

Abstract

The instability of an amide bond with dilute trifluoroacetic acid (TFA) is a rare chemical event. The native amide bonds are stable even in the neat TFA, which is one of the reagents that releases the peptides from the solid support in the solid-supported peptide synthesis method. In the repertoire of unnatural peptidomics, α-/β-hydrazino acids and their peptides are explored for the synthesis of N-amino peptide derivatives, and their amide bonds are stable in TFA (∼100%) as natural amide bonds. This report describes the synthesis of a β-hydrazino acid analogue as β-troponylhydrazino acid, containing a nonbenzenoid natural troponyl scaffold. The structural and conformational studies of their hybrid di-/tripeptides with the natural amino acid show that the 2-aminotroponyl residue is involved in hydrogen bonding. Surprisingly, the amide bond of β-troponylhydrazino peptides is cleavable with TFA (∼20%) through the formation of a new heterocyclic molecule N-troponylpyrazolidinone or troponylpyrazolidinone. Tropolone and related compounds are excellent biocompatible chromophores. Hence, β-troponylhydrazino acid could be employed for tuning the peptide structure and considered a promising chromophoric acid-sensitive protecting group of a free amine of amino acids/peptides. It could be applied for the estimation of the free amine group functionality by a UV–vis spectrophotometer.
Introduction
Natural amide bonds are pretty stable, with an estimated half-life of around ∼350–600 years for spontaneous hydrolysis at neutral pH and room temperature (RT).1,2 The natural amide bonds are resonance-stabilized. The carbonyl group of the natural amide is inert toward the nucleophilic addition reaction.3,4 The cleavage/hydrolysis of amide bonds could be achieved under extreme conditions as the heating under strongly acidic or basic conditions. However, the cyclic amides (lactams) are more cleavable as compared to the linear amides because of ring-strained amides.5−7 A large number of ring-strained lactams are synthesized and their poor stability is reported even under mild conditions because of the resonance decoupling through N–C=O torsion, which induces the strong electrophilicity at the C=O group as ketonic carbonyl.8 The cleavage of an amide bond without metal ions becomes a center point of discussion. Brown and co-workers have shown that the resonance decoupling enhances the hydrolysis rate in the strained amide bond because of the direct nucleophilic attack.3,9 For instance, the twisted amide of 1-aza-2-admantanone derivatives is a highly strained lactam ring and readily cleavable under mild conditions.10 This twisted amide also shows the dual reactivities such as (i) nucleophilic character of amine and (ii) electrophilicity of the carbonyl. The hydrolysis of linear amide bonds is also possible by decoupling the N–C=O resonance stability within the structurally modified amide bonds. However, the sequence-specific amide is cleaved/hydrolyzed with enzymes such as proteases. The zinc metal-dependent peptidase cleaves the specific amide bond through zinc ion mediation. These results encourage the synthetic chemists for the development of artificial peptidases.11 Mashima and co-workers have explored the role of zinc ions in the cleavage of amides bearing β-hydroxyethyl using Lewis acid Zn(OTf)2.12 Recently, the activation of specific amide bonds has been explored using a metal catalyst. For example, Garg, Houk, and co-workers have shown the conversion of an amide functional group into an ester group by cleaving the C–N bond of amide with the Ni-catalyst.13 The cleavage of the amide bond under near-physiological conditions is still challenging. Booker-Milburn and co-workers have reported the solvolysis of acyclic synthetic amide bonds at RT under neutral conditions via the formation of ketene intermediates.14 They have shown that an electron-withdrawing group, at the α-position, of amide carbonyl enhances the protonation of sterically hindered amide amine and facilitates the formation of ketene by cleaving the C–N bond of amide. In a recent report, the cleavage of the terminal amide bond occurs with ammonium salt/aqueous hydrazine under heating conditions via hydrazinolysis.15 The cleavage of modified N-terminal amide bonds, such as aminopyrazolonyloxy-containing acetamides, occurs under mild acidic conditions.16 Another reactivity of amide bond as transamidation also reported, such as the transamidation reaction of the amide bond using Zr/Hf-catalyst.17 For the development of peptide-based materials, various aromatic amino acids/peptides are synthesized and explored for novel peptidomimetics.18 In addition to benzenoid aromatic peptides, recently, nonbenzenoid aromatic amino acids/peptides have also been synthesized from the tropolone molecule and unnatural amino acid backbone for evaluating the role of tropolonyl carbonyl in the structural and functional changes of peptides (Figure 1a).19−21 The α-troponylalkyl amino acid and its peptides exhibit rare characteristic chemical properties as the cleavage of amide bond under mild acidic conditions [5% trifluoroacetic acid (TFA)] along with the reversible amidation and transamidation activities under basic conditions. However, their β-analogues as β-troponylalkyl amino acid derivatives are stable like other natural amide bonds, even with neat TFA (∼100%) (Figure 1b). In repertoire of unnatural peptidomimics, α-hydrazino acids and their peptides as N-amino peptide (NAP) derivatives are explored, and it was found that those peptides exhibit improved biostability and bioactivity as compared to control (Figure 1c).18,22 Thus, we designed a β-troponylhydrazino acid analogue to explore the role of the troponyl group for novel peptidomimetics (Figure 1d). This report describes the synthesis of β-troponylhydrazino acid and its hybrid peptides with the amino group of natural α-amino acid/peptide ester derivative (Figure 1b). For conformational studies, the DMSO-d6 titration experiment and X-ray studies are performed with representative peptides. For practical utilities, the stability of such peptides is also investigated under near-physiological pH conditions (mild acidic/alkaline conditions) by nuclear magnetic resonance (NMR) and electrospray ionization mass spectrometry (ESI-MS) techniques, which reveal the cleavage of their amide bonds with dilute TFA (20%).
Figure 1.

(i) Previously reported troponyl-/hydrazine-containing amides and (ii) rationally designed β-troponylhydrazinyl peptides and their instability under acidic conditions.
Results and Discussion
We used commercially available N-Boc-hydrazine (1) for N-alkylation with 3-bromopropionate ester under basic conditions that produced alkylated hydrazine derivative (2). This derivative was treated with O-tosylate tropolone (3) under reflux conditions for 4 days for N-troponylation of amine that is converted into a new unnatural amino acid as a 2-aminotroponyl hydrazine derivative (4). However, the O-tosylate tropolone (3) was derived from the commercially available tropolone molecule. For the synthesis of an amide bond, the ester group of derivative (4) was hydrolyzed into the carboxylate derivative under alkaline conditions, followed by coupling with various natural α-l-amino acid esters (Gly, Ala, Leu, Val, Ile, Phe, and Pro)/peptide ester (Leu–Phe) derivatives using peptide coupling reagents. Subsequently, the hybrid di-/tripeptide derivatives (5a–5g/5h) were isolated (Scheme 1). These peptides were well characterized by NMR and ESI high-resolution MS (ESI-HRMS) (see the Supporting Information). Pleasantly, we obtained the single crystal of one dipeptide (5a) from the organic solvent mixture (EtOAc/hexane), which was analyzed by an X-ray diffractometer, which confirmed the structures of peptide 5a as Boc-β-troponylhydrazino-glycine ester (Supporting Information, Table S4 and Figure S69). Its crystal data were deposited to the Cambridge Crystallographic Data Centre (CCDC) with number CCDC 2003629 (5a).
Scheme 1. Synthesis of N-Troponylated-β-Hydrazino Acid/Peptides.

For comparative studies, we synthesized three types of control β-hydrazino acid derivatives without containing a troponyl scaffold from the same β-hydrazino acid ester (2) and their hybrid peptides with natural α-amino acids (Scheme 2). The β-hydrazino acid ester (2) was treated with benzyl bromide under the basic condition, which produced N-benzyl-β-hydrazino ester (2-Bn). Similarly, N-hexyl-β-hydrazino ester (2-hexyl) was prepared by ester (2) and hexyl bromide under the basic condition. However, the N-amide derivative of β-hydrazino acid ester (2) was developed with picolinic acid under peptide coupling reaction conditions, which produced β-picolinylhydrazino ester (2-picolamide). These non-troponyl β-hydrazino ester derivatives (2-Bn/2-hexyl/2-picolamide) were hydrolyzed into the respective carboxylate with LiOH and then directly coupled with the amine group of α-amino acid ester under peptide coupling conditions. As resultants, the control di-/tripeptides β-benzylhydrazino peptides (6a/6b), β-picolinylhydrazino peptide (7), and β-hexylhydrazino peptide (8) were synthesized for further studies. The characterization data are described in the Experimental Section, while their NMR and mass spectra are provided in the Supporting Information (Figures S1–S34). We also attempted to synthesize a more resemble analogue as N-phenyl-β-hydrazino ester for control studies but could not achieve it.
Scheme 2. Synthesis of Control N-Alkylated-β-hydrazino Acid/Peptides.

Herein, we also attempted the involvement of amine protons (BocNH/amide NH) in intramolecular hydrogen bonding with carbonyl oxygen (troponyl/Boc/Amide) in solution state by DMSO-d6-titration 1H NMR experiments. In this experiment, the chemical shift of intramolecular hydrogen-bonded proton remains constant or exhibits a small downfield shift (N–H), while the chemical shift of intermolecular hydrogen-bonded proton exhibits a significant downfield shift with increasing concentration of DMSO-d6 (a strong hydrogen bond acceptor solvent).19 The strength of the intramolecular hydrogen bond is inversely proportional to the downfield shift of N–H by dimethyl sulfoxide (DMSO) addition. We assigned proton resonance signals of BocNH (NH1) and amide N–H (NH2) in troponylated dipeptides (5a–5e) and a control peptide (7). Pleasantly, we performed the DMSO titration experiment by recording the consecutive 1H NMR spectra of the respective peptides (5a–5e/7) in CDCl3 with successive addition of DMSO-d6 in a small amount. Their 1H NMR titration spectra are provided in the Supporting Information (Figures S35–40). We extracted the chemical shift value of BocNH and amide N–H with respect to the volume of DMSO-d6 addition and then generated a plot as a chemical shift (ppm) versus the DMSO-d6 volume (μL). These plots are depicted in Figure 2A,B, which exhibit a marginal downfield shift in the chemical shift of BocN–H/amide N–H in troponylated peptides (5a–5e) as compared to the BocN–H of control peptide (7). Importantly, we noticed that the extent of downfield shift in BocNH and amide N–H is almost equal in troponyl peptide (5a/5c). However, the extent of downfield shift in BocNH of the troponyl peptide (5b/5d/5e) is lower than its respective amide N–H, which is almost equal to that of control peptide (7). Hence, intramolecular hydrogen bond in troponyl peptides (5a–5e) due to BocNH is equal to or stronger than the respective amide N–H. Our 1H NMR titration results reveal two intramolecular hydrogen bonds in β-troponylhydrazino peptides while one control peptide. Herein, we propose the preferable intramolecular hydrogen bonding in β-troponylhydrazino peptides (5a–5e)/control peptide as shown in Figure 2C. In troponyl peptides (5a–5e), the intramolecular hydrogen bond between BocN–H···O=C (troponyl carbonyl), six-membered ring, could be slightly stronger than another intramolecular hydrogen bond between amide N–H···O=C (Boc carbonyl), nine-membered ring in solution state. In the literature, a nine-membered ring α-N-O turn is reported in peptide containing α-aminoxy acid.23
Figure 2.

(a) 1H NMR DMSO-d6 titration plot for BocNH (A) and amide NH (B) and (b) proposed conformation of β-troponylhydrazino peptides/control peptide (C).
To investigate the role of the troponyl carbonyl group in β-troponylhydrazino acid containing hybrid peptides, we attempted to crystalize hybrid peptides under various solvent systems. Pleasantly, we obtained the single crystal of one dipeptide (5a) and one tripeptide (5h). Their crystal data are submitted to CCDC with number CCDC 2003629 for peptide 5a and CCDC 2003628 for peptide 5h. We extracted their packing diagram in unit cell and supramolecular self-assembled structure using software Diamond 3.2. The structural analyses of dipeptide (5a) in solid state are depicted in Figure 3, which includes Oak Ridge thermal ellipsoid plot (ORTEP) diagram (Figure 3a), unit cell (Figure 3b), packing arrangement (Figure 3c), and supramolecular helical structure (Figure 3d). However, other crystal data are provided in the Supporting Information. Importantly, peptide 5a forms an intramolecular hydrogen bonding between troponyl carbonyl with amide N–H (C=O···H–N) and hydrazine NH with amide carbonyl (N–H···C=O), which leads to a novel supramolecular helical structure with a pitch of 6.02 (Å) (Figure 3d). Thus, troponyl carbonyl has a significant role in the conformational changes of peptides for interesting supramolecular self-assembled structures in solid state.
Figure 3.

Conformational analyses of crystal peptide 5a* in solid state: analysis of crystal 5a*: (a) ORTEP diagram; (b) unit cell packing; (c) packing arrangement; and (d) helical supramolecular self-assembled structure. (*Tertiary butyl ester of 5a.)
Similarly, the structural analysis data of tripeptide (5h) are depicted in Figure 4, which describe the ORTEP diagram (Figure 4a), unit cell (Figure 4b), packing diagram (Figure 4c), and exceptional supramolecular helical structure (Figure 4d). Other crystal data are provided in the Supporting Information. This peptide forms inter- and intramolecular hydrogen bonding and generates a new supramolecular self-assembled helical structure. Most importantly, we noticed two intramolecular hydrogen bondings—(a) troponyl carbonyl with adjacent amide NH (Leu) (C=O···H–N, 2.04 Å) as i + 9 helical structure and (b) hydrazine Boc carbonyl with N–H of the third residue (Phe) (C=O···H–N, 2.1 Å). Other carbonyl and NH of amide form intermolecular hydrogen bonding and assemble into a supramolecular helical supramolecular structure.
Figure 4.

Conformational analyses of crystal 5h in solid state: (a) ORTEP diagram, (b) unit cell, (c) hydrogen bonding pattern, and (d) helical supramolecular assembly.
We also obtained the single crystal of control nontroponyl hybrid peptide (6a), β-benzylhydrazino acid containing peptide, and analyzed its structural conformation by X-ray studies (Figure 5). Other crystal data are provided in the Supporting Information. Their crystal data are submitted to CCDC with number CCDC 2003626 for peptide 6a. The ORTEP/unit cell packing diagram of 6a is shown in Figure 5a,b,respectively. It has circular packing rearrangement by self-assembly through hydrogen bonding, as shown in Figure 5c. There are two types of intermolecular hydrogen bonding: BocN–H···O=CBoc (2.1 Å) and amide N–H···O=C amide (2.0 Å) (Figure 5c). These intermolecular hydrogen bonds of peptide 6a form a unique ladder type of supramolecular helical structure, as shown in Figure 5e. Thus, nontroponyl β-hydrazino acid containing peptide is also a building block of a new peptidomimetics.
Figure 5.

Conformational analyses of control peptide crystal (6a) in solid state: (a) ORTEP diagram, (b) unit cell, (c) packing arrangement, (d) intermolecular hydrogen bonding, and (e) supramolecular self-assembled helical structure.
For peptide coupling at the N-terminal of hybrid peptides, we attempted to remove the Boc group of di-/tripeptides (5a–5h) with versatile reagent 20–30% TFA in dichloromethane (DCM) (Scheme 3). Unexpectedly, we isolated a new pyrazolidinone derivative as troponyl pyrazolidinone or troponylpyrazolidinone (9) from all respective hybrid peptides (5a–5h) under similar conditions by unusual cleavage of the amide bond. The structure of troponylpyrazolidinone (9) is confirmed by NMR and HRMS. Their spectra are provided in the Supporting Information. We also obtained the single crystal of troponylpyrazolidinone (9), which was analyzed by an X-ray diffractometer. Crystal details and unit cells are provided in the Supporting Information (Figure S72). The X-ray analysis result confirms the structure of troponylpyrazolidinone (9). The X-ray data are also submitted to CCDC with number 2003627. The ORTEP diagram of one dipeptide (5a) and its cleaved product troponylpyrazolidinone (9) is depicted in Figure 6. We also noticed two types of intramolecular hydrogen bonds: (i) N–H of pyrrolidinone with tropolone carbonyl (1.8 Å) and (ii) N–H of pyrazolidinone with its carbonyl (2.5 Å). We also examined the instability of such amide bonds under different acids such as HCl (4.0 N), HClO4 (4.0 N), PTSA (10 equiv), and AcOH (4 N) by the ESI-MS technique (see the Supporting Information, Figure S68). For peptide 5a, our mass analysis results reveal the cleavage of the amide bond of 5a with acids (HCl, HClO4, and PTSA) and the formation of the same cyclic derivative troponylpyrazolidinone (9). We could not notice the cleavage of the Boc group and amide bond cleavage with AcOH, which is a relatively weak organic acid. To examine the role of tropolone residue for the cleavage of such amide bonds, we performed control studies with similar types of non-troponyl-β-alkylhydrazino acid containing hybrid peptides (6a, 6b, 7, and 8) and TFA (∼20%), which were analyzed by ESI-MS and NMR techniques (Figures S49–S52). Except Boc group deprotection, we could not find the cleavage of amide bond in control peptides. However, the control peptide 8 also forms a trifluoroacylated salt derivative by acylation at the pyridine ring of picolamide residue. Hence, troponyl residue has a critical role for the cleavage of amide bonds containing β-troponylhydrazino acid (peptides 5a–5h).
Scheme 3. Reaction of β-Troponylhydrazino Peptides/Nontropoyl-β-hydrazino Peptides with TFA.

Figure 6.

Conformational analyses of troponylpyrazolidinone crystal (9): (a) ORTEP diagram, (b) unit cell packing, and (C) intramolecular hydrogen bonding.
The cleavage of the amide bond in peptides (5a–5h), under acidic pH, was dependent only on the concentration of peptides and thus considered as a first-order kinetic reaction. Thus, we performed kinetic studies of dipeptide (5a/5b) cleavage with the time-dependent 1H NMR experiment under acidic conditions (20% TFA in CDCl3). Their NMR spectra are provided in the Supporting Information (Figures S41–S48). After addition of TFA, 1H NMR spectra arrays of dipeptide 5a exhibit the significant downfield shift of α-/β-hydrogen resonance signals such as the resonance signal δ2.7 (α-H) shifted to δ3.0 and δ3.8 (β-H) shifted to δ4.2. Simultaneously, the new signals appeared at δ3.2 and δ4.4, which presumably belong to new cyclic derivative troponylpyrazolidinone (9). The NMR spectral arrays also show an exponential decrease in the intensity α-H signal (δ3.0) while an exponential increase in the intensity of new signals (δ3.2) with respect to time. After completion of NMR experiments, we recorded their mass spectra, which are provided in the Supporting Information (Figures S49–S52). Their mass spectra confirm the removal of the Boc group, followed by the formation of cyclic compound 9. Thus, the amide bond of a dipeptide (5a) was cleaved with TFA (20%). We repeated the similar NMR kinetic experiments with another representative peptide (5b) and obtained almost similar results. To determine the kinetic parameters (equilibrium constant and half-life of amide bond cleavage), we extracted mole fractions of reactant (5a) and its product (9) at different intervals of time from their respective NMR (see the Supporting Information, Tables S1 and S2). Then, we generated a kinetic plot (mole fraction vs time) for the cleavage amide bond (5a) and formation of a new cyclic product (9) (Figure 7A). We also obtained similar results with peptide (5b), and its kinetic plot is provided in the Supporting Information (Figure S56A). Our kinetic results clearly indicate that the cleavage of amide bond (5a/5b) and the formation of cyclic derivative are the first-order kinetic reactions. By following our previous report,16 we extracted the equilibrium constant (ka) as 0.016 and 0.009 min–1 for peptides 5a and 5b, respectively (Figures 7B and S56B). We also compared our experimental kinetic results with the simulated kinetic model (COPASI) using reported software (see Supporting Information, Figures S57–S58).16,24 Then, we calculated the half-life of amide bond cleavage (5a/5b) from their respective logarithmic plots (mole fraction vs time) plot by following the previous reports (see the Supporting Information). We obtained the half-life 41.0 and 71.0 min for the cleavage of respective amide bonds 5a and 5b. We performed a similar experiment with control peptide (6a), but we noticed that only N-Boc group was deprotected. Thus, only β-troponylhydrazino acid-containing peptides are cleavable under acidic conditions, and the rate of cleavage in 5a/5b also depends on the substituent of α-amino acid.
Figure 7.
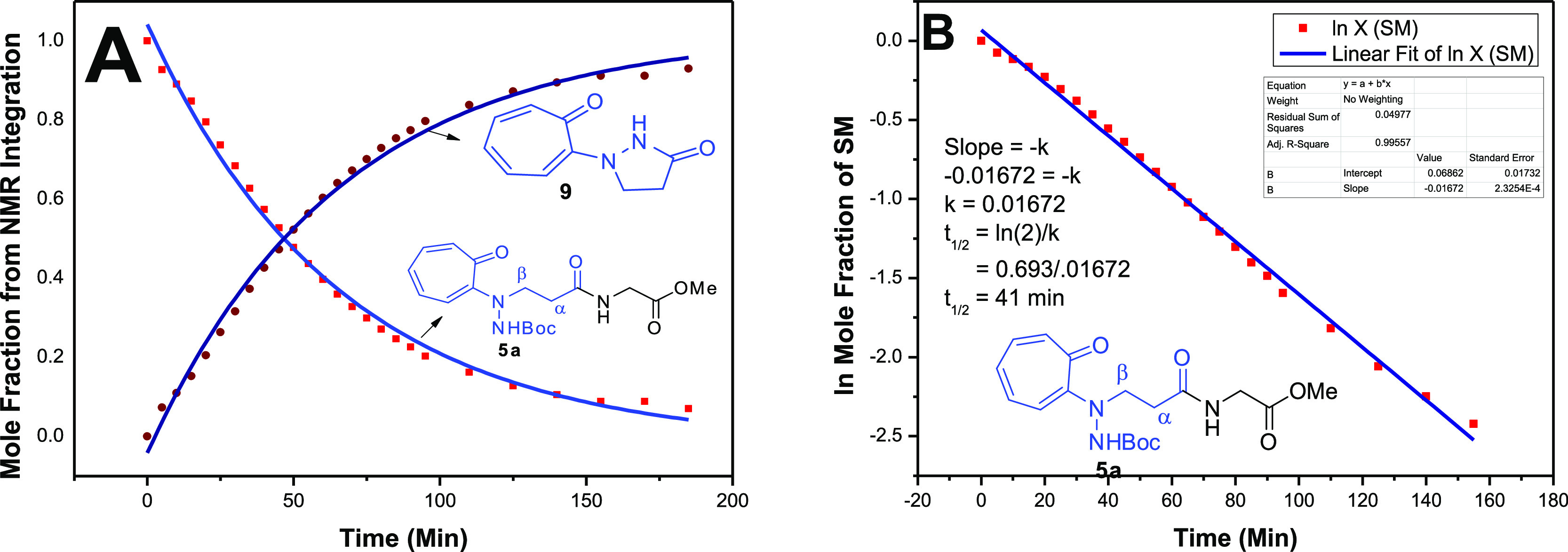
(a) Time-dependent 1H NMR kinetic plot mole fraction vs time (min) with hybrid peptide 5a (A) and its exponential plot for half-life calculation (B).
Because the troponyl derivative is a natural chromophore with characteristic absorption peaks,25 we thus planned to monitor the amide bond cleavage reaction of troponylated peptides (5a/5b) by UV–vis studies. We recorded the time-dependent UV–vis spectra of hybrid peptide 5a in acetonitrile (ACN) after the addition of 20% TFA (Figure 8). We noticed the significant changes in the UV spectra of peptide 5a after the addition of TFA, such as hypochromic (∼λ330nm)/hyperchromic shifts (∼λ400nm) with isosbestic point (∼λ355nm). Importantly, the electronic transition peak at ∼λ330nm was collapsed into one peak at ∼λ400nm after 100 min under acidic conditions that matched with the spectra of pure isolated troponylpyrazolidinone (9) and their isosbestic point at λ350nm (Supporting Information, Figure S64). We noticed similar results with dipeptide 5b from the time-dependent UV–vis studies and mass analyses under the same acidic conditions (Supporting Information, Figures S65/S66). Hence, troponylhydrazino hybrid peptides produced the same cyclic intermediate troponylpyrazolidinone (9) after the removal of N-Boc, followed by the cleavage of the amide bond under acidic conditions.
Figure 8.

Time-dependent UV spectra of dipeptide 6a under acidic conditions (20% TFA in ACN).
Finally, we propose the plausible mechanism for the cleavage of β-troponylhydrazino-containing amide bond (Figure 9). First, TFA (20%) removed the N-Boc group of the peptide (5) and produced the protonated hydrazinyl derivative (5-Boc)* under the acidic condition that facilitated the cleavage of an adjacent amide bond by the formation of a new cyclic molecule troponylpyrazolidinone (9). We assumed that the protonated hydrazinyl derivative (5-Boc)* activated its amide bond when the troponyl residue was present at the N-atom. Herein, the delocalization of cationic hydrazinyl proton possibly occurs through the troponyl ring as a tautomeric intermediate (T1 and T2), possessing a hydrazine amine (−NH2) nucleophile. This nucleophile is attacked at the protonated amide carbonyl group via the nucleophilic addition reaction and then a reactive cyclic-1,1-aminol intermediate (T3) is generated. Then, the protonation of the aminol amine group followed by elimination leads to the stable molecule N-troponylpyrazolidinone (9) and amine residue. In the case of control peptides, the delocalization of cationic hydrazinyl proton is not possible, and we could not notice the cleavage of amide bond or formation of troponylpyrazolidinone (9) under similar acidic conditions. Thus, tropolone played a crucial role in the cleavage of the amide bond in troponylated-β-hydrazinopeptide (5) under acidic conditions, possibly through the proposed mechanism.
Figure 9.

Proposed mechanism of β-troponylhydrazinyl peptide propenamide cleavage under mild acidic conditions.
Conclusions
β-Troponylhydrazino acid analogues and their hybrid peptides are synthesized from natural amino acid derivatives. Conformational analyses of β-hydrazino acid-containing peptides are demonstrated by extracting hydrogen bonding in representative peptides in solid state. The intramolecular hydrogen bonding of amide N–H has been shown in β-troponylhydrazino peptide by DMSO titration methods in solution state. X-ray studies reveal the role of the troponyl group in the self-assembled supramolecular structures in solid state. Most importantly, the troponyl-β-hydrazino acid-containing hybrid peptides show a unique feature as the cleavage of the amide bond through the formation of a new cyclic molecule troponylpyrazolidinone under mild acidic conditions. Time-dependent NMR studies determine the equilibrium constant and half-life of amide cleavage. The cleavage of the amide bond and formation of troponylpyrazolidinone are explained with the plausible mechanism. Hence, β-troponylhydrazino acid could be a promising chromophoric acid-sensitive protecting group of free amines. It could also be useful to estimate the free amine group by a UV–vis spectrophotometer.
Experimental Section
Materials
All required materials were obtained from commercial suppliers and used without any further purification. Dimethylformamide (DMF) was distilled over calcium hydride. Reactions were monitored by thin-layer chromatography (TLC) and visualized by UV and ninhydrin. Column chromatography was performed in 230–400 mesh silica. Mass spectra and HRMS were obtained using a Bruker micrOTOF-Q II spectrometer. 1H NMR and 13C NMR spectra were recorded on Bruker AV-400 or 700 MHz at 298 K. 1H and 13C NMR chemical shifts were recorded in ppm downfield from tetramethylsilane or residual solvent peak. Splitting patterns are abbreviated as s, singlet; d, doublet; dd, doublet of doublet; t, triplet; q, quartet; dq, doublet of quartet; and m, multiplet.
General Procedure for the Hydrolysis of Ester into Acid
Compounds having ester group (4), (2-Bn), (2-Hexyl), and (2-Picolamide) were dissolved in tetrahydrofuran containing 2 equiv of LiOH at 0 °C and then brought to RT with stirring. TLC monitored the completion of those ester hydrolysis reactions. The hydrolyses of N-alylated-β-hydrazino esters were completed within 3.0 h. The solvents are evaporated under vacuum to half of their volume and the pH was adjusted to 6–7 with 1 M HCl and then extracted thrice with EtOAc. The organic layers were combined, dried over Na2SO4, and concentrated under low pressure to afford the acid derivative products. Without any further characterization, we proceed for the next step (for amide coupling). These N-alkylated-β-hydrazino acids were coupled with natural α-amino acids in the presence of amide coupling reagents as HOAT (1.3 equiv), N-methyl morpholine (3 equiv) was EDC·HCl (1.3 equiv), and was dissolved in dry DMF (1.5 M). After stirring for 10 min, cooled it to 0 °C and added.
General Procedure for Peptide Synthesis
After hydrolysis of the ester group into the acid group, it is directly used for the peptide coupling reaction without any further purifications, where the corresponding amines (1.2 equiv) and HOAT (1.3 equiv) were dissolved in dry DMF (1.5 M). After stirring for 10 min, N-methyl morpholine (3 equiv) was added dropwise and cooled to 0 °C, and EDC·HCl was added (1.3 equiv). After 20 min, it was allowed to cool to RT, followed by heating at 55 °C for 14 h. The crude reaction mixture was evaporated under reduced pressure. The resultant crude was purified by column chromatography with MeOH in CH2Cl2 (1–3%). The obtained product was characterized using 1H/13C by NMR and HRMS by ESI-MS techniques. The characterization data of all synthesized hybrid peptides are provided in the following sections.
General Procedure for Boc Deprotection
TFA (30%) in CH2Cl2 (5 mL) was added to compound (5a) at RT and stirred for 3 h. The solvents were removed under vacuum, resulting in a red color residue (only in the case of troponyl derivative). The residual viscous oil was purified via chromatography with 3% MeOH in CH2Cl2. The obtained product was characterized using 1H/13C by NMR and HRMS by ESI-MS techniques.
tert-Butyl-2-(3-ethoxy-3-oxopropyl)hydrazine-1-carboxylate (2)
Experimental procedure and its characterization data for compounds (2) are previously reported.26 A solution of 3-bromo-propionic acid ethyl ester (2 mL, 15.7 mmol), DIPEA (2.6 mL, 15.7 mmol), and bochydrazine (1) (3.1 g, 23.55 mmol) in toluene was heated at 80 °C for 4 days. After removal of the solvent under reduced pressure, the crude product was purified by column chromatography (hexane/EtOAc 6:4) to give compound (2), which is a slightly yellow oil (1.58 g, 43%).
tert-Butyl-2-(3-ethoxy-3-oxopropyl)-2-(7-oxocyclohepta-1,3,5-trien-1-yl)hydrazine-1-carboxylate (4)
The Boc-protected β-hydrazino acid derivative 2 (4.0 g, 17.24 mmol) was dissolved in ethanol containing tetraethylammonium (7.20 mL, 51.72 mmol) and stirred at RT, followed by addition of 2-tosyloxy tropolone derivative 3 (9.5 g, 34.48 mmol). This reaction mixture was allowed to reflux for 4 days until the disappearance of the starting material (2). The reaction was monitored by TLC with 40% ethyl acetate in hexane. After completion of the reaction, the reaction mixture was concentrated under low pressure. The concentrated crude product was purified by silica gel column chromatography (230–400 mesh) in 20% ethyl acetate in hexane as the mobile phase. The purified product was obtained as a yellow color solid (2.0 g, 35% yield) and then characterized by NMR (1H/13C) and ESI-MS techniques. 1H NMR (400 MHz, deuterated solvent CDCl3): δ 7.58 (s, 1H), 7.20–6.95 (m, 4H), 6.84–6.74 (m, 1H), 4.15 (q, J = 7.1 Hz, 2H), 3.89 (t, J = 6.7 Hz, 2H), 2.76 (t, J = 6.8 Hz, 2H), 1.41 (s, 9H), 1.26 (t, J = 7.1 Hz, 3H). 13C NMR (176 MHz, CDCl3): δ 181.63, 172.14, 157.26, 155.27, 136.36, 135.78, 133.60, 127.81, 81.35, 60.66, 49.62, 32.76, 28.15, 14.13. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C17H24N2O5, 359.1572; found, 359.1577.
tert-Butyl-2-(3-((2-methoxy-2-oxoethyl)amino)-3-oxopropyl)-2-(7-oxocyclohepta-1,3,5-trien-1-yl)hydrazine-1-carboxylate (5a)
The dipeptide was synthesized by following the general procedure. The pure product (155 mg, 63% yield) was obtained as a yellow color solid. 1H NMR (400 MHz, deuterated solvent CDCl3): δ 7.97 (s, 1H), 7.81 (s, 1H), 7.28 (s, 1H), 7.23–7.03 (m, 3H), 6.86 (t, 1H), 4.04 (d, J = 5.0 Hz, 2H), 3.83 (t, J = 5.1 Hz, 2H), 3.73 (s, 3H), 2.65 (t, J = 5.9 Hz, 2H), 2.09 (s, 1H), 1.44 (s, 9H). 13C NMR (101 MHz, CDCl3): δ 182.44, 172.17, 170.74, 157.26, 155.58, 137.24, 136.27, 134.00, 129.44, 81.47, 52.23, 50.78, 41.22, 34.62, 28.23. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C18H25N3O6, 380.1816; found, 380.1817.
tert-Butyl-(S)-2-(3-((1-methoxy-1-oxopropan-2-yl)amino)-3-oxopropyl)-2-(7-oxocyclohepta-1,3,5-trien-1-yl)hydrazine-1-carboxylate (5b)
The dipeptide was synthesized by following the general procedure. Pure product (110 mg, 58% yield) was obtained as a yellow color solid. 1H NMR (700 MHz, deuterated solvent CDCl3): δ 7.77 (s, 2H), 7.26 (s, 1H), 7.21–7.16 (m, 1H), 7.13–7.05 (m, 2H), 6.86 (t, J = 9.3 Hz, 1H), 4.58–4.50 (m, 1H), 3.97–3.89 (m, 1H), 3.76 (s, 1H), 3.73 (s, 3H), 2.71–2.53 (m, 2H), 1.44 (s, 9H), 1.40 (d, J = 7.3 Hz, 3H). 13C NMR (176 MHz, CDCl3): δ 182.34, 173.82, 171.42, 157.26, 155.56, 137.09, 136.21, 133.95, 129.26, 81.49, 58.38, 52.35, 48.24, 34.63, 28.22, 17.53. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C19H27N3O6, 416.1792; found, 416.1828.
tert-Butyl-(S)-2-(3-((1-methoxy-4-methyl-1-oxopentan-2-yl)amino)-3-oxopropyl)-2-(7-oxocyclohepta-1,3,5-trien-1-yl)hydrazine-1-carboxylate (5c)
The dipeptide was synthesized by following the general procedure. The pure product (135 mg, 48% yield) was obtained as a yellow viscous liquid. 1H NMR (400 MHz, deuterated solvent CDCl3): δ 7.80 (s, 2H), 7.34 (s, 1H), 7.24–7.00 (m, 3H), 6.86 (t, 1H), 4.54 (q, J = 7.7 Hz, 1H), 3.97–3.83 (m, 1H), 3.71 (s, 4H), 2.75–2.48 (m, 2H), 1.76–1.64 (m, 1H), 1.63–1.57 (m, 2H), 1.44 (s, 9H), 0.91 (dd, J = 6.4, 4.2 Hz, 6H). 13C NMR (101 MHz, CDCl3): δ 182.52, 173.92, 171.82, 157.24, 155.68, 137.38, 136.22, 134.04, 129.59, 81.38, 52.17, 51.14, 40.61, 36.66, 34.70, 28.21, 24.83, 22.88, 21.63. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C22H33N3O6, 436.2442; found, 436.2451.
tert-Butyl-(S)-2-(3-((1-methoxy-3-methyl-1-oxobutan-2-yl)amino)-3-oxopropyl)-2-(7-oxocyclohepta-1,3,5-trien-1-yl)hydrazine-1-carboxylate (5d)
The dipeptide was synthesized by following the general procedure. The pure product (112 mg, 54% yield) was obtained as a yellow viscous liquid. 1H NMR (400 MHz, deuterated solvent CDCl3) δ 7.82 (s, 1H), 7.56 (s, 1H), 7.26 (s, 1H), 7.21–7.02 (m, 3H), 6.83 (t, 1H), 4.44 (t, 1H), 4.01–3.87 (m, 1H), 3.71 (s, 4H), 2.75–2.51 (m, 2H), 2.23–2.08 (m, 1H), 1.43 (s, 9H), 0.92 (dd, J = 6.6, 3.2 Hz, 6H). 13C NMR (101 MHz, CDCl3): δ 182.27, 172.79, 171.94, 157.31, 155.67, 137.06, 136.10, 133.95, 129.10, 81.39, 57.91, 52.01, 51.50, 34.91, 30.56, 28.21, 18.98, 18.00. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C21H31N3O6, 444.2105; found, 444.2147.
tert-Butyl-2-(3-(((2S,3R)-1-methoxy-3-methyl-1-oxopentan-2-yl)amino)-3-oxopropyl)-2-(7-oxocyclohepta-1,3,5-trien-1-yl)hydrazine-1-carboxylate (5e)
The dipeptide was synthesized by following the general procedure. The pure product (112 mg, 48% yield) was obtained as a yellow viscous liquid. 1H NMR (400 MHz, deuterated solvent CDCl3): δ 7.83 (s, 1H), 7.57 (s, 1H), 7.24 (s, 1H), 7.20–6.99 (m, 3H), 6.83 (t, J = 9.1 Hz, 1H), 4.47 (t, J = 6.4 Hz, 1H), 4.00–3.86 (m, 1H), 3.70 (s, 4H), 2.77–2.47 (m, 2H), 1.86 (s, 1H), 1.43 (s, 9H), 1.30–1.13 (m, 2H), 0.87 (t, J = 7.8 Hz, 6H). 13C NMR (101 MHz, CDCl3): δ 182.21, 172.80, 171.81, 157.29, 155.67, 137.00, 136.08, 133.95, 129.02, 81.39, 57.11, 51.95, 51.47, 37.22, 34.90, 28.20, 25.29, 15.48, 11.54. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C22H33N3O6, 458.2262; found, 458.2295.
tert-Butyl-(S)-2-(3-((1-methoxy-1-oxo-3-phenylpropan-2-yl)amino)-3-oxopropyl)-2-(7-oxocyclohepta-1,3,5-trien-1-yl)hydrazine-1-carboxylate (5f)
The dipeptide was synthesized by following the general procedure. The pure product (160 mg, 50% yield) was obtained as a yellow viscous liquid. 1H NMR (400 MHz, deuterated solvent CDCl3): δ 7.73 (s, 1H), 7.60 (s, 1H), 7.27–7.13 (m, 7H), 7.11–7.03 (m, 2H), 6.86 (t, 1H), 4.84 (q, J = 7.8 Hz, 1H), 3.78 (s, 2H), 3.71 (s, 3H), 3.16 (dd, J = 14.0, 5.5 Hz, 1H), 3.04 (dd, J = 14.0, 8.1 Hz, 1H), 2.66–2.47 (m, 2H), 1.44 (s, 9H). 13C NMR (101 MHz, CDCl3): δ 182.24, 172.48, 171.55, 157.15, 155.41, 137.14, 136.52, 136.18, 133.95, 129.16, 128.44, 126.87, 81.41, 53.69, 52.27, 50.42, 37.48, 34.53, 28.23. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C25H31N3O6, 470.2286; found, 470.2291.
Methyl-(3-(2-(tert-butoxycarbonyl)-1-(7-oxocyclohepta-1,3,5-trien-1-yl)hydrazineyl)propanoyl)-l-prolinate (5g)
The dipeptide was synthesized by following the general procedure. The pure product (96 mg, 43% yield) was obtained as a yellow viscous liquid. 1H NMR (400 MHz, deuterated solvent CDCl3): δ 7.92 (s, 1H), 7.18–6.99 (m, 4H), 6.75 (t, J = 9.0 Hz, 1H), 4.53–4.45 (m, 1H), 4.03 (s, 2H), 3.75 (s, 3H), 3.69–3.63 (m, 1H), 3.59–3.51 (m, 1H), 2.98–2.84 (m, 1H), 2.77–2.65 (m, 1H), 2.25–2.13 (m, 1H), 2.07–1.95 (m, 3H), 1.45 (s, 9H). 13C NMR (101 MHz, CDCl3): δ 181.09, 173.02, 170.67, 157.40, 155.09, 135.70, 133.96, 126.86, 117.90, 81.24, 58.64, 52.32, 50.68, 47.30, 33.27, 29.24, 28.23, 24.72. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C21H29N3O6, 442.1949; found, 442.1984.
tert-Butyl-2-(3-(((S)-1-(((R)-1-methoxy-1-oxo-3-phenylpropan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)amino)-3-oxopropyl)-2-(7-oxocyclohepta-1,3,5-trien-1-yl)hydrazine-1-carboxylate (5h)
The tripeptide was synthesized by following the general procedure. The pure product (320 mg, 42% yield) was obtained as a yellow color solid. 1H NMR (400 MHz, deuterated solvent CDCl3): δ 8.41 (s, 1H), 7.93 (s, 1H), 7.26–7.05 (m, 8H), 7.02 (d, J = 6.7 Hz, 2H), 6.94–6.82 (m, 1H), 4.78 (q, J = 7.4 Hz, 1H), 4.49–4.38 (m, 1H), 3.98 (s, 1H), 3.67 (s, 3H), 3.58 (d, J = 12.3 Hz, 1H), 3.09 (dd, J = 13.8, 5.6 Hz, 1H), 2.91 (dd, J = 13.8, 7.4 Hz, 1H), 2.69–2.54 (m, 2H), 1.78–1.65 (m, 1H), 1.64–1.52 (m, 2H), 1.46 (s, 9H), 0.89 (dd, J = 16.4, 6.4 Hz, 6H). 13C NMR (101 MHz, CDCl3): δ 181.86, 172.49, 172.17, 171.37, 157.55, 155.16, 137.20, 136.31, 136.23, 133.96, 129.18, 128.21, 126.74, 81.42, 53.17, 52.22, 48.73, 40.30, 37.57, 34.29, 28.22, 24.77, 23.02, 21.63. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C31H42N4O7, 605.2918; found, 605.2946.
1-(7-Oxocyclohepta-1,3,5-trien-1-yl)pyrazolidin-3-one (9)
Troponylpyrazolidinone was synthesized by following the general procedure. The product was purified by column chromatography with MeOH in CH2Cl2 (2%). The pure product (18 mg, 94%) was obtained as a red color solid and characterized by NMR (1H/13C) and MS. 1H NMR (400 MHz, deuterated solvent CDCl3): δ 12.74 (s, 1H), 7.21–7.06 (m, 2H), 6.94 (d, J = 11.6 Hz, 1H), 6.64 (t, J = 9.3 Hz, 1H), 6.36 (d, J = 10.8 Hz, 1H), 3.99 (t, J = 8.6 Hz, 2H), 2.80 (t, J = 8.6 Hz, 2H). 13C NMR (101 MHz, CDCl3): δ 178.12, 169.05, 150.01, 137.76, 135.62, 132.02, 124.59, 113.50, 47.68, 29.16. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C10H10N2O2, 191.0815; found, 191.0815.
tert-Butyl-2-benzyl-2-(3-ethoxy-3-oxopropyl)hydrazine-1-carboxylate (2-Bn)
The Boc-protected β-hydrazino acid derivative 2 (4.0 g, 17.24 mmol) was dissolved in ACN and K2CO3 (4.75 g, 34.48 mmol) was added to it and stirred at RT, followed by addition of benzyl bromide (3.07 mL, 25.86 mmol). This reaction mixture was allowed to stir at RT for 24 h until the disappearance of the starting material (2). The reaction was monitored by TLC with 40% ethyl acetate in hexane. After completion of the reaction, the reaction mixture was filtrated and concentrated under reduced pressure to obtain the crude product. The concentrated crude product was purified by silica gel column chromatography (230–400 mesh) in 10–20% ethyl acetate in hexane as the mobile phase. The desired product (2.8 g, 50%) was obtained as a white color solid and characterized by NMR (1H/13C) and MS methods. 1H NMR (400 MHz, deuterated solvent CDCl3): δ 7.31 (d, J = 4.3 Hz, 4H), 7.27 (d, J = 3.8 Hz, 1H), 5.57 (s, 1H), 4.13 (q, J = 7.1 Hz, 2H), 4.01 (s, 2H), 3.13 (s, 2H), 2.54 (t, J = 6.9 Hz, 2H), 1.39 (s, 9H), 1.24 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 172.40, 154.97, 136.81, 129.35, 128.23, 127.41, 79.95, 60.36, 51.56, 33.14, 28.27, 14.17. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C17H26N2O4, 345.1785; found, 345.1779.
tert-Butyl-(S)-2-benzyl-2-(3-((1-methoxy-1-oxo-3-phenylpropan-2-yl)amino)-3-oxopropyl)hydrazine-1-carboxylate (6a)
The dipeptide was synthesized by following the general procedure. The pure product (150 mg, 50% yield) was obtained as a white color solid. 1H NMR (400 MHz, deuterated solvent CDCl3): δ 8.33 (s, 1H), 7.35–7.18 (m, 10H), 5.43 (s, 1H), 4.86 (q, J = 7.4 Hz, 1H), 3.98 (d, J = 12.5 Hz, 1H), 3.85 (d, J = 12.8 Hz, 1H), 3.72 (s, 3H), 3.24 (dd, J = 13.8, 5.6 Hz, 1H), 3.13 (dd, J = 13.9, 7.9 Hz, 1H), 3.09–2.99 (m, 1H), 2.99–2.90 (m, 1H), 2.45–2.28 (m, 2H), 1.38 (s, 9H). 13C NMR (101 MHz, CDCl3): δ 172.43, 172.17, 155.60, 136.98, 135.53, 129.67, 129.34, 128.36, 127.71, 126.85, 80.37, 61.73, 53.73, 53.23, 52.16, 37.66, 33.77, 28.22. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C25H33N3O5, 456.2493; found, 456.2501.
tert-Butyl-2-benzyl-2-(3-(((S)-1-(((R)-1-methoxy-1-oxo-3-phenylpropan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)amino)-3-oxopropyl)hydrazine-1-carboxylate (6b)
The tripeptide was synthesized by following the general procedure. The pure product (400 mg, 55% yield) was obtained as a white color solid crystalline. 1H NMR (400 MHz, deuterated solvent CDCl3): δ 8.45 (s, 1H), 7.33 (q, J = 9.2, 7.9 Hz, 3H), 7.28–7.18 (m, 5H), 7.15 (d, J = 6.7 Hz, 3H), 5.40 (d, J = 10.0 Hz, 1H), 4.84 (q, J = 7.5 Hz, 1H), 4.48–4.38 (m, 1H), 3.91–3.75 (m, 2H), 3.66 (s, 3H), 3.15 (dd, J = 13.8, 5.6 Hz, 1H), 3.01 (dd, J = 13.8, 7.5 Hz, 1H), 2.96–2.82 (m, 2H), 2.36 (t, J = 5.5 Hz, 2H), 2.00–1.81 (m, 1H), 1.71–1.59 (m, 2H), 1.36 (s, 9H), 0.91 (dd, J = 9.9, 6.1 Hz, 6H). 13C NMR (101 MHz, CDCl3): δ 172.48, 172.30, 172.03, 156.22, 136.50, 134.54, 129.61, 129.44, 128.54, 128.37, 127.96, 126.80, 80.81, 62.66, 54.08, 53.19, 52.35, 52.17, 40.53, 38.02, 33.88, 28.15, 24.84, 23.10, 21.57. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C31H44N4O6, 569.3322; found, 569.3334.
tert-Butyl 2-(3-Ethoxypropyl)-2-hexylhydrazine-1-carboxylate (2-Hexyl)
The Boc-protected β-hydrazino acid derivative 2 (4.0 g, 17.24 mmol) was dissolved in ACN and K2CO3 (4.75 g, 34.48 mmol) was added to it and stirred at RT, followed by addition of 1-bromohexane (3.6 mL, 25.86 mmol). This reaction mixture was allowed to stir at 55 °C for 48 h until the disappearance of the starting material (2). The reaction was monitored by TLC with 40% ethyl acetate in hexane. After completion of the reaction, the reaction mixture was filtrated and concentrated under reduced pressure to obtain the crude product. The concentrated crude product was purified by silica gel column chromatography (230–400 mesh) in 10–20% ethyl acetate in hexane as the mobile phase. The desired product 2.1 g (40%) was obtained as a slightly yellow color liquid and characterized by NMR (1H/13C) and MS methods. 1H NMR (400 MHz, deuterated solvent CDCl3): δ 5.41 (s, 1H), 4.10 (q, J = 7.1 Hz, 2H), 2.98 (s, 2H), 2.67 (s, 2H), 2.50 (t, J = 6.5 Hz, 2H), 1.41 (s, 11H), 1.32–1.18 (m, 9H), 0.85 (t, J = 6.5 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 172.56, 155.06, 79.68, 60.37, 58.20, 32.74, 31.69, 28.28, 26.73, 22.54, 14.15, 14.00. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C16H32N2O4, 339.2254, found 339.2245.
tert-Butyl-(R)-2-hexyl-2-(3-((1-methoxy-1-oxo-3-phenylpropan-2-yl)amino)-3-oxopropyl)hydrazine-1-carboxylate (7)
The dipeptide was synthesized by following the general procedure. The pure product (150 mg, 45% yield) was obtained as a white viscous liquid. 1H NMR (400 MHz, deuterated solvent CDCl3): δ 8.45–8.30 (m, 1H), 7.34–7.16 (m, 5H), 5.35 (s, 1H), 4.81 (q, J = 7.8 Hz, 1H), 3.70 (s, 3H), 3.15 (dd, 2H), 2.98–2.76 (m, 2H), 2.74–2.53 (m, 2H), 2.40–2.24 (m, 2H), 1.47 (s, 9H), 1.34–1.23 (m, 8H), 0.90 (t, J = 6.7 Hz, 3H). 13C NMR (101 MHz, CDCl3): δ 172.35, 172.26, 155.83, 137.14, 129.30, 128.58, 128.29, 126.70, 80.28, 58.33, 54.41, 53.74, 52.04, 37.56, 33.73, 31.79, 29.69, 28.30, 26.82, 26.67, 22.58, 14.06. HRMS (ESI-TOF) m/z: [M + Na]+ calcd for C24H39N3O5, 472.2782; found, 472.2783.
tert-Butyl 2-(3-Ethoxy-3-oxopropyl)-2-picolinoylhydrazine-1-carboxylate (2-Picolamide)
The picolinoyl derivative peptide ester was synthesized by following the general procedure. The pure product (1.6 g, 55% yield) was obtained as a yellow viscous liquid. 1H NMR (400 MHz, deuterated solvent CDCl3): δ 8.54 (s, 1H), 7.90–7.69 (m, 3H), 7.41–7.28 (m, 1H), 4.16 (q, J = 7.0 Hz, 2H), 4.02 (t, J = 6.4 Hz, 2H), 2.77 (t, J = 6.7 Hz, 2H), 1.31–1.17 (m, 12H). 13C NMR (101 MHz, CDCl3): δ 171.87, 169.76, 153.00, 147.74, 136.91, 124.81, 124.02, 81.43, 60.65, 45.30, 32.14, 27.84, 14.14. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C16H23N3O5, 338.1706; found, 338.1710.
tert-Butyl-(S)-2-(3-((1-methoxy-1-oxo-3-phenylpropan-2-yl)amino)-3-oxopropyl)-2-picolinoylhydrazine-1-carboxylate (8)
The tripeptide was synthesized by following the general procedure. The pure product (120 mg, 40% yield) was obtained as a white viscous liquid. 1H NMR (400 MHz, deuterated solvent CDCl3): δ 8.54 (d, J = 3.7 Hz, 1H), 7.82–7.75 (m, 1H), 7.70 (d, J = 7.6 Hz, 1H), 7.42–7.33 (m, 1H), 7.30 (d, J = 7.0 Hz, 1H), 7.28–7.21 (m, 2H), 7.17 (d, J = 7.0 Hz, 2H), 6.66 (s, 1H), 4.88 (q, J = 6.6 Hz, 1H), 4.07 (s, 1H), 3.91 (d, J = 12.9 Hz, 1H), 3.72 (s, 3H), 3.18–3.08 (m, 2H), 2.69–2.58 (m, 2H), 1.25 (s, 9H). 13C NMR (101 MHz, CDCl3): δ 172.24, 170.83, 169.97, 147.93, 136.88, 136.04, 129.21, 128.61, 127.09, 124.76, 123.65, 81.54, 53.44, 52.31, 46.50, 37.80, 34.23, 27.90. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C24H30N4O6, 471.2220; found, 471.2238.
General Procedure for the Synthesis of N-Troponylpyrazolidinone (9)
The Boc-protected troponyl peptides (∼100 mmol), containing β-troponyl hydrazino acid, were added to 20–30% TFA in DCM (∼5.0 mL) and stirred for 2–3 h at RT. The reaction was monitored by TLC before characterization by ESI-MS. The reaction mixture was purified through the silica column chromatography technique. The yield of the isolated cyclic product was ∼94%.
Acknowledgments
N.R.D. thanks Dr Amarnath Bollu for analyzing the NMR kinetic data. B.B.P. thanks UGC-New Delhi for JRF/SRF Fellowship. N.K.S. thanks CSIR-HRDG-New Delhi, grant number 02(0166)/13/EMR, for financial support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c03729.
NMR (1H/13C) and ESI-HRMS spectra of new intermediates and β-hydrazine acids/peptides and time-dependent NMR in TFA (PDF)
Crystallographic data for compound 5a* (tert-butyl ester) (CIF)
Crystallographic data for compound 5h (CIF)
Crystallographic data for compound 6a (CIF)
Crystallographic data for compound 9 (CIF)
Author Contributions
§ N.R.D., S.M., and B.B.P. authors contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Barrett A.; Woessner J.; Rawlings N. D.. Enzymes can of course efficiently orchestrate amide cleavage under physiological conditions. Handbook of Proteolytic Enzymes, 2nd ed.; Academic Press: San Diego, 2004; pp 1–30. [Google Scholar]
- Radzicka A.; Wolfenden R. Rates of uncatalyzed peptide bond hydrolysis in neutral solution and the transition state affinities of proteases. J. Am. Chem. Soc. 1996, 118, 6105–6109. 10.1021/ja954077c. [DOI] [Google Scholar]
- Bennet A. J.; Somayaji V.; Brown R. S.; Santarsiero B. D. The influence of altered amidic resonance on the infrared and carbon-13 and nitrogen-15 NMR spectroscopic characteristics and barriers to rotation about the NC (O) bond in some anilides and toluamides. J. Am. Chem. Soc. 1991, 113, 7563–7571. 10.1021/ja00020a017. [DOI] [Google Scholar]
- Lide D. R.CRC Handbook of Chemistry and Physics, 3rd ed.; CRC, Boca Raton, Florida, 2000; pp 3–84. [Google Scholar]
- Clayden J. Organic chemistry: Stabilizers cause instability. Nature 2012, 481, 274–275. 10.1038/481274a. [DOI] [PubMed] [Google Scholar]
- Clayden J.; Moran W. J. The Twisted Amide 2-Quinuclidone: 60 Years in the Making. Angew. Chem., Int. Ed. Engl. 2006, 45, 7118–7120. 10.1002/anie.200603016. [DOI] [PubMed] [Google Scholar]
- Szostak M.; Aubé J. Medium-bridged lactams: a new class of non-planar amides. Org. Biomol. Chem. 2011, 9, 27–35. 10.1039/c0ob00215a. [DOI] [PubMed] [Google Scholar]
- Tani K.; Stoltz B. M. Synthesis and structural analysis of 2-quinuclidonium tetrafluoroborate. Nature 2006, 441, 731–734. 10.1038/nature04842. [DOI] [PubMed] [Google Scholar]
- Somayaji V.; Brown R. S. Distorted amides as models for activated peptide NC: O units produced during enzyme-catalyzed acyl transfer reactions. 1. The mechanism of hydrolysis of 3, 4-dihydro-2-oxo-1, 4-ethanoquinoline and 2, 3, 4, 5-tetrahydro-2-oxo-1, 5-ethanobenzazepine. J. Org. Chem. 1986, 51, 2676–2686. 10.1021/jo00364a012. [DOI] [Google Scholar]
- Komarov I. V.; Yanik S.; Ishchenko A. Y.; Davies J. E.; Goodman J. M.; Kirby A. J. The most reactive amide as a transition-state mimic For cis-trans interconversion. J. Am. Chem. Soc. 2015, 137, 926–930. 10.1021/ja511460a. [DOI] [PubMed] [Google Scholar]
- Balachandra C.; Sharma N. K. Novel fluorophores: Syntheses and photophysical studies of boron-aminotroponimines. Dyes Pigm. 2017, 137, 532–538. 10.1016/j.dyepig.2016.10.051. [DOI] [Google Scholar]
- Kita Y.; Nishii Y.; Onoue A.; Mashima K. Combined catalytic system of scandium triflate and boronic ester for amide bond cleavage. Adv. Synth. Catal. 2013, 355, 3391–3395. 10.1002/adsc.201300819. [DOI] [Google Scholar]
- Hie L.; Fine Nathel N. F.; Shah T. K.; Baker E. L.; Hong X.; Yang Y.-F.; Liu P.; Houk K. N.; Garg N. K. Conversion of amides to esters by the nickel-catalysed activation of amide C–N bonds. Nature 2015, 524, 79. 10.1038/nature14615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchby M.; Houlden C. E.; Haddow M. F.; Tyler S. N. G.; Lloyd-Jones G. C.; Booker-Milburn K. I. Switching pathways: room-temperature neutral solvolysis and substitution of amides. Angew. Chem., Int. Ed. Engl. 2012, 51, 548–551. 10.1002/anie.201107117. [DOI] [PubMed] [Google Scholar]
- Shimizu Y.; Noshita M.; Mukai Y.; Morimoto H.; Ohshima T. Cleavage of unactivated amide bonds by ammonium salt-accelerated hydrazinolysis. Chem. Commun. 2014, 50, 12623–12625. 10.1039/c4cc02014f. [DOI] [PubMed] [Google Scholar]
- Bollu A.; Sharma N. K. Cleavable Amide Bond: Mechanistic Insight into Cleavable 4-Aminopyrazolyloxy Acetamide at Low pH. J. Org. Chem. 2019, 84, 5596–5602. 10.1021/acs.joc.9b00535. [DOI] [PubMed] [Google Scholar]
- Stephenson N. A.; Zhu J.; Gellman S. H.; Stahl S. S. Catalytic transamidation reactions compatible with tertiary amide metathesis under ambient conditions. J. Am. Chem. Soc. 2009, 131, 10003–10008. 10.1021/ja8094262. [DOI] [PubMed] [Google Scholar]
- Avan I.; Hall C. D.; Katritzky A. R. Peptidomimetics via modifications of amino acids and peptide bonds. Chem. Soc. Rev. 2014, 43, 3575–3594. 10.1039/c3cs60384a. [DOI] [PubMed] [Google Scholar]
- Balachandra C.; Sharma N. K. Synthesis and conformational analysis of new troponyl aromatic amino acid. Tetrahedron 2014, 70, 7464–7469. 10.1016/j.tet.2014.08.014. [DOI] [Google Scholar]
- Balachandra C.; Sharma N. K. Instability of Amide Bond Comprising the 2-Aminotropone Moiety: Cleavable under Mild Acidic Conditions. Org. Lett. 2015, 17, 3948–3951. 10.1021/acs.orglett.5b01535. [DOI] [PubMed] [Google Scholar]
- Balachandra C.; Sharma N. K. Direct/Reversible Amidation of Troponyl Alkylglycinates via Cationic Troponyl Lactones and Mechanistic Insights. ACS Omega 2018, 3, 997–1013. 10.1021/acsomega.7b01540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang C. W.; Sarnowski M. P.; Elbatrawi Y. M.; Del Valle J. R. Access to enantiopure α-hydrazino acids for N-amino peptide synthesis. J. Org. Chem. 2017, 82, 1833–1841. 10.1021/acs.joc.6b02718. [DOI] [PubMed] [Google Scholar]
- Li X.; Yang D. Peptides of aminoxy acids as foldamers. Chem. Commun. 2006, 3367–3379. 10.1039/b602230h. [DOI] [PubMed] [Google Scholar]
- Kent E.; Hoops S.; Mendes P. Condor-COPASI: high-throughput computing for biochemical networks. BMC Syst. Biol. 2012, 6, 91. 10.1186/1752-0509-6-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breheret E. F.; Martin M. M. Electronic relaxation of troponoids: tropolone fluorescence. J. Lumin. 1978, 17, 49–60. 10.1016/0022-2313(78)90025-x. [DOI] [Google Scholar]
- Bordessa A.; Keita M.; Maréchal X.; Formicola L.; Lagarde N.; Rodrigo J.; Bernadat G.; Bauvais C.; Soulier J.-L.; Dufau L.; Milcentau T.; Crousse B.; Reboud-Ravaux M.; Ongeri S. α-and β-hydrazino acid-based pseudopeptides inhibit the chymotrypsin-like activity of the eukaryotic 20S proteasome. Eur. J. Med. Chem. 2013, 70, 505–524. 10.1016/j.ejmech.2013.09.059. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.