

Abstract
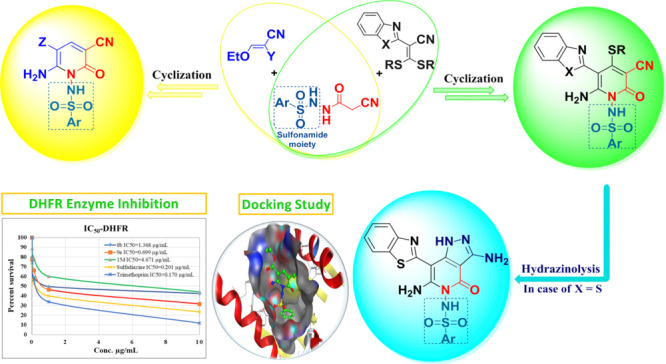
A new strategy for designing and assembling a novel class of functionalized pyridine-based benzothiazole and benzimidazole incorporating sulfonamide moieties was developed. The synthesis was carried out by reacting N-cyanoacetoarylsulfonylhydrazide with various electrophiles such as 2-(benzo[d]thiazol-2-yl)-3,3-bis(alkylthio)acrylonitriles and 2-(benzo[d]imidazol-2-yl)-3,3-bis(methylthio)-acrylonitriles, as well as 2-ethoxyl acrylonitrile derivatives. The synthesized compounds were tested for their antiviral and antimicrobial potency. Two of the synthesized compounds, 15c and 15d, showed more than 50% viral reduction against HSV-1 and CBV4, with significant IC50 and CC50 values. The two potent compounds 15c and 15d have also shown inhibitory activity against Hsp90α protein with IC50 values of 10.24 and 4.48 μg/mL, respectively. A combination of 15c and 15d with acyclovir has led to IC50 values that are lower than that of acyclovir alone. Molecular modeling studies were used to identify the interactions between the 15c and 15d compounds and the active site of Hsp90α enzyme. The antimicrobial investigation of the new compounds has also shown that 8b and 15d exhibited a higher inhibition zone (IZ) than sulfadiazine and gentamicin against Klebsiella pneumonia, whereas 9a showed higher IZ than ampicillin against Staphylococcus aureus. According to the enzyme assay study on dihydrofolate reductase, 9a was shown to be the most potent compound among all examined compounds.
1. Introduction
Recently, we conducted numerous research investigations to develop different innovative synthetic methods for the preparation of N-sulfonylamino and N-sulfonyl-based heterocyclic compounds that have come into application as new forms for antiviral and antimicrobial agents.1−4 Series of N-sulfonylpyrazole, synthesized by our group,5,6 were evaluated against the enzyme cathepsin B.16 and breast adenocarcinoma MCF-7 cell line.7,8 Similarly, the N-arylsulfonylpyrazole series was also identified to be active as inhibitors of the NS2B-NS3 enzyme.9 These promising results led our research team to further investigate new approaches for the synthesis of alternative scaffolds for use as promising chemotherapeutics. In 1990, we have reported the first synthesis of structurally simple 2-arylbenzothiazoles (A), by the reaction of o-aminothiophenol with arylmethylenecyanothioacetamide,10 whose derivatives displayed thereafter interesting pharmacological properties suggesting their potential as anticancer, antiviral, and antimicrobial agents, Figure 1. The aniline derivative, compound (B), had excellent in vitro cytotoxicity in nanomolar concentrations against breast cancer cell line.11 Compound (C) has demonstrated a superior in vivo efficacy against breast cell line; however, metabolic instability prevented its development as a chemotherapeutic agent, Figure 2. To overcome this problem, fluorinated analog (D) had been developed.12 Compound (E) showed excellent growth inhibition of kidney cancer cell line A498,13,14 whereas compound (F), naphthalimidearylbenzothiazole, was found to possess potent cytotoxicity against human hepatocarcinoma cell line SMMC-7721.15 These findings were later on filed as patents. As a result of these interesting findings, we plan in this study to complement our previous work on 2-arylbenzothiazoles by replacing the benzene ring with a pyridine ring-tagged sulfonamide moiety. The synthesis was carried out by reacting N-cyanoacetoarylsulfonylhydrazide with various electrophiles such as 2-(benzo[d]thiazol-2-yl)-3,3-bis(alkylthio)-acrylonitriles and 2-(benzo[d]imidazol-2-yl)-3,3-bis(methylthio)-acrylonitriles, as well as 2-ethoxyl acrylonitrile derivatives. Antiviral and antimicrobial activities, as well as the toxicity effect of the synthesized compounds have all been evaluated. Many of the synthesized compounds showed interesting activities when compared to that of current antimicrobial drugs.
Figure 1.

Chemical reaction illustrating the first method for the synthesis of 2-arylbenzothiazole.
Figure 2.

Chemical structure of drugs containing 2-arylbenzothiazole nucleus A–F.
2. Results and Discussion
2.1. Chemistry
The synthetic strategies adopted for producing the target pyridine-based benzothiazole and benzimidazole compounds are represented in Schemes 1–4. This work focused on the key compound bearing sulfonamide moiety, which was used as a potential precursor for the synthesis of the novel series of N-sulfonylaminopyridones tagged either a benzothiazole or benzoimidazole ring. The key compound, N-cyanoacetoarylsulfonylhydrazide was afforded via a convenient reaction that was developed earlier of cyanoacetohydrazide with arylsulfonyl chloride.6 The reaction of N-cyanoacetoarylsulfonylhydrazide 4a,b with 2-(benzo[d]thiazol-2-yl)-3,3-bis(alkylthio)-acrylonitrile 3 in the presence of potassium hydroxide in dry dimethylformamide (DMF) produced an adduct for which four possible isomeric structures were considered, as shown in Scheme 1. The X-ray crystal structure of 8b confirmed the presence of the compound in the solid state.16 Addition of the active methylene carbon atom of 4a,b to the double bond of 3a,b, followed by the elimination of RSH and the subsequent cyclization via the addition of the NH group to the cyano group of benzothiazole led to the formation of the thermodynamically controlled and kinetically favored product 8a–d. The 1H NMR spectra of 8b revealed the presence of an amino group at 8.83 ppm and a pyridine methylthio group at 2.45 ppm. In addition, NH2 stretching band was confirmed in the infrared (IR) spectra at a range of 3200–3208 cm–1.
Scheme 1. Synthesis of N-(5-(Benzo[d]thiazol-2-yl)-4-(alkylthio)-2-oxopyridin-1(2H)-yl)arylsulfonamide.

Scheme 4. Synthesis of 2-Oxopyridin-1(2H)-yl-benzenesulfonamide.
Furthermore, treatment of thioalkylpyridone derivatives 8a–d with hydrazine hydrate in the presence of a catalytic amount of piperidine in DMF resulted in the formation of the corresponding 1H-pyrazolo[4,3-c]pyrid-2-ones 9a,b, Scheme 2. This reaction occurred with the loss of the alkylthio group by nucleophilic attack of hydrazine and the subsequent cyclization through the attack on the cyano group in the pyridone ring to afford pyrazolopyridones 9a,b. The structure of compounds 9a,b was confirmed by their basic analysis spectroscopic data [Fourier transform infrared spectroscopy (FTIR), 1H NMR and mass]. The absence of cyano stretching band in the IR spectra as well as the disappearance of the SCH3 protons in 1H NMR validated the conversion of 8a–d to 9a,b. In addition, the 1H NMR spectra showed two peaks at 11.92–11.95 and 6.10–6.11 ppm and thus indicating the presence of NH2 and NH, respectively.
Scheme 2. Synthesis of N-(7-(Benzo[d]thiazol-2-yl)-4-oxo-1H-pyrazolo[4,3-c]pyridin-5(4H)-yl)arylsulfonamide.

The target benzoimidazole derivatives 13a,b was synthesized using the reaction sequence illustrated in Scheme 3. They are formed by the reaction of N-cyanoacetoarylsulfonylhydrazide 4a,b with 2-(1H-benzo[d]imidazole-2-yl)-3,3-bis(methylthio)-acrylonitrile derivatives 12 at room temperature. The structure of 13a,b was characterized by their basic analysis and spectroscopic data (1H NMR and IR). For example, 1H NMR spectra of 13a revealed singlet peaks at 2.35 and 2.90 ppm, which are assigned to CH3 and SCH3 protons, respectively, and multiplet peaks at range of 8.30–7.40 ppm for aromatic protons.
Scheme 3. Synthesis of N-(5-(1H-Benzo[d]imidazol-2-yl)-2-oxopyridin-1(2H)-yl)arylsulfonamide.

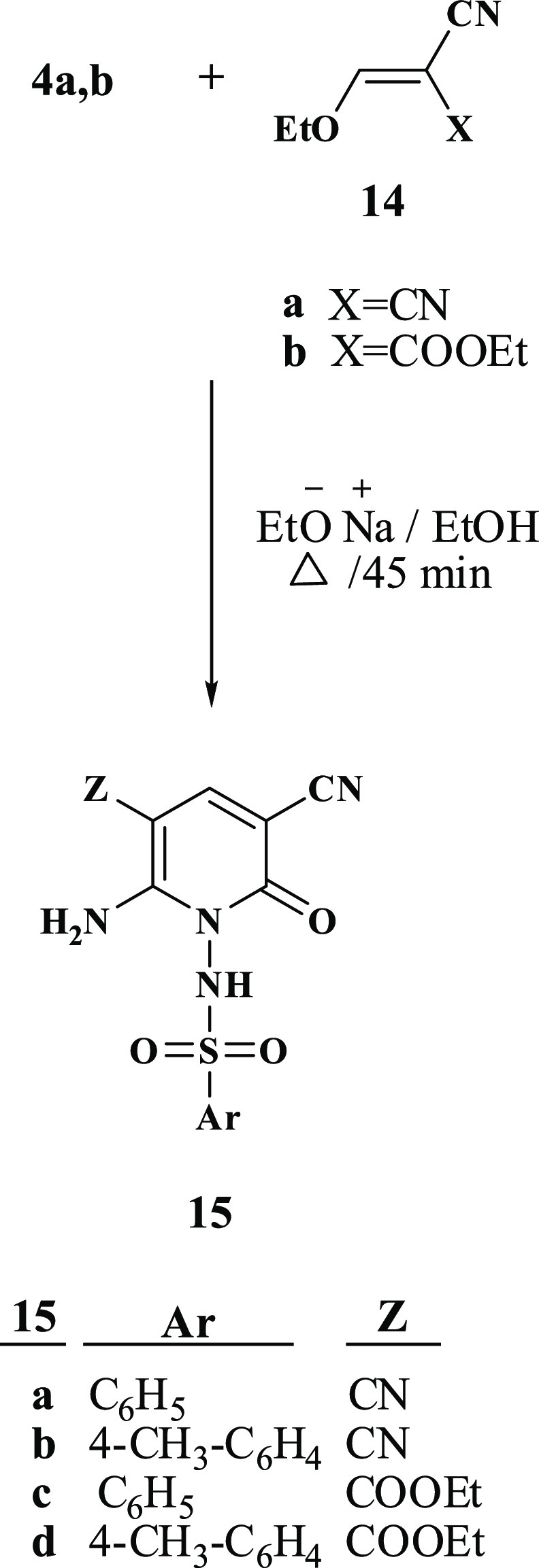
Moreover, our investigation was extended to include reacting N-cyanoacetoarylsulfonylhydrazide 4a,b with various acrylonitrile derivatives, Scheme 4. The reaction of N-cyanoacetoarylsulfonylhydrazide 4a,b with 2-ethyl acrylonitrile derivatives 14a,b in ethanolic sodium ethoxide afforded the corresponding N-arylsulfonylpyridones 15a–d. The synthetic route to target compounds 15a–d is assumed to occur via the addition of the active methylene group of 4a,b to the ylidene bond in 14a,b followed by the elimination of one ethanol molecule and the cyclization via the addition of the NH group to the cyano group. The elemental analysis and spectral data validated the proposed individual structures of compounds 15a–d. IR showed NH2 absorption band at a range of 3308–3286 cm–1. In addition, a singlet peak in 1H NMR at range of 7.95–7.94 ppm confirmed the presence of pyridine-H. In the case of compounds 15c,d, 1H NMR spectrum showed a triplet peak at 1.28 ppm and quartet at 4.24 ppm for CH2 and CH3, respectively, of the COOCH2CH3 group. Furthermore, 1H NMR spectra revealed the presence of an amino group at the range of 7.68–8.02 ppm.
2.2. Biological Results
2.2.1. Antiviral Evaluation
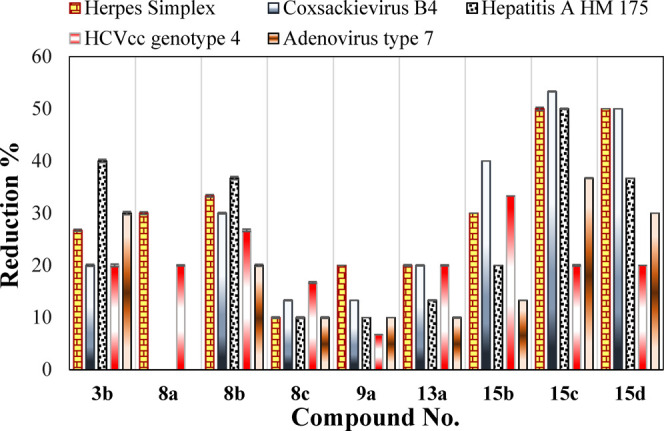
The antiviral activities of the newly synthesized were evaluated in vitro against a wide variety of viruses such as herpes simplex virus type 1 (HSV-1), coxsackievirus B4 (CBV4), hepatitis A virus HM 175 (HAV HM 175), ED-43/SG-Feo (VYG) replicon of hepatitis C virus genotype 4a (HCVcc), and adenovirus type 7 (HAdV7). As reported, no specific medications are available for HCVgenotype4, CBV4, and HAdV7 viruses and the commercial drugs are only used to treat the symptoms but not the illness itself except for HSV-1, acyclovir is used.17−19 In order to study their antiviral activities, the newly synthesized compounds were first subjected to a cytotoxicity evaluation as shown in the Supporting Information document using cell line FRHK-4, Hep2, BGM, Vero, and Huh 7.5 as the specific hosts to the various studied viruses. For comparison, acyclovir was used as standard drug against HSV-1. No significant difference was observed between the nontoxic doses of the different synthesized compounds, which ranged between 90 and 120 μg/mL. The synthesized compounds showed an apparent effect on viruses having different types of genome, that is, either RNA such as CBV4, HAV, and HCV or DNA such as HAdV7 and HSV-1, Table 1. Two compounds in particular, 15c and 15d, showed interesting antiviral effects that exceed 50% reduction against three of the studied viruses, Figure 3. Error bars in the Figure 3 represent standard division of the measured data. The 50% maximum cytotoxicity concentration (CC50), defined as the concentration in μg/mL required to reach 50% cytotoxicity of the treated uninfected cells, and the 50% maximal inhibitory concentration (IC50), defined as the concentration in μg/mL required to inhibit 50% of the tested viruses, were both evaluated for the two compounds, 15c and 15d, that exhibited greater than 50% viral reduction against the aforementioned tested viruses. In addition, the selectivity index (SI) of the promising compounds was calculated by evaluating the CC50/IC50 ratio, Table 2.
Table 1. Antiviral Activity of Nontoxic Doses of Synthesized Compounds against Herpes Simplex Virus, CBV4, HAV HM 175, HCVcc Genotype 4 & Adenovirus Type 7.
| mean
% of reduction |
|||||
|---|---|---|---|---|---|
| compd no.c | herpes simplex virus | CBV4 | HAV HM 175 | HCVcc genotype 4 | adenovirus type 7 |
| 3b | 26.7 ± 0.20 | 20.0 ± 0.20 | 40.0 ± 0.30 | 20.0 ± 0.25 | 30.0 ± 0.30 |
| 8a | 30.0 ± 0.25 | a | 20.0 ± 0.10 | ||
| 8b | 33.3 ± 0.25 | 30.0 ± 0.15 | 36.7 ± 0.30 | 26.7 ± 0.25 | 20.0 ± 0.20 |
| 8c | 10.0 ± 0.15 | 13.3 ± 0.10 | 10.0 ± 0.15 | 16.7 ± 0.15 | 10.0 ± 0.15 |
| 8d | |||||
| 9a | 20.0 ± 0.15 | 13.3 ± 0.10 | 10.0 ± 0.10 | 13.3 ± 0.10 | 10.0 ± 0.10 |
| 9b | |||||
| 13a | 20.0 ± 0.30 | 20.0 ± 0.10 | 13.3 ± 0.10 | 20.0 ± 0.15 | 10.0 ± 0.10 |
| 15a | |||||
| 15b | 30.0 ± 0.30 | 40.0 ± 0.20 | 20.0 ± 0.10 | 33.3 ± 0.10 | 13.3 ± 0.50 |
| 15c | 50.0 ± 0.35 | 53.3 ± 0.30 | 50.0 ± 0.30 | 20.0 ± 0.15 | 36.7 ± 0.30 |
| 15d | 50.0 ± 0.30 | 50.0 ± 0.35 | 36.7 ± 0.25 | 20.0 ± 0.20 | 30.0 ± 0.10 |
| acyclovir | 99.6 ± 2.80 | NTb | NT | NT | NT |
No activity.
NT = not tested.
All data were taken as the average of three independent measurements.
Figure 3.
Comparison between the percent of viral load reduction of synthesized compounds.
Table 2. Antiviral Activity in Terms of CC50, IC50, (μg/mL) and SI.
| compd no.a | CC50 (μg/mL) | IC50 (μg/mL) | SI |
|---|---|---|---|
| Herpes Simplex Virus | |||
| 15c | 250 | 90 | 2.77 |
| 15d | 220 | 90 | 2.44 |
| acyclovir | 2.8 | 0.7 | 4.00 |
| CBV4 | |||
| 15c | 260 | 88 | 2.95 |
| 15d | 240 | 90 | 2.66 |
| HAV HM 175 | |||
| 15c | 260 | 88 | 2.95 |
All data were taken as the average of three measurements.
The two compounds, 15c and 15d, showed, however, moderate levels of activity against HSV-1, CBV4, and HAV. In the case of HSV-1, both compounds 15c and 15d exhibited IC50 values of 90 μg/mL, whereas their CC50 were 250 and 220 μg/mL, respectively. Both compounds showed 50% viral reduction against HSV-1 while acyclovir showed 99.6% viral reduction. Additionally, compound 15c showed 50% reduction for CBV4 and 53% reduction for HAV, with IC50 88 μg/mL and SI 2.95 for both viruses while compound 15d showed reduction 50% for CBV4 alone with IC50 90 μg/mL and SI 2.66.
2.2.1.1. Structure Activities Relationship
Based on the results of the tested activities against HSV-1, CBV4, and HAV, as shown in Tables 1 and 2, the structure–activity relationships (SARs) have been established. We can observe that both compounds 15c and 15d possessing the ethoxycarbonyl group at C5 of the pyridine ring have almost equal activities toward HSV-1 and CBV4. However, replacement of the ethoxycarbonyl group with a cyano group led to the other two compounds, 15a and 15b, which showed lower to no activity against the aforementioned viruses. Alternatively, the presence of a benzothiazole ring at C5 and an alkylthio group at C4 of the pyridine ring, compounds 8a–d, resulted in lower activities for these compounds than those of the compounds containing ethoxycarbonyl group at C5, 15c and 15d.
2.2.1.2. Hsp90α Inhibition Assay
Enzyme Assay: It is well-known that the protein “heat shock protein 90” (Hsp90α), which exists in almost all cell types, is quite important in the viral protein processes such as protein folding, protein assembly, and protein replication.20 During infection, HSV-1 uses the protein Hsp90α as part of the viral polymerase process.21 Therefore, an inhibitor of Hsp90α would naturally be an inhibitor of the HSV-1 infection. This has encouraged us to examine the possibility that our newly synthesized compounds 15c and 15d that showed high potency against HSV-1 might act as novel Hsp90α inhibitors.
The Hsp90α (C-terminal) inhibitor screening assay kit was used to examine the effect of 15c and 15d against Hsp90α. A combination of 15c and 15d with the well-known drug acyclovir (standard drug), in 1:1 ratio, were also investigated. The IC50, the concentration of tested compounds that is required to inhibit 50% of the virus cell population, was evaluated, Table 3.
Table 3. IC50 Values of the Compounds 15c, 15d, and Acyclovir on the Hsp90α Protein.
| compd no.a | 100 (μg/mL) | 10 (μg/mL) | 1 (μg/mL) | 0.1 (μg/mL) | IC50 (μg/mL) |
|---|---|---|---|---|---|
| 15c | 72.742% | 48.686% | 26.135% | 8.4154% | 10.25 ± 0.30 |
| 15d | 89.892% | 57.424% | 25.873% | 10.459% | 4.48 ± 0.12 |
| acyclovir | 84.063% | 52.339% | 35.451% | 11.242% | 4.78 ± 0.18 |
| 15c/acyclovir | 85.554% | 60.166% | 26.411% | 3.432% | 5.22 ± 0.14 |
| 15d/acyclovir | 87.314% | 62.996% | 44.340% | 18.436% | 2.26 ± 0.16 |
All data were taken as the average of three measurements.
It is interesting to report that both compounds had promising inhibitory effect against Hsp90α as measured in μg/mL. According to the calculated IC50, compound 15d with IC50 value of 4.48 μg/mL showed more potency as the Hsp90α inhibitor than both acyclovir with IC50 value of 4.78 μg/mL and 15c with IC50 values of 10.24 μg/mL. Combining 15c as well as 15d with acyclovir has resulted in IC50 values of 5.22 and 2.26 μg/mL, respectively, which is a major reduction from those of the individual compounds. It is clear that the combination of both compounds 15c and 15d with acyclovir has increased their potency. The resultant data clearly indicates that compound 15c and its combination with acyclovir act as potential inhibitors for Hsp90α and accordingly inhibitors for HSV-1 as was clearly illustrated from the antiviral reduction investigation and are, therefore, highly recommended.
2.2.1.3. Molecular Modeling and Docking Study
To further understand the underlying mechanism of the anti-HSV-1 action of the potent compounds with viral reduction of 50% or more, 15c and 15d were tested by docking them onto the crystal structure of the Hsp90α protein (PDB ID: 3B25) obtained from protein data bank (PDB). To prepare the models, the bound ligand, B2K (4-methyl-6-(toluene-4-sulfonyl)-pyrimidin-2-ylamine) was removed to uncover the binding pattern of these compounds with the receptor. The molecular modeling environment (MOE) was used to perform the docking study. Three-dimensional molecular structures were then readied and docked onto the enzyme active site. The docking study showed that the compounds 15c and 15d had binding energies of −5.7789 and −6.3783, respectively. The benzene ring of 15c and 15d showed hydrophobic interaction with Asn 51, Figures 4 and 5. Furthermore, both 15c and 15d displayed two hydrogen bonding interactions with one interaction present between the nitrogen atom of the sulfonamide group and residue Met 98, and the other one present between the oxygen atom of the acetyl group and residue Lys 58, Figures 4 and 5. The bond length between the sulfonamide group and residue Met 98 of compounds 15c and 15d is 3.39 and 3.37 c5, respectively, while the bond length between the acetyl group and residue Lys 58 is 1.91 and 2.90 c5, respectively. The docking study revealed that the investigated compounds occupy the protein binding pockets of the Hsp90α inhibitor and thus suggests its inhibitory action of Hsp90α.
Figure 4.

Best docked pose of 15c inside the binding pocket of Hsp90α (PDB id 3B25).
Figure 5.

Best docked pose of 15d inside the binding pocket of Hsp90α (PDB id 3B25).
2.2.2. Antimicrobial Evaluation
The newly synthesized target compounds were evaluated for their in vitro antibacterial activity against Escherichia coli, Klebsiella pneumonia, and Pseudomonas aeruginosa as examples of Gram-negative bacteria and Staphylococcus aureus and Streptococcus pyogenes as examples of Gram-positive bacteria. They were also evaluated for their in vitro antifungal potential against Candida albicans fungal strain. The agar-diffusion method was used for the determination of the preliminary antibacterial and antifungal activities. Gentamicin, ampicillin, and nystatin were used as standard drugs against Gram negative bacterial, Gram positive bacterial, and fungal strains, respectively. Sulfadiazine as one of sulfonamide antibiotics with pyrimidine ring bearing the sulfamide group at position 2 has been also tested. The results were recorded for each tested compound as the average diameter of inhibition zones (IZs) of the microbial growth around the disks in mm ± SD, as summarized in Table 4.
Table 4. Antimicrobial IZ in mm ± Standard Deviation of Synthesized Compounds.
| diameter
of the IZ (mm) |
||||||
|---|---|---|---|---|---|---|
| Gram
(−ve) bacteria |
Gram
(+ve) bacteria |
fungi | ||||
| compd no. | E. coli (ATCC:3008) | K. pneumonia (ATCC:4415) | P. aeruginosa (ATCC:27853) | S. aureus (ATCC:6538) | S. mutans (ATCC:25175) | C. albicans (ATCC:10231) |
| 3b | a | |||||
| 8a | 19.0 ± 0.5 | 25.8 ± 0.5 | 19.8 ± 1.0 | |||
| 8b | 32.2 ± 2.5 | 19.3 ± 1.0 | 10.7 ± 0.5 | |||
| 8c | 24.5 ± 0.7 | 21.3 ± 1.1 | ||||
| 8d | 29.9 ± 0.6 | 12.5 ± 1.0 | ||||
| 9a | 34.0 ± 1.0 | 13.9 ± 0.5 | ||||
| 9b | 15.0 ± 0.5 | 21.3 ± 1.0 | 24.3 ± 0.5 | |||
| 13a | 30.0 ± 1.0 | 22.0 ± 0.5 | ||||
| 13b | 28.9 ± 0.7 | 21.0 ± 0.1 | ||||
| 15a | 19.6 ± 2.0 | |||||
| 15b | ||||||
| 15c | ||||||
| 15d | 19.6 ± 1.5 | 35.3 ± 1.5 | 11.3 ± 1.1 | 9.0 ± 1.0 | ||
| gentamicin | 25.0 ± 0.1 | 32.0 ± 0.1 | 35.0 ± 0.1 | NT | NT | NT |
| ampicillin | NTb | NT | NT | 32.0 ± 0.5 | 22.0 ± 0.5 | NT |
| nystatin | NT | NT | NT | NT | NT | 28.0 ± 0.2 |
| sulfadiazine | 19.8 ± 0.4 | 20.4 ± 0.9 | 24.0 ± 1.4 | 31.3 ± 2.1 | 16.3 ± 1.1 | NA |
No activity.
Not tested.
The minimum inhibitory concentration (MIC) measurements were determined for the most active compounds 8b, 9a, and 15d by using twofold serial dilution method (as explained later) and the results are all summarized in Table 5.
Table 5. MIC (μg/mL) of the Most Active Compounds 8b, 9a, and 15d.
| MIC μg/mL |
standards |
||||
|---|---|---|---|---|---|
| selected organism | 8b | 9a | 15d | sulfadiazine | gentamicin |
| E. coli (ATCC:3008) | NTa | NT | 1000 | 250 | 31.25 |
| K. pneumonia (ATCC:4415) | 125 | NT | 62.5 | 250 | 62.5 |
| P. aeruginosa (ATCC:27853) | NT | NT | NT | 250 | 125 |
| MIC μg/mL |
standards |
||||
|---|---|---|---|---|---|
| selected organism | 8b | 9a | 15d | sulfadiazine | ampicillin |
| S. aureus (ATCC:6538) | 1000 | 125 | NT | 500 | 62.5 |
| S. mutans (ATCC:25175) | 250 | 1000 | NT | 125 | 62.5 |
| MIC μg/mL |
standards |
||||
|---|---|---|---|---|---|
| selected organism | 8b | 9a | 15d | nystatin | |
| C. albicans (ATCC:10231) | NT | NT | NT | NT | 31.25 |
Not test.
In general, thiomethylpyridone derivatives 8a and 8b showed improved inhibitory effectiveness against the tested Gram-positive bacteria. Compound 8b was identified as the most potent agent among the thiomethylpyridone series. It was equipotent to gentamicin against K. pneumonia strain (IZ 32.2 ± 2.5 mm, MIC 125 μg/mL). On other hand, 1H-pyrazolopyridones 9a (IZ 34 ± 1 mm, MIC 125 μg/mL) was shown to be the most potent derivative among all synthesized compounds against S. aureus, Table 1. Pyrazolopyridone 9b had the highest antifungal activity among all of the synthesized compounds investigated in this study. Pyridone derivatives 13a,b revealed moderate antifungal activities against C. albicans and strong bacterial activities against S. aureus. Pyridone derivative 13a was more active than 13b against both S. aureus and C. albicans. Regarding the antibacterial activities of sulfonylpyridones 15a–d, pyridone 15d was the only compound that showed some activities against most of tested antibacterial strains. Compound 15d exhibited higher potency than gentamicin against K. pneumonia (IZ 35.3 ± 1.5 mm, MIC 1000 μg/mL). Compound 15a showed a moderate antifungal activity against C. albicans. Both 15b and 15c have no activity to all tested bacterial and fungal strains. In addition, all of the synthesized compounds showed no activity against P. aeruginosa.
2.2.2.1. Structure Activities Relationship
The study of SAR of the synthesized compounds demonstrated that unsubstituted benzene sulfonamide pyridone derivatives 8a, 8c, and 9a were more potent than the p-tolyl pyridone derivatives 8b, 8d, and 9b against S. mutans while p-tolyl pyridone derivatives 8b, 9b, and 15d were more active against Gram-negative bacteria specially K. pneumonia as compared to the unsubstituted benzene sulfonamide pyridone derivatives 8a, 9a, and 15a. Moreover, the unsubstituted benzene sulfonamide pyridone derivatives 8a, 9a, and 13a were more potent than the p-tolyl pyridone derivatives 8b, 9b, and 13b against S. aureus. In addition, both 8b- and 15d-bearing p-tolyl group showed a higher potency than the reference drug, gentamicin, against K. pneumonia. Compound 9a showed higher potency than ampicillin against S. aureus.
Comparing the antimicrobial activity of synthesized compounds (as reported above in terms of its IZ) with sulfadiazine, which is considered to be a well-known antibacterial sulfonamide drug indicated that some of the synthesized compounds showed superior activities. For example, p-tolyl pyridone derivatives 8b, 9b, and 15d showed a much higher potency against Gram-negative bacteria, K. pneumonia, than sulfadiazine (IZ 20.4 ± 0.9 mm). In addition, both 8a and 8c, unsubstituted benzene sulfonamide pyridone derivatives, were highly potent than sulfadiazine (IZ 16.3 ± 1.1 mm) against S. mutans. Furthermore, 1H-pyrazolopyridones 9a was highly effective while pyridone derivatives 8d, 13a, and 13b showed a relatively lower activity than sulfadiazine (IZ 31.3 ± 2.1 mm) against S. aureus. Although, sulfadiazine had no activity against C. albicans, some of synthesized compounds such as 9b, 13a, 13b, and 15a showed some activity.
These remarkable findings indicate the influence of the synthesized compounds and the effectiveness of these newly synthesized chemical structures in effectively inhibiting and combating the mentioned hazardous bacterial strains, confirming the appropriate approach of this work in envisaging and generating new effective antimicrobial drugs.
2.2.2.2. Dihydrofolate Reductase Activity Assay
Folate inhibitors are well-known agents in provoking the synthesis of folic acid and therefore are usually used for treating protozoal, bacterial, and fungal infections. This class of antimicrobial drugs may contain compounds from the subclasses of sulfonamides or dihydrofolate reductase (DHFR) inhibitors or a combination of both drugs.22 Generally, sulfonamide drugs inhibit conversion of p-aminobenzoic acid PABA to dihydrofolate (DHF) through the action of the enzyme dihydropteroate synthase, which can be converted to tetrahydrofolic acid through the action of the enzyme DHFR. DHFR inhibitors are commonly used for fighting malaria and other protozoal infections, as well as for treating fungal, bacterial, and mycobacterial infections.23 Trimethoprim is used as the DHFR inhibitor, in the case of bacterial infections, because it competes with the pteridine moiety of DHF in binding with the DHFR enzyme. It has been shown that drugs containing pyrimidine ring such as trimethoprim in association with sulfonamide drug are commonly used as an antibacterial drug because of its ability to bind to the active site of the enzyme.24 Inasmuch, it has been also reported that drugs such as trimetrexate having pyridopyrimidines inhibit the DHFR enzyme’s function during the development of the cancer cells.25 Therefore, it would be quite interesting to perform an enzyme assay on the DHFR enzyme. In these regards, the highly potent antimicrobial compounds, 8b, 9a, and 15d were tested in terms of their inhibitory activities toward the enzyme using the DHFR inhibitor screening kit. For the purpose of comparison, both sulfadiazine, as one of sulfonamide antibiotics, and trimethoprim were used as standard drugs, Table 6.
Table 6. IC50 Values (the Drug Concentrations That Inhibited 50% of Cell Proliferation) of the Compounds on the DHFRa.
| compd no. | 10 (μg/mL) | 1 (μg/mL) | 0.1 (μg/mL) | 0.01 (μg/mL) | IC50 (μg/mL) |
|---|---|---|---|---|---|
| 8b | 57.653% | 50.383% | 40.816% | 19.897% | 1.368 ± 0.04 |
| 9a | 68.367% | 53.571% | 33.800% | 22.576% | 0.699 ± 0.02 |
| 15d | 56.122% | 39.923% | 20.662% | 11.096% | 4.673 ± 0.13 |
| sulfadiazine | 76.658% | 60.204% | 46.811% | 28.699% | 0.201 ± 0.01 |
| trimethoprim | 88.070% | 66.114% | 43.022% | 25.511% | 0.170 ± 0.01 |
All data were taken as the average of three measurements.
The inhibitory potency IC50 values of tested compounds, 8b, 9a, and 15d, were compared to that of sulfadiazine. All tested compounds showed IC50 values higher than that of sulfadiazine (IC50 = 0.201 μg/mL) and trimethoprim (IC50 = 0.170 μg/mL), Figure 6. It was determined that compound 9a had IC50 value of 0.699 μg/mL followed by 8b with IC50 value of 1.368 μg/mL and 15d with IC50 value of 4.671 μg/mL making 9a the most potent agent against DHFR among the newly synthesized compounds.
Figure 6.

Percent cell survival vs the concentration of sulfadiazine, trimethoprim, 8b, 9a, and 15d.
3. Conclusions
The work in this article is focused on synthesizing a new class of functionalized benzothiazole and benzimidazole-based pyridine incorporating sulfonamide moieties, with remarkable antiviral and antimicrobial potency. The synthesis was carried out by reacting N-cyanoacetoarylsulfonylhydrazide with various electrophiles such as 2-(benzo[d]thiazol-2-yl)-3,3-bis(alkylthio)acrylonitriles and 2-(benzo[d]imidazol-2-yl)-3,3-bis(methylthio)-acrylonitriles as well as 2-ethoxyl acrylonitrile derivatives. The structure of the different synthesized compounds was confirmed by basic spectroscopic data and elemental analysis. The antiviral activities of the newly synthesized compounds were evaluated in vitro against a wide variety of viruses such as HSV-1, CVB4, HAV HM 175, HCVcc genotype 4a, and HAdV7. The CC50, IC50, and SI were also evaluated for the promising compounds. Two of the synthesized compounds, 15c and 15d showed more than 50% viral reduction against HSV-1 and CBV4, with significant IC50 and CC50 values. Even though these compounds have also shown inhibitory activity against Hsp90α protein, their combination with the known drug acyclovir has resulted in a very high potency against the Hsp90α and consequently against the HSV-1 virus. To evaluate the underlying principles behind the action of these new compounds in inhibiting HSV-1, a molecular docking study was performed with focus on compounds that showed the greatest potency as HSV-1 virus. The study showed that the investigated compounds occupied the protein binding pockets of Hsp90α with strong binding interactions. The newly synthesized target compounds were also evaluated for their in vitro antimicrobial activity against E. coli, K. pneumonia, P. aeruginosa, S. aureus, and S. mutans, as well as C. albicans. In general, the thiomethylpyridone derivatives showed improved inhibitory effectiveness against the tested Gram-positive bacteria while 1H-pyrazolopyridones showed potency more than ampicillin against S. aureus. According to the enzyme assay study on DHFR, compound 9a was the most potent among the tested potent compounds.
4. Experimental Part
4.1. Chemistry
Melting points were determined on digital melting point apparatus, SMP3, using one end open capillary tubes and are uncorrected. IR spectra were recorded on FTIR plus 460 or using KBr pellets. 1H NMR and 13C NMR were carried out in the Center of Drug Discovery Research at Ain Shams University and spectra were recorded on a Bruker ADVANCE (III) model (400 MHz) spectrometer in DMSO-d6 as a solvent using tetramethylsilane as an internal standard and chemical shifts are reported as δppm units. The elemental analyses were done at the microanalytical data unit at Cairo University and performed on a Vario EI III Elemental CHNS Analyzer. Progress of the reactions was monitored by thin-layer chromatography (TLC) using aluminum sheet coated with silica gel Merck 60F and was visualized by a UV lamp. The reagents and solvents were purchased in commercially available grade purity.
4.1.1. General Procedure for the Synthesis of 3b
A mixture of sodium ethoxide (0.08 mol) and 2-cyanomethylbenzothiazole (0.04 mol) in absolute ethanol (100 mL) was refluxed for 20 min. After cooling, carbon disulfide (0.04 mol) was added gradually and the solution was warmed for 20 min. The reaction mixture was stirred overnight at room temperature after the addition of methyl iodide (0.08 mol). The solution was poured onto ice water and the solid product formed was filtered. After drying, the solid product was dissolved in hot petroleum ether and then filtrated. The precipitate formed after evaporation of the solvent recrystallized from DMF.
4.1.1.1. 2-(Benzo[d]thiazol-2-yl)-3,3-bis(ethylthio)acrylonitrile (3b)
Pale yellow crystals, mp 93–95 °C, yield 72%; IR (KBr, cm–1): ν 3056 (ArCH), 2969 (CH2, CH3), 2213 (CN), 1502 (C=N); 1H NMR (300 MHz, DMSO-d6): δ 1.27–1.34 (m, 6H, 2SCH2CH3), 3.168–3.23 (m, 4H, 2SCH2CH3), 7.41–7.57 (m, 2H, benzothiazole-H), 8.03–8.17 (m, 2H, benzothiazole-H); Anal. Calcd for C14H14N2S3 (306.47): C %, 54.87; H %, 4.60; N %, 9.14. Found: C %, 54.85; H %, 4.58; N %, 9.16.
4.1.2. General Procedure for the Synthesis of 8a–d
2-(Benzo[d]thiazol-2-yl)-3,3-bis(alkylthio)acrylonitrile (0.01 mol) was added to a solution of N-cyanoacetoarylsulfonylhydrazides (0.01 mol) in dry DMF (30 mL) containing pulverized potassium hydroxide (0.01 mol). The reaction mixture was refluxed with stirring for 2 h (TLC monitoring). After cooling, the reaction mixture poured onto ice-cold water and neutralized with HCl. The solid product was filtered, washed with water, and dried. Further purification was done using a hot mixture of petroleum ether: ethyl acetate (50:50; v/v). The remaining solid compound was crystallized from DMF.
4.1.2.1. N-(6-Amino-5-(benzo[d]thiazol-2-yl)-3-cyano-4-(methylthio)-2-oxopyridin-1(2H)-yl)benzenesulfonamide (8a)
Yellow crystals; (DMF), yield (80%), mp 211 °C; IR (KBr, cm–1): ν 3392, 3200 (NH, NH2), 3067 (ArCH), 2925 (CH3), 2210 (CN), 1675 (CO), 1594 (C=N), 1353, 1172 (O=S=O); 1H NMR (400 MHz, DMSO-d6): δ 2.51 (s, 3H, SCH3), 7.49 (t, J = 7.6 Hz, 1H, benzothiazole-H), 7.54–7.63 (m, 3H, 3Ar-H), 7.73 (t, J = 8 Hz, 1H, benzothiazole-H), 7.83 (d, J = 7.6 Hz, 2H, Ar-H), 8.05 (d, J = 8 Hz, 1H, benzothiazole-H), 8.13 (d, J = 8 Hz, 1H, benzothiazole-H), 8.87 (br, 2H, NH2), 11.52 (s, 1H, NH); Anal. Calcd for C20H15N5O3S3 (469.56): C %, 51.16; H %, 3.22; N %, 14.91. Found: C %, 51.09; H %, 3.20; N %, 14.94.
4.1.2.2. N-(6-Amino-5-(benzo[d]thiazol-2-yl)-3-cyano-4-(methylthio)-2-oxopyridin-1(2H)-yl)-4-methylbenzenesulfonamide (8b)16
Yellow crystals; (DMF), yield (78%), mp 221 °C; IR (KBr, cm–1): ν 3393, 3208 (NH, NH2), 3072 (ArCH), 2922 (CH3), 2210 (CN), 1677 (CO), 1594 (C=N), 1350, 1170 (O=S=O); 1H NMR (400 MHz, DMSO-d6): δ 2.42 (s, 3H, CH3), 2.45 (s, 3H, SCH3), 7.42 (d, J = 8 Hz, 2H, Ar-H), 7.49 (t, J = 7.6 Hz, 1H, benzothiazole-H), 7.56 (t, J = 7.6 Hz, 1H, benzothiazole-H), 7.71 (d, J = 7.2 Hz, 2H, Ar-H), 8.06 (d, J = 8 Hz, 1H, benzothiazole-H), 8.13 (d, J = 8 Hz, 1H, benzothiazole-H), 8.84 (br, 2H, NH2), 11.44 (s, 1H, NH); Anal. Calcd for C21H17N5O3S3 (483.59): C %, 52.16; H %, 3.54; N %, 14.48. Found: C %, 52.11; H %, 3.48; N %, 14.50.
4.1.2.3. N-(6-Amino-5-(benzo[d]thiazol-2-yl)-3-cyano-4-(ethylthio)-2-oxopyridin-1(2H)-yl)benzenesulfonamide (8c)
Yellow crystal; (EtOH), yield (77%), mp 226–229 °C; IR (KBr, cm–1): ν 3395, 3206 (NH2), 3062 (ArCH), 2923 (CH2, CH3), 2208 (CN), 1677 (CO), 1593 (C=N) 1352, 1176 (O=S=O); 1H NMR (400 MHz, DMSO-d6): δ 1.18 (t, J = 6.8 Hz, 3H, SCH2CH3), 2.95 (q, J = 6.8 Hz, 2H, SCH2CH3), 7.48 (t, J = 7.6 Hz, 1H, benzothiazole-H), 7.53–7.63 (m, 3H, Ar-H), 7.74 (t, J = 7.6 Hz, 1H, benzothiazole-H), 7.82 (d, J = 8 Hz, 2H, Ar-H), 8.05 (d, J = 8 Hz, 1H, benzothiazole-H), 8.12 (d, J = 8 Hz, 1H, benzothiazole-H), 11.53 (s, 1H, NH); Anal. Calcd for C21H17N5O3S3 (483.59): C %, 52.16; H %, 3.54; N %, 14.48. Found: C %, 52.19; H %, 3.50; N %, 14.46.
4.1.2.4. N-(6-Amino-5-(benzo[d]thiazol-2-yl)-3-cyano-4-(ethylthio)-2-oxopyridin-1(2H)-yl)-4-methylbenzenesulfonamide (8d)
Yellow crystal; (EtOH), yield (71%), mp 255 °C; IR (KBr, cm–1): ν 3407, 3201 (NH, NH2), 3068 (ArCH), 2927 (CH2, CH3), 2209 (CN), 1676 (CO), 1592 (C=N), 1350, 1170 (O=S=O); 1H NMR (400 MHz, MeOH-d4): δ 1.30 (t, J = 8 Hz, 3H, SCH2CH3), 2.48 (s, 3H, CH3), 3.05 (q, J = 6.8 Hz, 2H, SCH2CH3), 7.41 (d, J = 8 Hz, 2H, C6H4), 7.48 (t, J = 7.2 Hz, 1H, benzothiazole-H), 7.56 (t, J = 7.2 Hz, 1H, benzothiazole-H), 7.77 (d, J = 8 Hz, 2H, Ar-H), 8.00–8.03 (m, 2H, benzothiazole-H); 13C NMR (100 MHz, DMSO-d4): 14.7, 21.6 (2CH3), 30.9 (CH2), 112.8 (CN), 89.9, 98.4, 144.7, 155.3, 162.9 (pyridone-C), 107.9, 122.2, 122.9, 124.1, 126.0, 126.7, 128.4, 129.9, 135.7, 136.5, 156.8 (Ar–C); Anal. Calcd for C22H19N5O3S3 (497.61): C %, 53.10; H %, 3.85; N %, 14.07. Found: C %, 53.14; H %, 3.81; N %, 14.09.
4.1.3. General Procedure for the Synthesis of 9a,b
A mixture of 8a–d (0.01 mol) and hydrazine hydrate (0.1 g, 0.02 mol) in dry DMF containing a catalytic amount of piperidine (3 drops) was heated for 2 h (TLC monitoring). The mixture was then concentrated under reduced pressure to afford a solid residue. Ethanol was added to solid and then filtered off and washed with ethanol. The solid product was recrystallized from methanol.
4.1.3.1. N-(3,6-Diamino-7-(benzo[d]thiazol-2-yl)-4-oxo-1H-pyrazolo[4,3-c]pyridin-5(4H)-yl)benzenesulfonamide (9a)
White solid; (MeOH), yield (62%), mp 299 °C; IR (KBr, cm–1): ν 3365, 3200 (NH, NH2), 3066 (ArCH), 1678 (CO), 1596 (C=N), 1339, 1165 (O=S=O); 1H NMR (400 MHz, DMSO-d6): δ 6.10 (br, 2H, NH2), 7.26 (t, J = 7.6 Hz, 1H, benzothiazole-H), 7.42 (t, J = 8 Hz, 1H, Ar-H), 7.57–7.60 (m, 2H, Ar-H), 7.70 (t, J = 7.6 Hz, 1H, benzothiazole-H), 7.80–7.83 (m, 3H, 2Ar-H & benzothiazole-H), 7.98 (d, J = 8 Hz, 1H, benzothiazole-H), 10.99 (s, 1H, NH), 11.95 (s, 1H, NH); Anal. Calcd for C19H15N7O3S2 (453.50): C %, 50.32; H %, 3.33; N %, 21.62. Found: C %, 50.30; H %, 3.32; N %, 21.60.
4.1.3.2. N-(3,6-Diamino-7-(benzo[d]thiazol-2-yl)-4-oxo-1H-pyrazolo[4,3-c]pyridin-5(4H)-yl)-4-methylbenzenesulfonamide (9b)
White solid; MeOH, yield (58%), mp 322 °C; IR (KBr, cm–1): ν 3410, 3351 (NH, NH2), 3184 (ArCH), 2922 (CH3), 1678 (CO), 1616 (C=N), 1337, 1157 (O=S=O); 1H NMR (400 MHz, DMSO-d6): δ 2.41 (s, 3H, CH3), 6.11 (br, 2H, NH2), 7.27 (t, J = 8 Hz, 1H, benzothiazole H), 7.37–7.42 (m, 3H, 2Ar-H & benzothiazole-H), 7.71 (d, J = 7.2 Hz, 2H, Ar-H), 7.82 (d, J = 7.6 Hz, 1H, benzothiazole-H), 7.99 (d, J = 7.6 Hz, 1H, benzothiazole-H), 10.85 (s, 1H, NH), 11.92 (s, 1H, NH); 13C NMR (100 MHz, DMSO-d4): 21.6 (CH3), 81.4, 89.6, 144.2, 152.0, 164.1 (pyridone-C), 120.2, 121.6, 123.1, 126.2, 128.1, 129.9, 133.3, 137.3, 126.7, 149.9, 151.3, 156.6 (Ar–C); Anal. Calcd for C20H17N7O3S2 (467.52): C %, 51.38; H %, 3.67; N %, 20.97. Found: C %, 51.39; H %, 3.65; N %, 20.96.
4.1.4. General Procedure for the Synthesis of 13a,b
To a stirred solution of N-cyanoacetoarylsulfonylhydrazides (0.01 mol) in dry dioxane (30 mL) containing pulverized potassium hydroxide (0.01 mol), 2-(1H-benzo[d]imidazol-2-yl)-3,3-bis(methylthio)acrylonitrile (0.01 mol) was added. The reaction mixture was left to stir overnight at room temperature (TLC monitoring) and then poured onto ice-cold water and neutralized with HCl. The separated product was filtered and washed with water. The crude product was further purified by using a hot mixture of petroleum ether/ethyl acetate (80:20; v/v) and the residual precipitate was recrystallized from ethyl acetate.
4.1.4.1. N-(6-Amino-5-(1H-benzo[d]imidazol-2-yl)-3-cyano-4-(methylthio)-2-oxopyridin-1(2H)-yl)benzenesulfonamide (13a)
Orange solid; (ethyl acetate), yield (69%), mp 226–230 °C; IR (KBr, cm–1): ν 3441 (NH, NH2), 3143 (ArCH), 2209 (CN), 1687 (CO), 1590 (C=N), 1370, 1170 (O=S=O); 1H NMR (400 MHz, DMSO-d6): δ 2.34 (s, 3H, SCH3), 7.22–7.86 (m, 7H, 5Ar-H & 2benzoimidazole-H); 7.97 (d, J = 8 Hz, 1H, benzoimidazole-H), 8.27 (d, J = 8 Hz, 1H, benzoimidazole-H); Anal. Calcd for C20H16N6O3S2 (452.51): C %, 53.08; H %, 3.56; N %, 18.57. Found: C %, 53.11; H %, 3.53; N %, 18.56.
4.1.4.2. N-(6-Amino-5-(1H-benzo[d]imidazol-2-yl)-3-cyano-4-(methylthio)-2-oxopyridin-1(2H)-yl)-4-methylbenzenesulfonamide (13b)
Orange solid; (ethyl acetate), yield (68%), mp 289 °C; IR (KBr, cm–1): ν 3411, (NH, NH2), 3143 (ArCH), 2926 (CH3), 2199 (CN), 1687 (CO), 1590 (C=N), 1370, 1170 (O=S=O); 1H NMR (400 MHz, DMSO-d6): δ 2.34 (s, 3H, CH3), 2.50 (s, 3H, SCH3), 7.43 (d, J = 8 Hz, 2H, C6H4), 7.42–7.62 (m, 2H, benzoimidazole-H), 7.67 (d, J = 8 Hz, 1H, benzoimidazole-H), 7.89 (d, J = 8 Hz, 2H, Ar-H), 8.29 (d, J = 8 Hz, 1H, benzoimidazole-H); Anal. Calcd for C21H18N6O3S2 (466.54): C %, 54.06; H %, 3.89; N %, 18.01. Found: C %, 54.09; H %, 3.90; N %, 18.01.
4.1.5. General Procedure for the Synthesis of 15a–d
2-(Ethoxymethylene)malononitrile or (Z)-ethyl 2-cyano-3-ethoxyacrylate (0.01 mol) was refluxed with N-cyanoacetoarylsulfonylhydrazides in sodium ethoxide solution (30 mL) for 30–45 min (TLC monitoring). After completion of the reaction, the mixture was concentrated under reduced pressure to afford a solid residue. Ethanol was added to solid and then filtered off and washed with ethanol. The solid product was recrystallized from proper solvents.
4.1.5.1. N-(6-Amino-3,5-dicyano-2-oxopyridin-1(2H)-yl)benzenesulfonamide (15a)
White solid; (EtOH), yield (77%), mp 339–340 °C; IR (KBr, cm–1): ν 3383, 3286 (NH, NH2), 2925 (ArCH), 2221 (CN), 1607 (CO), 1265, 1132 (O=S=O); 1H NMR (400 MHz, DMSO-d6): 7.36–7.38 (m, 3H, Ar-H), 7.69–7.71 (m, 2H, Ar-H), 7.95 (s, 1H, pyridone-H); Anal. Calcd for C13H9N5O3S (315.31): C %, 49.52; H %, 2.88; N %, 22.21. Found: C %, 49.50; H %, 2.89; N %, 22.25.
4.1.5.2. N-(6-Amino-3,5-dicyano-2-oxopyridin-1(2H)-yl)-4-methylbenzenesulfonamide (15b)
White solid; EtOH, yield (75%), mp 332 °C; IR (KBr, cm–1): ν 3383, 3308, 3202 (NH, NH2), 3065 (ArCH), 2927 (CH3), 2218 (CN), 1682 (CO), 1341, 1131 (O=S=O); 1H NMR (400 MHz, DMSO-d6): δ 2.33 (s, 3H, CH3), 7.16 (d, J = 7.6 Hz, 2H, Ar-H), 7.59 (d, J = 7.6 Hz, 2H, Ar-H), 7.94 (s, 1H, pyridone-H); 13C NMR (100 MHz, DMSO-d4): 21.3 (CH3), 116.9, 117.9 (2CN), 72.0, 87.2, 145.3, 145.9, 159.8 (pyridone-C), 126.5, 128.5, 136.3, 139.3 (Ar–C); Anal. Calcd for C14H11N5O3S (329.33): C %, 51.06; H %, 3.37; N %, 21.27. Found: C %, 51.09; H %, 3.34; N %, 21.23.
4.1.5.3. Ethyl 2-Amino-5-cyano-6-oxo-1-(phenylsulfonamido)-1,6-dihydropyridine-3-carboxylate (15c)
Off-white solid; EtOH, yield (74%), mp 246 °C; IR (KBr, cm–1): ν 3585, 3411, 3271 (NH, NH2), 3059 (ArCH), 3981 (CH2, CH3), 2221 (CN), 1.679, 1641 (2CO), 1586 (C=N), 1381, 1134 (O=S=O); 1H NMR (400 MHz, DMSO-d6): δ 1.28 (t, J = 7.2 Hz, 3H, SCH2CH3), 4.21 (q, J = 7.2 Hz, 2H, SCH2CH3), 7.37–7.71 (m, 6H, 5Ar-H & NH), 8.02 (s, 1H, pyridone-H), 8.63 (s, 2H, NH2); Anal. Calcd for C15H14N4O5S (362.36): C %, 49.72; H %, 3.89; N %, 15.46. Found: C %, 49.70; H %, 3.92; N %, 15.45.
4.1.5.4. Ethyl 2-Amino-5-cyano-1-(4-methylphenylsulfonamido)-6-oxo-1,6-dihydropyridine-3-carboxylate (15d)
Off-white solid; EtOH, yield (71%), mp 239–241 °C; IR (KBr, cm–1): ν 4580, 3410, 3276 (NH, NH2), 3053 (ArCH), 2980 (CH3, CH2), 2221 (CN), 1680, 1642 (2CO), 1587 (C=N), 1375, 1133 (O=S=O); 1H NMR (400 MHz, DMSO-d6): δ 1.28 (t, J = 7.2 Hz, 3H, SCH2CH3), 2.33 (s, 3H, CH3), 2.21 (q, J = 7.6 Hz, 2H, SCH2CH3), 7.16 (d, J = 7.6 Hz, 2H, Ar-H), 7.59 (d, J = 7.6 Hz, 2H, Ar-H), 7.64 (br, 1H, NH), 8.03 (s, 1H, pyridone-H), 8.63 (s, 2H, NH2); 13C NMR (100 MHz, DMSO-d4): 14.6, 21.3 (2CH3), 60.5 (CH2), 118.5 (CN), 86.3, 89.8, 144.2, 158.7, 165.3 (pyridone-C), 126.5, 128.5, 139.3, 145.4, 160.4 (Ar–C); Anal. Calcd for C16H16N4O5S (376.39): C %, 51.06; H %, 4.38; N %, 14.89. Found: C %, 51.05; H %, 4.35; N %, 14.85.
4.2. Antiviral Activity
4.2.1. Cytotoxicity Test
Measuring of the antiviral activities were carried out at the National Research Center, Giza, Egypt. The antiviral experiments were done according to the literature.3,26,27 First, 50 mg of each sample was dissolved in 1 mL of DMSO. Decontamination of samples was done by adding 0.024 mL of 100× of antimycotic–antibiotic mixture to 1 mL of each sample. To estimate the nontoxic dose of the tested samples, bi-fold dilutions were done to 0.1 mL of original dissolved samples and 0.1 mL of each dilutions were inoculated in Hep-2, Vero, BGM, FRHK4, and Huh 7.5 cell lines, which obtained from the Holding Company for Biological Products & Vaccines VACSERA, Egypt, and previously cultured in 96 multi well plates (Greiner Bio-One, Germany). Cytotoxicity assay was done using cell morphology evaluation by inverted light microscope and cell viability test applying the trypan blue dye exclusion method.
4.2.2. Cell Morphology Evaluation by Inverted Light Microscopy
Vero, FRHK4, BGM, Hep-2, and Huh 7.5 cell cultures (2 × 105 cells/mL) were prepared individually in 96-well tissue culture plates (Greiner Bio-One, Germany). The cell cultures were then incubated for 24 h at 37 °C in a humidified 5% (v/v) CO2 atmosphere cell monolayers. The medium was then removed from each well and replenished with 0.1 mL of bi-fold dilutions of different samples tested and prepared in Dulbecco’s modified Eagle’s medium (DMEM) (GIBCO BRL). 0.1 mL of DMEM without samples was added as cell control. Subsequently, all cultures were incubated for 72 h at 37 °C in a humidified 5% (v/v) CO2 atmosphere. Cell morphology was observed daily for microscopically detectable morphological alterations, such as cell rounding, loss of confluence and shrinking, and vacuolization and cytoplasm granulation. Morphological deviations were recorded.26
4.2.3. Cell Viability Assay
This assay was done by the trypan blue dye exclusion method.27 Vero, FRHK4, BGM, Hep-2, and Huh 7.5 cell cultures (2 × 105 cells/mL) were grown in 12-well tissue culture plates (Greiner Bio-One, Germany). After 24 h of incubation, the same procedure described above for tested sample cytotoxicity was carried out by applying 0.1 mL of tested sample dilutions (bifold dilutions) per well. The medium was removed after 72 h and cells were trypsinized and an equal volume of 0.4% (w/v). Trypan blue dye aqueous solution was added to the cell suspension. By using the phase contrast microscope, viable cells were counted.
4.2.4. Determination of Adenovirus 7, HAV HM 175, CBV4, and HSV-1 Titers Using Plaque Assay
Nontoxic dilutions were mixed (0.1 mL) with 0.1 mL of different doses of HSV-1, HAdV7, HAV HM 175, and CBV4 (1 × 105, 1 × 106, 1 × 107). The mixture was incubated in 37 °C for 1/2 h. The inoculation of (0.1 mL) 10-fold dilutions of treated and untreated CBV4, adenovirus 7, HSV-1, and HAV HM 175 was added separately into BGM, Hep-2, Vero, and FRHK4 cell lines, respectively, in 12 multi well plates. The palates were left for 1 h of incubation for adsorption at 37 °C in a 5% CO2–water vapor atmosphere without constant rocking. Afterward, the plates were rocked occasionally to keep the cells from drying. After adsorption, 1 mL of 2× media DMEM, Gibco-BRL and 1 mL 1% agarose were added to each well. The plates were incubated at 37 °C in a 5% CO2-water vapor atmosphere. After incubation, the cells were stained with 0.4% crystal violet after formalin fixation, and the number of plaques were counted. The viral titers were then calculated, and expressed as plaque-forming units per milliliter (pfu/mL).28 CC50 and IC50 were done for the promising compounds that showed viral reduction of 50% or more. The 50% cytotoxic concentration (CC50) of the test extract was defined as the concentration that reduce the OD492 of treated uninfected cells to 50% of that of untreated uninfected cells. IC50 is the concentration at which the compound plaque reduction rate reaches halfway between the baseline and the maximum. All data were taken as the average of three measurements (triplicate).
4.2.5. Antiviral Bioassay of Tested Compounds against ED-43/SG-Feo (VYG) Replicon of HCVgenotype 4a
ED-43/SG-Feo (VYG) replicon of HCV genotype 4a was treated with the nontoxic dose of the tested compounds. HCV RNA was quantified in algal extracts treated Huh 7.5 infected cells using qRT-PCR (TaqMan probe kit, QIAGEN) and according to the manufacturer’s instructions to show a dose-dependent decrease in sub genomic RNA copies according to the literature.29
4.3. Hsp90α (C-Terminal) Inhibitor Screening Assay
The Hsp90α (C-terminal) inhibitor screening assay was carried out using Hsp90α (C-terminal) Inhibitor Screening Assay Kit actalog 50317, size: 384 reactions, according to the method described in the catalog and literature.3 Enzyme assay experiments were carried out at the Tissue Culture Unit, the Egyptian Company for Production of Vaccines, Sera and Drugs (VACSERA), Giza, Egypt. All samples and controls were tested in triplicate. First, both Hsp90α and PPID were thawed on ice. Aliquot protein into single use aliquots. Store remaining undiluted protein in aliquots at −80 °C immediately. Note: Hsp90α and PPID proteins are very sensitive to freeze/thaw cycles. Dilute 3× Hsp90α assay buffer 2 with water to 1× Hsp90α assay buffer 2. Dilute Hsp90α in 1× Hsp90α assay buffer 2 at 1.5 ng/μL. Add 4 μL of diluted HSPα protein to each well designated for the “Positive Control”, “Test Inhibitor”, and “Blank”. To the wells labeled “Substrate Control”, add 4 μL of 1× Hsp90 assay buffer 2. Discard any unused diluted protein after use. Add 2 μL of inhibitor solution to each well-designated “Test Inhibitor”. For the “Positive Control”, “Substrate Control”, and “Blank”, add 2 μL of the same solution without inhibitor (inhibitor buffer). Dilute PPID in 1× Hsp90α assay buffer 2 at 10 ng/μL. Keep diluted protein on ice until use. Add 4 μL of 1× Hsp90α assay buffer 2 to the well-designated “Blank”. Initiate reaction by adding 4 μL of diluted PPID to each well designated for the “Substrate control”, “Positive Control”, and “Test Inhibitor”. Incubate at room temperature for 30 min. Dilute 3× detection buffer with water to 1× detection buffer. Dilute glutathione acceptor beads 250-fold with 1× detection buffer. Add 10 μL per well. Shake plate briefly. Incubate at room temperature for 30 min. Dilute streptavidin-conjugated donor beads 125-fold with 1× Detection Buffer. Add 10 μL per well. Incubate at room temperature for 1 h. Read Alpha-counts. The percentage inhibition was calculated for the different concentrations tested against the control, and the IC50 values against Hsp90α protein were calculated from the concentration inhibition response curve.
4.4. Molecular Modeling
Docking study was performed using the crystal structure of Hsp90α (PDB code: 3B25) in complex to B2K (4-methyl-6-(toluene-4-sulfonyl)-pyrimidin-2-ylamine).30 The PDB file was downloaded from the PDB. Structure of chain A was processed using the structure preparation application in MOE (Molecular Operating Environment, 2014). The ligand molecule was removed from the protein active site. Then, the missing hydrogens was added using the Protonate three-dimensional application of MOE and properly assign the ionization states. To discover the favorable binding conformation, the default procedure in the MOE Dock application was used. Primary placement poses created by the alpha triangle matcher were rescored and filtered using the London dG scoring method to pick exhibiting maximal hydrophobic, ionic, and hydrogen-bond contacts to the protein. This was followed by a refinement stage. The generated poses were energy minimized using the MMFF94× force field. Finally, the optimized poses were ranked using the GBVI/WSA DG free-energy estimates. Docking poses were inspected and interactions with binding pocket residues were analyzed.
4.5. Antimicrobial Activity
Antimicrobial assessment and the MIC performed at the Microbiology Unit in the Biochemistry Central Laboratory, Faculty of Science, Cairo University, Cairo, Egypt. The synthesized compounds were individually tested against Gram-positive and Gram-negative bacterial pathogens and fungi. Agar well diffusion method was used to determine the activity of tested compounds.31,32 The compounds were tested at a concentration of 15 mg/mL against both bacterial and fungal strains. Microbial suspension was prepared in sterilized saline equivalent to McFarland 0.5 standard solution (1.5 × 105 CFU mL–1). Its turbidity was adjusted to the optical density equals to 0.13 using spectrophotometer at 625 nm. After adjusting the turbidity of the inoculum suspension within 15 min, a sterile cotton swab was dipped into the adjusted suspension and was flooded on the dried agar surface. The agar was then allowed to dry for 15 min. Wells of 6 mm diameter was made in the solidified media with the help of sterile borer. The solution of the tested compound (100 μL) was added to each well with micropipette. The plates were then incubated at 37 °C. The IZ was measured in millimeter (mm) after 24 h incubation at 30 °C in the case of bacterial plates while in the case of fugal plates, the incubation was for 48 h. This experiment was carried out in triplicate and the results were recorded for each tested compound as mm ± SD.
4.6. MIC Measurement
Stock solutions of the tested compounds, ampicillin, gentamicin, and nystatin were prepared in DMSO at concentration of 1000 μg/mL followed by serial twofold dilution at concentrations of (500, 250,125, 62.5, 31.25 μg/mL), which was then mixed with sterile nutrient agar from Sigma-Aldrich, USA, in a sterile plate, followed by the inoculation of a defined microbial inoculum onto the agar plate surface. The plates were then left to incubate at 37 °C in a humid chamber. After 24 h, the MIC endpoints were read and recorded as the lowest concentration of an antimicrobial agent that completely inhibits growth under suitable incubation conditions.32
4.7. DHFR Inhibitor Screening Assay
Enzyme assay experiments were carried out at the Tissue Culture Unit, the Egyptian Company for Production of Vaccines, Sera and Drugs (VACSERA), Giza, Egypt. The assay for the inhibitory effect was applied as indicated in the BioVision manufacture’s protocol. Dilute methotrexate 100-fold was prepared by diluting 2 μL of methotrexate with 198 μL of DHFR buffer. Each tested sample was then dissolved into 100× of an appropriate solvent. 2 μL of the tested sample, diluted methotrexate or DHFR assay buffer was added into wells assigned as sample screening (S), inhibitor control (IC), or enzyme control (EC), respectively. Dilute DHFR 400-fold was then prepared by diluting 2 μL of DHFR with 798 μL of DHFR buffer. Enough enzyme mixture for the number of wells were also prepared to be analyzed. Diluted DHFR (98 μL) was added into desired well(s) having the tested samples, EC or IC with volume equivalent to 100 μL. Background control (BC) was prepared by adding 100 μL of DHFR assay buffer to desired well(s). A stock solution of nicotinamide adenine dinucleotide phosphate (NADPH), a 40-fold dilution was prepared by diluting 10 μL of NADPH stock solution with 390 μL DHFR assay buffer, vortex briefly and kept at 0 °C. Diluted NADPH (40 μL) was added to each well containing the tested samples, EC, IC, or BC. Samples were then mixed well and incubated at room temperature for 10–15 min and was kept away from light. A 15-fold dilution of the DHFR substrate was prepare by diluting 40 μL of DHFR stock substrate with 560 μL of DHFR assay buffer, vortexed briefly and kept at 0 °C. Diluted DHFR substrate (60 μL) was then added to each well containing the tested samples, EC, IC, or BC. After samples were mixed well, the total volume reached to 200 μL. The absorbance was measured then at 340 nm in kinetic mode for 10–20 min at room temperature. Choose two time points (t1 & t2) in the linear range of the plot and obtain the corresponding values for the absorbance (OD1 and OD2). Calculate the slope for all test inhibitor samples [S] and [EC] by dividing the net DOD (A1 – A2) values with the time Dt (t2 – t1). Subtract the solvent control or inhibitor BC readings from its paired sample readings.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c03773.
All spectral analysis such as IR, 1H NMR, and 13C NMR spectra and nontoxic doses for the newly synthesized compounds on FRHK-4, Hep2, BGM, Vero, and Huh 7.5 cell lines (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Elgemeie G. H.; Jones P. G. N-[3-Cyano-2-oxo-5,6,7,8-tetrahydroquinoline-1(2H)-yl]-4-methylbenzenesulfonamide. Acta Crystallogr., Sect. E: Struct. Rep. Online 2002, 58, o1250–o1251. 10.1107/s1600536802018731. [DOI] [Google Scholar]
- Azzam R. A.; Elgemeie G. H. Synthesis and antimicrobial evaluation of novel N-substituted 4-ethylsulfanyl-2-pyridones and triazolopyridines. Med. Chem. Res. 2019, 28, 62–70. 10.1007/s00044-018-2264-z. [DOI] [Google Scholar]
- Azzam R. A.; Osman R. R.; Elgemeie G. H. Efficient synthesis and docking studies of novel benzothiazole-based pyrimidinesulfonamide scaffolds as new antiviral agents and Hsp90α Inhibitors. ACS Omega 2020, 5, 1640–1655. 10.1021/acsomega.9b03706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgemeie G. H.; Azzam R. A.; Elsayed R. E. Sulfa drug analogs: new classes of N-sulfonyl aminated azines and their biological and preclinical importance in medicinal chemistry (2000–2018). Med. Chem. Res. 2019, 28, 1099–1131. 10.1007/s00044-019-02378-6. [DOI] [Google Scholar]
- Elgemeie G. E. H.; Hanfy N.; Hopf H.; Jones P. G. 5-Amino-1-phenylsulfonyl-4-pyrazolin-3-one. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 1998, 54, 136–138. 10.1107/s0108270197012420. [DOI] [PubMed] [Google Scholar]
- Elgemeie G. H.; Hanfy N. Novel synthesis of 5-amino-1-arylsulfonyl-4-pyrazolin-3-ones as a new class of N-sulfonylated pyrazoles. J. Chem. Res. 1999, 23, 385–386. 10.1039/a800465j. [DOI] [Google Scholar]
- Myers M. C.; Napper A. D.; Motlekar N.; Shah P. P.; Chiu C.-H.; Beavers M. P.; Diamond S. L.; Huryn D. M.; Smith A. B. Identification and characterization of 3-substituted pyrazolyl esters as alternate substrates for cathepsin B: The confounding effects of DTT and cysteine in biological assays. Bioorg. Med. Chem. Lett. 2007, 17, 4761–4766. 10.1016/j.bmcl.2007.06.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan G. S.; Kadry H. H.; Abou-Seri S. M.; Ali M. M.; Mahmoud A. E. E.-D. Synthesis and in vitro cytotoxic activity of novel pyrazolo[3, 4-d]pyrimidines and related pyrazole hydrazones toward breast adenocarcinoma MCF-7 cell line. Bioorg. Med. Chem. 2011, 19, 6808–6817. 10.1016/j.bmc.2011.09.036. [DOI] [PubMed] [Google Scholar]
- Sidique S.; Shiryaev S. A.; Ratnikov B. I.; Herath A.; Su Y.; Strongin A. Y.; Cosford N. D. P. Structure–activity relationship and improved hydrolytic stability of pyrazole derivatives that are allosteric inhibitors of West Nile Virus NS2B-NS3 proteinase. Bioorg. Med. Chem. Lett. 2009, 19, 5773–5777. 10.1016/j.bmcl.2009.07.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgemeie G. E. H.; Elghandour A. H. H. Activated nitriles in heterocyclic synthesis: novel synthesis of pyridine, benzo[1,4]thiazine and benzo[1,3]thiazole derivatives. Phosphorus, Sulfur Silicon Relat. Elem. 1990, 48, 281–284. 10.1080/10426509008045911. [DOI] [Google Scholar]
- Shi D.-F.; Bradshaw T. D.; Wrigley S.; McCall C. J.; Lelieveld P.; Fichtner I.; Stevens M. F. G. Antitumor benzothiazoles. 3. Synthesis of 2-(4-aminophenyl)benzothiazoles and evaluation of their activities against breast cancer cell lines in vitro and in vivo. J. Med. Chem. 1996, 39, 3375–3384. 10.1021/jm9600959. [DOI] [PubMed] [Google Scholar]
- Bradshaw T.; Stevens M. F.; Westwell A. The discovery of the potent and selective antitumour agent 2-(4-amino-3-methylphenyl) benzothiazole (DF 203) and related compounds. Curr. Med. Chem. 2001, 8, 203–210. 10.2174/0929867013373714. [DOI] [PubMed] [Google Scholar]
- Kamal A.; Reddy C. R.; Prabhakar S.. Imidazolylphenyl)benzothiazole derivatives as anticancer agents and process for the preparation thereof. WO110959, 2012.
- Kamal A.; Reddy C. R.; Prabhakar S.. 2-Phenyl benzothiazole linked imidazole compounds as potential anticancer agents U.S. Patent 9,187,467B2, 2015.
- Li X.; Liu X.; Wang C.; et al. 2-(4-Aminophenyl)benzothiazole-containing naphthalimide derivatives useful in treatment of cancers and their preparation. CN103450176 A, 2013.
- Azzam R. A.; Elgemeie G. H.; Elsayed R. E.; Jones P. G. Crystal structure of N-[6-amino-5-(benzo[d]thia-zol-2-yl)-3-cyano-4-methyl-sulfanyl-2-oxo-1,2-di-hydro-pyridin-1-yl]-4-methyl-benzene-sulfonamide di-methyl-formamide monosolvate. Acta Crystallogr., Sect. E: Crystallogr. Commun. 2017, 73, 1820–1822. 10.1107/s2056989017015778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triantafilou K.; Triantafilou M. Coxsackievirus B4-induced cytokine production in pancreatic cells is mediated through toll-like receptor 4. J. Virol. 2004, 78, 11313–11320. 10.1128/jvi.78.20.11313-11320.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg A.-K.; Olsson A.; Korsgren O.; Frisk G. Antiviral treatment of Coxsackie B virus infection in human pancreatic islets. Antivir. Res. 2007, 74, 65–71. 10.1016/j.antiviral.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Chen S.; Tian X. Vaccine development for human mastadenovirus. J. Thorac. Dis. 2018, 10, S2280. 10.21037/jtd.2018.03.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloney A.; Workman P. Hsp90 as a new therapeutic target for cancer therapy: the story unfolds. Expet Opin. Biol. Ther. 2002, 2, 3–24. 10.1517/14712598.2.1.3. [DOI] [PubMed] [Google Scholar]
- Geller R.; Taguwa S.; Frydman J. Broad action of Hsp90 as a host chaperone required for viral replication. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 698–706. 10.1016/j.bbamcr.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derouin F.; Chastang C. In vitro effects of folate inhibitors on Toxoplasma gondii. Antimicrob. Agents Chemother. 1989, 33, 1753–1759. 10.1128/aac.33.10.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan B.; Tonddast-Navaei S.; Roy A.; Zhou H.; Skolnick J. Chemical space of Escherichia coli dihydrofolate reductase inhibitors: New approaches for discovering novel drugs for old bugs. Med. Res. Rev. 2019, 39, 684–705. 10.1002/med.21538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsaad N.; van Altena R.; Pranger A. D.; van Soolingen D.; de Lange W. C. M.; van der Werf T. S.; Kosterink J. G. W.; Alffenaar J.-W. C. Evaluation of co-trimoxazole in the treatment of multidrug-resistant tuberculosis. Eur. Respir. J. 2013, 42, 504–512. 10.1183/09031936.00114812. [DOI] [PubMed] [Google Scholar]
- Du X. H.; Zhuang W. C. Neural network model for predicting anticancer activity of pyridopyrimidines derivatives. Adv. Mater. Res. 2014, 905, 96–100. 10.4028/www.scientific.net/amr.905.96. [DOI] [Google Scholar]
- Simões C. M. O.; Amoros M.; Girre L. Mechanism of antiviral activity of triterpenoid saponins. Phytother Res. 1999, 13, 323–328. . [DOI] [PubMed] [Google Scholar]
- Walum E.; Stenberg K.; Jenssen D.. Understanding Cell Toxicology. Principles and Practice; Ellis Horwood: New York, 1990; pp 389–390. [Google Scholar]
- Schmidtke M.; Knorre C.; Blei L.; Stelzner A.; Birch-Hirschfeld E. Penetration and antiviral activity of Coxsackievirus B3 (CVB3)-specific phosphorothioate oligodeoxynucleotides (PS-ODN). Nucleosides Nucleotides 1998, 17, 1557–1566. 10.1080/07328319808004686. [DOI] [Google Scholar]
- Saeed M.; Scheel T. K. H.; Gottwein J. M.; Marukian S.; Dustin L. B.; Bukh J.; Rice C. M. Efficient replication of genotype 3a and 4a hepatitis C virus replicons in human hepatoma cells. Antimicrob. Agents Chemother. 2012, 56, 5365–5373. 10.1128/aac.01256-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju H.-Q.; Xiang Y.-F.; Xin B.-J.; Pei Y.; Lu J.-X.; Wang Q.-L.; Xia M.; Qian C.-W.; Ren Z.; Wang S.-Y.; Wang Y.-F.; Xing G.-W. Synthesis and in vitro anti-HSV-1 activity of a novel Hsp90 inhibitor BJ-B11. Bioorg. Med. Chem. Lett. 2011, 21, 1675–1677. 10.1016/j.bmcl.2011.01.098. [DOI] [PubMed] [Google Scholar]
- Scòtt A. C.Practical Medical Microbiology, 13th ed.; Collee J. G., Ed.; Churchill Livingstone: Edinburgh, 1989; Vol. 161. [Google Scholar]
- Azzam R. A.; Elsayed R. E.; Elgemeie G. H. Design, synthesis, and antimicrobial evaluation of a new series of N-sulfonamide 2-pyridones as dual inhibitors of DHPS and DHFR enzymes. ACS Omega 2020, 5, 10401–10414. 10.1021/acsomega.0c00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.