

Abstract
The analysis of low-volume samples provides valuable insight into complex biological systems. However, the proteomic and metabolomic analysis of low-volume samples remains challenging due to the lack of simple, efficient, and reproducible microsampling techniques. We have developed an electrospray-assisted device for quantitative low-volume sample extraction, referred to here as “Spray-Capillary”. Stable electrospray was achieved through a chemically etched tip from a long (e.g., 50 cm) capillary with a conductive sheath flow. This electrospray provided the driving force to quantitatively draw low-volume samples into the capillary. We evaluated the precision and accuracy of sample injection volumes using our spray-capillary as the electrospray voltage, capillary ID, and column length were varied. Our results demonstrate that spray-capillary allows for reproducible and quantitative microsampling with low injection flow rates (as low as 15 pL/s). Furthermore, spray-capillary can be directly coupled with capillary zone electrophoresis (CZE) for separation. Overall, spray-capillary is a simple microsampling device that holds great potential for high-throughput quantitative omics analysis of ultralow-volume samples.
Graphical abstract
Efforts have been made to improve the sensitivity and throughput of low quantity sample analysis in mass spectrometry (MS)-based omics including the development of specialized sample preparation devices,1,2 high-resolution separation methods,3,4 efficient spray-MS interfaces (i.e., novel ambient ionization techniques),5−11 and advancements in MS instrumentation.12 The sensitivity of low quantity sample analysis has been dramatically improved using these techniques, but many challenges remain in quantitative low-volume sample injection and extraction.13
Micropipettes have been the most commonly applied tools for the manipulation of low-volume samples.14−22 Briefly, one end of the capillary tubing is pulled to make a micropipette to aspirate samples into the capillary through a driving force. Two approaches have been applied as the driving force for the operation of micropipettes: pump-based extraction and electroosmotic force. In the pump-based micropipette sample handling method, a syringe is connected to a vacuum or mechanical pump to pull the sample into the capillary. Using this method, low sample injection volume can be accurately controlled.23 Coupling the pump-based extraction approach with offline capillary electrophoresis (CE)-MS for complex sample analysis has led to promising results.24−26 For example, the Nemes group constructed a pump-based micropipette method to study live Xenopus laevis and Zebrafish embryos.27 When an electroosmotic driving force is utilized, an electrode is inserted into a sampling capillary and current is applied to induce electroosmotic flow. Recently, a micropipetting method based on using electroosmotic flow was developed by the Laskin group and applied to the analysis of a single cell.28 During the collection process, +2 V was maintained to prevent the sample buffer from getting into the laser-pulled tips before the cell was penetrated. Low-volume cellular contents were extracted using −2 V after penetration. This work demonstrated that electroosmotic force could be utilized as the driving force for microsampling low-volume samples such as single onion cells. Another approach utilized an electroosmotic pump for ultralow-volume sample extraction from a Zebrafish embryo.22
Other microsampling approaches include hydrodynamic methods,29−35 fluidic force microscopy,36 capillary force,37 and electrowetting.38 A microfluidic chip-based platform, nanoPOTS,13 has been recently developed for low sample volume processing in single-cell proteomics analysis. Nano-POTS reduces total processing volumes from the conventional hundreds of microliters to <200 nL within a single droplet reactor. However, a customized automated droplet-based microfluidic system has to be incorporated for the ultralow-volume manipulation in nanoPOTS.
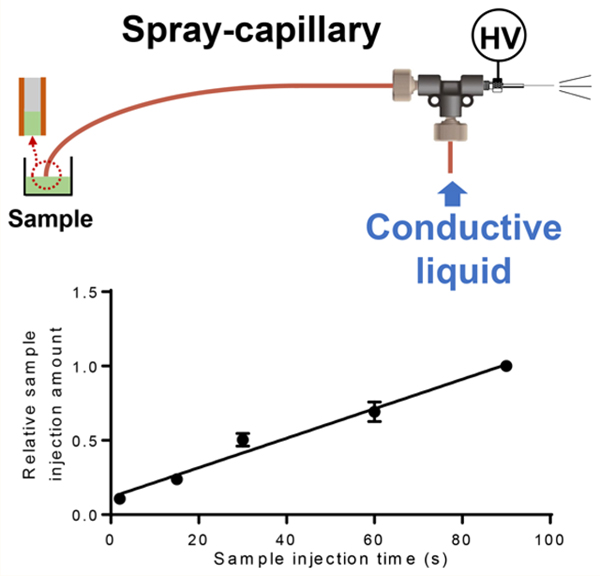
Dr. Wysocki’s group has demonstrated a continuous electrospray using a platinum wire inserted into a laser pulled glass capillary with preinjected samples.39 Inspired by this research, we developed an electrospray-assisted microsampling method, referred to here as “spray-capillary”. The spray-capillary is a simple hydrodynamic device that utilizes electrospray ionization (ESI) as the driving force for the injection of low-volume samples. During the ESI process, a liquid jet is created when the electrostatic force of the sample overcomes the surface tension, which reduces the pressure around the spray tip and creates a pressure difference between opposing ends of the ESI capillary. The spray-capillary device utilizes this pressure difference to serve as the driving force to move the liquid inside the capillary toward the spray tip. A conductive porous tip, similar to the sheathless interface proposed by Moini,5 was utilized to generate ESI as the driving force to quantitatively draw low-volume samples into a long (e.g., 50 cm) spray-capillary (Figure 1). The performance of spray-capillary was evaluated to determine the robustness, reproducibility, and flexibility through both offline and online coupling with MS detection. Furthermore, we demonstrated that the spray-capillary can be directly inserted into a background electrolyte (BGE) solution after sample injection for online CE-MS analysis, which holds great potential for high-throughput omics analysis of ultralow-volume samples.
Figure 1.

(A) Schematic of the spray-capillary CZE-MS platform and detailed diagram of the sheathless CZE-MS interface. Operation steps are as follows: (1) sample is injected into the separation column; (2) high voltage was initiated for CZE separation; (3) N2 was introduced into the BGE vial to perform gas elution. Reproducibility was demonstrated for (B) run-to-run, (C) day-to-day, and (D) batch-to-batch sample injections.
EXPERIMENTAL SECTION
Chemicals and Reagents.
Angiotensin II (AngII, A9525), Syntide 2 (Syn-2, SCP0250), HPLC water (270733), ACS-reagent acetonitrile (ACN, 360457), formic acid (FA; ≥95%, F0507), and hydrofluoric acid (HF, ≥48%, 30107) were purchased from Sigma-Aldrich (St. Louis, MO). Fused-silica capillaries were purchased from Polymicro Technologies (Phoenix, AZ). AngII and Syn-2 stock solutions were prepared in HPLC water. The standard peptide mixture used here was a solution of 10 μM AngII and 10 μM Syn-2 (0.1% FA in 45% ACN in water).
Spray-Capillary Device Fabrication and Operation.
A long capillary (e.g., 360 μm O.D., 50 μm I.D., 50 cm in length) was used to make the spray-capillary. To produce precise, clean and reproducible interfaces, both ends of the capillary (the MS end and the sample inlet end, Figure 1A) were cut using a Shortix capillary column cutter (purchased from Agilent, San Jose, CA), and were evaluated using an inverted microscope. The MS end of the spray-capillary was fabricated similarly to the previously reported sheathless interface.5 Briefly, the outside polymer coating of the MS end (∼3 cm) was removed by flame. The exposed silica was etched using a 49% HF solution at room temperature to generate a porous segment for electric contact. (Caution: HF is an extremely dangerous chemical and should be handled properly in a ventilated chemical hood.) During the etching process, the capillary was continuously flushed with water at a flow rate of 0.2−0.4 μL/min to prevent etching of the inner wall by the HF solution. After etching, the thickness of the capillary wall was approximately 5 μm. The tip shape and porous condition of the MS end were evaluated using an inverted microscope. The MS end of the spray-capillary was inserted into a PEEK tee connector through a short stainless steel tube (4 cm, 1/16” O.D., 0.04” i.d.) so that ∼1.5 cm of the porous segment emerged from the metal tube. A continuous flow of conductive liquid (0.1% FA, 1 μL/min) driven by the syringe pump was introduced into the tee to create the electric contact for generating ESI. The ESI voltage was applied to the stainless steel tube connector through an alligator clip. The sample inlet end of the spray-capillary was either placed directly into a sample vial for sample injection or into a BGE vial with a metal wire used to apply high voltages for CE separation.
Pressure-Based Sample Elution Setup and Fabrication.
After spray-capillary sample injection, an in-house sample injector was utilized to apply pressure-based sample elution for the follow-up MS detection. A PEEK tee connector was used in the in-house sample injector (details in Figure S1). The sample inlet end of the spray-capillary was inserted into a glass sample vial with a rubber cap to seal the inlet when pressure was applied. Nitrogen was introduced into the electrospray buffer vial (i.e., 0.1% FA) through the gas inlet on the PEEK tee to move the injected sample toward the MS end of the spray-capillary. A gas gauge (0−3 psi) was used to control nitrogen pressure for sample elution.
CE Separation of Standard Peptide Mixtures.
A spray-capillary (360 μm O.D., 50 μm I.D., 50 cm in length) was used for the CE separation of standard peptide mixtures. The MS end of the spray-capillary, with a 3 cm porous section, was inserted into the sheathless interface, as described above for spray-capillary-based sample injection. Then the sample inlet end was inserted into the sample vial. After sample injection, the sample inlet end of the spray-capillary was inserted into the BGE solution (0.1% FA). The CE separation was then conducted by applying 15 kV (300 V/cm) at the sample inlet end. High voltage (HV) was applied to the sample injector via a stainless-steel wire. The sample injector was assembled in-house according to Figure S1. (Attention is needed when performing experiments with high voltage. Only manual operation of HV power supply is allowed here, and all the other conductive parts in the system are grounded as needed. Proper warning signs were provided and displayed.) After a 15 min CE separation, N2 was introduced at low pressure to elute peptides. A total of 2.6 kV was used for the electrospray ionization at the sheathless interface.
Mass Spectrometry Analysis.
An LTQ Orbitrap Elite mass spectrometer was utilized for related spray-capillary experiments. The temperature of the inlet capillary was 275 °C. For pressure elution experiments, both 2+ and 3+ AngII ions were targeted with two scan ranges: 523−527 m/z for 2+ ions and 348−352 m/z for 3+ ions. Both scans were acquired at the resolving power of 120000 at m/z = 400. For the CE-MS experiments, full MS scans were acquired at the resolving power of 120000 at m/z = 400, and the AGC target was set as 1E6 with the maximum ion injection time of 1000 ms. The scan range is 150−2000 m/z. All data files were collected in profile mode. Peak extraction was done in XCalibur using RAW files. Results were processed and plotted using GraphPad Prism. In addition, chromatograms extracted from raw data were processed using boxcar smoothing algorithms.
RESULTS AND DISCUSSION
Development and Characterization of the Spray-Capillary Device.
Before sample injection, the sample inlet end of the spray-capillary was placed in the sample vial with the electrospray buffer (0.1% FA). Low-pressure nitrogen was introduced into the sample vial to fill the capillary until a droplet was observed at the MS end. Then the sample inlet end of the spray-capillary was immersed in the sample vial with standard peptide samples; the sample injection process was initiated by application of high voltage on the metal tee of the sheathless interface to generate continuous ESI (Figure 1). The sample-injection flow rate was monitored and recorded using a digital camera. A segment of the polymer coating on the sample inlet end of the capillary was removed by flame so that the capillary was transparent, and the contents could be visually observed. For the proof-of-principle experiments, H2O was used as the column liquid and an organic solution (90% n-butanol) was used as sample so a clear boundary could be observed, as suggested in a previous study.28 The sample injection flow rate was calculated by measuring the injection rate of the sample/column liquid interface movement in the spray-capillary (Figure S2).
A 50 cm spray-capillary (360 μm O.D. × 50 μm I.D.) was used for proof-of-concept experiments. Initially, to determine the primary driving force for sample injection into the spray-capillary device, a grounded copper plate was used as the spray target instead of the MS inlet to eliminate the vacuum force from the MS inlet capillary. The sample injection flow rate was estimated using the aforementioned monitoring method (H2O as the column liquid). The results, shown in Figure S3, indicate that ESI can be used alone as the driving force for sample injection, with an injection flow rate of 160.7 pL/s. The vacuum of the ESI inlet can increase the sample injection flow rate to about 263.8 pL/s. However, the vacuum from the MS inlet alone does not initiate the injection procedure as there was no obvious movement of the sample boundary observed under the camera.
We further evaluated the reproducibility of sample injection using the same spray-capillary and using reproductions. The 50 cm spray-capillary (360 μm O.D. × 50 μm I.D.) was used here with the injection voltage of 4 kV. Four individual sample injections were evaluated using the same capillary and conditions. The average sample injection flow rate in this capillary was estimated to be 255.2 pL/s and an RSD value of 4.9% was obtained demonstrating reasonable reproducibility among runs (Figure 1B). Similarly, we evaluated the day-to-day reproducibility and calculated an RSD of 3.29% (Figure 1C). Triplicate experiments were also performed using two different spray-capillaries to determine batch-to-batch reproducibility, which yielded an RSD value of 5.8% (Figure 1D).
The effect of the viscosity of the column liquid on injection flow rate was also tested to evaluate the influence of friction on liquid motion in the capillary. For evaluation, three types of column liquids with different viscosities (H2O, 50% ACN, 90% n-butanol, theoretical viscosities40 listed in Figure S4) were evaluated using the spray-capillary device; injection flow rates were calculated. Samples were chosen so that a clear boundary could be observed between the column liquid and the sample (Figure S4A). Our results (Figure S4B) suggested an inverse proportionality between flow rate and viscosity such that higher sample injection flow rates were observed for low viscosity column liquids. The increase in friction of the higher viscosity liquid may result in slower liquid motion, as suggested by Poiseuille’s law.22,41
Characterization of the Spray-Capillary Device Coupled with MS Detection.
Next, we investigated the feasibility of the spray-capillary to be directly coupled with MS for quantitative sample injection and analysis. After the spray-capillary sample injection, a pressure-based sample elution step was incorporated to flush the sample through the capillary for MS detection. Briefly, nitrogen (∼2.5 psi) was introduced into a glass sample vial filled with electrospray buffer (0.1% FA in 45% ACN), as described in the Experimental Section. For MS detection, 2.3−2.6 kV was used for the electrospray ionization on the metal tee of the sheathless interface.
A 100 μM solution of AngII (0.1% FA in 45% ACN) was used to characterize the efficiency of the spray-capillary (360 μm o.d., 50 μm i.d., 50 cm in length) coupled with MS detection. Sample was injected into the spray-capillary using an ESI voltage of 4 kV for 2, 15, 30, 60, and 90 s (Figure 2A). An injection time correlation plot (Figure 2B) was constructed based on the integrated peak areas of extracted ion chromatograms (EICs) of AngII 2+ ions. Good linearity (R2 = 0.98) indicates the proposed method is quantifiable for a wide range of injection times. The total injection volume ranges from 520 pL to 23.7 nL, which was estimated based on our precalculated capillary flow rates (∼260 pL/s) from camera monitoring. We also evaluated the reproducibility of the spray-capillary coupled with MS detection. Triplicate experiments with 60 s injection time were demonstrated in Figure 2C with an average RSD value of 5.79%, which is comparable to previous camera-based evaluation.
Figure 2.

Spray-capillary coupled with MS for quantitative sample injection. (A) EICs of AngII (m/z = 523.77−523.80) were evaluated with different injection times; (B) Calibration curve was constructed as a function of injection time (N = 3 for each experiment condition); (C) The injection reproducibility was demonstrated here using three replicated injections with 60 s injection times using the EICs of AngII (m/z = 523.77−523.80).
Several other parameters can be tuned to vary the rate of sample injection, such as capillary inner diameter, capillary length, and ESI voltage. (1) Capillary inner diameter: we evaluated the performance of the spray-capillary device (50 cm length) with 20 μm, 50 μm, and 75 μm I.D. The sample injection time was 60 s for all spray-capillaries. Our results suggested that the sample injection volume increased as the spray-capillary inner diameter increased (Figure 3A). Figure 3B shows a relatively high reproducibility (RSD = 11.3%) of detected MS signals for spray-capillary injections with 20 μm I.D. We also calculated the sample injection rate using the spray-capillary with 20 μm I.D. using camera monitoring. The estimated injection rate is 15.0 pL/s without vacuum and 48.2 pL/s when the spray-capillary was placed in front of the MS inlet. Some discontinuity in the peak shape and deviations from linearity was observed as the spray-capillary parameters were varied, which may be cause by the fluctuations of the N2 source. The flow resistance at the interface between the sample and running buffer under pressure may also contribute to some of the observed variation. (2) Spray-capillary length: Spray-capillaries (360 μm O.D. × 50 μm I.D.) with different lengths (30, 40, and 50 cm) were used to determine the effect of spray-capillary length on injection volume. To minimize potential errors, we used the same spray-capillary for all the experiments by trimming the capillary from the sample inlet end to produce the desired capillary lengths while the same MS inlet end was used. The strength of the MS signal decreased as the length of the spray-capillary increased, Figure 3C. This inverse trend may be the result of the longer plug of the column liquid present in longer capillaries, which leads to a stronger friction force and lower injection flow rate. (3) ESI voltage: A 50 cm spray-capillary with 360 μm O.D. × 50 μm I.D. was used to perform a series of experiments at varying electrospray voltages (between 2.6 and 4.5 kV; Figure 3D). The voltage of 2.6 kV was the lowest ESI voltage tested because electrospray was not stable at lower voltages with the 50 μm I.D. spray-capillary. The voltage of 4.5 kV was the highest ESI voltage selected because arcing started to affect the ESI process when the voltage was raised beyond this limit. A linear relationship was found between injection volume and electrospray voltage. The formation of the cone-jet depends on the balance between the surface tension of the spraying liquid and the electric field force.42 Therefore, a linear relationship between sample injection volume and ESI voltage is expected because, when electrospray voltage increases, the electric field force increases as well, resulting in a higher sample injection rate.
Figure 3.

Evaluation of spray-capillary performance as (A, B) capillary inner diameter, (C) capillary length, and (D) electrospray voltage were varied. Three spray-capillary injections using 20 μm I.D. spray-capillary were plotted in (B). N = 3 and the sample injection time of 60 s were used for each experiment condition.
Other factors that may affect the spray-capillary injection process such as EOF, capillary action, and sample adherence to the surface were evaluated based on the following set of experiments (Figure S5). (1) Background experiments (evaluation of random sample injection such as capillary action or sample adherence to the surface): the sample inlet end of spray-capillary was inserted into sample vials (10, 30, and 60 s, N = 3) with no voltage applied; (2) EOF experiments (evaluation of the effect of EOF on sampling): the sample inlet end of the spray-capillary was inserted into sample vials (10, 30, and 60 s, N = 3) with the application of 100 and 500 V. These voltage values were chosen because no electrospray is formed under these conditions. This experiment can provide us with a quantitative measurement of EOF during the injection, if any EOF exists. We found that the capillary action or sample adherence to the surface does contribute a relatively small amount of the sample that is injected into the capillary during the spray-capillary process (less than 10%). The EOF effect on the tip contributes minimally to the sample injection process. Overall, our results suggest that ESI is the main driving force for our spray-capillary experiments and EOF by itself does not significantly affect the sample aspiration.
Direct Coupling of the Spray-Capillary with a CE-MS Platform.
One advantage of the spray-capillary device is its ability to directly serve as the CE separation column after sample injection. The device can be directly coupled to the MS to produce an online spray-capillary CE-MS platform. We tested the performance of the spray-capillary CE-MS using a standard peptide mixture (10 μM AngII and 10 μM Syn-2). A bare spray-capillary (50 cm in length, 360 μm O.D., 50 μm I.D.) was used for these experiments. For sample injection, the ESI voltage was set at 3 kV, and the sample injection time was varied from 5 to 60 s (N = 3 for each condition). Baseline separation was achieved for these two peptides under these conditions. We also observed reasonably good reproducibility for both CE-MS elution time (RSD = 9.3%) and extract ion intensities of individual peptides (RSD = 6.64% for Syn-2 and RSD = 9.71% for AngII; Figure 4). In addition, a good linear relationship between detected signals and sample injection time were detected for both AngII (R2 = 0.93) and Syn-2 (R2 = 0.97), indicating that spray-capillary is capable of quantitative sample injection when coupled with the CE-MS platform (Figure 4A).
Figure 4.

Application of spray-capillary for low-volume sample injection and CE-MS analysis. (A) Calibration curve of standard peptides separation using the spray-capillary CE-MS platform (N = 3 for each experiment condition). (B) Reproducibility of peptide separation using the spray-capillary CZE-MS platform. Sample injection time is 60 s for all replicates. (C) EICs of Syn-2 (m/z = 503.30−503.37) with different sample injection times. (D) EICs of AngII (m/z = 523.77−523.80) with different sample injection times.
In our proof-of-principle experiments, the calculated theoretical plate number of Angiotensin II was approximately 30,000, which is lower than recent CE studies using the same peptide (∼300,000).10 To improve the separation resolution for complex sample analysis, several improvements to the CE separation can be made. For example, experiments that utilize a longer capillary with a smaller inner diameter combined with higher applied voltages have resulted in higher sensitivity and resolution separations of peptides and metabolites. These experiments have also demonstrated utility when the sample is limited, such as in the analysis of single cells.43 In addition, high-quality capillary coatings, such as linear polyacrylamide (LPA), have been used to improve separation power by eliminating EOF.44,45 We note that the performance of the spray-capillary may be evaluated and improved using the modifications described above to achieve better quantitative results.
The detection limit for reproducible sample injection volume was estimated based on data collected using 3 kV ESI voltage and 5 s sample injection. The sample injection flow rate (57.8 pL/s) was estimated using the previously described video-monitoring approach. For the 3 kV and 5 s sample injection, the total injected sample volume was 290 pL, and the injection amount of each peptide was about 2.9 fmol. Based on the approach described by Faserl et al.,46 the estimated average detection limits was calculated through extrapolation and was found to be 17 and 65 amole for Syn-2 and AngII, respectively (Figure S6). The highly reproducible separation performance of the standard peptide mixture indicates the spray-capillary holds the potential to analyze complex biological samples with optimized CE conditions such as longer columns or coated capillaries.
CONCLUSION
The spray-capillary is a simple quantitative microsampling approach that is capable of handling low-volume samples. In this study, we estimated the sample injection flow rate of the spray-capillary using a camera-monitoring approach. The performance of the spray-capillary was further evaluated using MS detection after sample injection. Different parameters such as viscosities of the column liquid, vacuum force, capillary inner diameter, capillary length, and electrospray voltage were evaluated. We demonstrated that the spray-capillary can directly serve as a CE capillary after sample injection for online separation and MS detection with no additional devices. However, there is room for improvement and optimization of the CE separation and spray-capillary device. The separation power of the system is relatively low compared to other CE-MS works and requires further optimization (e.g., background electrolyte, separation voltage, capillary coating). Additionally, a fine ESI emitter tip is required for a stable electrospray and quantitative sample injection. This spray-capillary tip is extremely fragile and requires careful handling. In addition, characterization of the spray-capillary device would benefit from a complete mathematical evaluation for better optimization of the system. Despite the shortcomings of the current spray-capillary CZE-MS platform, we believe that the performance can be elevated with the usage of high-quality, coated CE capillary (LPA-coating, PEI-coating) and the automation of the operation workflow, which would open some new avenues for analyzing biological samples. For example, a microscale fractionation method can be applied to fractionate the elution from a nanoflow RPLC separation of complex biological samples, and the low-volume fractions can be injected into the spray-capillary for the online CE-MS analysis. Moreover, spray-capillary holds great potential for the high-throughput omics analysis of ultralow-volume samples such as single-cell mass spectrometry.
Supplementary Material
ACKNOWLEDGMENTS
This work was partly supported by Grants from NIH NIAID R01AI141625, NIAID CSGADP Pilot Project (NIH 5U01AI101990-04, BRI No. FY15109843), NIH NIGMS R01GM118470, OCAST HR16-125, and OU FIP Program.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.9b04131.
Figures S1−S6 (PDF)
Video of the micro-sampling process using the spray-capillary (sample: 90% n-butanol; column liquid: H2O) (MP4)
The authors declare no competing financial interest.
REFERENCES
- (1).Altelaar AF; Heck AJ Curr. Opin. Chem. Biol 2012, 16 (1−2), 206–13. [DOI] [PubMed] [Google Scholar]
- (2).Huang EL; Piehowski PD; Orton DJ; Moore RJ; Qian WJ; Casey CP; Sun X; Dey SK; Burnum-Johnson KE; Smith RD Endocrinology 2016, 157 (3), 1307–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Shen Y; Tolic N; Masselon C; Pasa-Tolic L; Camp DG; Hixson KK; Zhao R; Anderson GA; Smith RD Anal. Chem 2004, 76 (1), 144–154. [DOI] [PubMed] [Google Scholar]
- (4).Shen YF; Zhao R; Berger SJ; Anderson GA; Rodriguez N; Smith RD Anal. Chem 2002, 74 (16), 4235–4249. [DOI] [PubMed] [Google Scholar]
- (5).Moini M. Anal. Chem 2007, 79 (11), 4241–4246. [DOI] [PubMed] [Google Scholar]
- (6).Sun LL; Zhu GJ; Zhao YM; Yan XJ; Mou S; Dovichi NJ Angew. Chem., Int. Ed 2013, 52 (51), 13661–13664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Wojcik R; Dada OO; Sadilek M; Dovichi NJ Rapid Commun. Mass Spectrom 2010, 24 (17), 2554–2560. [DOI] [PubMed] [Google Scholar]
- (8).Sun LL; Zhu GJ; Zhang ZB; Mou S; Dovichi NJ J. Proteome Res 2015, 14 (5), 2312–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Wilm M; Mann M. Anal. Chem 1996, 68 (1), 1–8. [DOI] [PubMed] [Google Scholar]
- (10).Choi SB; Zamarbide M; Manzini MC; Nemes P. J. Am. Soc. Mass Spectrom 2017, 28 (4), 597–607. [DOI] [PubMed] [Google Scholar]
- (11).Yang Y; Huang Y; Wu J; Liu N; Deng J; Luan T. TrAC, Trends Anal. Chem 2017, 90, 14–26. [Google Scholar]
- (12).Kelly RT; Tolmachev AV; Page JS; Tang K; Smith RD Mass Spectrom. Rev 2009, 29 (2), 294–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Zhu Y; Piehowski PD; Zhao R; Chen J; Shen YF; Moore RJ; Shukla AK; Petyuk VA; Campbell-Thompson M; Mathews CE; Smith RD; Qian WJ; Kelly RT Nat. Commun 2018, 9, na. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Saha-Shah A; Weber AE; Karty JA; Ray SJ; Hieftje GM; Baker LA Chem. Sci 2015, 6 (6), 3334–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Morris CA; Friedman AK; Baker LA Analyst 2010, 135 (9), 2190–202. [DOI] [PubMed] [Google Scholar]
- (16).Laforge FO; Carpino J; Rotenberg SA; Mirkin MV Proc. Natl. Acad. Sci. U. S. A 2007, 104 (29), 11895–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Actis P; Maalouf MM; Kim HJ; Lohith A; Vilozny B; Seger RA; Pourmand N. ACS Nano 2014, 8 (1), 546–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Shi W; Sa N; Thakar R; Baker LA Analyst 2015, 140 (14), 4835–42. [DOI] [PubMed] [Google Scholar]
- (19).Masujima T. Anal. Sci 2009, 25 (8), 953–960. [DOI] [PubMed] [Google Scholar]
- (20).Gholipour Y; Erra-Balsells R; Hiraoka K; Nonami H. Anal. Biochem 2013, 433 (1), 70–8. [DOI] [PubMed] [Google Scholar]
- (21).Zhang LW; Foreman DP; Grant PA; Shrestha B; Moody SA; Villiers F; Kwak JM; Vertes A. Analyst 2014, 139 (20), 5079–5085. [DOI] [PubMed] [Google Scholar]
- (22).Chen A; Lynch KB; Ren J; Jia Z; Yang Y; Lu JJ; Liu S. Anal. Chem 2017, 89 (20), 10806–10812. [DOI] [PubMed] [Google Scholar]
- (23).Zhang LW; Vertes A. Anal. Chem 2015, 87 (20), 10397–10405. [DOI] [PubMed] [Google Scholar]
- (24).Aerts JT; Louis KR; Crandall SR; Govindaiah G; Cox CL; Sweedler JV Anal. Chem 2014, 86 (6), 3203–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Saha-Shah A; Karty JA; Baker LA Analyst 2017, 142 (9), 1512–1518. [DOI] [PubMed] [Google Scholar]
- (26).Onjiko RM; Moody SA; Nemes P. Proc. Natl. Acad. Sci. U. S. A 2015, 112 (21), 6545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Lombard-Banek C; Moody SA; Manzini MC; Nemes P. Anal. Chem 2019, 91 (7), 4797–4805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Yin RC; Prabhakaran V; Laskin J. Anal. Chem 2018, 90 (13), 7937–7945. [DOI] [PubMed] [Google Scholar]
- (29).Onjiko RM; Portero EP; Moody SA; Nemes P. Anal. Chem 2017, 89 (13), 7069–7076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Huang XH; Coleman WF; Zare RN J. Chromatogr 1989, 480, 95–110. [Google Scholar]
- (31).Rose DJ; Jorgenson JW Anal. Chem 1988, 60 (7), 642–648. [Google Scholar]
- (32).Honda S; Iwase S; Fujiwara S. J. Chromatogr A 1987, 404, 313–320. [Google Scholar]
- (33).Nemes P; Rubakhin SS; Aerts JT; Sweedler JV Nat. Protoc 2013, 8 (4), 783–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Krylov SN; Starke DA; Arriaga EA; Zhang ZR; Chan NWC; Palcic MM; Dovichi NJ Anal. Chem 2000, 72 (4), 872–877. [DOI] [PubMed] [Google Scholar]
- (35).Zhang L; Khattar N; Kemenes I; Kemenes G; Zrinyi Z; Pirger Z; Vertes A. Sci. Rep 2018, 8 (1), 12227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Guillaume-Gentil O; Rey T; Kiefer P; Ibanez AJ; Steinhoff R; Bronnimann R; Dorwling-Carter L; Zambelli T; Zenobi R; Vorholt JA Anal. Chem 2017, 89 (9), 5017–5023. [DOI] [PubMed] [Google Scholar]
- (37).Prager DJ; Bowman RL; Vurek GG Science 1965, 147 (3658), 606–608. [DOI] [PubMed] [Google Scholar]
- (38).Actis P; Maalouf MM; Kim HJ; Lohith A; Vilozny B; Seger RA; Pourmand N. ACS Nano 2014, 8 (1), 546–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Zhou M; Huang C; Wysocki VH Anal. Chem 2012, 84 (14), 6016–23. [DOI] [PubMed] [Google Scholar]
- (40).Wohlfarth C; Wohlfahrt B. 2 Pure Compounds: References. Pure Organometallic and Organononmetallic Liquids, Binary Liquid Mixtures; Springer, 2001; pp 144–149. [Google Scholar]
- (41).Giddings JC Unified Separation Science; Wiley: New York, 1991. [Google Scholar]
- (42).Wilm MS; Mann M. Int. J. Mass Spectrom. Ion Processes 1994, 136 (2−3), 167–180. [Google Scholar]
- (43).Onjiko RM; Portero EP; Moody SA; Nemes P. Anal. Chem 2017, 89 (13), 7069–7076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Zhu G; Sun L; Dovichi NJ Talanta 2016, 146, 839–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Gomes FP; Yates JR 3rd Mass Spectrom. Rev 2019, 38 (6), 445–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Faserl K; Sarg B; Kremser L; Lindner H. Anal. Chem 2011, 83 (19), 7297–7305. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.