

Abstract
Human dihydroorotate dehydrogenase (DHODH) is a viable target for the development of therapeutics to treat cancer and immunological diseases, such as rheumatoid arthritis (RA), psoriasis and multiple sclerosis (MS). Herein, a series of acrylamide-based novel DHODH inhibitors as potential RA treatment agents were designed and synthesized. 2-Acrylamidobenzoic acid analog 11 was identified as the lead compound for structure–activity relationship (SAR) studies. The replacement of the phenyl group with naphthyl moieties improved inhibitory activity significantly to double-digit nanomolar range. Further structure optimization revealed that an acrylamide with small hydrophobic groups (Me, Cl or Br) at the 2-position was preferred. Moreover, adding a fluoro atom at the 5-position of the benzoic acid enhanced the potency. The optimization efforts led to potent compounds 42 and 53‒55 with IC50 values of 41, 44, 32, and 42 nmol/L, respectively. The most potent compound 54 also displayed favorable pharmacokinetic (PK) profiles and encouraging in vivo anti-arthritic effects in a dose-dependent manner.
KEY WORDS: DHODH, De novo pyrimidine biosynthesis, DHODH inhibitors, Acrylamide derivatives, Rheumatoid arthritis
Abbreviations: AML, acute myeloid leukemia; BPO, benzoyl peroxide; CIA, collagen-induced arthritis; DCE, 1,2-dichloroethane; DCM, dichloromethane; DHODH, dihydroorotate dehydrogenase; DMAP, 4-dimethylaminopyridine; DMARDs, disease-modifying antirheumatic drugs; DMF, N,N-dimethylformamide; DMSO, dimethyl sulfoxide; EA, ethyl acetate; FMN, flavin mononucleotide; HPLC, high performance liquid chromatography; HRMS, high-resolution mass spectrometry; IBD, inflammatory bowel disease; LAH, lithium aluminium hydride; LCMS, liquid chromatography mass spectrometry; MeOH, methanol; MS, multiple sclerosis; NBS, N-bromosuccinimide; NCS, N-chlorosuccinimide; NSAIDs, non-steroidal anti-inflammatory drugs; PDA, photodiode array detector; PE, petroleum ether; PhMe, toluene; PK, pharmacokinetic; RA, rheumatoid arthritis; SEL, systemic lupus erythematosus; TEA, triethylamine; TFA, trifluoroacetic acid; THF, tetrahydrofuran; TsCl, tosyl chloride
Graphical abstract
Dihydroorotate dehydrogenase (DHODH) is an attracting target for the treatment of immunological diseases. A series of acrylamide-based novel DHODH inhibitors as potential rheumatoid arthritis (RA) treatment agents were designed and synthesized, among which 54 is the most potent one with good pharmacokinetic profiles and favorable in vivo anti-arthritic effects.

Highlights
-
•
A series of acrylamide-based novel human dihydroorotate dehydrogenase inhibitors as potential rheumatoid arthritis treatment agents were designed and synthesized.
-
•
The structure-based design and optimization led to potent compounds 42 and 53‒55 with IC50 values of 41, 44, 32 and 42 nmol/L, respectively.
-
•
The most potent compound 54 displayed favorable pharmacokinetic profiles and encouraging in vivo anti-arthritic effects in a dose-dependent manner.
1. Introduction
Rheumatoid arthritis (RA) is the most common autoimmune inflammatory arthritis in adults characterized by synovial inflammation, which can cause cartilage and bone damage and may lead to disability and increase mortality ultimately1, 2, 3, 4, 5. Approximately 0.5%–1% of the world population is affected by the dilemma with a higher incidence among woman and elderly people6, 7, 8. Existing drugs can improve symptoms and slow the progress of RA, but so far, no cure therapy is available9. Analgesics and non-steroidal anti-inflammatory drugs (NSAIDs) are widely used to control symptoms of RA10, but with limited effect. The use of NSAIDs is restricted mainly for their inability to improve the long-term course of the disease, and have gastrointestinal and cardiac toxic effects11,12. The primary treatment for RA relies on disease-modifying antirheumatic drugs (DMARDs) such as methotrexate, sulfasalazine, and leflunomide, which can provide long-term remission. However, many of the DMARDs have severe side effects10. Glucocorticoids are mainly used in the short-term treatment, and long-term use results in osteoporosis and susceptibility to infections. Although biosimilars such as infliximab are undoubtedly effective, concerns about their safety have persisted, especially for the risk of infection13. Consequently, the development of novel DMARDs with fewer side effects has great value14,15.
Flavin-dependent mitochondrial enzyme dihydroorotate dehydrogenase (DHODH) catalyzes the oxidation of dihydroorotate to orotate in the rate-limiting step in de novo pyrimidine biosynthesis. They are divided into two classes based on amino acid sequence, cellular localization, and cofactor dependence16. Class 1 DHODHs are cytosolic enzymes in prokaryotes using fumarate or NAD+ as electron acceptor17,18. Human DHODH, located in the inner mitochondrial membrane, belongs to class 2 enzymes according to the cofactors involved, with ubiquinone as terminal electron acceptor19,20. Pyrimidines are vital for the cell proliferation and metabolism since they are important components in the biosynthesis of DNA, RNA, glycoprotein, and membrane lipids21. For humans, in most normal cells, pyrimidines are mainly obtained by salvage pathways21. Conversely, rapidly proliferating cells, such as tumor cells and the activated lymphocytes, heavily rely on de novo pyrimidines biosynthesis to get sufficient nutrition for survival22. This de novo biosynthesis pathway can be blocked by inhibiting DHODH, which makes it an important target for the development of therapeutics used for the treatment of cancer and immunological diseases, such as RA, psoriasis and multiple sclerosis (MS)20,23.
So far, a large number of DHODH inhibitors have been reported, among which leflunomide (1)24, 25, 26 and brequinar (3)27,28 have been extensively studied (Fig. 1). Leflunomide is the first U.S. Food and Drug Administration (FDA) approved DHODH inhibitor in 1998 for the treatment of RA29. This drug was the prodrug of teriflunomide (A771726, 2), which was also approved for the treatment of MS30. However, its long half-life (approximately two weeks) and its active metabolites, which may even be detected within two years of discontinuing treatment31, can cause many side effects, such as symptomatic liver damage and interstitial lung disease. Drug candidate brequinar failed in phase II clinical trials because of its narrow therapeutic window in human solid tumor study32,33. The immunosuppressive activity of brequinar and its potential application in the treatment of organ graft rejection were also illustrated34. Thereafter, many leflunomide and brequinar analogs were reported as potent DHODH inhibitors. Vidofludimus (4SC-101, 4) is the most studied one and displays potent efficacy towards autoimmune diseases like RA, MS, inflammatory bowel disease (IBD) and systemic lupus erythematosus (SLE). It is in phase II clinical trial35, 36, 37, 38, 39, 40. Recently, more clinical candidates, including ASLAN003 (5), PTC299 (6), and BAY-2402234 (7), were discovered for acute myeloid leukemia (AML) since Sykes et al.41 reported that inhibition of DHODH can overcome differentiation blockade in AML. There were also promising DHODH inhibitors in the preclinical stage such as S312 (8) which is reported to display broad-spectrum antiviral activities against various RNA viruses including influenza A virus, Zika virus, Ebola virus, and particularly against SARS-CoV-242, 43, 44, 45, 46, 47, 48, 49. Besides, the new roles of DHODH in various cancer types were also uncovered in recent years50, 51, 52, 53, 54, 55. DHODH has been proved to be a validated target for the treatment of various diseases such as autoimmune disorders, tumors, virus, and AML. The severe side effects of leflunomide/teriflunomide and the lack of newly marketed DHODH inhibitors for years drive to develop novel chemotypes of DHODH inhibitors.
Figure 1.

Representative structures of DHODH inhibitors.
Co‐crystal structures of DHODH in complex with inhibitors reveal that inhibitors share a common binding region, where the cofactor ubiquinone is believed to bind56. The binding site is made up of a hydrophobic entrance (site A) and a rather polar end (site B)57. The site A offers hydrophobic interactions with non-polar groups such as para-(trifluoromethyl)phenyl in teriflunomide 2 or biphenyl in brequinar 3 (Supporting Information Fig. S1). Additional salt bridges or hydrogen bonds are usually formed between site B and the polar end of the ligand. For instance, a salt bridge and a hydrogen bond are formed between the carboxylic acid of brequinar and site B. For teriflunomide, the cyano and the enolic OH are involved in the formation of two hydrogen bonds with site B. Understanding of this classic binding mode of DHODH inhibitors is very helpful in de novo design and discovery of DHODH inhibitors.
Toward this end, we wish to report new and potent DHODH inhibitors. The new structures are developed based on our previously discovered DHODH inhibitor intermediate 2-acetamidobenzoic acid58. Its derivative 2-acrylamidobenzoic acid analog 11 was identified as the new starting point for structural optimization. After four rounds of structure-guided optimization, potent compounds 42, 53 and 54 were discovered with IC50 values at the double-digit nanomolar range. The most potent compound 54 (IC50 = 32 nmol/L) has proven to be a promising candidate due to its favorable pharmacokinetic (PK) profiles and encouraging in vivo anti-arthritic effects in a dose-dependent manner.
2. Results and discussion
2.1. Chemistry
The synthesis routes for the preparation of ethers 9, 10, 36 and 37 were outlined in Scheme 1. The amidation of 2-bromoacetyl chloride and 2-aminobenzoic acid yielded intermediate 9b, followed by an etherification under alkaline condition to generate target compounds 9, 10 and 36. Synthesis of intermediate 37b was achieved by a Bargellini reaction to provide a method for the formation of the gem-dimethyl incorporated carboxylic acid derivative. Then, target compound 37 was achieved by amidation of 37b with 2-aminobenzoic acid.
Scheme 1.

Synthesis of compounds 9, 10, 36, and 37. Reagents and conditions: (a) TEA, DCM, 5 h, 79%; (b) appropriate phenol, KOH, DMF, Ar, 120 °C; or for 9, 4-nitrophenol, K2CO3, DMF, 70 °C, 40%; (c) acetone, CHCl3, NaOH, reflux, 90%; (d) (i) (COCl)2, DMF, DCM; (ii) 2-aminobenzoic acid, TEA, DCM, 70% over 2 steps.
The key intermediates of compounds 11‒35 and 38‒55 were prepared as described in Scheme 2. The precursors 11c‒15c, 16e, 17c‒21c, 22e, 23e, 24c‒26c, 27g, 28f, 30c, 31c, 32e, 33e, and 34e were synthesized by Wittig reactions of corresponding aromatic aldehydes with ethyl 2-(triphenylphosphoranylidene)propionate followed by ester hydrolysis using NaOH in THF/MeOH/H2O. Precursors 29b and 35d were prepared by ester hydrolyzation of 29a and 35c. Cinnamic acid derivative 38b was prepared through the Knoevenagel condensation of 38a with malonic acid in the presence of pyridine. Intermediates 39b and 40b (Scheme 2) were prepared by Wittig reactions of 2-naphthaldehyde with chloro- or bromophosphoylides formed in situ. Further saponification reactions of 39b and 40b gave the acid analogs 39c and 40c. The ethyl substituted analog 41c was obtained by Wittig reaction of 2-naphthaldehyde with triethyl 2-phosphonobutyrate in the presence of NaH, followed by ester hydrolysis.
Scheme 2.

Synthesis of compounds 11‒35 and 38‒55. Reagents and conditions: (a) ethyl 2-(triphenylphosphoranylidene)propionate, DCM, r.t., 4 h, 90%‒96%; for 39b and 40b ethyl (triphenylphosphoranylidene)acetate, NXS (NCS for 39b, NBS for 40b), DCM, Ar, −20 °C; K2CO3, r.t. 10 h; for 41b triethyl 2-phosphonobutyrate, NaH, THF, r.t. 60%; (b) NaOH, MeOH, THF, 40 °C, 5 h, 90%‒95%; (c) malonic acid, pyridine, DMF, 90 °C, 5 h, 80%; (d) (i) (COCl)2, DMF, DCM 0 °C; r.t. 4 h; (ii) appropriate arylamine, TEA, DMAP, DCM for 11‒31, 33‒35, 38‒49, 51, and 53‒55; or 4-aminonicotinic acid, pyridine for 50; or for 32 (i) TsCl, DMAP, TEA, DCE, 35 °C, 1 h; (ii) 2-aminobenzoic acid, 60 °C, 10 h, 50% over 2 steps; (e) MeOH, (COCl)2, DMF, DCM, r.t. 30%.
The synthetic strategies used for the synthesis of arylaldehyde precursors 16c, 22c, 23c, 27e, 28d, 32c and 33c were described in Scheme 3. The fluoro derivative 28b was obtained from 28a by a Balz–Schiemann reaction using tetrafluoroboric acid. Benzyl alcohol 28c was synthesized by bromination of 28b with NBS, followed by substitution of the bromides using aqueous CaCO3. Intermediates 16b, 22b, 23b, 32b and 33b were obtained from corresponding acid derivatives by reduction with LAH. Esterification of 27a gave the ester intermediate 27b and following halogen-exchange reaction provided chloroarene 27c, which was further reduced to 27d. MnO2 oxidation of the benzyl alcohols afforded arylaldehydes 16c, 22c, 23c, 27e, 28d, 32c, and 33c. Generation of ether 34c was accomplished effectively via a SNAr displacement reaction of the activated aryl fluoride of 34b by phenol in the presence of potassium carbonate. The strategies to the formation of precursors 29a, 35c and 49b were described in Scheme 4. Chlorine derivative 29a was synthesized from the corresponding bromide 30b via a halogen-exchange reaction. An efficient Cu-catalyzed O-arylation of phenol with bromoarene 35b, which was prepared by Wittig reaction of 35a with ethyl 2-(triphenylphosphoranylidene) propionate, was carried out to afford precursor 35c. The corresponding methyl ester 49a was hydrolyzed by treating with NaOH to provide intermediate 49b. As described in Scheme 2, acrylic acid analogs were transformed into target compounds 11‒35, 38‒51, and 53‒55 by the amidation with appropriate arylamines. The ester 52 was easily prepared from corresponding acid derivative 24 under standard esterification condition.
Scheme 3.

Synthesis of arylaldehyde precursors. Reagents and conditions: (a) (i) HBF4, NaNO2 (aq.), 0 °C; (ii) PhMe, 90 °C; reflux, 30% over 2 steps; (b) (i) NBS, BPO, CCl4, Ar, reflux; (ii) CaCO3, dioxane/H2O, reflux; 60% over 2 steps; (c) LAH, THF, Ar, 0 °C; r.t.; (d) H2SO4, MeOH, 0 °C; reflux, 97%; (e) CuCl, DMF, Ar, reflux, overnight, 97%; (f) LAH, THF, Ar, 0 °C; r.t.; (g) MnO2, DCM, r.t.; (h) K2CO3, DMSO, Ar, 110 °C, overnight, 90%.
Scheme 4.

Synthesis of precursors 29a, 35c, and 49b. Reagents and conditions: (a) CuCl, DMF, Ar, reflux, overnight, 97%; (b) ethyl 2-(triphenylphosphoranylidene)propionate, DCM, r.t., 4 h, 96%; (c) phenol, CuCl, 1-butyl-1H-imidazole, K2CO3, o-xylene, reflux, 85%; (d) NaOH, MeOH/H2O, Ar, reflux, 32%.
2.2. Lead identification
Compound 9, discovered in our previously designed DHODH inhibitor58, consists of a hydrophilic head and a hydrophobic tail (Fig. 2) and may be a potential DHODH inhibitor in view of the classic binding mode of reported DHODH inhibitors. The proposed binding mode of compound 9 with DHODH was explored via molecular docking using the co-crystal structure of a close analog of brequinar (Fig. 3A). The 4-nitrophenoxy group of 9 is located at the hydrophobic entrance, interacting with hydrophobic residues like M43 and F98. The 2-aminobenzoic acid of 9 occupies the polar end of the binding site. The carboxyl group forms hydrogen bonds with Q47 and R136. Compound 9 and its analog 10 displayed moderate inhibitory rate of 45.27% and 31.70% at 10 μmol/L in enzyme assay in vitro, respectively (Fig. 2). Because highly active DHODH inhibitors are usually planar to accommodate the shape of the binding pocket59, the flexible linker of compound 10 was then replaced by a more rigid and flat one to give compound 11. As a result, compound 11 showed an improved IC50 value of 0.814 μmol/L. Compared with 9, the methyl substituent on the 2-methyl-acrylamide linker could form additional hydrophobic interactions with residues M43 and L46 (Fig. 3B). Besides, the more rigid acrylamide linker in 11 makes the molecule more planar and appropriate than 9 to accommodate the slim binding pocket. Those factors may contribute to the largely increased potency of 11, which was then selected as a lead for further optimization.
Figure 2.

Design of lead compound 11 and strategies of modification.
Figure 3.

Proposed binding mode of compound 9 (A) and 11 (B) with DHODH. DHODH (PDB ID: 1D3G) is shown as a transparent gray cartoon. The critical residues and the substrate FMN are presented as gray sticks. Compound 9 is colored cyan, while compound 11 is colored yellow and superimposed to 9 in (B). Hydrogen bonds are shown as black dashed lines.
2.3. Structure–activity relationship (SAR)
The structure of 11 can be divided into a phenyl part (A) and a benzoic acid part (B), connected via 2-methyl-acrylamide (Fig. 2). Systematic structural modification of both parts was carried out. The structures and the DHODH inhibitory activities of the synthesized compounds are listed in Table 1, Table 2, Table 3, Table 4.
Table 1.
Structures and enzyme activities of compounds 11−23.
| Compd. | Ar | Inhibition (%) at 10 μmol/L | IC50 (μmol/L) |
|---|---|---|---|
| 11 | 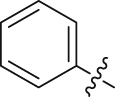 |
85.64 | 0.814 ± 0.049 |
| 12 | 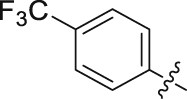 |
82.32 | 1.044 ± 0.040 |
| 13 | 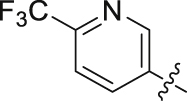 |
69.32 | 6.113 ± 0.102 |
| 14 | 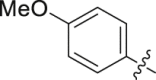 |
88.71 | 0.561 ± 0.014 |
| 15 | 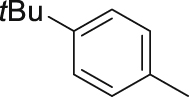 |
86.56 | 0.836 ± 0.021 |
| 16 | 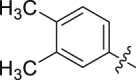 |
92.91 | 0.394 ± 0.004 |
| 17 | 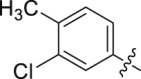 |
91.02 | 0.384 ± 0.016 |
| 18 | 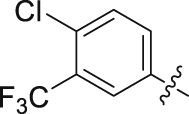 |
84.62 | 0.894 ± 0.063 |
| 19 | 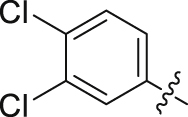 |
91.65 | 0.529 ± 0.024 |
| 20 | 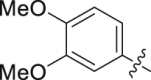 |
91.49 | 0.612 ± 0.011 |
| 21 | 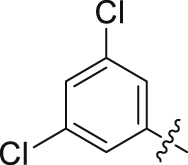 |
89.39 | 0.724 ± 0.028 |
| 22 | 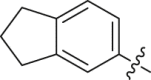 |
86.93 | 0.252 ± 0.017 |
| 23 | 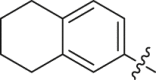 |
85.24 | 0.115 ± 0.001 |
| Brequinar | – | – | 0.008 ± 0.001 |
‒Not applicable.
Table 2.
Structures and enzyme activities of compounds 24−35.
| Compd. | Ar | Inhibition (%) at 10 μmol/L | IC50 (μmol/L) |
|---|---|---|---|
| 24 | 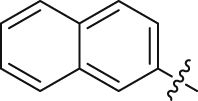 |
93.92 | 0.075 ± 0.002 |
| 25 | 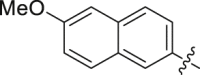 |
88.18 | 0.129 ± 0.008 |
| 26 | 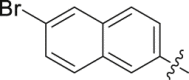 |
85.41 | 0.448 ± 0.008 |
| 27 | 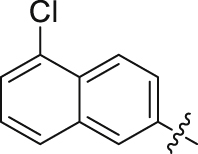 |
96.84 | 0.256 ± 0.009 |
| 28 | 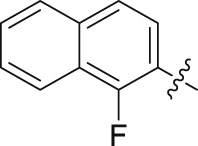 |
89.35 | 0.057 ± 0.002 |
| 29 | 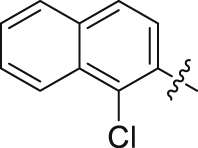 |
96.36 | 0.078 ± 0.003 |
| 30 | 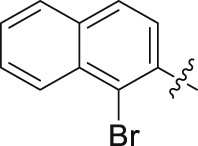 |
90.17 | 0.058 ± 0.001 |
| 31 | 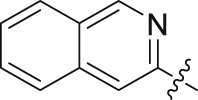 |
87.44 | 0.646 ± 0.009 |
| 32 | 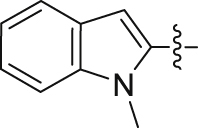 |
84.31 | 0.317 ± 0.008 |
| 33 | 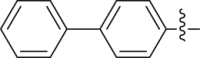 |
89.75 | 0.626 ± 0.013 |
| 34 | 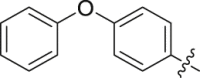 |
90.36 | 0.630 ± 0.014 |
| 35 | 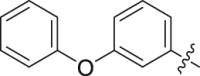 |
89.55 | 0.597 ± 0.006 |
Table 3.
Structures and enzyme activities of compounds 36–41.
| Compd. | Linker | Inhibition (%) at 10 μmol/L | IC50 (μmol/L) |
|---|---|---|---|
| 24 | 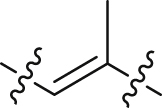 |
93.92 | 0.075 ± 0.002 |
| 36 | 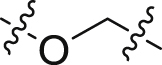 |
76.08 | 1.865 ± 0.048 |
| 37 | 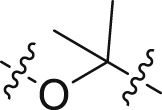 |
20.28 | >10 |
| 38 | 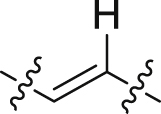 |
87.45 | 0.712 ± 0.012 |
| 39 | 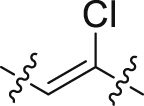 |
88.58 | 0.061 ± 0.001 |
| 40 | 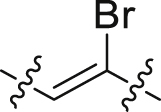 |
88.63 | 0.085 ± 0.004 |
| 41 | 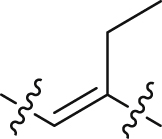 |
82.78 | 0.606 ± 0.031 |
Table 4.
Structures and enzyme activities of compounds 42–55.
| Compd. | Ar | R | Inhibition (%) at 10 μmol/L | IC50 (μmol/L) |
|---|---|---|---|---|
| 24 | 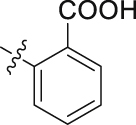 |
H | 93.92 | 0.075 ± 0.002 |
| 42 | 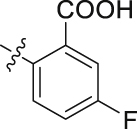 |
H | 90.04 | 0.041 ± 0.001 |
| 43 | 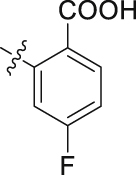 |
H | 34.93 | >10 |
| 44 | 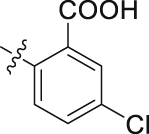 |
H | 82.98 | 0.623 ± 0.001 |
| 45 | 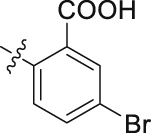 |
H | 80.76 | 1.821 ± 0.020 |
| 46 | 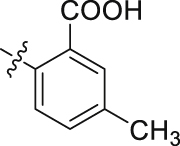 |
H | 82.88 | 0.688 ± 0.006 |
| 47 | 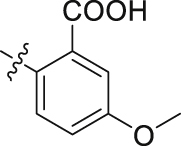 |
H | 48.24 | >10 |
| 48 | 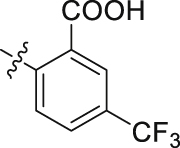 |
H | 87.36 | 0.874 ± 0.005 |
| 49 | 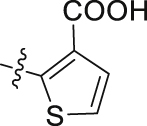 |
H | 79.32 | 2.133 ± 0.110 |
| 50 | 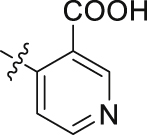 |
H | 74.11 | 1.691 ± 0.014 |
| 51 |  |
H | 66.93 | 5.350 ± 0.172 |
| 52 |  |
H | 29.57 | >10 |
| 53 | |
F | 97.96 | 0.044 ± 0.001 |
| 54 | |
Cl | 98.72 | 0.032 ± 0.001 |
| 55 | |
Br | 98.62 | 0.042 ± 0.001 |
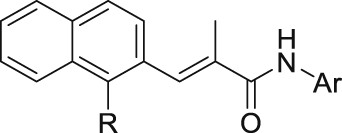
Initial optimization efforts focused on the modification of phenyl part (A) to explore the effects of substituent size, lipophilicity, and polarity on the potency, as shown in Table 1. Introducing a single substituent in the para-position (12, 14, and 15) gave compounds with potencies similar to that of 11. Compound 14 bearing 4-OMe group possessed a slightly higher activity (IC50 = 0.561 μmol/L) compared with compound 12 bearing CF3 and compound 15 bearing tBu group. However, the replacement of the phenyl group of 12 with a 3-pyridine (compound 13) resulted in a significant loss of activity. Benzene is more hydrophobic than pyridine (logP: 2.13 vs. 0.65). Thus, a benzene ring in 12 would be more preferred at the hydrophobic entrance of the binding site than the less hydrophobic pyridine ring in 13, which may be the reason for the decreased bioactivity of 13 compared to 12. It was found that 3,4-disubstituted analogs were well tolerated, as exemplified by analogs 16‒20, with all compounds having comparable or improved potencies (IC50 values ranging from 0.384 to 0.894 μmol/L) relative to the lead compound 11. A slight loss of activity was observed when the 3,4-dichlorophenyl (19) was replaced by a 3,5-dichlorophenyl group (21). Based on the data obtained from the preliminary SAR described above, 2,3-dihydro-1H-inden-5-yl and 5,6,7,8-tetrahydronaphthalen-2-yl analogs (compounds 22 and 23) were developed. This effort delivered more potent DHODH inhibitors with IC50 values of 0.252 and 0.115 μmol/L, which implied that the hydrophobic dual-ring structure was benefit for the increasing of the inhibitory activity.
To further improve the potency, we synthesized several naphthalene substituted derivatives 24‒27 (Table 2) inspired by the structures of compounds 22 and 23. Introduction of the naphthyl group (24) provided a further enhancement in potency compared with 5,6,7,8-tetrahydronaphthalen-2-yl analog 23. However, adding small hydrophobic substituent groups (OMe, Br, or Cl) to the 5-position or 6-position of the naphthyl resulted in a loss of inhibitory activity (compounds 25‒27 vs. 24), probably due to steric hindrance. To explore the action mechanism of the new inhibitors reported here, the binding mode of compound 24 was simulated by molecular docking (Fig. 4A). The proposed binding mode revealed that the naphthyl group of 24 is located at the hydrophobic entrance and incorporation of a small hydrophobic group to the 1-position of the naphthyl might occupy the hydrophobic subsite S1 to give more potent compounds. Encouragingly, the introduction of suitable substituents (F, Cl or Br) to the 1-position of the naphthyl really led to equal or more potent compounds 28‒30. Replacement of the naphthyl group (24) with isoquinolinyl (31) and 1-methyl-1H-indolyl (32) caused 8-fold and 4-fold potency loss, respectively. The inhibitory activities decreased sharply when biphenyl (33) or phenoxyphenyl (34 and 35) was introduced, which might be due to steric hindrance. From the overall SAR of these compounds, it was clear that the hydrophobicity and size of the group at part A is crucial for maintaining the potency against DHODH.
Figure 4.

Proposed binding modes of compounds 24 (A) and 54 (B) with DHODH. DHODH (PDB ID: 1D3G) is shown as a transparent gray cartoon. The critical residues and the substrate FMN are presented as gray sticks. Compound 24 is colored purple while compound 54 is colored blue. The binding pocket of 24 is displayed as gray surface with subsites S1–S3 marked using red dashed lines. Hydrogen bonds are shown as black dashed lines.
The structure–activity relationships of the linkers (part C) were also investigated with compounds 24 and 36‒41 (Table 3). Replacing the acrylamide with more flexible ethers (36 and 37), which cannot bind to the planar and hydrophobic entrance (site A) very well, resulting in a dramatic decrease in potency. Then we turned our attention back to acrylamide analogs. The proposed binding mode revealed that the methyl substituent on the linker (24) is nicely accommodated in the subsite S2 (Fig. 4A). The removal of the methyl in 24 had a negative impact on potency, with the corresponding derivative 38 possessing a much weaker inhibitory activity (IC50 = 0.712 μmol/L). The chloro and bromo analogs 39 and 40 showed activities similar to compound 24. The larger ethyl group substituent (41) was detrimental. This preliminary SAR evaluation of the linkers suggested that an acrylamide with a small hydrophobic group (Me, Cl or Br) at the 2-position was favorable.
The role of substituents in the hydrophilic head region (part B) was further investigated (Table 4). Further inspection of the binding mode of 24 (Fig. 4A) suggested that introduction of a small hydrophobic group at the 5-position of the benzoic acid group might occupy the small hydrophobic subsite S3 to further improve the potency. Rewardingly, a 1-fold improvement in potency for inhibition of DHODH was obtained (IC50 = 0.041 μmol/L) when the 5-fluorobenzoic acid was introduced (compounds 42 vs. 24). Moving the fluoro group to the 4-position of the benzoic acid group resulted in a dramatic decrease in potency. Then structural optimization was focused on the 5-position of the benzoic acid (44‒48). An over 10-fold loss in potency was observed when the fluoro group in 42 was replaced by larger substituents (Cl, Br, Me and CF3). The potency was further reduced when methoxy group was introduced into 47, suggesting that only small substituents hydrogen and fluorine were tolerated. The thienyl analog 49 and the pyridyl analog 50 showed only modest activities with IC50 values of 2.133 and 1.691 μmol/L, respectively. Finally, to determine the key role of the carboxylic acid moiety, we synthesized the corresponding amide (51) and methyl ester (52) derivatives. The amide analog 51 only showed weak inhibition with an IC50 value of 5.350 μmol/L, while compound 52 was completely inactive, illustrating the requirement of the carboxylic acid moiety to maintain potent inhibition.
As a final optimization step, we combined our SAR knowledge on the three parts of the acrylamide scaffold. That is, naphthyl or 1-halogens (F, Cl or Br) substituted naphthyl moiety in part A, 2-position small hydrophobic group substituted acrylamide in part C and 5-fluorobenzoic acid moiety in part B have been turned out to be more favorable building blocks. These structural variations were combined in molecules 53‒55 as extensively optimized and highly potent DHODH inhibitors, which afforded the most potent compound 54 with an IC50 value of 0.032 μmol/L. The proposed binding mode of 54 is similar to that of 24 (Fig. 4B).
2.4. In vivo PK study of compound 54
A PK study of the most potent inhibitor compound 54 was performed by administering rats a 1 mg/kg intravenous (i.v.) dose and a 10 mg/kg oral dose (p.o.) of compound 54. After i.v. dosing, 54 exhibited a terminal half-life of 18.10 h, a steady-state volume of distribution of 0.069 L/kg, a high exposure (AUC0‒∞) of 381,207.98 h·mg/L and a low plasma clearance of 0.0026 L/h/kg (Table 5). After oral administration, 54 exhibited a large exposure (AUC0‒∞) of 2,557,087.39 h·mg/L and a maximum plasma concentration (Cmax) of 77,781.76 μg/L. The time to reach the maximum concentration (Tmax) of the inhibitor was 4.67 h. Moreover, compound 54 has a satisfactory oral bioavailability of 67.08%.
Table 5.
PK profiles of 54 after intravenous (i.v.) and oral (p.o.) administrationa.
| Parameter | i.v. | p.o. |
|---|---|---|
| Dose (mg/kg) | 1 | 10 |
| CL (L/h/kg) | 0.0026 | NC |
| t1/2 (h) | 18.10 | 21.47 |
| Cmax (μg/L) | 31,524.54b | 77,781.76 |
| Tmax (h) | NC | 4.67 |
| AUC0‒∞ (h·μg/L) | 381,207.98 | 2,557,087.39 |
| Vdss (L/kg) | 0.069 | NC |
| F (%) | NC | 67.08 |
Compound was dosed to equal number of Sprague–Dawley rats in i.v. and p.o. administration respectively (n = 3).
Cmax=C0 (t = 0) for i.v. administration; NC: not calculated.
2.5. In vivo antiarthritic effect of compound 54
The in vivo antiarthritic effect of compound 54 was investigated in an animal model of collagen-induced arthritis (CIA) in Wistar rats, the most commonly studied autoimmune model of RA60. In this study, autoimmune arthritis was induced by immunization with type II collagen. About 2 weeks after the first immunization, the limbs of rats became swollen and redness, indicating the onset of arthritis. The rats of treatment groups were administered orally once a day with methotrexate (0.3 mg/kg) or 54 at 10, 30 mg/kg for 28 days, while the rats in model group were treated with vehicle (0.5% CMC-Na solution). The antiarthritic effects of compound 54 were mainly evaluated by measurement of hind paws volume and morphological observation of the joint tissue of rats (Fig. 5).
Figure 5.

The effects of compound 54 on CIA rats in vivo. Arthritis was induced in Wistar rats by twice immunization with type II collagen on Days 1 and 7. Compound 54 was orally administered at 10 and 30 mg/kg for 28 days. Methotrexate, as the positive control, was administered at the dose of 0.3 mg/kg. (A) Curves of body weight in different groups. Measurement of body weight was taken every three days. (B) Arthritis scores in different treatments. (C) The representative photomicrographs of hind paws in different treatment groups taken after 28-day trial. (D) Representative H&E stained sections of joint tissue of each group. All quantitative data were expressed as mean ± SEM, and statistical differences among various groups were determined using a one-way ANOVA, with a post hoc test (LSD) performed for multiple comparisons. #P < 0.05, ##P < 0.01, ###P < 0.001 vs. control; ∗P < 0.05, ∗∗P < 0.01, ∗∗∗P < 0.001 vs. RA.
During the experiment, the control group (normal rats) had normal eating patterns and shiny hair. By contrast, the rats in the model group exhibited rough and dull hair. Mean body weights of all groups kept growing during the course of the experiment, but model group had a slower rate of growth after Day 14 (Fig. 5A). It was found that CIA rats exhibited a significant increase in paw volume over the control group. As shown in Fig. 5B, the arthritis scores of model group reached a peak on Day 20 and then stabilized around 2.3 after a transitory fall. Compared to model rats, the volumes of paws in CIA rats were significantly decreased after treatment with methotrexate or compound 54 in a dose-dependent manner with the arthritis scores of the compound 54 treated rats stabilized around 1 (10 mg/kg) and 0.6 (30 mg/kg) lastly.
Ankle joints were examined further for histopathological evidence of bone erosion after four weeks of therapy using hematoxylin and eosin (H&E) staining to clarify the effects of compound 54. Results of the histologic assessment were shown in Fig. 5D. The rats in CIA model group (Fig. 5B) displayed prominent signs of arthritis, such as synovial hyperplasia, inflammatory cell infiltration, moderate cartilage damage, new blood vessels formation and bone damage. Above-mentioned symptoms of arthritis in CIA rats treated with compound 54 were significantly alleviated in a dose-dependent manner. Briefly, compound 54 displayed substantially in vivo antiarthritic effects in a dose-related manner. It might serve as a promising lead compound for further development as immunosuppressant and antiproliferative agent targeting DHODH.
2.6. Acute toxicity study
Adult ICR mice (male, 28–32 g, n = 3) was used to evaluate the acute toxicity of 54. A single-dose of compound 54 (300 and 500 mg/kg, respectively) was intragastrically administrated. The mice were fasted for 12 h before the compound administration. The general condition, behavior and mortality were observed continuously after compound treatment for 14 days. All mice were sacrificed on the 14th day for gross examination of heart, liver, spleen, lung and kidney while the liver and kidney were reserved for histological examination. No obvious abnormal in behaviors or mortality were observed. There is no significant change in body weight compared to the control group at the end of the experiment for mice treated with both doses of 54 (Supporting Information Fig. S2). The morphological examination did not find remarkable injury in the liver and kidney for both groups treated with 54 (Supporting Information Fig. S3). These results indicated that 54 can be tolerated at the dose as high as 500 mg/kg in mice.
3. Conclusions
The structure-based design and optimization of a series of acrylamide derivatives as novel and potent DHODH inhibitors have been carried out. Starting from the promising 2-acetamidobenzoic acid scaffold of compounds 9 and 10, compound 11 was identified as the lead compound for further optimization. The structure–activity relationship indicated that naphthyl or 1-position halogens (F, Cl or Br) substituted naphthyl moiety in part A, 2-position small hydrophobic group substituted acrylamide in part C and 5-position fluorobenzoic acid substituted moiety in part B have been turned out to be more favorable building blocks. Overall, these optimization efforts led to potent compounds 42 and 53‒55 with IC50 values of 41, 44, 32 and 42 nmol/L, respectively. The most potent compound 54 also displays favorable PK profiles and encouraging in vivo antiarthritic effects in a dose-dependent manner.
4. Experimental
4.1. General methods of chemistry
Unless otherwise indicated, all commercially available solvents and reagents were purchased directly from commercial suppliers and used as received without further purification. Melting points (m.p.) were recorded on Büchi B540 apparatus (Büchi Labortechnik AG, Flawil, Switzerland) and are uncorrected. 1H NMR, 19F NMR and 13C NMR spectra were recorded on Bruker AM-400 (1H at 400 MHz, 13C at 100 MHz, 19F at 376 MHz) spectrometer (Bruker Corp., Karlsruhe, Germany) with CDCl3 or DMSO-d6 as the solvent and TMS as the internal standard. Chemical shifts are reported in δ (parts per million) values. The following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, coupling constant (Hz) and integration. High-resolution electron mass spectra (ESI-TOF) were performed on a Micromass Xevo G2 TOF MS (Waters Corp., Milford, MA, USA). High Resolution Mass Spectrometry (HRMS) EI was recorded under electron impact (70 eV) condition using a MicroMass GCT Premier (Waters Corp., Milford, MA, USA). Analytical thin-layer chromatography (TLC) was carried out on precoated plates (silica gel 60 F254) and spots were visualized with ultraviolet (UV) light. Purity was determined using HPLC analysis (Shimadzu Corp., Kyoto, Japan), equipped with Shimadzu shim-pack GIST C18 reversed phase column with 250 mm × 4.6 mm × i.d. 5 μm particle size, and a photodiode array detector (PDA). The HPLC separations were carried out using gradient separation at a flow rate of 1 mL/min. The mobile phase was a mixture of MilliQ ultrapure 0.01% TFA solution (A) and acetonitrile (B). The following elution gradient totally lasted 18 min: initial mobile-phase composition, 95:5 (v/v) phase A: B; 0–10 min, linear change from 5% to 95% B; 10–15 min, 95% B; 15–18 min, linear change from 95% to 5% B. The column and injection chamber were maintained at 40 and 25 °C, respectively. The sample injection volume was 5 μL and the detector was set at 254 nm. The purities of compounds for biological testing were assessed by NMR and HPLC to be ≥ 95%.
4.2. Synthesis of key intermediates and target compounds
4.2.1. 2-(2-Bromoacetamido)benzoic acid (9b)
2-Aminobenzoic acid (1.37 g, 10 mmol) was dissolved in dichloromethane (20 mL) and triethylamine (2 mL, 15 mmol). 2-Bromoacetyl chloride (1 mL, 12 mmol) was added dropwise at 0 °C. The resulting solution was further stirred at room temperature for 5 h. The reaction mixture was washed with 1 mol/L HCl and brine. The resulting suspension of solid was filtered and washed with dichloromethane. The filtrate was dried over Na2SO4, and concentrated in reduced pressure to remove most of the solvent. The resulting suspension of solid was filtered and washed with dichloromethane. The resulted solid was collected to give 9b in yield of 70%.
4.2.2. 2-(2-(4-Nitrophenoxy)acetamido)benzoic acid (9)
K2CO3 (483 mg, 3.5 mmol) was carefully added to a solution of 9b (257 mg, 1 mmol) and 4-nitrophenol (139 mg, 1 mmol) in DMF (5 mL). The mixture was stirred at 70 °C until the reaction was complete. After being cooled down to room temperature, diluted with water (10 mL), acidified with 1 mol/L HCl, the resulting solid was filtered off, washed with EtOH, and further crystallized from DCM/MeOH to give target compound 9 in yield of 40%.
4.2.3. General procedure for 10 and 36
KOH (168 mg, 3 mmol) was carefully added to a solution of 9b (257 mg, 1 mmol) and appropriate phenol (1 mmol) in DMF (5 mL) under an atmosphere of Ar. The reaction mixture was heated at 120 °C for 4 h. After being cooled down to room temperature, diluted with water (10 mL), acidified with 1 mol/L HCl, the resulting solid was filtered off, washed with EtOH and further crystallized from DCM–MeOH to give target compounds 10 and 36.
4.2.4. 2-Methyl-2-(naphthalen-2-yloxy)propanoic acid (37b)
NaOH (2.4 g, 60 mmol) was added to the solution of naphthalen-2-ol in acetone (10 mL). Then a mixture of chloroform (1.2 mL) and acetone (2 mL) was added dropwise under reflux condition. The mixture was stirred under reflux condition until the reaction was complete as indicated by TLC. After being cooled down to room temperature, diluted with water (10 mL), acidified with 1 mol/L HCl. The resulted solid was filtered off, washed with EtOH and further crystallized from DCM–MeOH to give 37b in yield of 90%.
4.2.5. 2-(2-Methyl-2-(naphthalen-2-yloxy)propanamido)benzoic acid (37)
Compound 37b (460 mg, 2 mmol) was suspended in anhydrous DCM (6 mL), and the solution was cooled to 0 °C. The (COCl)2 (0.5 mL, 6 mmol) followed by a drop of DMF was added dropwise. After the addition, the reaction mixture was stirred at room temperature for 3 h. The solvent was evaporated in vacuum to give the intermediate acyl chloride. 2-Aminobenzoic acid (274 mg, 2 mmol) was dissolved in dichloromethane (4 mL) and triethylamine (303 mg, 3 mmol). The obtained acyl chloride was dissolved in 2 mL DCM and added to the reaction mixture dropwise at 0 °C. The resulted solution was further stirred at room temperature overnight. The reaction mixture was washed with 1 mol/L HCl and brine. The resulted suspension of solid was filtered and washed with water. The resulted solid was further crystallized from DCM–MeOH to give 37 in yield of 70%.
4.2.6. General procedure for 16b, 22b, 23b, 32b, and 33b
The appropriate acid (10 mmol) was dissolved in THF (25 mL) under Ar atmosphere. Then LAH (20 mmol) was added carefully at 0 °C. After keeping at this temperature for 10 min, the mixture was stirred at room temperature for another 4 h. Then the reaction was quenched with ice water (0.76 g), 15% NaOH (0.76 g), and water (2.28 g) in ice bath. Then water (25 mL) was added and the resulted mixture was filtered off. The filtrate was concentrated under reduced pressure to remove THF. The residue was dissolved in EA and washed with water. The organic layer was dried (Na2SO4) and concentrated under reduced pressure to give target precursor, which was used in the next step without further purification.
4.2.7. Methyl 5-chloro-2-naphthoate (27d)
Concentrated sulfuric acid (1 mL) was dropwise added to a mixture of 5-bromo-2-naphthoic acid (3 mmol) in MeOH (10 mL) in ice bath. The reaction mixture was heated to reflux for 2 h. After cooling to room temperature, the resulted solid was separated by filtration and dried to give 27b in yield of 97%.
27b (4.5 mmol) and CuCl (4.5 mmol) were added to DMF (12 mL) under Ar atmosphere, and the mixture was heated to reflux overnight. After cooling to room temperature, the mixture was diluted with H2O (25 mL) and filtered. The filter cake was dissolved in DCM (10 mL) and filtered. The filtrate was concentrated under reduced pressure to give 27c in yield of 97%. GG‒MS m/z [M]+ 222.
27c (2 mmol) was dissolved in THF (10 mL) under Ar atmosphere. Then LAH (2.6 mmol) was added carefully at 0 °C. After keeping at this temperature for 10 min, the mixture was stirred at room temperature for another 12 h. Then the reaction was quenched with ice water (0.1 g), 15% NaOH (0.1 g), and water (0.3 g) in ice bath. Then water (10 mL) was added and the resulted mixture was filtered off. The filtrate was concentrated under reduced pressure. The residue was dissolved in EA and washed with water. The organic layer was dried (Na2SO4) and concentrated under reduced pressure to give 27d, which was used in the next step without further purification. GG‒MS m/z [M]+ 194.
4.2.8. (1-Fluoronaphthalen-2-yl)methanol (28c)
HBF4 (15 mL) was dropwise added to a round-bottomed flask with 2-methylnaphthalen-1-amine (10 mmol) at room temperature. Then a solution of NaNO2 (40 mmol, 10 mL) was dropwise added in ice bath. The mixture was stirred for 0.5 h in such condition. The resulted solid was separated by filtration and dried. Then the solid was suspended in PhMe (50 mL). The resulted mixture was reacted at 90 °C for 1 h, followed by under reflux condition for 2 h. After cooling to room temperature, the solution was washed by saturated sodium bicarbonate solution and saturated salt water in order. The organic layer was dried (Na2SO4), concentrated under reduced pressure and purified by silica gel chromatography (PE:EA = 7:1) to give 1-fluoro-2-methylnaphthalene in yield of 67%. yellow oil. GG‒MS m/z [M]+ 160.
NBS (10 mmol) and BPO (0.1 mmol) were added to a solution of 1-fluoro-2-methylnaphthalene (5 mmol) in CCl4 (10 mL) under Ar atmosphere. The mixture was heated to reflux for 5 h. After cooling to room temperature, the reaction mixture was filtered. The filtrate was concentrated under reduced pressure. The resulted residue was dissolved in dioxane (10 mL). Then water (10 mL) and CaCO3 (20 mmol) were added. The mixture was heated to reflux for 10 h. After cooling to room temperature, the mixture was filtered and extracted with EA. The organic layer was dried (Na2SO4), filtrated, concentrated under reduced pressure and purified by silica gel chromatography (PE:EA = 3:1) to give 28c in yield of 80%. Colorless oil. GG‒MS m/z [M]+ 176.
4.2.9. General procedure for 16c, 22c, 23c, 27e, 28d, 32c, and 33c
MnO2 (35.0 mmol) was added to a solution of corresponding benzyl alcohol (5.0 mmol) in DCM (15 mL). The mixture was stirred at room temperature for 4 h and filtrated. The filtrate was concentrated in reduced pressure and purified by silica gel chromatography (PE:EA = 7:1) or without further purification to give target intermediate.
4.2.10. 4-Phenoxybenzaldehyde (34c)
Phenol (7.5 mmol), 4-fluorobenzaldehyde (5.0 mmol), and K2CO3 (10.0 mmol) were suspended in DMSO (5 mL) under Ar atmosphere. The mixture was heated to 110 °C overnight. After cooling to room temperature, the reaction mixture was diluted with water and extracted with EA. The organic layer was washed with saturated NaCl, dried (Na2SO4), concentrated under reduced pressure, and purified by silica gel chromatography (PE:EA = 5:1) to give 34c in yield of 80%.
4.2.11. General procedure for 11b‒15b, 16d, 17b‒21b, 22d, 23d, 24b‒26b, 27f, 28e, 30b, 31b, 32d, 33d, 34d, and 35b
Corresponding aromatic aldehyde (5 mmol) was dissolved in 15 mL DCM. Then ethyl 2-(triphenylphosphoranylidene)propionate (3.258 g, 9 mmol) was added. The reaction was allowed to stir at room temperature for 4 h. The reaction mixture was concentrated in reduced pressure and further purified by silica gel chromatography (PE:EA = 7:1) to give target compounds in yield of 90%–96%.
4.2.12. Ethyl (E)-3-(1-chloronaphthalen-2-yl)-2-methylacrylate (29a)
30b (0.936 g, 4.0 mmol) and CuCl (0.6 g, 6.0 mmol) were added to DMF (16 mL) under Ar atmosphere, and the mixture was heated to reflux overnight. After cooling to room temperature, the mixture was diluted with H2O (30 mL) and filtered. The filter cake was dissolved in DCM (10 mL) and filtered. The filtrate was concentrated under reduced pressure to give 29a in yield of 96%.
4.2.13. Ethyl (E)-2-methyl-3-(3-phenoxyphenyl)acrylate (35c)
35b (5.0 mmol), phenol (6.0 mmol), K2CO3 (10.0 mmol), CuCl (0.25 mmol), 1-butyl-1H-imidazole (2.5 mmol), and o-xylene (5 mL) were added to a round-bottom flask. The mixture was heated to 140 °C for 24 h under Ar atmosphere. After cooling to room temperature, the reaction mixture was filtrated. The filtrate was concentrated in reduced pressure and further purified by silica gel chromatography (PE:EA = 7:1) to give 35c in yield of 85%.
4.2.14. 2-Aminothiophene-3-carboxylic acid (49b)
NaOH (40.0 mmol) was added to a mixture of 49a (2.0 mmol) in water (10 mL) and MeOH (10 mL). The mixture was heated to reflux under Ar atmosphere for 2 h. After cooling to room temperature, the solution was concentrated in vacuum to remove MeOH. The residue was acidified with 1 mol/L HCl and extracted with EA. The organic layer was washed with saturated NaCl, dried (Na2SO4), filtrated, concentrated in vacuum, and purified by silica gel chromatography (DCM:MeOH = 10:1) to give 49b in yield of 46%.
4.2.15. General procedure for 39b and 40b
NXS (NCS for 39b, NBS for 40b, 9.0 mmol) was slowly added to the solution of ethyl (triphenylphosphoranylidene)acetate (3.132 g, 9.0 mmol) in DCM (15 mL) at −20 °C under Ar atmosphere. The mixture was stirred at −20 °C for 1 h and then warmed to room temperature. 2-Naphthaldehyde (780 mg, 5 mmol) and K2CO3 (1.725 g, 12.5 mmol) were added to the mixture. After stirring at room temperature for 10 h, the solution was washed with water, dried (Na2SO4), filtrated, concentrated under reduced pressure, and further purified by silica gel chromatography (PE:EA = 7:1) to give target compounds 39b and 40b.
4.2.16. Ethyl (E)-2-(naphthalen-2-ylmethylene)butanoate (41b)
NaH (4.2 mmol), followed by dry THF (24 mL), was added to a three-necked flask under Ar atmosphere. After the addition of triethyl 2-phosphonobutyrate (3.9 mmol), the mixture was stirred at room temperature for 0.5 h. Then a solution of 2-naphthaldehyde (3.0 mmol) in THF (12 h) was dropwise added. After stirring at room temperature for 12 h, the reaction mixture was acidified by 2 mol/L HCl and concentrated in vacuum to remove THF. The residue was diluted with water and extracted with EA. The organic layer was washed with saturated NaCl, dried (Na2SO4), filtrated, concentrated under reduced pressure, and purified by silica gel chromatography (PE:EA = 7:1) to give 41b in yield of 60%.
4.2.17. General procedure for 11c‒15c, 16e, 17c‒21c, 22e, 23e, 24c‒26c, 27g, 28f, 29b, 30c, 31c, 32e, 33e, 34e, 35d, 39c, 40c, and 41c
Corresponding ester (2.0 mmol) was dissolved in a mixture of THF (6 mL) and MeOH (6 mL). Then 2 mol/L NaOH (8 mL) was added. The mixture was allowed to react at 40 °C for about 5 h. After cooling to room temperature, the mixture was acidified with 1 mol/L HCl and filtered. The solid was washed with some water and dried to give target precursors.
4.2.18. (E)-3-(Naphthalen-2-yl)acrylic acid (38b)
2-Naphthaldehyde (2.0 mmol) and malonic acid (6.0 mmol) were suspended in DMF (1.2 mL). After the addition of pyridine (2.0 mmol), the mixture was heated to 90 °C for 5 h. Then the reaction mixture was cooled to room temperature, diluted with water (1.2 mL), and acidified with 1 mol/L HCl. The resulted solid was separated by filtration, washed with water, and dried to give 38b in yield of 80%, which was used in next step without further purification.
4.2.19. General procedure for compounds 11‒31, 33‒35, 38‒49, 51, and 53‒55
Appropriate acid (2 mmol) was suspended in anhydrous DCM (6 mL), and the solution was cooled to 0 °C. The (COCl)2 (0.5 mL, 6 mmol) followed by a drop of DMF was added dropwise. After the addition, the reaction mixture was stirred at room temperature for 3 h. The solvent was evaporated in vacuum to give the intermediate acyl chloride. Appropriate arylamine (2 mmol) was dissolved in dichloromethane (4 mL) and triethylamine (303 mg, 3 mmol). The obtained acyl chloride was dissolved in 2 mL DCM and added to the reaction mixture dropwise at 0 °C. The resulted solution was further stirred at room temperature overnight. The reaction mixture was washed with 1 mol/L HCl and brine. The resulted suspension of solid was filtered and washed with water. The resulted solid was further crystallized from DCM–MeOH to give target compounds.
4.2.20. (E)-2-(2-Methyl-3-(1-methyl-1H-indol-2-yl)acrylamido)benzoic acid (32)
32e (1.0 mmol), TsCl (1.1 mmol), dry DCE (5 mL), DMAP (0.1 mmol), and TEA (1.5 mmol) were added to round bottom flask. After the addition, the reaction mixture was stirred at 35 °C for 1 h. Then 2-aminobenzoic acid was added and the resulted mixture was allowed to react at 60 °C for another 10 h. The solution was acidified with 1 mol/L HCl, filtered off, washed with water, dried and further purified by silica gel chromatography (DCM:MeOH = 5:1) to give 32 in yield of 50%.
4.2.21. (E)-4-(2-Methyl-3-(naphthalen-2-yl)acrylamido)nicotinic acid (50)
Compound 24c (2 mmol) was suspended in anhydrous DCM (6 mL), and the solution was cooled to 0 °C. Then (COCl)2 (0.5 mL, 6 mmol) followed by a drop of DMF was added dropwise. After the addition, the reaction mixture was stirred at room temperature for 3 h. The solvent was evaporated in vacuum to give the intermediate acyl chloride. 4-Aminonicotinic acid (2 mmol) was dissolved in pyridine (4 mL). The obtained acyl chloride was dissolved in 2 mL pyridine and added to the reaction mixture dropwise at 0 °C. The resulted solution was further stirred at room temperature overnight. The reaction mixture was diluted with water (10 mL) and filtered. The resulted solid was washed with water, dried, and further purified by silica gel chromatography (DCM:MeOH = 3:1) to give 50 in yield of 30%.
4.2.22. Methyl (E)-2-(2-methyl-3-(naphthalen-2-yl)acrylamido)benzoate (52)
Compound 24 (1 mmol) was suspended in anhydrous DCM (5 mL), and the solution was cooled to 0 °C. Then (COCl)2 (3 mmol) followed by a drop of DMF and MeOH (1.5 mmol) were added dropwise. After the addition, the reaction mixture was stirred at room temperature for 12 h. The reaction mixture was concentrated in reduced pressure and further purified by silica gel chromatography (PE:EA = 3:1) to give 52 in yield of 40%.
4.3. Molecular docking
The X-ray crystal structure of DHODH in complex with a close analog of 3 (brequinar) (PDB ID: 1D3G) was downloaded from the Protein Data Bank and used for protein preparation using the Protein Preparation Wizard in Maestro (Schrödinger, Inc., version 10.2, New York, NY, USA). The grid-enclosing box was centered on the centroid of 3 and defined so as to enclose residues located within 20 Å around the ubiquinone binding site, and a scaling factor of 1.0 was set to van der Waals (VDW) radii of those receptor atoms with the partial atomic charge less than 0.25. Compounds were prepared using the LigPrep module with a pH of 7.0 ± 2.0 for Epik. Standard precision (SP) mode in Glide with default settings was used to dock the ligands. The docked poses were ranked by Gscore, and the one with the lowest binding energy was selected for binding mode analysis.
4.4. In vitro enzyme assay
Expression and purification of recombined DHODH were followed the protocols of previously published literatures44,56. The DHODH inhibition assays were carried out using the DCIP-based assay43,44. The assay buffer contained 50 mmol/L HEPES pH 8.0, 150 mmol/L KCl, 0.1% Triton X-100. The purified DHODH was diluted into a final concentration of 10 nmol/L with the assay buffer. UQ0 and DCIP were added to the assay buffer to final concentrations of 100 and 120 μmol/L, respectively. The dihydroorotate was added to a final concentration of 500 μmol/L to initiate the reaction. Inhibition studies were performed in this assay with additional various amounts of inhibitors. Brequinar was measured as the positive control. The inhibition rate was calculated from (1‒Vi/V0) × 100. For the determination of the IC50 values, eight to nine different concentrations were applied. Each inhibitor concentration point was tested in triplicate. IC50 values were calculated using the sigmoidal fitting option of the program Origin 8.0 (OriginLab Corp., Northampton, MA, USA).
4.5. In vivo PK assay
Compound 54, the most potent inhibitor, was selected to perform the single dose PK studies. Male Sprague–Dawley rats were raised according to the guide for the care and use of laboratory animals. Blood samples of the rats were collected at the intervals of 5 min, 15 min, 30 min, 1 h, 2 h, 4 h, 8 h and 24 h. Heparin sodium was applied as the anticoagulant of the blood samples. Based on the LC/MS/MS quantitative data, PK parameters were analyzed using WinNonlin 7.0 (Certara Corp., Princeton, NJ, USA) with noncompartment model. The PK study was conducted by using Medicilon's preclinical pharmacokinetic study services (Shanghai Medicilon Inc., Shanghai, China).
4.6. In vivo efficacy study
Adult male Wistar rats (130–150 g) were purchased from Shanghai SLRC Laboratory animal company and housed in standard pathogen-free cages. The environment was climate-controlled (20–25 °C, 50%–60% relative humidity) with a 12-h light and 12-h dark cycle as described previously44. Sterilized food and water were provided naturally. All procedures involving animals conform to the Chinese government guidelines for animal experiments and were conducted with approval of the Institutional Animal Care and Use Committee of East China University of Science & Technology, Shanghai, China. Bovine type II collagen (CII, Chondrex Inc., Redmond, WA, USA) was emulsified in the same manner as that conducted previously to get the CIA model61. Specifically, 40 rats were randomly assigned into five groups: normal control (solvent, i.p., n = 10), CIA model (solvent, i.p., n = 10), methotrexate-treated (0.3 mg/kg, i.p., n = 10), compound 54-treated (10 mg/kg, i.p., n = 10) and compound 54-treated (30 mg/kg, i.p., n = 10) groups. Methotrexate was used as a positive control in this study. The main workflows of this experiment were consistent with that reported recently44. During the 28 days of the experiment, compound 54 (10 and 30 mg/kg) and methotrexate (0.3 mg/kg) were administered orally once a day. At the same time, the normal control group and model group were administered the same amount of 0.5% CMC-Na solution.
In this study, the clinical-mimic arthritis was observed and the severity was assessed by the criteria as below: 0, normal, without any swelling; 1, mild swelling; 2, swelling; 3, significant swelling; 4, severe swelling. When arthritic signs appeared, the incidence and severity of the arthritis in rats with different treatments were evaluated via arthritis scores recorded every day under blinded conditions. Measurement of body weight was taken every 3 days. All Wistar rats were killed after 28-day trails. Pathological detections against the whole knee joints of the rats were conducted using the same method as described previously44. Mean ± standard error of the mean (mean ± SEM) was applied to present the data in this study. SPSS was used to evaluate the statistical significance of differences among the groups. P ≤ 0.05 was the threshold to be statistically significant62.
Acknowledgments
This work was supported by the National Key Research and Development Program (2017YFD0200505 to Xiaoyong Xu, 2016YFA0502304 to Honglin Li, China); the National Natural Science Foundation of China (81825020 to Honglin Li, 81803437 to Shiliang Li); the National Science & Technology Major Project “Key New Drug Creation and Manufacturing Program” (2018ZX09711002, China); the Fundamental Research Funds for the Central Universities; and the Shanghai Foundation of Science and Technology (15431902100 to Xiaoyong Xu). Shiliang Li is also sponsored by Shanghai Sailing Program (No. 18YF1405100, China). Honglin Li is also sponsored by the National Program for Special Supports of Eminent Professionals and National Program for Support of Top-Notch Young Professionals, China. We thank Dr. Wei Wang for English proofreading of this manuscript.
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
This paper is dedicated to Prof. Youyou Tu, the 2015 Nobel Prize laureate of Physiology or Medicine on the occasion of her 90th birthday.
Supporting information to this article can be found online at https://doi.org/10.1016/j.apsb.2020.10.008.
Contributor Information
Rui Wang, Email: ruiwang@ecust.edu.cn.
Lili Zhu, Email: zhulfl@ecust.edu.cn.
Honglin Li, Email: hlli@ecust.edu.cn.
Xiaoyong Xu, Email: xyxu@ecust.edu.cn.
Author contributions
Fanxun Zeng, Shiliang Li, GuantianYang and Yating Luo performed research and drafted the manuscript, they contributed equally to the study. Fanxun Zeng, Guantian Yang and Letian Zhang synthesized the compounds. Shiliang Li and Honglin Li designed the compounds, performed the computational work and interpreted the data. Tiantian Qi and Lili Zhu tested the in vitro bioactivities of the compounds; Yating Luo, Yingfan Liang, Tingyuan Yang, and Rui Wang were in charge of the in vivo antiarthritic effect of compound 54 and the acute toxicity study. Xiaoyong Xu and Honglin Li conceived and supervised the overall project. All authors revised and approved the manuscript being submitted.
Conflicts of interest
The authors have no conflicts of interest to declare.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Firestein G.S., McInnes I.B. Immunopathogenesis of rheumatoid arthritis. Immunity. 2017;46:183–196. doi: 10.1016/j.immuni.2017.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aletaha D., Neogi T., Silman A.J., Funovits J., Felson D.T., Bingham C.O. 2010 Rheumatoid arthritis classification criteria: an American college of rheumatology/European League against rheumatism collaborative initiative. Arthritis Rheum. 2010;62:2569–2581. doi: 10.1002/art.27584. [DOI] [PubMed] [Google Scholar]
- 3.Firestein G.S. Evolving concepts of rheumatoid arthritis. Nature. 2003;423:356–361. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
- 4.Helmick C.G., Felson D.T., Lawrence R.C., Gabriel S., Hirsch R., Kwoh C.K. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States: Part I. Arthritis Rheum. 2008;58:15–25. doi: 10.1002/art.23177. [DOI] [PubMed] [Google Scholar]
- 5.McInnes I.B., Thompson L., Giles J.T., Bathon J.M., Salmon J.E., Beaulieu A.D. Effect of interleukin-6 receptor blockade on surrogates of vascular risk in rheumatoid arthritis: MEASURE, a randomised, placebo-controlled study. Ann Rheum Dis. 2015;74:694–702. doi: 10.1136/annrheumdis-2013-204345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ziarko M., Siemiatkowska K., Sienski M., Samborski W., Samborska J., Mojs E. Mental health and rheumatoid arthritis: toward understanding the emotional status of people with chronic disease. BioMed Res Int. 2019;2019 doi: 10.1155/2019/1473925. 1473925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takeuchi T. Biomarkers as a treatment guide in rheumatoid arthritis. Clin Immunol. 2018;186:59–62. doi: 10.1016/j.clim.2017.07.019. [DOI] [PubMed] [Google Scholar]
- 8.Lamichhane D., Collins C., Constantinescu F., Walitt B., Pettinger M., Parks C. Coffee and tea consumption in relation to risk of rheumatoid arthritis in the women's health initiative observational cohort. J Clin Rheumatol. 2019;25:127–132. doi: 10.1097/RHU.0000000000000788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abhishek N., Parveen S., Vishnupriya V., Gayathri R. Treatment models for rheumatoid arthritis—a review. J Pharmaceut Sci Res. 2016;8:520–524. [Google Scholar]
- 10.Bullock J., Rizvi S.A.A., Saleh A.M., Ahmed S.S., Do D.P., Ansari R.A. Rheumatoid arthritis: a brief overview of the treatment. Med Princ Pract. 2018;27:501–507. doi: 10.1159/000493390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Vollenhoven R.F. Treatment of rheumatoid arthritis: state of the art 2009. Nat Rev Rheumatol. 2009;5:531–541. doi: 10.1038/nrrheum.2009.182. [DOI] [PubMed] [Google Scholar]
- 12.Singh J.A., Saag K.G., Bridges S.L., Akl E.A., Bannuru R.R., Sullivan M.C. 2015 American college of rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Rheum. 2016;68:1–26. doi: 10.1002/art.39480. [DOI] [PubMed] [Google Scholar]
- 13.Ramiro S., Sepriano A., Chatzidionysiou K., Nam J.L., Smolen J.S., van der Heijde D. Safety of synthetic and biological DMARDs: a systematic literature review informing the 2016 update of the EULAR recommendations for management of rheumatoid arthritis. Ann Rheum Dis. 2017;76:1101–1136. doi: 10.1136/annrheumdis-2016-210708. [DOI] [PubMed] [Google Scholar]
- 14.Clark J.D., Flanagan M.E., Telliez J.B. Discovery and development of Janus kinase (JAK) inhibitors for inflammatory diseases: miniperspective. J Med Chem. 2014;57:5023–5038. doi: 10.1021/jm401490p. [DOI] [PubMed] [Google Scholar]
- 15.Lou Y., Han X., Kuglstatter A., Kondru R.K., Sweeney Z.K., Soth M. Structure-based drug design of RN486, a potent and selective Bruton's tyrosine kinase (BTK) inhibitor, for the treatment of rheumatoid arthritis. J Med Chem. 2014;58:512–516. doi: 10.1021/jm500305p. [DOI] [PubMed] [Google Scholar]
- 16.Vyas V.K., Ghate M. Recent developments in the medicinal chemistry and therapeutic potential of dihydroorotate dehydrogenase (DHODH) inhibitors. Mini Rev Med Chem. 2011;11:1039–1055. doi: 10.2174/138955711797247707. [DOI] [PubMed] [Google Scholar]
- 17.Nørager S., Jensen K.F., Björnberg O., Larsen S. E. coli Dihydroorotate dehydrogenase reveals structural and functional distinctions between different classes of dihydroorotate dehydrogenases. Structure. 2002;10:1211–1223. doi: 10.1016/s0969-2126(02)00831-6. [DOI] [PubMed] [Google Scholar]
- 18.Krungkrai J. Purification, characterization and localization of mitochondrial dihydroorotate dehydrogenase in Plasmodium falciparum, human malaria parasite. Biochim Biophys Acta. 1995;1243:351–360. doi: 10.1016/0304-4165(94)00158-t. [DOI] [PubMed] [Google Scholar]
- 19.Jiang W., Locke G., Harpel M.R., Copeland R.A., Marcinkeviciene J. Role of Lys 100 in human dihydroorotate dehydrogenase: mutagenesis studies and chemical rescue by external amines. Biochemistry. 2000;39:7990–7997. doi: 10.1021/bi000630d. [DOI] [PubMed] [Google Scholar]
- 20.Munier-Lehmann H., Vidalain P.O., Tangy F., Janin Y.L. On dihydroorotate dehydrogenases and their inhibitors and uses. J Med Chem. 2013;56:3148–3167. doi: 10.1021/jm301848w. [DOI] [PubMed] [Google Scholar]
- 21.Löffler M., Fairbanks L.D., Zameitat E., Marinaki A.M., Simmonds H.A. Pyrimidine pathways in health and disease. Trends Mol Med. 2005;11:430–437. doi: 10.1016/j.molmed.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 22.Li L., Ng S.R., Colón C.I., Drapkin B.J., Hsu P.P., Li Z. Identification of DHODH as a therapeutic target in small cell lung cancer. Sci Transl Med. 2019;11 doi: 10.1126/scitranslmed.aaw7852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Christian S., Merz C., Evans L., Gradl S., Seidel H., Friberg A. The novel dihydroorotate dehydrogenase (DHODH) inhibitor BAY 2402234 triggers differentiation and is effective in the treatment of myeloid malignancies. Leukemia. 2019;33:2403–2415. doi: 10.1038/s41375-019-0461-5. [DOI] [PubMed] [Google Scholar]
- 24.Alcorn N., Saunders S., Madhok R. Benefit-risk assessment of leflunomide. Drug Saf. 2009;32:1123–1134. doi: 10.2165/11316650-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 25.White R.M., Cech J., Ratanasirintrawoot S., Lin C.Y., Rahl P.B., Burke C.J. DHODH modulates transcriptional elongation in the neural crest and melanoma. Nature. 2011;471:518–522. doi: 10.1038/nature09882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Silva H.T., Morris R.E. Leflunomide and malononitrilamides. Am J Med Sci. 1997;313:289–301. doi: 10.1097/00000441-199705000-00008. [DOI] [PubMed] [Google Scholar]
- 27.Makowka L., Sher L.S., Cramer D.V. The development of brequinar as an immunosuppressive drug for transplantation. Immunol Rev. 1993;136:51–70. doi: 10.1111/j.1600-065x.1993.tb00654.x. [DOI] [PubMed] [Google Scholar]
- 28.Maroun J., Ruckdeschel J., Natale R., Morgan R., Dallaire B., Sisk R. Multicenter phase II study of brequinar sodium in patients with advanced lung cancer. Canc Chemother Pharmacol. 1993;32:64–66. doi: 10.1007/BF00685878. [DOI] [PubMed] [Google Scholar]
- 29.Gummert J.F., Ikonen T., Morris R.E. Newer immunosuppressive drugs: a review. J Am Soc Nephrol. 1999;10:1366–1380. doi: 10.1681/ASN.V1061366. [DOI] [PubMed] [Google Scholar]
- 30.Chan A., de Seze J., Comabella M. Teriflunomide in patients with relapsing–remitting forms of multiple sclerosis. CNS Drugs. 2016;30:41–51. doi: 10.1007/s40263-015-0299-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Díaz-Borjón A. Guidelines for the use of conventional and newer disease-modifying antirheumatic drugs in elderly patients with rheumatoid arthritis. Drugs Aging. 2009;26:273–293. doi: 10.2165/00002512-200926040-00001. [DOI] [PubMed] [Google Scholar]
- 32.Cody R., Stewart D., DeForni M., Moore M., Dallaire B., Azarnia N. Multicenter phase II study of brequinar sodium in patients with advanced breast cancer. Am J Clin Oncol. 1993;16:526–528. doi: 10.1097/00000421-199312000-00014. [DOI] [PubMed] [Google Scholar]
- 33.Dexter D.L., Hesson D.P., Ardecky R.J., Rao G.V., Tippett D.L., Dusak B.A. Activity of a novel 4-quinolinecarboxylic acid, NSC 368390 [6-fluoro-2-(2′-fluoro-1,1′-biphenyl-4-yl)-3-methyl-4-quinolinecarboxylic acid sodium salt], against experimental tumors. Canc Res. 1985;45:5563–5568. [PubMed] [Google Scholar]
- 34.Cramer D.V., Chapman F.A., Jaffee B.D., Jones E.A., Knoop M., Hreha-Eiras G. The effect of a new immunosuppressive drug, brequinar sodium, on heart, liver, and kidney allograft rejection in the rat. Transplantation. 1992;53:303–307. doi: 10.1097/00007890-199202010-00009. [DOI] [PubMed] [Google Scholar]
- 35.Herrlinger K., Diculescu M., Fellermann K., Hartmann H., Howaldt S., Nikolov R. Efficacy, safety and tolerability of vidofludimus in patients with inflammatory bowel disease: the ENTRANCE study. J Crohn’s Colitis. 2013;7:636–643. doi: 10.1016/j.crohns.2012.09.016. [DOI] [PubMed] [Google Scholar]
- 36.Rusai K., Schmaderer C., Baumann M., Chmielewski S., Prókai Á., Kis E. Immunosuppression with 4SC-101, a novel inhibitor of dihydroorotate dehydrogenase, in a rat model of renal transplantation. Transplantation. 2012;93:1101–1107. doi: 10.1097/TP.0b013e31824fd861. [DOI] [PubMed] [Google Scholar]
- 37.Muehler A., Peelen E., Kohlhof H., Groppel M., Vitt D. Vidofludimus calcium, a next generation DHODH inhibitor for the treatment of relapsing-remitting multiple sclerosis. Mult Scler Relat Disord. 2020;43:102129. doi: 10.1016/j.msard.2020.102129. [DOI] [PubMed] [Google Scholar]
- 38.Fitzpatrick L.R., Small J.S., Doblhofer R., Ammendola A. Vidofludimus inhibits colonic interleukin-17 and improves hapten-induced colitis in rats by a unique dual mode of action. J Pharmacol Exp Therapeut. 2012;342:850–860. doi: 10.1124/jpet.112.192203. [DOI] [PubMed] [Google Scholar]
- 39.Kulkarni O.P., Sayyed S.G., Kantner C., Ryu M., Schnurr M., Sárdy M. 4SC-101, a novel small molecule dihydroorotate dehydrogenase inhibitor, suppresses systemic lupus erythematosus in MRL-(Fas) lpr mice. Am J Pathol. 2010;176:2840–2847. doi: 10.2353/ajpath.2010.091227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fitzpatrick L.R., Deml L., Hofmann C., Small J.S., Groeppel M., Hamm S. 4SC-101, a novel immunosuppressive drug, inhibits IL-17 and attenuates colitis in two murine models of inflammatory bowel disease. Inflamm Bowel Dis. 2010;16:1763–1777. doi: 10.1002/ibd.21264. [DOI] [PubMed] [Google Scholar]
- 41.Sykes D.B., Kfoury Y.S., Mercier F.E., Wawer M.J., Law J.M., Haynes M.K. Inhibition of dihydroorotate dehydrogenase overcomes differentiation blockade in acute myeloid leukemia. Cell. 2016;167:171–186. doi: 10.1016/j.cell.2016.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Diao Y., Lu W., Jin H., Zhu J., Han L., Xu M. Discovery of diverse human dihydroorotate dehydrogenase inhibitors as immunosuppressive agents by structure-based virtual screening. J Med Chem. 2012;55:8341–8349. doi: 10.1021/jm300630p. [DOI] [PubMed] [Google Scholar]
- 43.Li S., Luan G., Ren X., Song W., Xu L., Xu M. Rational design of benzylidenehydrazinyl-substituted thiazole derivatives as potent inhibitors of human dihydroorotate dehydrogenase with in vivo anti-arthritic activity. Sci Rep. 2015;5:14836–14855. doi: 10.1038/srep14836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu J., Han L., Diao Y., Ren X., Xu M., Xu L. Design, synthesis, X-ray crystallographic analysis, and biological evaluation of thiazole derivatives as potent and selective inhibitors of human dihydroorotate dehydrogenase. J Med Chem. 2015;58:1123–1139. doi: 10.1021/jm501127s. [DOI] [PubMed] [Google Scholar]
- 45.Song W., Li S., Tong Y., Wang J., Quan L., Chen Z. Structure-based design of potent human dihydroorotate dehydrogenase inhibitors as anticancer agents. MedChemComm. 2016;7:1441–1448. [Google Scholar]
- 46.Madak J.T., Cuthbertson C.R., Miyata Y., Tamura S., Petrunak E.M., Stuckey J.A. Design, synthesis, and biological evaluation of 4-quinoline carboxylic acids as inhibitors of dihydroorotate dehydrogenase. J Med Chem. 2018;61:5162–5186. doi: 10.1021/acs.jmedchem.7b01862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sainas S., Pippione A.C., Lupino E., Giorgis M., Circosta P., Gaidano V. Targeting myeloid differentiation using potent 2-hydroxypyrazolo [1,5-a] pyridine scaffold-based human dihydroorotate dehydrogenase inhibitors. J Med Chem. 2018;61:6034–6055. doi: 10.1021/acs.jmedchem.8b00373. [DOI] [PubMed] [Google Scholar]
- 48.Lolli M.L., Sainas S., Pippione A.C., Giorgis M., Boschi D., Dosio F. Use of human dihydroorotate dehydrogenase (hDHODH) inhibitors in autoimmune diseases and new perspectives in cancer therapy. Recent Pat Anti-Cancer Drug Discov. 2018;13:86–105. doi: 10.2174/1574892812666171108124218. [DOI] [PubMed] [Google Scholar]
- 49.Xiong R., Zhang L., Li S., Sun Y., Ding M., Wang Y. Novel and potent inhibitors targeting DHODH are broad-spectrum antivirals against RNA viruses including newly-emerged coronavirus SARS-CoV-2. Protein Cell. 2020;11:723–739. doi: 10.1007/s13238-020-00768-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brown K.K., Spinelli J.B., Asara J.M., Toker A. Adaptive reprogramming of de novo pyrimidine synthesis is a metabolic vulnerability in triple-negative breast cancer. Canc Discov. 2017;7:391–399. doi: 10.1158/2159-8290.CD-16-0611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mathur D., Stratikopoulos E., Ozturk S., Steinbach N., Pegno S., Schoenfeld S. PTEN regulates glutamine flux to pyrimidine synthesis and sensitivity to dihydroorotate dehydrogenase inhibition. Canc Discov. 2017;7:380–390. doi: 10.1158/2159-8290.CD-16-0612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koundinya M., Sudhalter J., Courjaud A., Lionne B., Touyer G., Bonnet L. Dependence on the pyrimidine biosynthetic enzyme DHODH is a synthetic lethal vulnerability in mutant KRAS-driven cancers. Cell Chem Biol. 2018;25:705–717. doi: 10.1016/j.chembiol.2018.03.005. [DOI] [PubMed] [Google Scholar]
- 53.Bajzikova M., Kovarova J., Coelho A.R., Boukalova S., Oh S., Rohlenova K. Reactivation of dihydroorotate dehydrogenase-driven pyrimidine biosynthesis restores tumor growth of respiration-deficient cancer cells. Cell Metabol. 2019;29:399–416. doi: 10.1016/j.cmet.2018.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hosseini M., Dousset L., Mahfouf W., Serrano-Sanchez M., Redonnet-Vernhet I., Mesli S. Energy metabolism rewiring precedes UVB-induced primary skin tumor formation. Cell Rep. 2018;23:3621–3634. doi: 10.1016/j.celrep.2018.05.060. [DOI] [PubMed] [Google Scholar]
- 55.Yi J., He Z., Xu S., Feng S. Efficacy and safety of leflunomide in IgA nephropathy: a systematic review and meta-analysis. Int Urol Nephrol. 2019;51:1987–1998. doi: 10.1007/s11255-019-02255-6. [DOI] [PubMed] [Google Scholar]
- 56.Liu S., Neidhardt E.A., Grossman T.H., Ocain T., Clardy J. Structures of human dihydroorotate dehydrogenase in complex with antiproliferative agents. Structure. 2000;8:25–33. doi: 10.1016/s0969-2126(00)00077-0. [DOI] [PubMed] [Google Scholar]
- 57.Fritzson I., Svensson B., Al-Karadaghi S., Walse B., Wellmar U., Nilsson U.J. Inhibition of human DHODH by 4-hydroxycoumarins, fenamic acids, and N-(alkylcarbonyl)anthranilic acids identified by structure-guided fragment selection. ChemMedChem. 2010;5:608–617. doi: 10.1002/cmdc.200900454. [DOI] [PubMed] [Google Scholar]
- 58.Zeng F., Qi T., Li C., Li T., Li H., Li S. Synthesis, structure–activity relationship and binding mode analysis of 4-thiazolidinone derivatives as novel inhibitors of human dihydroorotate dehydrogenase. MedChemComm. 2017;8:1297–1302. doi: 10.1039/c7md00081b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Heikkila T., Thirumalairajan S., Davies M., Parsons M.R., McConkey A.G., Fishwick C.W. The first de novo designed inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. Bioorg Med Chem Lett. 2006;16:88–92. doi: 10.1016/j.bmcl.2005.09.045. [DOI] [PubMed] [Google Scholar]
- 60.Brand D.D., Latham K.A., Rosloniec E.F. Collagen-induced arthritis. Nat Protoc. 2007;2:1269–1275. doi: 10.1038/nprot.2007.173. [DOI] [PubMed] [Google Scholar]
- 61.Yue R., Zhao L., Hu Y., Jiang P., Wang S., Xiang L. Rapid-resolution liquid chromatography TOF-MS for urine metabolomic analysis of collagen-induced arthritis in rats and its applications. J Ethnopharmacol. 2013;145:465–475. doi: 10.1016/j.jep.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 62.Rosillo M.Á., Alcaraz M.J., Sánchez-Hidalgo M., Fernández-Bolaños J.G., Alarcón-De-La-Lastra C., Ferrándiz M.L. Anti-inflammatory and joint protective effects of extra-virgin olive-oil polyphenol extract in experimental arthritis. J Nutr Biochem. 2014;25:1275–1281. doi: 10.1016/j.jnutbio.2014.07.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.