

Abstract
Longitudinal bone growth is driven by endochondral ossification, a process in which cartilage tissue is generated by growth plate chondrocytes and then remodeled into bone by osteoblasts. In the postnatal growth plate, as hypertrophic chondrocytes approach the chondro-osseous junction, they may undergo apoptosis, or directly transdifferentiate into osteoblasts. The molecular mechanisms governing this switch in cell lineage are poorly understood. Here we show that the physiological downregulation of Sox9 in hypertrophic chondrocyte is associated with upregulation of osteoblast-associated genes (such as Mmp13, Cola1, Ibsp) in hypertrophic chondrocytes, before they enter the metaphyseal bone. In transgenic mice that continued to express Sox9 in all cells derived from the chondrocytic lineage, upregulation of these osteoblast-associated genes in the hypertrophic zone failed to occur. Furthermore, lineage tracing experiments showed that, in transgenic mice expressing Sox9, the number of chondrocytes transdifferentiating into osteoblasts was markedly reduced. Collectively, our findings suggest that Sox9 downregulation in hypertrophic chondrocytes promotes expression of osteoblast-associated genes in hypertrophic chondrocytes and promotes the subsequent transdifferentiation of these cells into osteoblasts.
Introduction
Some bones in the mammalian skeleton, such as the skull vault bones and other flat bones, form through intramembranous bone formation, which involves direct differentiation of mesenchymal stem cells into bone-forming osteoblasts. In contrast, long bones such as the humerus, femur and tibia form by endochondral ossification [1]. In the embryo, these structure form first as mesenchymal stem cell (MSC) condensations. The MSCs differentiate into cartilage-forming chondrocytes, and their proliferation results in expansion of this cartilage mold. Then, at the center of the cartilage structures, cells stop proliferating and undergo hypertrophic differentiation. Some of these hypertrophic chondrocytes undergo apoptosis [2], leaving a scaffold of cartilage matrix for invasion of blood vessels, osteoblasts, and osteoclasts, which remodel the cartilage matrix into bone. The same process of endochondral ossification persists into postnatal life but becomes confined to an area near the ends of long bones known as the growth plate. In the growth plate, chondrogenesis continues through chondrocyte proliferation, hypertrophy, and cartilage matrix secretion, and the newly formed cartilage is remodeled into bone. The net result is bone elongation.
Previously it was thought that all hypertrophic chondrocytes undergo apoptosis as they approach the chondro-osseous junction of the growth plate [3, 4] and that the osteoblasts below the chondro-osseous junction were all derived directly from MSCs or bone marrow stromal/mesenchymal progenitor cells (BMSCs) in the metaphysis [5, 6]. However, several recent lineage-tracing studies showed that many osteoblasts in the trabecular bone near the growth plate originated from chondrogenic cells in the growth plate [7–9], suggesting that many terminal hypertrophic chondrocytes transdifferentiated into osteoblasts rather than undergoing apoptosis. The molecular mechanisms that regulate chondrocyte-osteoblast transdifferentiation remain poorly elucidated.
The Sry-related transcription factor Sox9 is a key regulator of early chondrogenesis. It is expressed in mesenchymal condensations as well as in all chondroprogenitor cells during development [10]. Sox9 regulates a large number of genes important for chondrogenesis [11, 12], including extracellular matrix protein genes Col2a1, Col9a1, Col11a2, and Acan [13]. Previous studies have shown that, in the growth plate, Sox9 is expressed in the resting, proliferative, and prehypertrophic zone, but is downregulated in the hypertrophic zone [14]. Cartilage-specific Sox9 deletion resulted in shortened hypertrophic zones and early ossification [15]. In contrast, overexpression of Sox9 in hypertrophic zone driven by a BAC-Col10a1 promoter resulted in expanded hypertrophic zones due to retarded vascular invasion and delayed ossification [16]. These previous studies strongly suggested that Sox9 plays an important role in the regulation of chondrocyte hypertrophic maturation and ossification. In the current study, we used a mouse model in which the physiological downregulation of Sox9 in hypertrophic chondrocytes was prevented and found evidence that the normal downregulation of Sox9 serves specifically to promote transdifferentiation of chondrocytes into osteoblasts at the cartilage-bone junction.
Materials and Methods
Animals
All animal procedures were approved by the National Institute of Child Health and Human Development Animal Care and Use Committee. C57BL/6 mice were obtained from Charles River Laboratory. Constitutive Col2a1cre mice were previously described [17]. Tamoxifen-inducible Col2a1cre mice (Stock number: 006774) and CAG-GFP mice (Stock number: 008705) were obtained from Jackson Laboratory, and CAG-Sox9-GFP mice were kindly provided by Dr. Richard Behringer from University of Texas MD Anderson Cancer Center [18]. All animals used in this study were pathogen-free and they were housed ≤6 per cage and provided with food (regular chow) and water ad libitum. For studying the effect of Sox9 on osteoblastic gene expression, heterozygous CAG-Sox9-GFP mice were crossed with homozygous constitutive Col2a1cre and gene expression were compared between Col2a1cre+/−CAG-Sox9-GFP+/− and Col2a1cre+/− only. For lineage tracing experiments, Tamoxifen-inducible Col2a1cre mice were crossed with either CAG-Sox9-GFP mice or CAG-GFP mice, and the double heterozygous mice (Col2a1creER+/−CAG-SOX9-GFP+/− or Col2a1creER+/−CAG-GFP+/−) were used for tamoxifen injection. A single dose of tamoxifen (50 μg/g body weight) was injected intraperitoneally at 4 days of age, and the mice were killed at 2- and 4-weeks of age for histological examination. A combination of male and female mice were used for all experiments except that cultured chondrocyte were isolated only from 1-wk old wilde-type male mice.
Growth Plate Microdissection
After animals were sacrificed, tibial epiphyses were excised. For laser capture microdissection (LCM), cartilage was embedded in optimum cutting temperature (O.C.T.) compound (Electron Microscopy Sciences, Hatfield, PA), frozen on dry ice, and stored at –80°C. LCM of growth plate cartilage was performed as previously described [19].
RNA Extraction and Purification
RNA was extracted using an RNeasy Micro Kit (QIAGEN, Valencia, CA). All RNA samples had a 260/280 nm ratio between 1.8 and 2.1. RNA integrity was determined using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA) and only high-quality RNA (RIN>7) was used for quantitative real-time RT-PCR (qPCR).
Quantitative PCR
Real-time RT-PCR was used to assess mRNA levels in specific zones of individual growth plates at various ages. Total RNA (50–100 ng) was reverse-transcribed using SuperScript IV Reverse Transcriptase (Invitrogen). Quantitative real-time PCR was performed as previously described [20] using commercially available FAM- or VIC-labeled Taqman assays (Applied Biosystems, Foster City, CA, USA). A list of all Taqman probes used is given in the supplemental information. Reactions were performed in triplicate on cDNA derived from individual animals using the ABI QuantStudio 6 Flex System instrument (Applied Biosystems). The relative quantity of each mRNA was calculated using the formula Relative Expression = 2−ΔCt × 106, where Ct represents the threshold cycle and ΔCt = (Ct of gene of interest) − (Ct of 18S rRNA). Values were multiplied by 106 for convenience of comparison.
In situ hybridization
In situ hybridization was performed as described previously [17]. Briefly, riboprobes for collagen X and for Sox9 were generated by PCR using mouse growth plate cDNA as template and primers that contained an SP6 promoter. Single-stranded digoxigenin-labeled riboprobe for in situ hybridization was transcribed using a DIG RNA Labeling Kit (Roche Diagnostics) following the manufacturer’s protocol. Riboprobes were purified by Micro Bio-Spin Columns P-30 Tris RNase free (Bio-Rad). Paraffin-embedded sections of epiphyseal cartilage from 1-week-old mice were hybridized to digoxigenin-labeled riboprobes. For detection, tissue sections were incubated with anti-digoxigenin alkaline phosphatase Fab fragments (Roche) for 2 hours at room temperature and treated with NBT/BCIP (Sigma) in the dark until a colorimetric change was detected. Sections were counter stained with 10% eosin and visualized using a ScanScope CS digital scanner (Aperio Technologies, Inc) under bright field microscopy.
Fluorescence Microscopy
After animals were sacrificed, tibial and femoral epiphyses were excised and fixed overnight in formalin at 4°C and decalcified in 10% (w/v) EDTA, pH 7.4. Decalcified bones were then either used for making paraffin-embedded sections (Histoserv, Germantown, Maryland) or frozen sections. To make frozen sections, samples were cryoprotected by immersing in phosphate-buffered saline (PBS) with 30% (w/v) sucrose overnight and embedded in O.C.T. and frozen on dry ice for sectioning. To detect GFP or Sp7 signal in cartilage and bone, 10μm frozen sections were baked at 65°C, post-fixed in 1% formaldehyde for 10 min, antigen retrieval was performed with treatment of 100 μg/ml proteinase K in PBS for 30 min at room temperature (r.t.) for GFP, or by heating the slides in citrate buffer (pH6.0) to 99°C for 20 min for Sp7 (or double staining of Sp7 and GFP), blocked in 10% goat serum in Tris-buffered saline with 0.1% tween (0.1% TBS-T) and 0.3 M glycine (1 hour, r.t.). Slides were incubated with unconjugated chicken anti-GFP IgY (Thermo Fisher Scientific, 2mg/ml, 1:200) or rabbit anti-Sp7 IgG (ab22552, abcam, 1:500) overnight at 4°C, washed, and AlexaFluor488-conjugated goat anti-chicken IgG(H+L) or AlexaFluor647-conjugated goat anti-rabbit IgG (both from Thermo Fisher Scientific, 2 mg/ml, 1:1000) was used as the secondary antibody. After washing, slides were incubated in DAPI (300nM, in 0.1% TBS-T) for 10 min at r.t. to visualize the cell nuclei and mounted with Prolong Diamond Anti Fade Mountant (Thermo Fisher Scientific) overnight at 4°C. Immunostaining of Sox9 and active Caspase-3 were performed on paraffin-embedded sections. Slides were baked at 65°C, deparaffinized with xylene and rehydrated with ethanol steps. Antigen retrieval was performed with treatment of 100 μg/ml proteinase K in PBS for 30 min at room temperature (r.t.) for caspase-3, or by heating the slides in citrate buffer (pH 6.0) to 99°C for 20 min for Sox9. Slides were then incubated with rabbit anti-Sox9 IgG (Millipore, AB5535) or anti-active Caspase-3 (Abcam, ab2302) as primary antibody and AlexaFluor647-conjugated goat anti-rabbit IgG (Thermo Fisher) as secondary antibody. Slides were scanned with a Keyence BZ-X700 fluorescence microscope (Keyence Corp, Osaka, Japan) at 20X magnification. For quantification of GFP-positive cells, for each animal, fluorescent images were taken from 3 non-consecutive sections and the number of GFP-positive cells in the first 10 μm of metaphysis (measuring from the cartilage-bone junction) were counted across the whole metaphysis, excluding cells in the periosteum. We also counted the number of GFP-positive chondrocyte columns in the growth plate. The observers were blinded to the identity of the animal while taking the GFP counts. The number of GFP-positive cells in the metaphysis was then normalized to the width of the metaphyseal bone (number of GFP-positive cells in trabecular bone per 500 μm). In a separate analysis, the number of GFP-positive cells in the metaphysis was normalized to the number of GFP-positive chondrocyte columns (number of GFP-positive chondrocyte columns per 500 μm, measured parallel to the chondro-osseous junction). Averages were taken for measurements from the 3 sections of the same animal, and 3 animals were used for each time point and genotype.
micro–computed Tomography (μCT)
To study bone mineral density and bone structure, after animals were sacrificed, whole tibias were excised and fixed in formalin for 3 days at 4°C and stored in phosphate-buffered saline. Bone morphology of tibia of 4-week-old Col2a1cre+/−CAG-Sox9-GFP+/− and Col2a1cre+/−−only mice were analyzed by μCT scanning using a SkyScan 1172 (Skyscan, Kontich, Belgium) at 5μm resolution. 3-dimensional reconstruction of scanned datasets were performed using NRecon (Bruker, Billerica, MA) and rotated with DataViewer (Bruker). Regions of interest (ROIs) were selected with reference to a growth plate reference slice. The trabecular and cortical regions were defined as positions along the long axis of the tibia relative to the growth plate reference. The trabecular region commenced 0.15 mm (31 image slices) from the growth plate level in the direction of the metaphysis and extended from this position for a further 1 mm (206 image slices). The cortical region commenced 3 mm (616 image slices) from the growth plate level in the direction of the metaphysis and extended from this position for a further 1 mm (206 image slices). Morphometric analysis of trabecular and cortical bone were performed according to method notes MN094 and MN095 (Bruker user manual), respectively, to obtain measurements such as bone mineral density (BMD) for trabecular and cortical bone, trabecular bone volume per tissue volume (BV/TV), trabecular number (Tb.N), trabecular spacing (Tb.Sp), and cortical thickness (Ct. Th).
Statistics
All data in this study, unless otherwise specify, are presented as mean ± SEM. ANOVA analyses were performed in SigmaPlot 11. One-way ANOVA was used when measuring the effect of a single factor, such as comparing the level of gene expression in the presence and absence of Sox9 siRNA. P-values were corrected for multiple comparisons, whenever applicable, using the Holm-Sidak method.
Result
Spatial downregulation of Sox9 in hypertrophic zone
Many previous studies have already characterized the spatial expression of Sox9 in the growth plate, predominantly using in situ hybridization [9, 16]. In the current study we used laser capture microdissection (LCM) to isolate chondrocytes from different zones of the growth plate and trabecular bone in 1wk old mice, and quantify the spatial expression of Sox9 by real-time RT-PCR (Fig S1A,D). We confirmed that Sox9 was expressed at similar levels in the resting, proliferative, and prehypertrophic zone, but was downregulated approximately 5-fold in the hypertrophic zone. Sox9 expression was essentially absent in the trabecular bone adjacent to the growth plate. Similarly, Col2a1 expression were downregulated slightly in the hypertrophic zone and essentially absent in the bone. We also performed in situ hybridization of Sox9, and found substantial expression as expected in the resting, proliferative, and prehypertrophic zones but minimal signal in the hypertrophic zone (Fig S1B,C). Our data thus provide a quantitative assessment of Sox9 expression across the growth plate, and demonstrate that Sox9 expression is downregulated, but not completely absent, in the hypertrophic zone.
A mouse model to prevent Sox9 downregulation in hypertrophic chondrocytes
To investigate the physiological role of Sox9 downregulation and test whether it affects chondrocyte-osteoblast transdifferentiation, we sought a mouse model that maintains expression of Sox9 in chondrocytes in the hypertrophic zone at levels similar to that of the proliferative zone, and ideally, maintains that expression if the chondrocytes transdifferentiate into osteoblast. One previous study used the Col10a1 promoter to drive Sox9 transgene expression and showed severely delayed ossification [16]. However, we reasoned that the hypertrophic chondrocytes from this mouse model potentially overexpressed Sox9 at levels greater than physiologically relevant levels, because Col10a1 expression in the hypertrophic zone is >100-fold higher than Sox9 expression anywhere in the growth plate (see Fig.S1A). More importantly, the Col10a1-driven Sox9 transgene would likely be switched off if these chondrocytes transdifferentiated into osteoblasts, in which the Col10a1 promoter is no longer transcriptionally active; therefore this mouse is not an ideal model to determine whether preventing downregulation of Sox9 would block transdifferentiation.
We decided to use a previously described mouse model [18] in which a Sox9 transgene and enhanced green fluorescence protein (EGFP) are driven by a human cytomegalovirus enhancer and chicken β-actin (CAG) promoter, with a floxed monomeric red fluorescence protein (mRFP1) inserted between the promoter and the Sox9 transgene. We crossed this mouse (CAG-Sox9-GFP mouse) with mice with a constitutive Col2a1cre transgene to remove the mRFP1 and induce expression of the Sox9 transgene and EGFP. We found that the Sox9 transgene was expressed in the entire growth plate at levels similar to endogenous Sox9 (Fig. 1A,B,C). Unexpectedly, the expression of the hSox9 transgene was lower in the proliferative zone than in the hypertrophic zone, but this relatively low expression in proliferative zone did not affect the suitability of the model, which was designed to study persistent Sox9 expression in hypertrophic zone. In addition, fluorescence microscopy confirmed the presence of EGFP signal in both proliferative and hypertrophic chondrocytes, which served as a surrogate signal for transgene expression (Fig.1B–E). More importantly, we also confirmed with immunostaining that Sox9 protein expression was normally downregulated in the hypertrophic zone (Fig.1H,I) but persisted in the Col2cre SOX9-GFP mice (Fig.1F,G). Thus, mouse model successfully prevented the physiological downregulation of Sox9 in the hypertrophic zone
Figure 1. A transgenic mouse model expressing human SOX9.
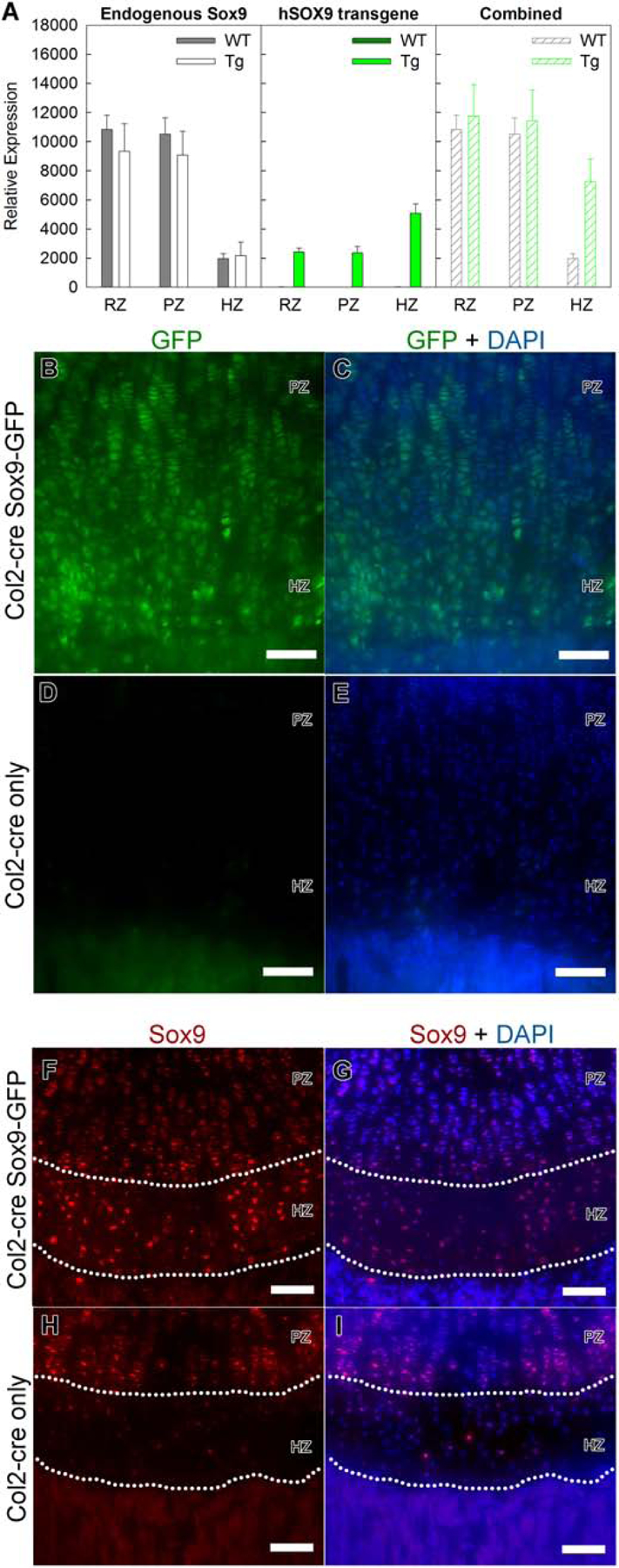
A detailed study of the phenotype of this mouse line has been described in a prior publication [18] (A) qPCR was used to quantitatively assess expression of endogenous Sox9, transgenic hSOX9, or their combined expression in resting (RZ), proliferative (PZ) and hypertrophic zone (HZ) of growth plates from Col2-cre Sox9-GFP mice (TG) and Col2-cre littermates (WT). (Number of animals, N=6) (B–E) Fluorescent microscopy images showing the presence of GFP signal in 1 wk tibial growth plate in the Col2-cre Sox9-GFP transgenic mouse (B,C), but not in wild-type littermates (D,E, Col2-cre only). DAPI was used for counterstaining (C,E). (F–I) Fluorescence immunohistochemistry confirmed that Sox9 expression was present throughout the growth plate, including the hypertrophic zone (the area between the dotted curves), in Col2-cre Sox9-GFP transgenic mouse (F,G). In contrast, Sox9 expression decreased notably in the hypertrophic zone of the growth plate in wild-type littermates (H,I, Col2-cre only). DAPI was used for counterstaining (G,I) PZ, proliferative zone; HZ, hypertrophic zone; scale bar, 100 μm.
Previous work on this CAG-Sox9-GFP mouse when crossed into a different Col2a1cre line showed slightly expanded hypertrophic zone and delayed ossification at embryonic days E15.5 and E17.5 [18]. In the current study, when we examined the postnatal growth plate of these Sox9-GFP transgenic mice at 1- and 4-weeks of age, we also found a slight expansion of the hypertrophic zone and collagen-X expressing area compared to wild-type, with no noticeable difference in chondrocyte appearance (Fig.S2A–H). However, persistent expression of Sox9 in the hypertrophic zone did not seem to have a major impact on bone growth as there was no difference in tibia length at 4 weeks of age (Fig.S2I,J).
Sox9 transgene expression suppressed upregulation of osteoblast-associated genes in hypertrophic chondrocytes
Previous studies suggest that some genes, such as Col1a1 and Mmp13, which are highly expressed in bone, are also expressed in the lower hypertrophic zone and that overexpression of Sox9 using the Col10a1 promoter caused downregulation of these genes in the hypertrophic zone [15, 16]. We therefore sought to determine whether this regulation of gene expression occurs in the postnatal growth plate with expression of Sox9 at modest levels, comparable to those found in the proliferative zone.
To this end, we first used LCM and real-time PCR to study a panel of genes associated with osteoblast differentiation in the 1-wk old mouse growth plate. For many genes that are typically considered to be expressed in bone, including Mmp9, Mmp13, Sp7, Col1a1, and Ibsp, we found expression levels in the hypertrophic zone to be intermediate between the lower levels in the remainder of the growth plate and the higher levels in the adjacent bone (Fig 2, wild-type only), suggesting that the transcription profile of hypertrophic chondrocytes had begun the transition toward a profile characteristic of an osteoblast. This increase in expression of osteoblastic-associated genes in the hypertrophic zone was unlikely to be caused by inaccurate dissection because LCM allowed us to precisely separate different zones of the cartilage, and not all of the osteoblastic genes that we studied (for example, Bglap, a mature osteoblast marker gene) showed an increase in hypertrophic zone.
Figure 2. Expression of specific osteoblast-associated genes were upregulated in wild-type hypertrophic zone compared to proliferative zone and were suppressed by transgenic expression of SOX9.
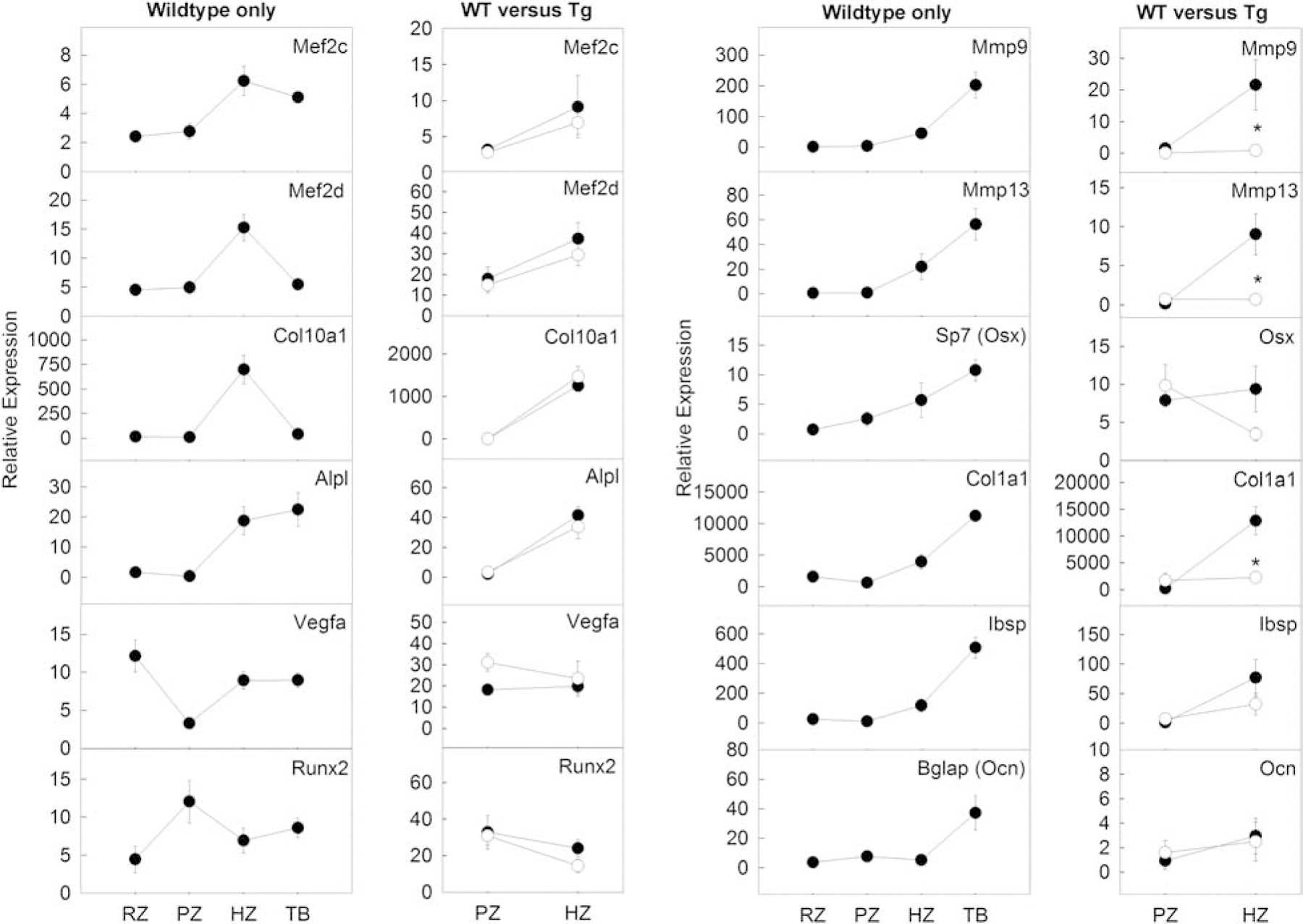
1st and 3rd panels: LCM and qPCR was used to quantitatively assess gene expression in specific genes known to be expressed in the hypertrophic zone (Mef2c, Mef2d, Col10a1, Alpl) or predominantly in trabecular bone (Mmp9, Mmp13, Vegfa, Sp7, Runx2, Col1a1, Ibsp, Bglap). For many of the genes expected to be high in the trabecular bone, their expression was already beginning to increase in the hypertrophic zone. RZ, resting zone; PZ, proliferative zone; HZ, hypertrophic zone; TB, trabecular bone. 2nd and 4th panels: expression of these genes in the proliferative zone and hypertrophic zone was assessed in Col2a1-cre Sox9-GFP transgenic mice (Tg, open circle) versus Col2a1-cre only littermates (WT, closed circle). For some of these genes (Mmp9, Mmp13, Col1a1, Ibsp), the physiological upregulation of gene expression in the hypertrophic zone was suppressed by the presence of the Sox9 transgene. (Number of animals, N=6)
We next asked whether transgenic Sox9 expression affected the expression of these osteoblastic genes in the hypertrophic zone. We therefore microdissected the proliferative and hypertrophic zones of the Col2a1cre CAG-Sox9-GFP mouse and compared gene expression with Col2a1cre wild-type littermates. Interestingly, we found that the normal increases in Mmp9, Mmp13, Sp7, and Col1a1 expression in the hypertrophic zone were suppressed in the Col2a1cre GAG-Sox9-GFP mice (Fig. 2, WT versus Tg). Ibsp also showed a similar trend but did not reach statistical significance. In contrast, for genes that do not normally show expression specific to bone versus cartilage, such as Mef2c, Mef2d, Col10a1, Runx2, Vegfa, and Alp1, the Sox 9 transgene expression in hypertrophic zone had little effect.
Continuous Sox9 expression suppresses chondrocyte-osteoblast transdifferentiation
We next sought to test the hypothesis that Sox9 downregulation is important for chondrocyte-osteoblast transdifferentiation. To this end, we crossed the CAG-Sox9-GFP mice with a tamoxifen-inducible Col2a1-cre (Col2a1ERcre CAG-Sox9-GFP). Injecting tamoxifen in these mice allowed us to trace the lineage of chondrocytes that had undergone cre recombination and express the Sox9 transgene with EGFP. As a control, we crossed the Col2a1ERcre with the CAG-GFP mice that similarly expressed GFP upon cre recombination but without the Sox9 transgene (Col2a1ERcre CAG-GFP mice). A single dose of tamoxifen was given to both lines of mice at 4 days of age and histological sections of the tibial growth plate were assessed at 2- and 4-wk of age (Fig.3A). As expected, columnar clones of chondrocytes were labeled for GFP in both Col2a1ERcre CAG-GFP mice and Col2a1ERcre CAG-Sox9-GFP mice (Fig.3B–Q). In addition, GFP positive cells were found also in the trabecular bone below the chondro-osseous junction. Because cre expression is driven by a Col2a1 promoter, these GFP-positive are likely to be derived from growth plate chondrocytes in which cre-mediated recombination had occurred. Interestingly, at 2 weeks of age, we found significantly more GFP-positive cells in the trabecular bone, in the Col2a1ERcre CAG-GFP mice than in the Col2a1ERcre CAG-Sox9-GFP mice (Fig.3B–I), suggesting the expression of the Sox9 transgene suppressed this transition of chondrocytes into the trabecular bone area. Similarly, at 4 weeks of age, more GFP-positive cells in the trabecular bone were found in the Col2a1ERcre CAG-GFP mice compared to the Sox9 transgenic mice (Fig.3J–Q). Because the number of GFP-positive cells in Col2a1ERcre CAG-Sox9-GFP mice at both 2- and 4-weeks were substantially lower than in the Col2a1ERcre CAG-GFP mice (Fig.3R), the decrease appears not to represent a developmental delay but rather a general inhibition. We also considered the possibility that the difference in number of GFP-positive cells in the trabecular bone could be due to a difference in efficiency of cre-mediated recombination in the chondrocytes between the two lines, but the number of GFP positive chondrocyte columns in Col2a1ERcre CAG-GFP and Col2a1ERcre CAG-Sox9-GFP mice (Fig.3S) were not significantly different, and normalizing the number of GFP-positive cells in the trabecular bone to the number of GFP-positive chondrocyte columns did not eliminate the difference in GFP-positive cells in the trabecular area (Fig.3T) between the two lines of mice, suggesting there was indeed a difference in these GFP positive chondrocytes transitioning into the metaphysis. Next, to test whether these chondrocyte-derived, GFP-positive cells in the trabecular area were osteoblasts, we used immunostaining to detect both GFP and Sp7 expression. Importantly, we found that some of the GFP positive cells in the trabecular bone of the Col2a1ERcre CAG-GFP mice also stained positive for Sp7 (Fig.4A–C), suggesting that they were osteoblasts which had transdifferentiated from chondrocytes. In contrast, the number and percentage of GFP-positive cells in the trabecular bone of the Col2a1ERcre CAG-Sox9-GFP mice that express Sp7 was significantly lower (Fig.4D–F, G), suggesting that persistent Sox9 expression not only suppressed the transition of chondrocytes from the hypertrophic zone into the adjacent bone, but also affected their transdifferentiation into osteoblasts.
Figure 3. Transgenic Sox9 expression suppressed transition of chondrocytes into trabecular bone.
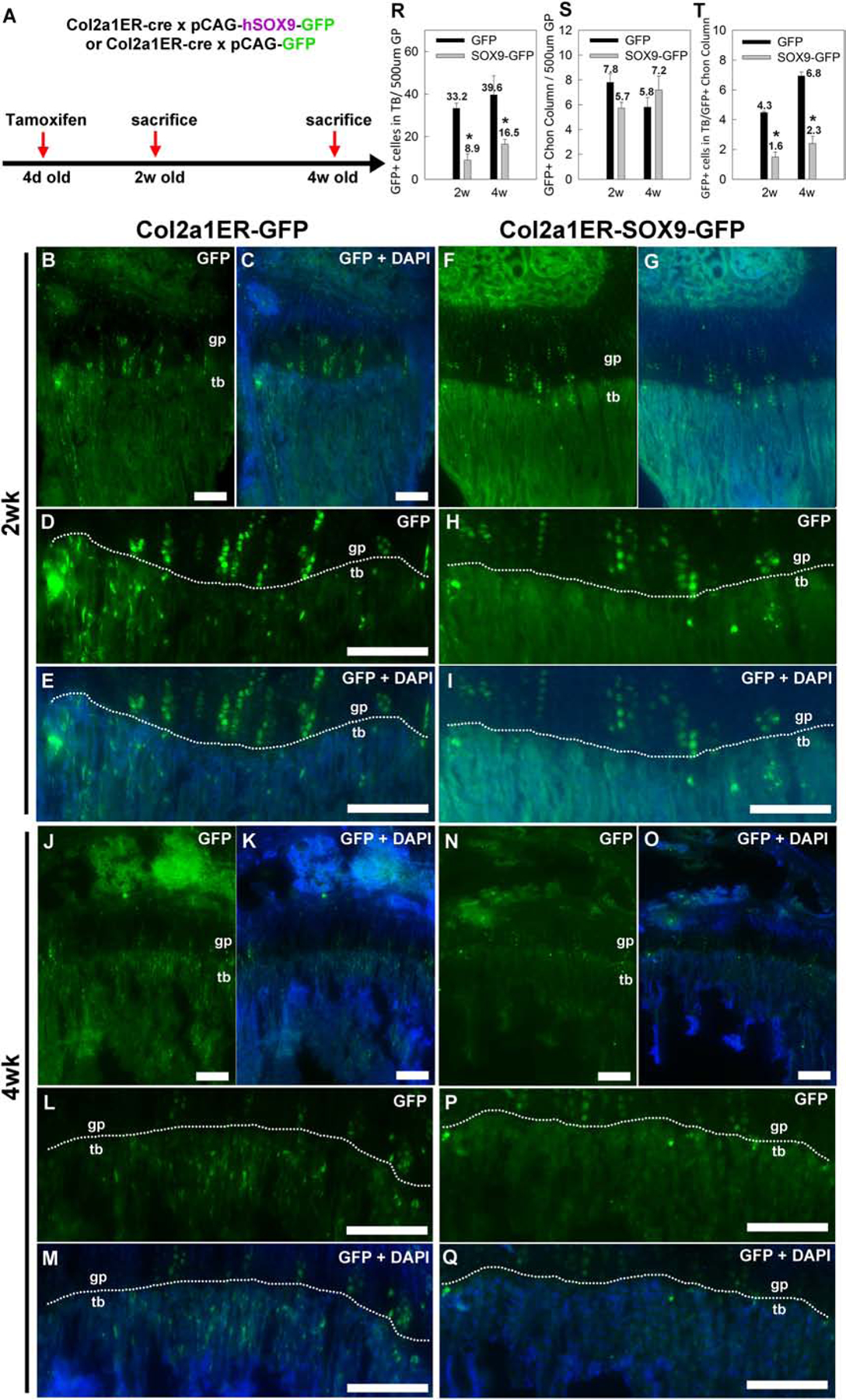
(A) Lineage tracing experiments were performed by crossing mice with tamoxifen-inducible cre expression driven by a collagen 2 promoter (Col2a1ER-cre) with either the transgenic Sox9-GFP mice (pCAG-Sox9-GFP) or transgenic mice expressing only GFP (pCAG-GFP). A single tamoxifen injection was performed at 4 days of age and mice were sacrificed at 2 or 4 weeks of age for sectioning. (B–I) Fluorescent microscopy showed that, at 2 weeks of age, GFP-positive chondrocyte columns were present in the growth plate (gp, area above the white dotted line) in both Col2a1ER-GFP and Col2a1ER-SOX9-GFP mice. However, in mice with Sox9 misexpression, there were significantly fewer GFP-positive cells in the metaphyseal trabecular bone (tb, area below the white dotted line), per 500 μm (width) of growth plate (R). The number of GFP-positive chondrocyte columns per 500 μm (width) of growth plate was not significantly affected (S) and therefore the number of GFP-positive cells in the metaphyseal trabecular bone per GFP-positive column (T). (J–T) The findings were similarly when bones were collected at 4 weeks of age. DAPI was used for counterstaining (C,E,G,I,K,M,O,Q). Scale bar, 100 μm. (Number of animals, N=5) *, P<0.05, ANOVA,.
Figure 4. Transgenic Sox9 expression suppressed transdifferentiation of chondrocytes into osteoblasts.
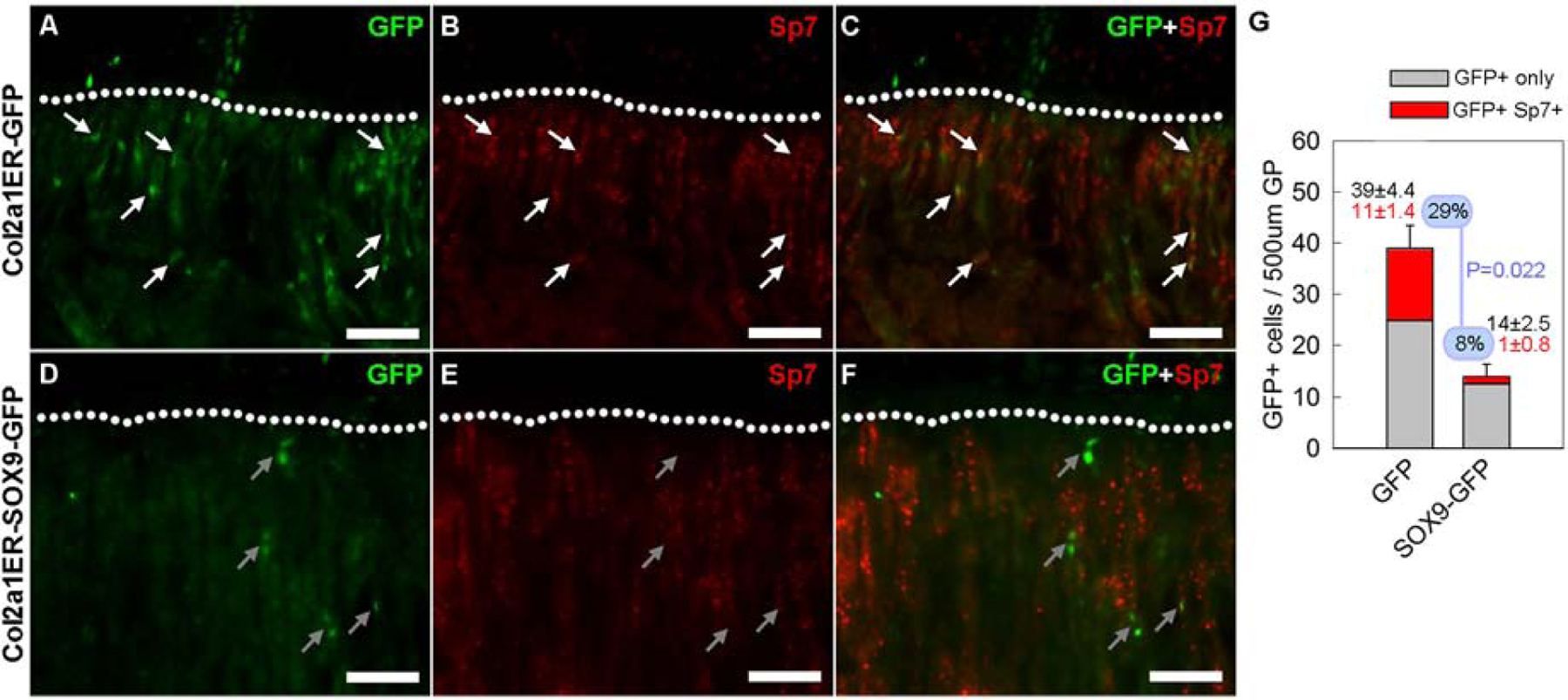
Lineage tracing experiments were performed as described in Figure 3A. At 4 weeks of age, bones were sectioned, and fluorescent microscopy was used to detect GFP and Sp7 expression. (A–C) In Col2a1ER-GFP mice, many of the GFP-positive cells that moved into the trabecular bone (area below the dotted curve) also expressed Sp7 (red, yellow when overlapping with green signal of GFP), as indicated by the white arrows. (D–F) In contrast, in Col2a1ER-SOX9-GFP mice, GFP-positive cells in the trabecular bone rarely stained positive for Sp7, as indicated by the gray arrows. (G) The number of GFP-positive, Sp7-negative (GFP+ only)and the number of GFP-positive and Sp7-positive (GFP+ Sp7+) cells in the metaphysis were counted. In mice with Sox9 misexpression, there were fewer total GFP-positive cells, fewer GFP-positive, Sp7-positive cells. The fraction of GFP-positive cells that were also Sp7-positive was also decreased (8% versus 29%, Col2a1ER-SOX9-GFP versus Col2a1ER-GFP mice, P=0.022, N=4).
Because an effect on transdifferentiation might affect the structure of the metaphyseal bone, we compared the bone mineral density and bone structure between Col2a1cre CAG-Sox9-GFP and their wild-type littermates using μCT. Overall we did not find any statistically significant effect of Sox9 misexpression on the structure of the metaphyseal bone, although it is conceivable that there could be a trend toward decreased trabecular bone in the Sox9-GFP mice (Fig.S3).
Finally, if persistent Sox9 expression in hypertrophic chondrocytes only suppressed transdifferentiation into osteoblasts, we would expect an accumulation of GFP-positive terminal hypertrophic chondrocytes in the growth plate, which we did not see. Because hypertrophic growth plate chondrocytes can also undergo apoptosis, we used immunostaining to detect active caspase-3 (a marker for apoptosis) in Col2a1cre CAG-Sox9-GFP mice. Interestingly, we found a significant increase in the number of active caspase-3 positive cells in the terminal hypertrophic zone of the Col2a1cre CAG-Sox9-GFP mice compared with wild-type littermates (Fig.5), suggested that failure to downregulate Sox9 in the terminal hypertrophic zone may also lead to increased apoptosis.
Figure 5. Transgenic Sox9 expression increased apoptosis in terminally differentiated hypertrophic chondrocytes.
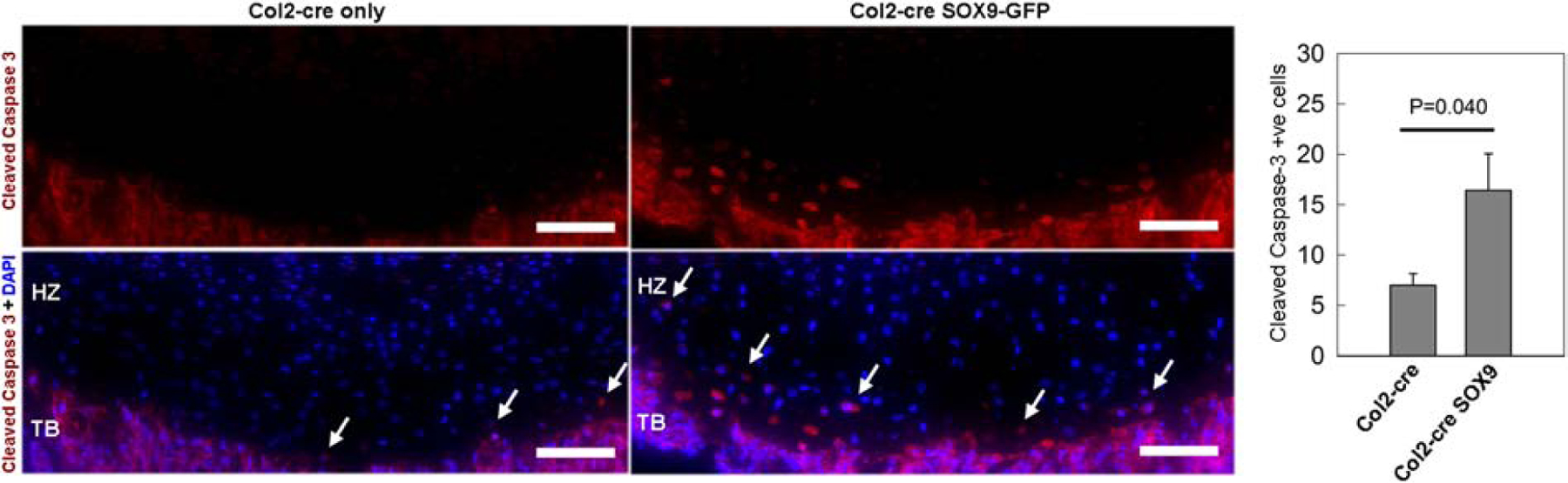
Immunostaining followed by fluorescent microscopy detected a high number of cells in the growth plate near the chondro-osseous junction with active caspase-3 (red fluorescent cells, white arrows), which is a marker for apoptosis, in 1-wk Col2-cre Sox9-GFP transgenic mouse compared with wild-type littermates (Col2-cre only). DAPI was used for counterstaining. Scale bar, 100 μm. Bar-graph: The number of these labeled cells in the hypertrophic zone (Cleaved Caspase-3+ve cells) per 500 μm was significantly greater in 1-wk Col2-cre Sox9-GFP transgenic mouse compared with wild-type littermates (Col2-cre only). P-value for t-test, N=5.
Discussion
In the current study, we investigated the importance of physiological downregulation of Sox9 in promoting chondrocyte-osteoblast transdifferentiation. We first used LCM followed by real-time PCR to better quantify the spatial regulation of Sox9 expression across growth plate zones and the trabecular bone and confirmed that Sox9 expression is significantly downregulated in the hypertrophic zone and almost completely switched off in the adjacent trabecular bone. Multiple previous studies have already demonstrated this downregulation of Sox9 and our data only served as a validation with a quantitative methodology [15, 16]. We found that certain genes that are normally expressed in osteoblasts, including Mmp9, Mmp13, Sp7, Col1a1, and Ibsp, already show some increased expression in the hypertrophic zone compared to the proliferative zone, suggesting that the cartilage-bone transition begins above the chondro-osseous junction, in the lower hypertrophic zone. This finding, in the postnatal growth plate, is consistent with previous studies of embryonic epiphyseal cartilage showing expression of osteoblast-associated gene in the lower hypertrophic zone [15, 16]. We next utilized a mouse model with persistent expression of a Sox9 transgene in all cells derived from the chondrocytic lineage, essentially abolishing Sox9 downregulation in the hypertrophic zone. In this mouse, we found that persistent Sox9 expression in the hypertrophic zone suppressed the upregulation of these osteoblast-associated genes, including Mmp9, Mmp13, Sp7, and Col1a1 (and possibly also Ibsp). Our data therefore suggest that these genes are negatively regulated by Sox9 and that their normal upregulation in the hypertrophic zone, and subsequently in bone, may be attributable to Sox9 downregulation. These findings are consistent with a previous study overexpressing Sox9 in the hypertrophic zone using the Col10a1 promoter and showing downregulation of Mmp9, Mmp13, and Col1a1[16]. In the current study, we found similar suppression of osteoblastic gene expression using a different transgenic SOX9 model. Because we also showed that these osteoblast-associated genes are normally upregulated beginning in the hypertrophic zone, the evidence suggests that this normal upregulation of osteoblast-associated gene expression is due to downregulation of SOX9 in hypertrophic zone. Finally, lineage tracing indicated that Sox9 transgene expression in chondrocytes caused a decrease in the number of chondrocyte-derived cells in the trabecular bone, and that most of the remaining chondrocyte-derived cells do not express Sp7, a transcription factor commonly expressed in early osteoblasts. These findings suggest that Sox9 downregulation normally promotes chondrocyte-osteoblast transdifferentiation. However, some GFP-positive cells were still found in the metaphyses of mice expressing Sox9, suggesting that normal Sox9 downregulation promotes transdifferentiation but is not an absolute requirement for chondrocytes to move into the trabecular bone area.
Although persistent expression of Sox9 in hypertrophic chondrocytes impaired transdifferentiation into osteoblasts, it did not significantly affect the structure of the metaphyseal bone (Fig.S3). A likely explanation is that osteoblasts in the metaphysis derive not only from transdifferentiation of chondrocytes but also from direct differentiation of mesenchymal stem cells. Our findings suggest that the latter process is the dominant source; many osteoblasts (as defined by Sp7 expression) in the metaphyses of juvenile mice did not derive from transdifferentiation (as defined by GFP expression, see Fig.4) and therefore likely derived directly from mesenchymal stem cells. Because transdifferentiation only provides some of the osteoblasts, the decrease in transdifferentation due to Sox9 mis-expression may not have a major impact on total osteoblast numbers and therefore on bone structure.
The mechanisms by which Sox9 suppresses transdifferentiation of chondrocytes into osteoblasts remains unclear. One possibility is that Sox9 expression in the terminally-differentiated hypertrophic chondrocyte directly alters its tendency to transdifferentiation versus apoptosis. However, another possibility is that Sox9 expression in the earlier hypertrophic chondrocytes might impair terminal hypertrophic differentiation and that this failure to undergo normal terminal differentiation may secondarily impair the ability of the chondrocyte to transdifferentiate into an osteoblast. As chondrocytes move down the hypertrophic zone, they adopt a transcriptional profile required for cell size expansion, cartilage matrix calcification, and induction of vascular invasion [21–23]. When these chondrocytes subsequently arrive at the chondro-osseous junction, presumably a sophisticated but rapid switch in the transcriptome is again needed to induce transdifferentiation into osteoblasts and thus to assume new cellular functions, such as secreting bone matrix rather than cartilage matrix. Downregulation of Sox9, which is a transcription factor that controls a large number of genes involved in chondrogenesis [12], could potentially affect the maturation of hypertrophic chondrocytes which might secondarily affect the subsequent differentiation to osteoblasts. A previous study by Dy et al. [15] demonstrated that conditional knockout of Sox9 using an aggrecan promoter led to expression of osteoblastic genes and ossification higher up in the growth plate, thus suggesting the importance of Sox9 in regulation of chondrocyte hypertrophic maturation and ossification. Our current study extends the previous findings involving ablation of Sox9 by using a complementary approach; by preventing the physiological downregulation of Sox9 in all cells from the chondrocytic lineage, and showing impaired transdifferentiation into osteoblasts.
Importantly, our current study differs from and complements prior studies overexpressing Sox9 in cartilage [16, 18, 24]. The current study involved persistent expression of Sox9 in the hypertrophic zone at levels similar to those found normally in the proliferative zone and then focused on the process of transdifferentiation of chonodrocytes into osteoblasts in the postnatal growth plate. In contrast to our study, Akiyama et al. inserted a Sox9 cDNA into one Col2a1 allele, which is expected to cause a high level of overexpression, and found decreased chondrocyte proliferation, delayed hypertrophic chondrocyte differentiation, and delayed endochondral bone formation [24]. Similarly, Hattori et al. studied mice that overexpressed Sox9 under the control of a BAC-Col10a1 promoter and found impaired terminal differentiation of hypertrophic chondrocytes, impaired vascular invasion into hypertrophic cartilage and impaired cartilage resorption [16]. In both studies, the Sox9 transgene driven by the Col2a1 or Col10a1 promoter allows overexpression in some or all parts of the growth plate, but then would likely be downregulated if transdifferentiation occurred. Our current mouse model, utilizing cre recombination, allows continued expression of the Sox9 transgene in cells derived from chondrocytes, and therefore may serve as a better model to investigate the role of Sox9 in transdifferentiation. Furthermore, the severe phenotype observed in both studies may have resulted from strong overexpression of Sox9 driven by the Col2a1 or Col10a1 promoters. In contrast, our model, which involved Sox9 expression at near-physiological levels, showed a much milder effects on growth plate histology and chondrocyte hypertrophy. The only noticeable effect of persistent Sox9 expression was suppression of osteoblastic genes and decreased transition of chondrocytes into the metaphysis. Thus, our findings suggest that the normal downregulation of Sox9 in the hypertrophic zone is likely not involved in chondrocyte hypertrophy but rather is an important cellular event priming chondrocyte-osteoblast transdifferentiation. In a third prior study, Kim et al, used a mis-expression approach similar to that used in the current study and found impaired chondrocyte hypertrophy, delayed terminal differentiation, and delayed ossification in the embryonic skeleton [18]. However, none of these three prior studies used lineage tracing to elucidate the effects of Sox9 downregulation on transdifferentiation.
In summary, our findings suggest that multiple osteoblast-associated genes normally begin to be upregulated in the hypertrophic zone of the growth plate and that this upregulation is driven by downregulation of Sox9 expression in the hypertrophic chondrocytes. In addition, our findings indicate that this Sox9 downregulation also serves to promote transdifferentiation of growth plate chondrocytes into osteoblasts.
Supplementary Material
Highlights:
a mouse model was used such that all chondrocyte-derived cells persistently expressed Sox9, which normally became downregulated in hypertrophic chondrocytes
Persistent Sox9 expression ablated normal upregulation of osteoblastic genes in the hypertrophic zone
Lineage tracing showed that transition of chondrocytes into the trabecular bone area was suppressed by persistent Sox9 expression
Sox9-expressing chondrocytes that transitioned into the trabecular bone did not transdifferentiate properly into osteoblasts
Acknowledgments
We thank Jeffrey Hanson from the LCM core laboratory of the National Cancer Institute for guidance related to the microdissection of growth plate zones by LCM. We thank Dr. Antony Cougnoux from the Section on Molecular Dysmorphology and Lynne Holtzclaw from The Microscopy and Imaging Core, NICHD, for their advice and help on acquiring and optimizing fluorescent images. We thank Danielle Donahue from the Mouse Imaging Facility, National Institute of Neurological Disorders and Stroke, for her training and assistance on acquiring and analyzing μCT data of the mouse tibia. We thank Professor Haruhiko Akiyama from the Department of Orthopaedics, Gifu University, Japan and Professor Roger Y. Tsien from the Department of Chemistry & Biochemistry, UCSD, for their permission to use the Sox9 transgenic mouse line. We also thank Dr. Richard Behringer from MD Anderson Cancer Center for providing the Sox9 transgenic mouse line.
Grant Support: This work was supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure: The authors have declared that no conflict of interest exists.
Reference
- [1].Kronenberg HM, Developmental regulation of the growth plate, Nature 423(6937) (2003) 332–6. [DOI] [PubMed] [Google Scholar]
- [2].Ahmed YA, Tatarczuch L, Pagel CN, Davies HM, Mirams M, Mackie EJ, Physiological death of hypertrophic chondrocytes, Osteoarthritis and cartilage 15(5) (2007) 575–86. [DOI] [PubMed] [Google Scholar]
- [3].Adams CS, Shapiro IM, The fate of the terminally differentiated chondrocyte: evidence for microenvironmental regulation of chondrocyte apoptosis, Critical reviews in oral biology and medicine : an official publication of the American Association of Oral Biologists 13(6) (2002) 465–73. [DOI] [PubMed] [Google Scholar]
- [4].Gibson G, Active role of chondrocyte apoptosis in endochondral ossification, Microscopy research and technique 43(2) (1998) 191–204. [DOI] [PubMed] [Google Scholar]
- [5].Frenette PS, Pinho S, Lucas D, Scheiermann C, Mesenchymal stem cell: keystone of the hematopoietic stem cell niche and a stepping-stone for regenerative medicine, Annual review of immunology 31 (2013) 285–316. [DOI] [PubMed] [Google Scholar]
- [6].Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR, Multilineage potential of adult human mesenchymal stem cells, Science (New York, N.Y.) 284(5411) (1999) 143–7. [DOI] [PubMed] [Google Scholar]
- [7].Ono N, Ono W, Nagasawa T, Kronenberg HM, A subset of chondrogenic cells provides early mesenchymal progenitors in growing bones, Nature cell biology 16(12) (2014) 1157–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Yang L, Tsang KY, Tang HC, Chan D, Cheah KS, Hypertrophic chondrocytes can become osteoblasts and osteocytes in endochondral bone formation, Proceedings of the National Academy of Sciences of the United States of America 111(33) (2014) 12097–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Zhou X, von der Mark K, Henry S, Norton W, Adams H, de Crombrugghe B, Chondrocytes transdifferentiate into osteoblasts in endochondral bone during development, postnatal growth and fracture healing in mice, PLoS genetics 10(12) (2014) e1004820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Akiyama H, Kim JE, Nakashima K, Balmes G, Iwai N, Deng JM, Zhang Z, Martin JF, Behringer RR, Nakamura T, de Crombrugghe B, Osteo-chondroprogenitor cells are derived from Sox9 expressing precursors, Proceedings of the National Academy of Sciences of the United States of America 102(41) (2005) 14665–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Akiyama H, Chaboissier MC, Martin JF, Schedl A, de Crombrugghe B, The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6, Genes & development 16(21) (2002) 2813–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ohba S, He X, Hojo H, McMahon AP, Distinct Transcriptional Programs Underlie Sox9 Regulation of the Mammalian Chondrocyte, Cell reports 12(2) (2015) 229–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Oh CD, Lu Y, Liang S, Mori-Akiyama Y, Chen D, de Crombrugghe B, Yasuda H, SOX9 regulates multiple genes in chondrocytes, including genes encoding ECM proteins, ECM modification enzymes, receptors, and transporters, PloS one 9(9) (2014) e107577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhao Q, Eberspaecher H, Lefebvre V, De Crombrugghe B, Parallel expression of Sox9 and Col2a1 in cells undergoing chondrogenesis, Developmental dynamics : an official publication of the American Association of Anatomists 209(4) (1997) 377–86. [DOI] [PubMed] [Google Scholar]
- [15].Dy P, Wang W, Bhattaram P, Wang Q, Wang L, Ballock RT, Lefebvre V, Sox9 directs hypertrophic maturation and blocks osteoblast differentiation of growth plate chondrocytes, Developmental cell 22(3) (2012) 597–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hattori T, Muller C, Gebhard S, Bauer E, Pausch F, Schlund B, Bosl MR, Hess A, Surmann-Schmitt C, von der Mark H, de Crombrugghe B, von der Mark K, SOX9 is a major negative regulator of cartilage vascularization, bone marrow formation and endochondral ossification, Development (Cambridge, England) 137(6) (2010) 901–11. [DOI] [PubMed] [Google Scholar]
- [17].Lui JC, Garrison P, Nguyen Q, Ad M, Keembiyehetty C, Chen W, Jee YH, Landman E, Nilsson O, Barnes KM, Baron J, EZH1 and EZH2 promote skeletal growth by repressing inhibitors of chondrocyte proliferation and hypertrophy, Nature communications 7 (2016) 13685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Kim Y, Murao H, Yamamoto K, Deng JM, Behringer RR, Nakamura T, Akiyama H, Generation of transgenic mice for conditional overexpression of Sox9, Journal of bone and mineral metabolism 29(1) (2011) 123–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Garrison P, Yue S, Hanson J, Baron J, Lui JC, Spatial regulation of bone morphogenetic proteins (BMPs) in postnatal articular and growth plate cartilage, PloS one 12(5) (2017) e0176752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lui JC, Forcinito P, Chang M, Chen W, Barnes KM, Baron J, Coordinated postnatal down-regulation of multiple growth-promoting genes: evidence for a genetic program limiting organ growth, FASEB journal : official publication of the Federation of American Societies for Experimental Biology 24(8) (2010) 3083–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Poole AR, Matsui Y, Hinek A, Lee ER, Cartilage macromolecules and the calcification of cartilage matrix, The Anatomical record 224(2) (1989) 167–79. [DOI] [PubMed] [Google Scholar]
- [22].Belluoccio D, Etich J, Rosenbaum S, Frie C, Grskovic I, Stermann J, Ehlen H, Vogel S, Zaucke F, von der Mark K, Bateman JF, Brachvogel B, Sorting of growth plate chondrocytes allows the isolation and characterization of cells of a defined differentiation status, Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 25(6) (2010) 1267–81. [DOI] [PubMed] [Google Scholar]
- [23].Lui JC, Andrade AC, Forcinito P, Hegde A, Chen W, Baron J, Nilsson O, Spatial and temporal regulation of gene expression in the mammalian growth plate, Bone 46(5) (2010) 1380–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Akiyama H, Lyons JP, Mori-Akiyama Y, Yang X, Zhang R, Zhang Z, Deng JM, Taketo MM, Nakamura T, Behringer RR, McCrea PD, de Crombrugghe B, Interactions between Sox9 and beta-catenin control chondrocyte differentiation, Genes & development 18(9) (2004) 1072–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.