

Abstract
Unnatural chemically modified nucleotide sugars UDP-4-N3-GlcNAc and UDP-4-N3-GalNAc were chemically synthesized for the first time. These unnatural UDP sugar products were then tested for incorporation into hyaluronan, heparosan, or chondroitin using polysaccharide synthases. UDP-4-N3-GlcNAc served as a chain termination substrate for hyaluronan or heparosan synthases; the resulting 4-N3-GlcNAc-terminated hyaluronan and heparosan were then successfully conjugated with Alexa Fluor 488 DIBO alkyne, demonstrating that this approach is generally applicable for labeling and detection of suitable glycosaminoglycans.
Graphical Abstract
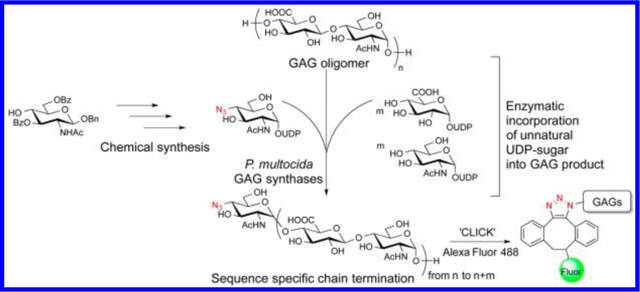
Glycosaminoglycans (GAGs) are a family of structurally complex heteropolysaccharides composed of repeating disaccharide units containing hexosamine residue.1 GAGs are prevalent as free glycans or in the core structures of glycans in glycoproteins and glycolipids and control a wide range of physiological and pathological events including cell-cell interactions,2 enzyme inhibition,3 cell proliferation,4 and growth factor receptor5 during various metabolic processes. Heparin/heparan sulfate, hyaluronan, and chondroitin/dermatan sulfate are the three most studied and abundant classes of GAG polymers. Nucleotide sugars are key intermediates in carbohydrate metabolism and glycoconjugate biosynthesis.6 Uridine diphosphate (UDP) monosaccharides are common sugar nucleotide donors being transferred to the nonreducing terminus of carbohydrate chains by glycosylation reactions with glycosyltransferases in the GAGs biosynthetic pathway.6,7
GlcNAc and GalNAc are naturally occurring saccharide residues prevalent in GAGs, with N-acetylglucosamine (GlcNAc) found in heparin/heparan sulfate/hyaluronan and N-acetylgalactosamine (GalNAc) found in chondroitin/dermatan sulfate, that play essential roles in biological processes.6,8 Thus, the corresponding naturally occurring UDP-GlcNAc/GalNAc as well as structural analogues are of great interest for enzymatic reactions in carbohydrate synthesis and would be a good approach to understand the mechanism of GlcNAc/GalNAc-related pathways.9 Because of its unique reactivity in biological systems, azide-derivatized UDP sugar becomes an exciting target and has been well established.10
The azido moiety is inert to natural processing or reactivity within biological systems but can be readily covalently tagged with imaging probes or epitope using an azide-specific reaction, such as Staudinger ligation11 with phosphines and the [3 + 2] cycloaddition with alkynes,12 which permits the detection of specific glycoconjugate types on cells or in living organisms or the selective capture of glycoproteins from cell or tissue lysates. At this time, various chemical or enzymatic approaches have been developed to synthesize azide-derivatized UDP sugars. Chen et al.10b reported a one-pot three enzyme system to produce UDP-6N3GlcNAc; Wang et al.10a developed a chemoenzymatic approach involving Escherichia coli GlcNAc-1-P uridyltransferase (GlmU) catalysis to obtain UDP-6N3GalNAc and UDP-GalNAz. However, because of the more challenging modification for the C4 position in either the chemical or enzymatic method, very few C4-modified azido-UDP sugars have been reported even though the 1 → 4 linkage is particular to GAGs.13
In our recent work, we reported the first chemoenzymatic synthesis of the UDP-4-fluoro (F) GlcNAc/−4-FGalNAc analogues using recombinantly expressed N-acetylglucos-amine-1-phosphate uridylyltransferase (GlmU) and the application of UDP-4-FGlcNAc as a glycosylation terminator in the chemoenzymatic synthesis of heparan sulfate/heparin.14 Here, we present the first synthesis of 4-N3-derivatized GlcNAc and GalNAc UDP sugars that were tested for their ability to mimic two natural donor substrates required in GAG synthesis, resulting in metabolic conversion to azide-functionalized GAGs as potential substrates for Huisgen 1,3-dipolar cycloaddition.12 The corresponding unnatural GlcNAc-1-phosphate substrate specificity of the enzyme GlmU was also investigated.
Our first objective was the synthesis of 4-N3-GlcNAc-/4-N3-GalNAc-1-phosphate analogues. These precursors could then be used as substrates for either chemical or enzymatic reactions to access the target UDP-4-N3-GlcNAc or UDP-4-N3-GalNAc donors. On the basis of our previous studies,14 we started the synthesis from the known compounds benzyl 2-acetamido-2-deoxy-β-d-gluco-/galacto-pyranoside 1a/1b (Scheme 1).14,15 The placement of the C-4 azide functionality was initiated through the reaction of Tf2O in pyridine with 1a/1b to afford the triflates 2a/2b. Nucleophilic displacement of triflate groups in 2a/2b with sodium azide in dimethylformamide afforded 4-N3-GalNAc/4-N3-GlcNAc derivatives 3a/3b in 85–90% yields with the inversion of the C-4 configuration.16 The anomeric benzyl group was oxidatively removed, affording 4a/4b followed by phosphorylation17 with tetrabenzyl pyrophosphate to obtain the phosphorylated intermediate 5a/5b in good yield (80%−84%) and with excellent α-selectivity. Deprotection of benzyl groups was achieved under hydrogenation to obtain the target benzoate-protected substrates 6a/6b in good yields (85–90%). Hydrogenation needed to be strictly controlled as longer reaction times would cause azido reduction.
Scheme 1. Synthesis of 4-N3-GalNAc-1-phosphate 6a and 4-N3-GlcNAc-1-phosphate 6ba.
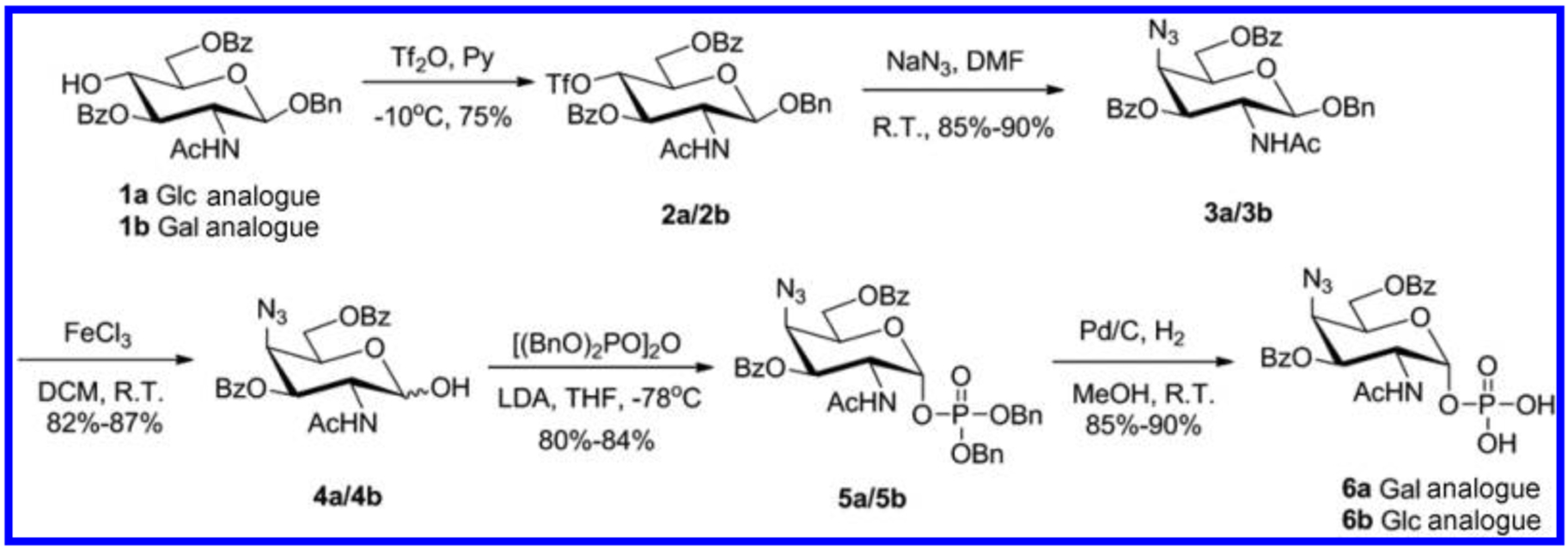
aAlthough only one stereochemical series is shown (starting from β-d-glucopyranoside 1a to 6a), both series were synthesized and the yields for both are provided.
With both protected 4-N3-N-acetylhexosamine-1-phosphates in hand, we next focused our attention on their deprotection and on determining whether they could be enzymatically converted with GlmU uridyltransferase to the two unnatural nucleotide sugar donors.14 Treatment of 6a/6b by sodium methoxide afforded the deprotected 4-N3-GalNAc-1-phosphate/4-N3-GlcNAc-1-phosphate analogues. Unfortunately, GlmU recognized neither of the analogues, and thus, no corresponding UDP sugars were observed by LC-MS (Scheme 2A). Wang et al.10a investigated the UDP-GlcNAc/UDP-GalNAc substrate specificity of GlmU and suggested that GlmU was intolerant of any functional group on the C4 position larger than a hydroxyl functionality. We also reported the successful GlmU recognition for 4-F-GlcNAc-1-phosphate/4-F-GalNAc-1-phosphate analogues.14 Because the fluorine atom is considerably smaller and the azido group is considerably larger than a hydroxy group, our observations of the failure of GlmU to utilize the 4-N3- and 4-F-N-actylhexosamine-1-phosphate analogues are consistent with those of Wang et al.10a
Scheme 2.
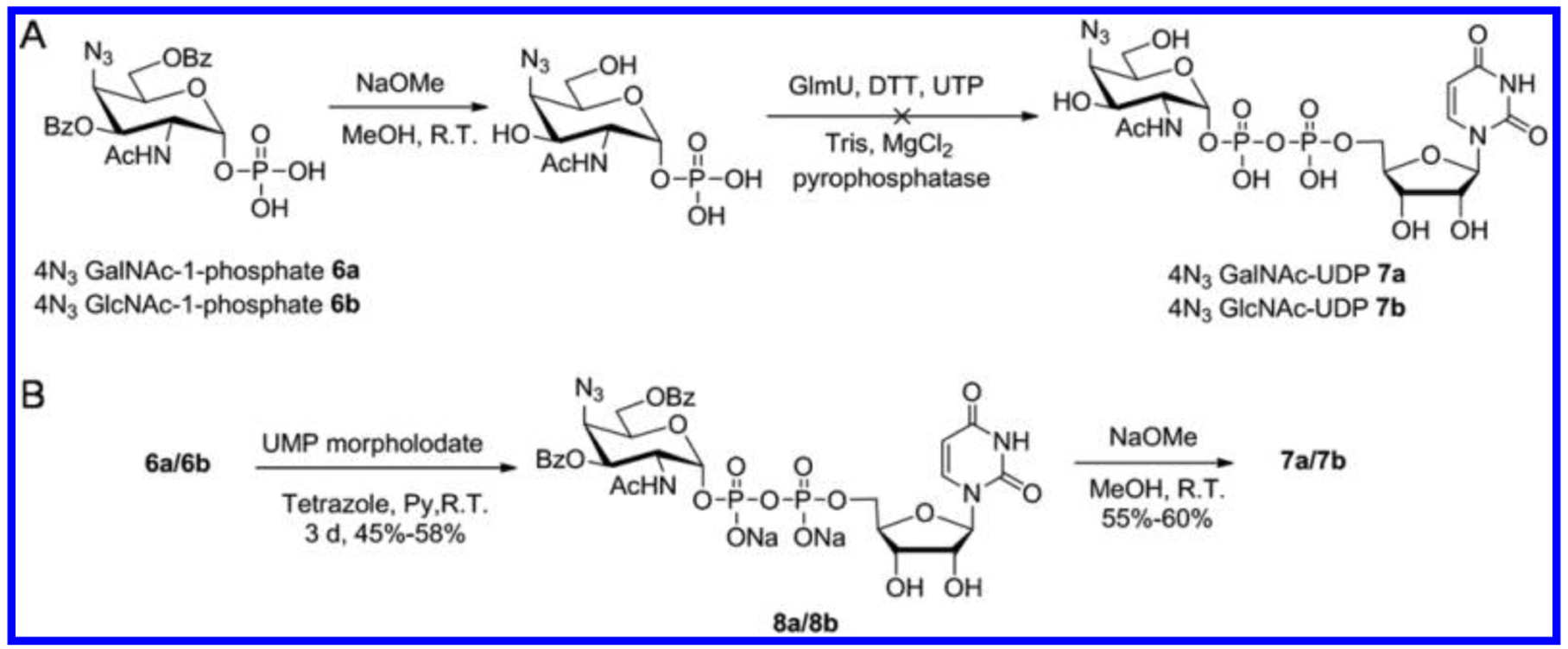
GlmU Attempt and Chemical Synthesis of UDP-4-N3-GalNAc and UDP-4-N3-GlcNAc Donors
Next, we turned our attention to the chemical approach to preparing the unnatural UDP sugar donors (Scheme 2B). The protected 4-N3-N-acetylhexosamine-1-phosphates 6a/6b were converted to their pyridinium salts and then stirred with UMP-morpholidate and tetrazole in pyridine for 3 days to afford the benzoate-protected UDP sugars.18 By treating the reaction crude with (Na+) resin, all the phosphate salts were converted to their sodium form and purified on a Biogel P2 column, affording the protected UDP derivatives 8a/8b in modest yields (45–58%). Deprotection of 8a/8b was carried out under standard Zemplén conditions, affording the corresponding donors UDP-4-N3-GalNAc 7a and UDP-4-N3-GlcNAc 7b.
The utility of the two chemically synthesized unnatural UDP donors as substrates in GAG synthesis was next examined. In the pilot trials, the incorporation of these new synthetic analogues was attempted using indirect radiochemical sugar assays that rely on coincorporation of a labeled natural sugar when the analogue is used by the Pasteurella GAG synthases,19 but the potential signals were very close to background levels. Therefore, a mass spectrometry-based assay was employed to monitor the incorporation of any analogue directly. Basically, a short GAG acceptor terminating in GlcA was reacted with the analogue, and the reaction mixture was analyzed for the presence of elongated species by MALDI-ToF MS. A single azido-monosaccharide derived from UDP-4-N3-GlcNAc was incorporated into a growing GAG chain of heparosan (HEP) with Pasteurella multocida heparosan synthases (PmHS1 or 2) or hyaluronan (HA) with Pasteurella multocida hyaluronan synthase (PmHAS) based on the very close agreement between the predicted and observed masses of the elongated species (Table 1).
Table 1.
Mass Spectrometric Analysis of GAG Synthase Reaction Productsa
| acceptor | donor | PmHAS | PmCS | PmHS1 | PmHS2 |
|---|---|---|---|---|---|
| HA4b | none | 775.05 | 775.04 | ||
| + UDP-GlcNAc | 978.09 | 978.08 | |||
| + UDP-4-N3-GlcNAc | 1003.12 | NDe | |||
| HEP6c | none | 1097.10 | |||
| + UDP-GlcNAc | 1300.10 | ||||
| + UDP-4-N3-GlcNAc | 1325.08 | ||||
| HEP3-Benzd | None | 747.28 | 747.33 | ||
| + UDP-GlcNAc | 950.39 | 950.43 | |||
| + UDP-4-N3-GlcNAc | NDe | 975.50 |
The theoretical azide mass addition compared to that of native sugars is 25.02 Da.
Hyaluronan tetrasaccharide.
Heparosan hexasaccharide.
Heparosan trisaccharide-benzaldehyde (aldehyde at the reducing end).
No desired product.
In parallel tests with UDP-4-N3-GalNAc and the Pasteurella multocida chondroitin synthase, PmCS, the analogue was not incorporated into the chain of the chondroitin acceptor (not shown). This is somewhat surprising as PmHAS and PmCS are very similar at the amino acid sequence level (~87% identical) and the tested reaction conditions were equivalent, but currently there is no structural information for these two enzymes; thus, some differences in their respective active sites seem a likely explanation for the observed usage of the 4-azido precursors.
We successfully prepared two azido-tagged GAGs (see the Experimental Section for details), either heparosan (HEP) or hyaluronan (HA), by employing the UDP-4-N3-GlcNAc as a chain-terminating donor. Thus, these end-labeled GAGs both possess a 4-N3-GlcNAc residue at their nonreducing end. We next examined whether these modified chains were “Click”-reactive. Reactions between the 4-N3-GlcNAc-terminated HEP or HA (~10–100 kDa) and Alexa Fluor 488 DIBO alkyne20 were performed, and the products were analyzed by polyacrylamide gel electrophoresis (PAGE) analysis and fluorescence detection (488 nm excitation; 494 nm emission) (Figure 1, lanes 1 and 2). As negative controls, either the starting material 4-N3-GlcNAc-terminated HA or HEP (~10–100 kDa) without added Alexa Fluor 488 DIBO alkyne were also loaded on the gel (lanes 3 and 4 in Figure 1). The gel was subjected to electrophoresis for 4 h to run the unreacted, excess Alexa Fluor 488 DIBO alkyne off the gel to reduce the level of residual fluorescent reagent in the GAG area. All four lanes containing GAG samples were then visualized by Alcian blue staining; each lane displayed a blue-stained band corresponding to the expected molecular weight and polydispersity of the input GAG samples (Figure 1, left panel). As expected, only the GAG bands present in lanes 1 and 2 (Figure 1, right panel) displayed fluorescence, confirming that 4-N3-GlcNAc-terminated HEP and HA had been successfully click-tagged with the alkyne dye.
Figure 1.
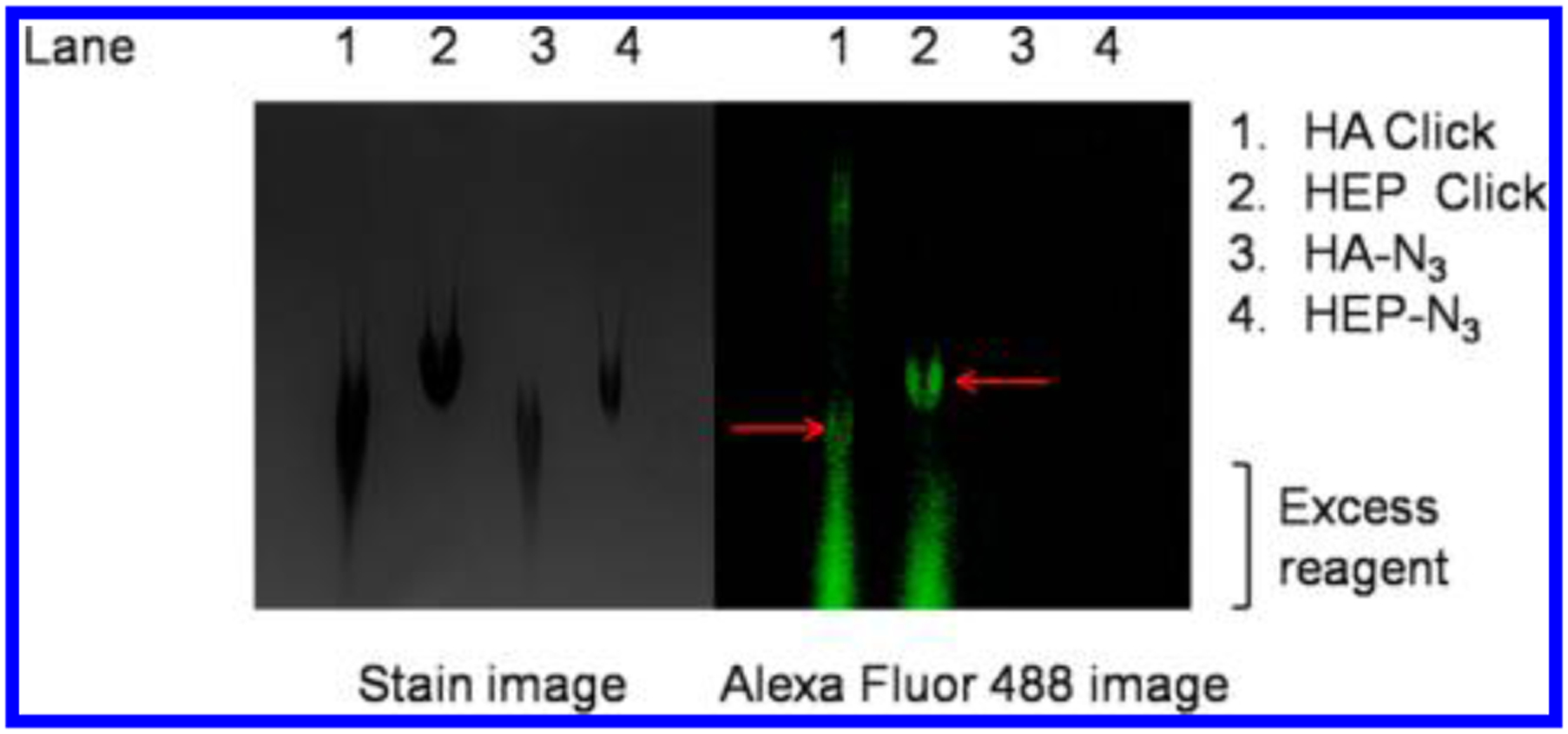
PAGE analysis of click reaction between 4-N3-GlcNAc-terminated HEP and HA and Alexa Fluor 488 DIBO alkyne. Products are marked with red arrows. The excess reagent runs as a smear running faster than the GAGs (denoted with a bracket).
In summary, this is the first synthesis of UDP-4-N3-GlcNAc/UDP-4-N3-GalNAc analogues reported using an efficient all-chemical approach. The final unnatural UDP sugars were then tested for incorporation into HA, HEP, and chondroitin using polysaccharide synthases, and UDP-4-N3-GlcNAc was added to the nonreducing end of the sugar chain and served as a chain termination substrate in HA and HEP. Compared to the chemical method to introduce azido groups into polysaccharides, this enzymatic technique provided a “green” approach to access regioselectively azide-functionalized GAGs, which would bring better control of the final structure and composition of these macromolecules. The resulting 4-N3-GlcNAc-terminated HEP and HA were then conjugated with Alexa Fluor 488 DIBO alkyne, demonstrating that this technology is generally applicable for labeling and detection of suitable GAGs.
EXPERIMENTAL SECTION
General Information.
All reagents were purchased from commercial vendors and, unless otherwise noted, used without further purification. Flash chromatography (FC) was performed using silica gel (200–300 mesh) according to standard protocols. Reactions were monitored by thin-layer chromatography (TLC) on silica gel F254 plates. Mass data were acquired by MALDI-ToF-MS or electrospray ionization (ESI)-high-resolution (HR) MS on an LTQ Orbitrap XL FT-MS spectrometer. 1H, 13C, 1H-1H COSY, and 1H-13C HSQC NMR spectra were recorded on an 800 MHz (200 MHz for 13C NMR) or 600 MHz (150 MHz for 13C NMR) spectrometer.
Synthesis of Benzyl 2-Acetamido-4-azido-3,6-di-O-benzoyl-2,4-dideoxy-β-d-galacto/gluco-pyranosides (3a/3b).
The dibenzoylated compound 1a/1b (520 mg, 1.0 mmol, 1.0 equiv) was dissolved in a DCM (5 mL) and pyridine (0.2 mL) solvent system. The solution was cooled to 0 °C, and trifluoromethane sulfonic anhydride (0.34 mL, 2.0 mmol, 2.0 equiv) was added dropwise; the resulting mixture was stirred at 0 °C for 3 h. The mixture was diluted with 20 mL of EtOAc, washed with H2O, and dried over anhydrous Na2SO4. After filtering, the solvent was removed under vacuum to afford the triflate derivative 2a/2b as yellow oils, which were used without further purification.
The triflate derivative 2a/2b from above was dissolved in DMF (5 mL); sodium azide (195 mg, 3.0 mmol, 3.0 equiv) was added and stirred at room temperature overnight. The solvent was removed under vacuum, and the residue was dissolved in 20 mL of EtOAc and washed with H2O. After drying over anhydrous MgSO4, the solvent was removed, and the resulting crude mixture was purified by column chromatography (1:2 hexanes/EtOAc) to yield 3a/3b as white solids (348 mg, 64% and 364 mg, 67%, respectively; two steps).
Benzyl 2-Acetamido-4-azido-3,6-di-O-benzoyl-2,4-dideoxy-β-d-galactopyranosides (3a).
White solid. 1H NMR (600 MHz, CDCl3) δ 8.13–8.10 (m, 2H), 8.05–8.02 (m, 2H), 7.64–7.58 (m, 2H), 7.53–7.49 (m, 2H), 7.48–7.43 (m, 2H), 7.34–7.29 (m, 3H), 7.29–7.27 (m, 2H), 5.33 (m, 1H), 4.89 (d, J = 12.50, 1H), 4.72 (dd, J = 12.20, 2.27, 1H), 4.64–4.58 (m, 2H), 4.56 (d, J = 8.47, 1H), 4.30–4.25 (m, 1H), 3.88 (dd, J = 10.20, 1H), 3.65–3.62 (m, 1H), 1.84 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 173.6, 170.1, 169.6, 140.2, 137.2, 136.8, 133.4, 133.3, 133.1, 133.0, 132.0, 131.9, 131.8, 131.7, 131.4, 103.0, 77.6, 75.9, 73.7, 66.8, 64.3, 57.6, 26.6. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C29H29N4O7 545.2031; found 545.2022.
Benzyl 2-Acetamido-4-azido-3,6-di-O-benzoyl-2,4-dideoxy-β-d-glucopyranosides (3b).
White solid. The NMR and MS data were consistent with the literature.16
Synthesis of 1-Phospho-2-acetamido-4-azido-3,6-di-O-benzoyl-α-d-galacto/glucopyranosides (5a/5b).
To a solution of 3a/3b (207 mg, 0.38 mmol) in dry DCM (5 mL) was added FeCl3 (136 mg, 0.84 mmol) under N2 protection. After stirring for 3 h, the reaction was quenched by the addition of aq NH4Cl. The aqueous layer was extracted with DCM (3 × 15 mL). The combined organic phase was washed with brine, dried over Na2SO4, and concentrated under a vacuum. The resulting crude product 4a/4b was used in the following step without further purification.
Crude 4a/4b was dissolved in dry THF (5 mL), and the solution was cooled to −78 °C. To the cooled solution was added lithium diisopropylamide solution (LDA, 2 M in THF) (0.42 mL, 0.84 mmol) dropwise. After 30 min, tetrabenzyl pyrophosphate (307 mg, 0.57 mmol) was added to the reaction mixture. The reaction was stirred at −78 °C for 30 min and slowly warmed to 0 °C over 3 h. Then, the reaction was quenched by the addition of aq NH4Cl. The aqueous layers were extracted with EtOAc (3 × 15 mL), and the combined organic phase was washed with brine, dried over Na2SO4, and concentrated under vacuum. The crude product was purified by FC (silica gel, 1:2 hexanes/EtOAc) to give compounds 5a/5b as yellow oils (187 mg, 69% and 198 mg, 73%, respectively; two steps).
1-Phospho-2-acetamido-4-azido-3,6-di-O-benzoyl-α-d-galactopyranosides (5a).
Yellow oil. 1H NMR (800 MHz, CDCl3) δ 8.06–8.04 (m, 2H), 7.99–7.97 (m, 2H), 7.60–7.57 (m, 1H), 7.54–7.51 (m, 1H), 7.47–7.44 (m, 2H), 7.39–7.36 (m, 2H), 7.35–7.28 (m, 10H), 5.71 (dd, J = 5.82, 3.29, 1H), 5.60 (d, J = 9.36, 1H), 5.45 (dd, J = 3.29, 11.28, 1H), 5.07–4.97 (m, 4H), 4.89–4.84 (m, 1H), 4.45–4.41 (m, 1H), 4.40–4.36 (m, 1H), 4.35–4.32 (m, 1H), 4.20–4.18 (m, 1H), 1.65 (s, 3H); 13C NMR (200 MHz, CDCl3) δ 170.2, 166.4, 165.9, 135.4, 135.3, 135.2, 135.1, 134.0, 133.4, 130.2, 129.7, 129.3, 128.9, 128.8, 128.7, 128.7, 128.5, 128.3, 128.2, 128.1, 97.0 (d, J = 6.13), 70.2, 70.0 (d, J = 5.11), 69.9 (d, J = 4.86), 68.8, 62.9, 60.5, 47.9 (d, J = 6.90), 22.9. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C36H36N4O10P 715.2164; found 715.2153.
1-Phospho-2-acetamido-4-azido-3,6-di-O-benzoyl-α-d-glucopyranosides (5b).
Yellow oil. 1H NMR (600 MHz, CDCl3) δ 8.07–8.04 (m, 2H), 8.04–8.02 (m, 2H), 7.61–7.57 (m, 2H), 7.48–7.44 (m, 4H), 7.41–7.33 (m, 10H), 5.85 (d, J = 9.60, 1H), 5.73 (dd, J = 3.20, 5.98, 1H), 5.42 (dd, J = 9.28, 10.88, 1H), 5.15–5.03 (m, 4H), 4.53–4.46 (m, 2H), 4.41 (dd, J = 2.03, 12.27, 1H), 3.94–3.87 (m, 2H), 1.63 (s, 3H);13C NMR (150 MHz, CDCl3) δ 173.7, 170.0, 169.4, 138.7, 138.6, 138.5, 138.4, 137.2, 136.7, 133.3, 133.1, 132.8, 132.3, 132.2, 132.1, 132.0, 131.9, 131.8, 131.5, 99.8 (d, J = 5.73), 74.8, 73.7, 73.4 (d, J = 4.46), 73.3 (d, J = 4.71), 65.9, 63.5, 55.4 (d, J = 7.50), 26.1. HRMS (ESI-TOF) m/z: [M + H]+ calcd for C36H36N4O10P 715.2164; found 715.2147.
Synthesis of Disodium Uridine 5′-(2-Acetamido-2,4-dideoxy-4-azido-3,6-di-O-benzoyl-α-d-galacto/glucopyranosyl) Diphosphate (8a/8b).
The protected phosphate 5a/5b (10 mg, 0.013 mmol) was dissolved in MeOH (2 mL). Pd/C (10 wt % on activated carbon) (6 mg) was added, and the mixture was stirred under a hydrogen atmosphere for 2 h. The palladium was then filtered off, and the solvent was removed in vacuo to produce crude 6a/6b in 90 and 85% yields, respectively, which were used in the next step without further purification.
A solution of monophosphate 6a/6b (8 mg, 0.013 mmol) in MeOH (2 mL) was treated with Et3N (9 μL, 0.07 mmol) and then concentrated to yield crude bis(triethylammonium) phosphate. Without purification, this crude material was repeatedly coevaporated with dry pyridine (3 × 3 mL). Uridine 5′-monophosphomorpholidate 4-morpholine-N,N′-dicyclohexylcarboxamidine salt (14 mg, 0.02 mmol) was coevaporated with pyridine (3 × 3 mL) in a separate vessel and then transferred in 2 mL of pyridine via a cannula into the reaction flask. The combined reagents were coevaporated with pyridine (2 × 2 mL); then, 1H-tetrazole (4 mg, 0.05 mmol) in pyridine (2 mL) was added, and the reaction mixture was stirred at rt for 3 d. The reaction mixture was then concentrated in vacuo and converted to Na+ form by passing through a Dowex (Na+) column. The resulting fraction was concentrated in a vacuum, and the residue was loaded onto a Bio Gel P2 column (1 × 120 cm) eluted with H2O. Fractions were collected, and those containing the product as determined by MS were combined and freeze-dried to afford 8a/8b as white powders (5 mg, 45% and 7 mg, 58%, respectively).
Disodium Uridine 5′-(2-Acetamido-2,4-dideoxy-4-azido-3,6-di-O-benzoyl-α-d-galactopyranosyl) Diphosphate (8a).
White solid. 1H NMR (600 MHz, D2O) δ 7.98–7.90 (m, 3H), 7.86 (d, J = 8.15, 1H), 7.81 (d, J = 8.15, 1H), 7.63–7.54 (m, 3H), 7.48–7.41 (m, 3H), 5.87–5.81 (m, 1H), 5.76–5.74 (m, 1H), 5.65 (d, J = 8.22, 1H), 5.61–5.58 (m, 1H), 5.45–5.41 (m, 1H), 4.53–4.43 (m, 2H), 4.30–4.19 (m, 3H), 4.18–4.12 (m, 2H), 4.00–3.89 (m, 2H), 1.85 (s, 3H); 13C NMR (150 MHz, D2O) δ 178.2, 170.7, 169.7, 169.4, 155.1, 145.2, 144.9, 144.6, 137.7, 137.3, 133.0, 132.9, 132.2, 132.1, 106.1, 105.2, 97.8, 91.6, 87.1, 86.5, 77.6, 77.2, 75.1, 73.3, 73.1, 72.3, 71.4, 67.0, 63.8, 25.4. HRMS (ESI-TOF) m/z: [M - H]− calcd for C31H33N6O18P2 839.1332; found 839.1340.
Disodium Uridine 5′-(2-Acetamido-2,4-dideoxy-4-azido-3,6-di-O-benzoyl-α-d-glucopyranosyl) Diphosphate (8b).
White solid. 1H NMR (600 MHz, D2O) δ 7.99–7.91 (m, 4H), 7.84–7.80 (m, 2H), 7.64–7.60 (m, 2H), 7.48–7.44 (m, 3H), 5.85–5.81 (m, 1H), 5.78 (d, J = 3.05, 1H), 5.73 (d, J = 8.08, 1H), 5.59 (dd, J = 3.12, 6.74, 1H), 5.44 (m, 1H), 4.63–4.53 (m, 2H), 4.45 (dt, J = 3.05, 10.49, 1H), 4.31–4.27 (m, 1H), 4.24–4.18 (m, 3H), 4.17–4.11 (m, 2H), 1.78 (s, 3H); 13C NMR (150 MHz, D2O) δ 177.8, 171.4, 171.0, 169.2, 154.8, 144.9, 144.7, 137.7, 137.5, 133.0, 132.8, 132.2, 132.1, 132.0, 106.0, 105.5, 97.7, 92.4, 86.1, 77.4, 77.1, 76.1, 72.9, 72.8, 72.6, 68.4, 66.8, 63.1, 55.2, 25.0. HRMS (ESI-TOF) m/z: [M - H]− calcd for C31H33N6O18P2 839.1332; found 839.1335.
Synthesis of Disodium Uridine 5′-(2-Acetamido-2,4-dideoxy-4-azido-α-d-galacto/glucopyranosyl) Diphosphate (7a/7b).
To a solution of 8a/8b (6 mg, 0.007 mmol) in anhydrous MeOH (1 mL) was added sodium methoxide solution (20 μL, 5.5 M in methanol), and the resulting solution was left to stir overnight. The solution was then neutralized with prewashed and acidified Amberlyte IR-120 hydrogen ion-exchange resin, and the reaction mixture was filtered through cotton wool to remove the resin. The reaction mixture was concentrated under reduced pressure, and the residue was loaded onto a Bio Gel P2 column (1 × 75 cm) and eluted with H2O to give 7a/7b as white solids (2 mg, 55% and 3 mg, 60%, respectively).
Disodium Uridine 5′-(2-acetamido-2,4-dideoxy-4-azido-α-d-galactopyranosyl) Diphosphate (7a).
White solid. 1H NMR (600 MHz, D2O) δ 7.82 (d, J = 8.06, 1H, uridine-H″−6), 5.86–5.82 (m, 2H, uridine-H″−5, rib-H′−1), 5.38–5.35 (m, 1H, H-1), 4.26–4.20 (m, 2H, rib-H′−2, rib-H′−5a), 4.17–4.08 (m, 4H, H-2, H-5, rib-H′−3, rib-H′−4), 4.07–4.00 (m, 2H, H-3, rib-H′−5b), 3.67–3.63(m, 1H, H-6a), 3.62–3.56 (m, 2H, H-4, H-6b), 1.95 (s, 3H); 13C NMR (150 MHz, D2O) δ 185.8, 172.9, 171.7, 151.9, 141.7, 102.6, 94.6, 88.5, 83.1, 73.6, 70.5, 69.5, 68.0, 65.1, 62.8, 60.7, 21.9. HRMS (ESI-TOF) m/z: [M - H]− calcd for C17H25N6O16P2 631.0808; found 631.0808.
Disodium Uridine 5′-(2-Acetamido-2,4-dideoxy-4-azido-α-d-glucopyranosyl) Diphosphate (7b).
White solid. 1H NMR (600 MHz, D2O) δ 7.86 (d, J = 7.95, 1H, uridine-H″−6), 5.88–5.84 (m, 2H, uridine-H″−5, rib-H′−1), 5.42 (dd, J = 3.18, 7.32, 1H, H-1), 4.27–4.22 (m, 2H, rib-H′−2, rib-H′−5a), 4.19–4.13 (m, 2H, rib-H′−3, rib-H′−4), 4.10–4.06 (m, 1H, rib-H′−5b), 3.95(dt, J = 2.84, 10.70, 1H, H-2), 3.79–3.66 (m, 4H, H-3, H-5, H-6a, H-6b), 3.48 (m, 1H, H-4), 1.97 (s, 3H, H-NAc); 13C NMR (150 MHz, D2O) δ 178.5, 169.9, 145.1, 106.0, 98.0, 92.3, 86.5, 77.2, 74.9, 73.7, 72.7, 68.2, 64.9, 63.8, 56.9, 25.6. HRMS (ESI-TOF) m/z: [M - H]− calcd for C17H25N6O16P2 631.0808; found 631.0809.
GAG Synthase Assays.
Either the azido-containing UDP-hexosamine analogue or authentic UDP-hexosamine (0.5 mM) was incubated in a reaction (25 μL) containing 50 mM Tris, pH 7.2, 1 mM MnCl2, radiolabeled UDP-[3H]GlcA (0.05 mM, 0.1 μCi; PerkinElmer), and 25 μg of purified recombinant enzyme (PmHS1 or PmHS2; PmHAS or PmCS) (as well as 1 M ethylene glycol for PmHAS and PmCS) at 30 °C for 5.25 h. The reaction mixtures were quenched with sodium dodecyl sulfate (2% final concn) and analyzed by descending paper chromatography (overnight in 65:35 ethanol/1 M ammonium acetate buffer; Whatman 3MM paper). As a negative control for assay background, a reaction with no UDP-hexosamine was tested in parallel (GAG chain polymerization can only occur when UDP-GlcA and a functional UDP-hexosamine are present simultaneously).
Mass Spectroscopy.
HA21a or heparosan oligosaccharides21b (0.1–1 μg) were reacted with a 2–10-fold molar excess of UDP-GlcNAc, UDP-GalNAc, the azido-containing analogue, or no analogue in a buffer containing 50 mM Tris, pH 7.2, 1 mM MnCl2, and purified recombinant synthase (5–10 μg) for 16 h at 30 °C. The reactions were assessed by MALDI-ToF-MS with the matrix 6-aza-2-thiothymine at a concentration of 5 mg/mL in 50% acetonitrile and 0.1% trifluoroacetic acid (TFA). HA oligosaccharides were employed as mass calibrants.
Azido-Tagged GAG Preparations.
GAGs (HA, 68 kDa or HEP, 40.1 kDa) were azido end-labeled at the nonreducing terminus using the donor sugar UDP-4-N3-GlcNAc in a reaction buffer containing 50 mM HEPES, pH 7.2, 1 mM MnCl2, and 1 μg/μL of purified enzyme (PmHAS or PmHS1).
Click Chemistry.
The 4-N3-GlcNAc-terminated HEP or HA polymers (30 μg) were mixed with a 10-fold excess Alexa Fluor 488 DIBO alkyne in 15 μL of deionized water and incubated at room temperature in the dark for 5 h. The crude reaction was then directly subjected to PAGE analysis.
Polyacrylamide Gel Electrophoresis (PAGE).
Phenol red dye was added to the sample for visualization of the ion front during electrophoresis. A 10 μg aliquot of each sample was analyzed on a 4–15% Mini-Protean TGX Precast Gel ran in Tris-Glycine buffer (at constant 30 mA for 4 h). The gel was first visualized by UV fluorescence followed by Alcian Blue staining (0.5% w/v Alcian blue dye and 2% v/v aqueous acetic acid) and destaining in water.22
Supplementary Material
ACKNOWLEDGMENTS
The authors are grateful for funding from the National Institutes of Health in the form of grants HL62244 and HL094463.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.7b01787.
HRMS, 1H, 13C, COSY and HSQC NMR spectra of the components (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).(a) DeAngelis PL; Liu J; Linhardt RJ Glycobiology 2013, 23, 764–777. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mende M; Bednarek C; Wawryszyn M; Sauter P; Biskup MB; Schepers U; Bräse S Chem. Rev 2016, 116, 8193–8255. [DOI] [PubMed] [Google Scholar]
- (2).Furukawa J; Okada K; Shinohara Y Glycoconjugate J. 2016, 33, 707–715. [DOI] [PubMed] [Google Scholar]
- (3).Ledoux D; Merciris D; Barritault D; Caruelle J-P FEBS Lett. 2003, 537, 23–29. [DOI] [PubMed] [Google Scholar]
- (4).Sirko S; von Holst A; Wizenmann A; Götz M; Faissner A Development 2007, 134, 2727–2738. [DOI] [PubMed] [Google Scholar]
- (5).Schultz V; Suflita M; Liu X; Zhang X; Yu Y; Li L; Green DE; Xu Y; Zhang F; DeAngelis PL; Liu J; Linhardt RJ J. Biol. Chem 2017, 292, 2495–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Wagner GK; Pesnot T; Field RA Nat. Prod. Rep 2009, 26, 1172–1194. [DOI] [PubMed] [Google Scholar]
- (7).Masuko S; Bera S; Green DE; Weïwer M; Liu J; DeAngelis PL; Linhardt RJ J. Org. Chem 2012, 77, 1449–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Liu J; Linhardt RJ Nat. Prod. Rep 2014, 31, 1676–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Liu R; Xu Y; Chen M; Weïwer M; Zhou X; Bridges AS; DeAngelis PL; Zhang Q; Linhardt RJ; Liu JJ Biol. Chem 2010, 285, 34240–34249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) Guan W; Cai L; Fang J; Wu B; George Wang P Chem. Commun 2009, 6976–6948. [DOI] [PubMed] [Google Scholar]; (b) Chen Y; Thon V; Li Y; Yu H; Ding L; Lau K; Qu J; Hie L; Chen X Chem. Commun 2011, 47, 10815–10817. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Nifker G; Levy-Sakin M; Berkov-Zrihen Y; Shahal T; Gabrieli T; Fridman M; Ebenstein Y ChemBioChem 2015, 16, 1857–1860. [DOI] [PubMed] [Google Scholar]
- (11).(a) Zhu S; Guo Z Org. Lett 2017, 19, 3063–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) He Y; Hinklin RJ; Chang J; Kiessling LL Org. Lett 2004, 6, 4479–4482. [DOI] [PubMed] [Google Scholar]
- (12).(a) Sletten EM; Bertozzi CR Org. Lett 2008, 10, 3097–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Huisgen R Angew. Chem 1963, 75, 604–637. [Google Scholar]
- (13).Weerapana E; Glover KJ; Chen MM; Imperiali BJ Am. Chem. Soc 2005, 127, 13766–13767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Schultz VL; Zhang X; Linkens K; Rimel J; Green DE; DeAngelis PL; Linhardt RJ J. Org. Chem 2017, 82, 2243–2248. [DOI] [PubMed] [Google Scholar]
- (15).Christensen H; Christiansen MS; Petersen J; Jensen HH Org. Biomol. Chem 2008, 6, 3276–3283. [DOI] [PubMed] [Google Scholar]
- (16).Weerapana E; Glover KJ; Chen MM; Imperiali BJ Am. Chem. Soc 2005, 127, 13766–13767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Muller T; Danac R; Ball L; Gurr SJ; Fairbanks AJ Tetrahedron: Asymmetry 2007, 18, 1299–1307. [Google Scholar]
- (18).Dinev Z; Wardak AZ; Brownlee RTC; Williams SJ Carbohydr. Res 2006, 341, 1743–1747. [DOI] [PubMed] [Google Scholar]
- (19).Otto NJ; Green DE; Masuko S; Mayer A; Tanner ME; Linhardt RJ; DeAngelis PL J. Biol. Chem 2012, 287, 7203–7212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).(a) Link AJ; Tirrell DA J. Am. Chem. Soc 2003, 125, 11164–11165. [DOI] [PubMed] [Google Scholar]; (b) Ning X; Guo J; Wolfert MA; Boons G-J Angew. Chem., Int. Ed 2008, 47, 2253–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21). The structures of these oligosaccharide acceptors are presented in the Supporting Information. For more information, see:; (a) Jing W; DeAngelis PL J. Biol. Chem 2004, 279, 42345–42349. [DOI] [PubMed] [Google Scholar]; (b) Sismey-Ragatz AE; Green DE; Otto NJ; Rejzek M; Field RA; DeAngelis PL J. Biol. Chem 2007, 282, 28321–28327 HEP3-benzaldehyde is a gift from Heparinex, Oklahoma City, OK.. [DOI] [PubMed] [Google Scholar]
- (22).Edens RE; Al-Hakim A; Weiler JM; Rethwisch DG; Fareed J; Linhardt RJ J. Pharm. Sci 1992, 81, 823–827. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.