Cell. Mol. Life Sci. 2014, 72, 137–151. 10.1007/s00018-014-1661-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacic V.; Uversky V. N.; Dunker A. K.; Lonardi S. Composition Profiler: a tool for discovery and visualization of amino acid composition differences. BMC Bioinf. 2007, 8, 211. 10.1186/1471-2105-8-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bairoch A.; Apweiler R.; Wu C. H.; Barker W. C.; Boeckmann B.; Ferro S.; Gasteiger E.; Huang H.; Lopez R.; Magrane M.; Martin M. J.; Natale D. A.; O’Donovan C.; Redaschi N.; Yeh L. S. The Universal Protein Resource (UniProt). Nucleic Acids Res. 2005, 33, D154–D159. 10.1093/nar/gki070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sickmeier M.; Hamilton J. A.; LeGall T.; Vacic V.; Cortese M. S.; Tantos A.; Szabo B.; Tompa P.; Chen J.; Uversky V. N.; Obradovic Z.; Dunker A. K. DisProt: the database of disordered proteins. Nucleic Acids Res. 2007, 35, D786–D793. 10.1093/nar/gkl893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunker A. K.; Lawson J. D.; Brown C. J.; Williams R. M.; Romero P.; Oh J. S.; Oldfield C. J.; Campen A. M.; Ratliff C. M.; Hipps K. W.; Ausio J.; Nissen M. S.; Reeves R.; Kang C.; Kissinger C. R.; Bailey R. W.; Griswold M. D.; Chiu W.; Garner E. C.; Obradovic Z. Intrinsically disordered protein. J. Mol. Graphics Modell. 2001, 19, 26–59. 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- Uversky V. N. The alphabet of intrinsic disorder. ntrinsically Disord. Proteins 2013, 1, e25496 10.4161/idp.25496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N. What does it mean to be natively unfolded?. Eur. J. Biochem. 2002, 269, 2–12. 10.1046/j.0014-2956.2001.02649.x. [DOI] [PubMed] [Google Scholar]
- Cerk I. K.; Salzburger B.; Boeszoermenyi A.; Heier C.; Pillip C.; Romauch M.; Schweiger M.; Cornaciu I.; Lass A.; Zimmermann R.; Zechner R.; Oberer M. A peptide derived from G0/G1 switch gene 2 acts as noncompetitive inhibitor of adipose triglyceride lipase. J. Biol. Chem. 2014, 289, 32559–32570. 10.1074/jbc.m114.602599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N.Size-exclusion chromatography in structural analysis of intrinsically disordered proteins. In Intrisically Disordered Protein Analysis, Methods in Molecular Biology; Uversky V. N., Dunker A. K., Eds.; Springer: New York, 2012; Vol. 896, pp 179–94. [DOI] [PubMed] [Google Scholar]
- Gast K.; Fiedler C.. Dynamic and static light scattering of intrinsically disordered proteins. In Intrinsically Disordered Protein Analysis, Methods in Molecular Biology; Uversky V. N., Dunker A. K., Eds.; Springer: New York, 2012; Vol. 896, pp 137–61. [DOI] [PubMed] [Google Scholar]
- Permyakov S. E.; Millett I. S.; Doniach S.; Permyakov E. A.; Uversky V. N. Natively unfolded c-terminal domain of caldesmon remains substantially unstructured after the effective binding to calmodulin. Proteins: Struct., Funct., Genet. 2003, 53, 855. 10.1002/prot.10481. [DOI] [PubMed] [Google Scholar]
- Erickson H. P. Size and shape of protein molecules at the nanometer level determined by sedimentation, gel filtration, and electron microscopy. Biol. Proced. Online 2009, 11, 32–51. 10.1007/s12575-009-9008-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabbani G.; Ahmad E.; Khan M. V.; Ashraf M. T.; Bhat R.; Khan R. H. Impact of structural stability of cold adapted Candida antarctica lipase B (CaLB): in relation to pH, chemical and thermal denaturation. RSC Adv. 2015, 5, 20115–20131. 10.1039/c4ra17093h. [DOI] [Google Scholar]
- Marsh J. A.; Forman-Kay J. D. Sequence determinants of compaction in intrinsically disordered proteins. Biophys. J. 2010, 98, 2383–2390. 10.1016/j.bpj.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W.; Borgia A.; Buholzer K.; Grishaev A.; Schuler B.; Best R. B. Probing the action of chemical denaturant on an intrinsically disordered protein by simulation and experiment. J. Am. Chem. Soc. 2016, 138, 11702–11713. 10.1021/jacs.6b05443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radivojac P.; Iakoucheva L. M.; Oldfield C. J.; Obradovic Z.; Uversky V. N.; Dunker A. K. Intrinsic disorder and functional proteomics. Biophys. J. 2007, 92, 1439–1456. 10.1529/biophysj.106.094045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disfani F. M.; Hsu W.-L.; Mizianty M. J.; Oldfield C. J.; Xue B.; Dunker A. K.; Uversky V. N.; Kurgan L. a computational tool for sequence-based prediction and characterization of short disorder-to-olrder transitioning binding regions in proteins. Bioinformatics 2012, 28, i75–i83. 10.1093/bioinformatics/bts209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drozdetskiy A.; Cole C.; Procter J.; Barton G. J. JPred4: a protein secondary structure prediction server. Nucleic Acids Res. 2015, 43, W389–W394. 10.1093/nar/gkv332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callebaut I.; Labesse G.; Durand P.; Poupon A.; Canard L.; Chomilier J.; Henrissat B.; Mornon J. P. Deciphering protein sequence information through hydrophobic cluster analysis (HCA): current status and perspectives. Cell. Mol. Life Sci. 1997, 53, 621–645. 10.1007/s000180050082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habchi J.; Tompa P.; Longhi S.; Uversky V. N. Introducing protein intrinsic disorder. Chem. Rev. 2014, 114, 6561–6588. 10.1021/cr400514h. [DOI] [PubMed] [Google Scholar]
- Lieutaud P.; Ferron F.; Uversky A. V.; Kurgan L.; Uversky V. N.; Longhi S. How disordered is my protein and what is its disorder for? A guide through the “dark side” of the protein universe. intrinsically Disord. Proteins 2016, 4, e1259708 10.1080/21690707.2016.1259708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield N. J. Using circular dichroism spectra to estimate protein secondary structure. Nat. Protoc. 2007, 1, 2876–2890. 10.1038/nprot.2006.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. 10.1016/s0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- Micsonai A.; Wien F.; Bulyáki É.; Kun J.; Moussong É.; Lee Y.-H.; Goto Y.; Réfrégiers M.; Kardos J. BeStSel: a web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra. Nucleic Acids Res. 2018, 46, W315–W322. 10.1093/nar/gky497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piovesan D.; Walsh I.; Minervini G.; Tosatto S. C. E. FELLS: fast estimator of latent local structure. Bioinformatics 2017, 33, 1889–1891. 10.1093/bioinformatics/btx085. [DOI] [PubMed] [Google Scholar]
- Mier P.; Paladin L.; Tamana S.; Petrosian S.; Hajdu-Soltész B.; Urbanek A.; Gruca A.; Plewczynski D.; Grynberg M.; Bernadó P.; Gáspári Z.; Ouzounis C. A.; Promponas V. J.; Kajava A. V.; Hancock J. M.; Tosatto S. C. E.; Dosztanyi Z.; Andrade-Navarro M. A. Disentangling the complexity of low complexity proteins. Briefings Bioinf. 2020, 21, 458–472. 10.1093/bib/bbz007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 2002, 11, 739–756. 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara K.; Shiina M.; Higo J.; Ogata K.; Nakamura H. Phosphorylation of an intrinsically disordered region of Ets1 shifts a multi-modal interaction ensemble to an auto-inhibitory state. Nucleic Acids Res. 2018, 46, 2243–2251. 10.1093/nar/gkx1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowell C.; Chen J.; Chen J. Potential conformational heterogeneity of P53 bound to S100B(beta beta). J. Mol. Biol. 2013, 425, 999–1010. 10.1016/j.jmb.2013.01.001. [DOI] [PubMed] [Google Scholar]
- Scopes R. K. Measurement of protein by spectrophotometry at 205 nm. Anal. Biochem. 1974, 59, 277–282. 10.1016/0003-2697(74)90034-7. [DOI] [PubMed] [Google Scholar]
- Anthis N. J.; Clore G. M. Sequence-specific determination of protein and peptide concentrations by absorbance at 205 nm. Protein Sci. 2013, 22, 851–858. 10.1002/pro.2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews P.Estimation of molecular size and molecular weights of biological compounds by gel filtration. In Methods of Biochemical Analysis; Glick D., Ed.; John Wiley and Sons: New York, 1970; Vol. 18, pp 1–53. [PubMed] [Google Scholar]